
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2(1H)-Pyrazinones from acyclic building blocks: methods of synthesis and further derivatizations
Gerard Riesco-Llach a,
Marta Planas*a,
Lidia Feliu*a and
John A. Jouleb
a,
Marta Planas*a,
Lidia Feliu*a and
John A. Jouleb
aLIPPSO, Department of Chemistry, Universitat de Girona, Maria Aurèlia Capmany 69, Girona, 17003, Spain. E-mail: lidia.feliu@udg.edu; marta.planas@udg.edu
bThe School of Chemistry, The University of Manchester, Manchester M13 9PL, UK. E-mail: John.Joule@manchester.ac.uk
First published on 4th January 2023
Abstract
Pyrazinones (2(1H)-pyrazinones) are found as components of a range of natural substances and are involved in the preparation of a great number of bioactive molecules. Synthesis of such compounds, and analogues, requires knowledge of the heterocyclic properties of pyrazinones and, in particular, methods for their ring construction. This review deals with the strategies that have been developed for the synthesis of pyrazinones from acyclic precursors, especially α-amino acid-derived units, from the first examples in 1905 up to the most recent in 2021.
1 Introduction
Pyrazinones (strictly 2(1H)-pyrazinones) 1 have received considerable attention because they are present in a large number of natural products showing interesting biological activities (Fig. 1). These natural products include deoxyaspergillic acid (2a) and flavacol (2b), isolated from Aspergillus flavus,1,2 phevalin (2c), tyrvalin (2d), leuvalin (2e) and phileucin (2f), from Streptomyces or Staphylococcus species,3–5 as well as arglecin (3a), argvalin (3b), and maremycin F (4), isolated from Streptomyces.6–8 Other examples are sorazinone B (5) and enhypyrazinone A (6), which have been more recently isolated from myxobacteria species.9,10 The pyrazinone skeleton is also found in the bromotyrosine alkaloids ma'edamines A and B (7a,b), isolated from an Okinawan sponge Suberea sp.,11 dragmacidin D (8), and the more elaborated dragmacidins E and F,12–14 which are present in several marine sponges species like Dragmacidon, Halicortex, Spongosorites and Hexadella, as well as the peptidyl metabolite JBIR-56 (9), isolated from a marine Streptomyces.15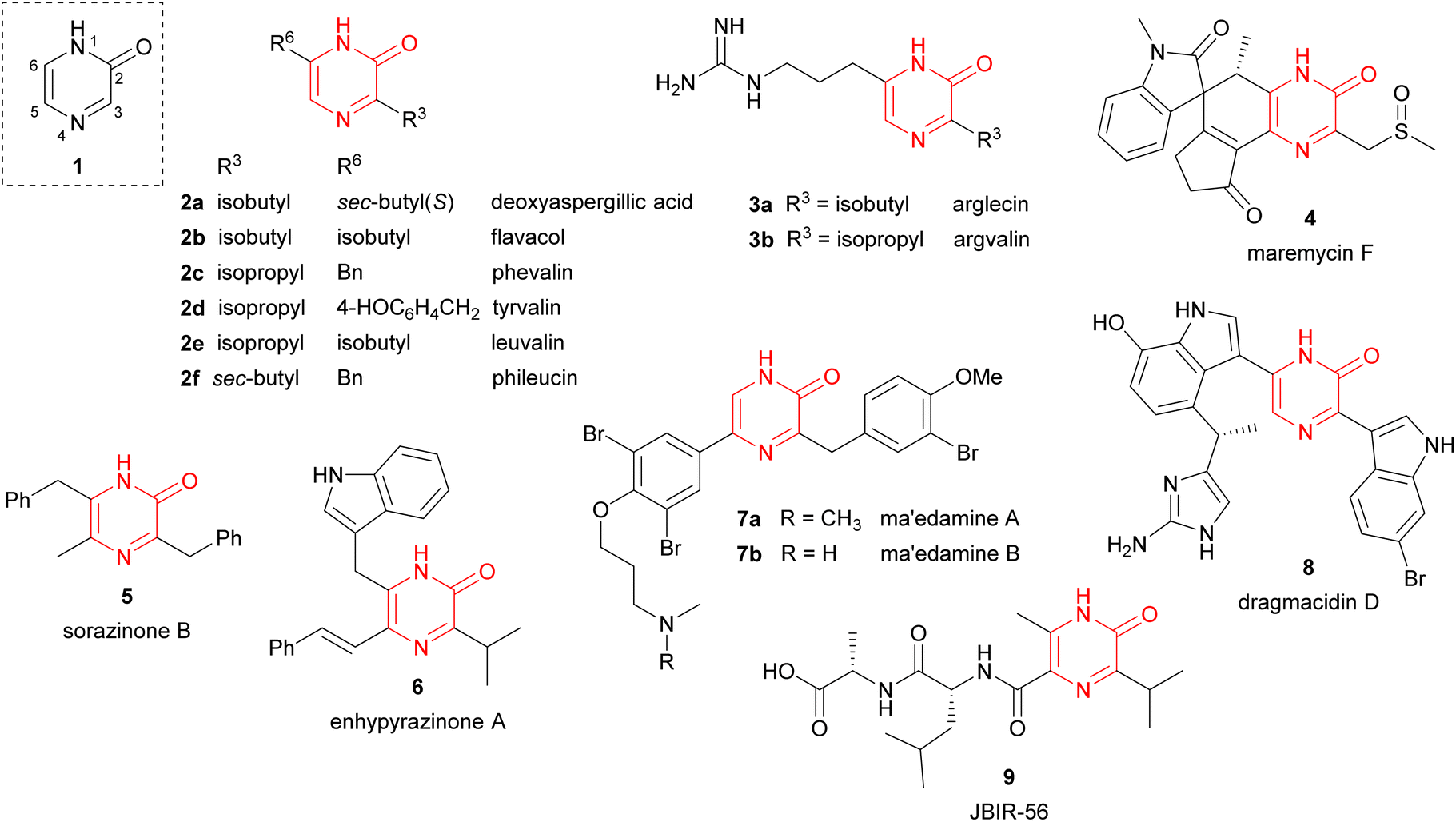 | ||
| Fig. 1 2(1H)-Pyrazinone and representative natural derivatives. | ||
Apart from constituting the central core in different natural products, the 2(1H)-pyrazinone scaffold has also been described as a key building block of great usefulness for many synthetic applications in drug design and medicinal chemistry programs, including the preparation of pharmacologically active derivatives (Fig. 2). In this regard, favipiravir (10) is an approved drug to target viral infections of influenza A and B, and avian influenza, and has recently been investigated for the treatment of Ebola virus and COVID-19.16,17 Another bioactive compound bearing a pyrazinone core is BMS-764459 (11), which has proven to be a potent antagonist of the receptor of corticotropin-releasing factor-1 (CRF1), elevated levels of which are associated with depression and anxiety-related disorders.18 The fungicide 12, patented by DuPont, disrupts the polymerization of tubulin altering the cellular proliferation in fungal infections caused by phytopathogens and exhibits strong activity against human rhabdomyosarcoma cells.19,20
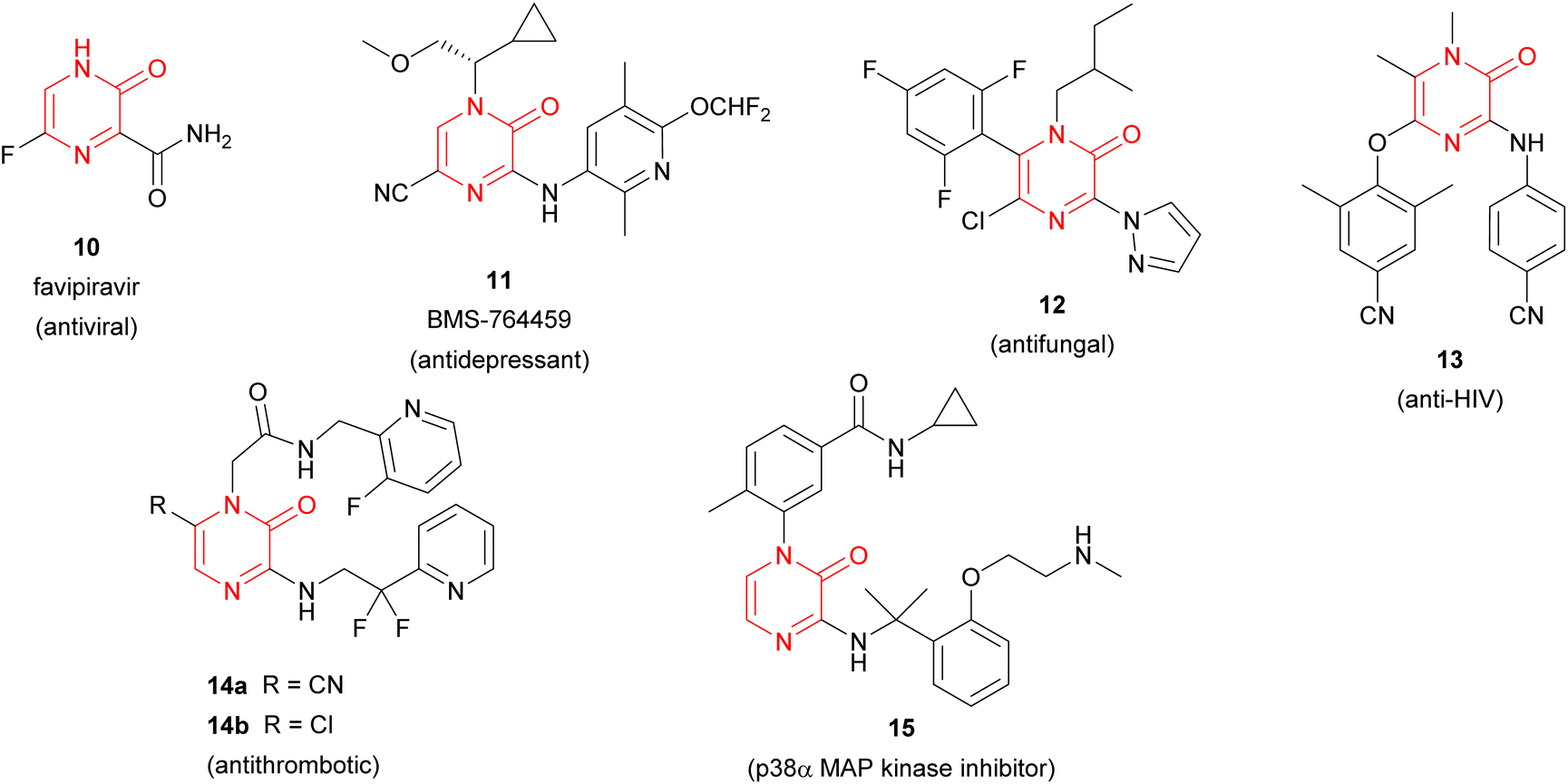 | ||
| Fig. 2 Synthetic 2(1H)-pyrazinones with pharmacological activity. | ||
Furthermore, synthetic pyrazinone derivatives have been described as inhibitors of different enzymes, which intervene in a wide range of diseases (Fig. 2). In this context, Heeres et al. developed a series of pyrazinone-containing derivatives among which 13 exhibits the best inhibitory activity against the reverse transcriptase, essential in the replication of human immunodeficiency virus (HIV).21 Besides, the inhibitory activity against thrombin of pyrazinone derivatives has been extensively studied with the identification of different candidates like 14a or 14b,22 which target trypsin-like serine protease thrombin (Factor IIa) involved in the blood coagulation system. More recently, Raubo et al. synthesised 15, an inhibitor of p38α mitogen-activated protein kinase, suitable for an inhaled method of administration.23 This enzyme plays an important role in the regulation of pro-inflammatory signalling, relevant in different pathologies such as rheumatoid arthritis, Crohn's disease, or chronic obstructive pulmonary disease (COPD).
Owing to the importance of pyrazinones, many efforts have been devoted to the development of methods for their preparation and for the synthesis of derivatives. In particular, our interest in 2(1H)-pyrazinones lies in their use in dipolar cycloadditions of 1H-pyrazinium-3-olates (also known as 3-oxidopyraziniums; referred to in chemical abstracts as “pyrazinium, 3,4-dihydro-1-R-3-oxo-, inner salts”) 16 derived from 2(1H)-pyrazinones via selective imine-N-alkylation (→ 17) followed by simple deprotonation (Scheme 1). These species undergo cycloadditions under mild conditions on reaction with dipolarophiles, forming 3,8-diazabicyclo[3.2.1]octanones 18.24–29 For a general study of the process typified by 16 → 18, and for synthesis of pyrazinone-containing natural products, and analogues, access to variously substituted 2(1H)-pyrazinones is required.
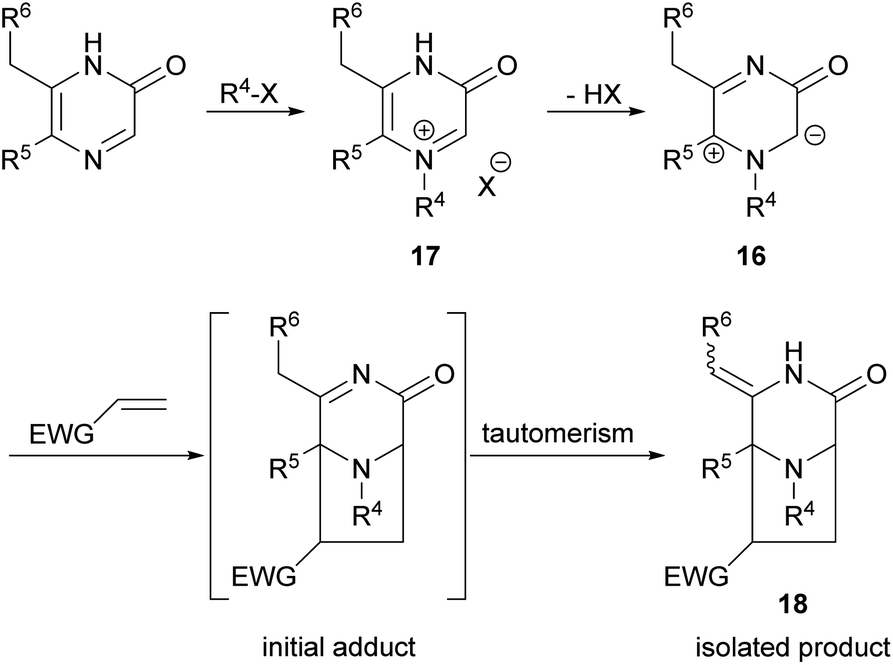 | ||
| Scheme 1 1,3-Dipolar cycloadditions of 3-oxidopyraziniums 16. | ||
In the early 20th century, a few papers described the first methods for the synthesis of pyrazinones.30–34 From the 1940s onwards, the preparation of pyrazinones was more extensively exploited due to their presence in natural products of biological interest. Since then, many authors have sought to overcome the limitations of the early methods and have designed new and more versatile strategies to obtain a wide variety of pyrazinones following easier and more practicable approaches.
Despite the large number of articles published in this context, to the best of our knowledge, there is no review that covers the available methods for the synthesis of pyrazinones. Some of these methods involve the modification of an existing pyrazine scaffold, however in this review we deal with methods which start from the structural components of a pyrazinone, i.e. routes involving ring formation, though in some cases initial products require further modification.
2 Synthetic approaches
2.1 Synthesis from an α-amino ketone or α-amino aldehyde and an α-haloacetyl halide or α-aminoacetyl halide
Prior to 1942 only a few methods for the preparation of substituted 2(1H)-pyrazinones had been described and they led to pyrazinones with identical groups at C-3 and C-6.30–34 Then, in 1942 Tota and Elderfield reported the preparation of 5,6-disubstituted and 3,5,6-trisubstituted 2(1H)-pyrazinones, in which the substituents can be different (Scheme 2a).35 This was the first example in what has become a group of methods for the synthesis of 2(1H)-pyrazinones involving: (i) the condensation of two compounds, one providing N-1, C-5 and C-6, and the other, C-2 and C-3; (ii) reaction with a nitrogen reagent to supply N-4, and (iii) ring closure through the formation of the bond between N-4 and C-5. | ||
| Scheme 2 Synthesis of 2(1H)-pyrazinones from an α-amino ketone or α-amino aldehyde and an α-haloacetyl halide or α-aminoacetyl halide. | ||
In particular, the two starting compounds used by Tota and Elderfield were the hydrochloride of a 1,2-disubstituted α-amino ketone (affording N-1, C-5 and C-6) and a 2-bromo- or 2-chloro-substituted acid halide (supplying C-2 and C-3) (Scheme 2a). After their condensation, treatment of the resulting ketoamide with ammonia in the presence of sodium iodide afforded a dihydropyrazine, which yielded the corresponding pyrazinone 19 by air oxidation. The best results were obtained when using the most reactive bromoacetyl bromide. A drawback of this procedure is the self-condensation of the starting free α-amino ketone, which occurs very easily. To avoid it, the authors mixed the α-amino ketone and the α-bromoacetyl bromide in cold chloroform in the presence of a small amount of water and excess solid calcium carbonate.
Later, Baxter et al. published a modification of this method with the aim of preparing pyrazinones unsubstituted at position 5 (Scheme 2b).36 This route involved the hydrochloride of an α-amino aldehyde or, alternatively, an α-amino aldehyde protected as an aminoacetal or aminothioacetal. 3,6-Dimethyl-2(1H)-pyrazinone (20) was only obtained when starting from 1,1-bis(ethylthio)propan-2-amine, which required an additional step to release the aldehyde functionality before reaction with ammonia. 6-Phenyl-2(1H)-pyrazinone (21) was also prepared following a similar approach, but in this case, the two starting components were an amino acetaldehyde and phthalimidoacetyl chloride, the latter providing N-4 after deprotection with hydrazine (Scheme 2c).37 Ring closure and aromatisation were achieved by refluxing in acetic acid followed by oxidation with manganese dioxide.
2.2 Synthesis from an α-amino ketone or tryptamine and a glyoxylic acid derivative
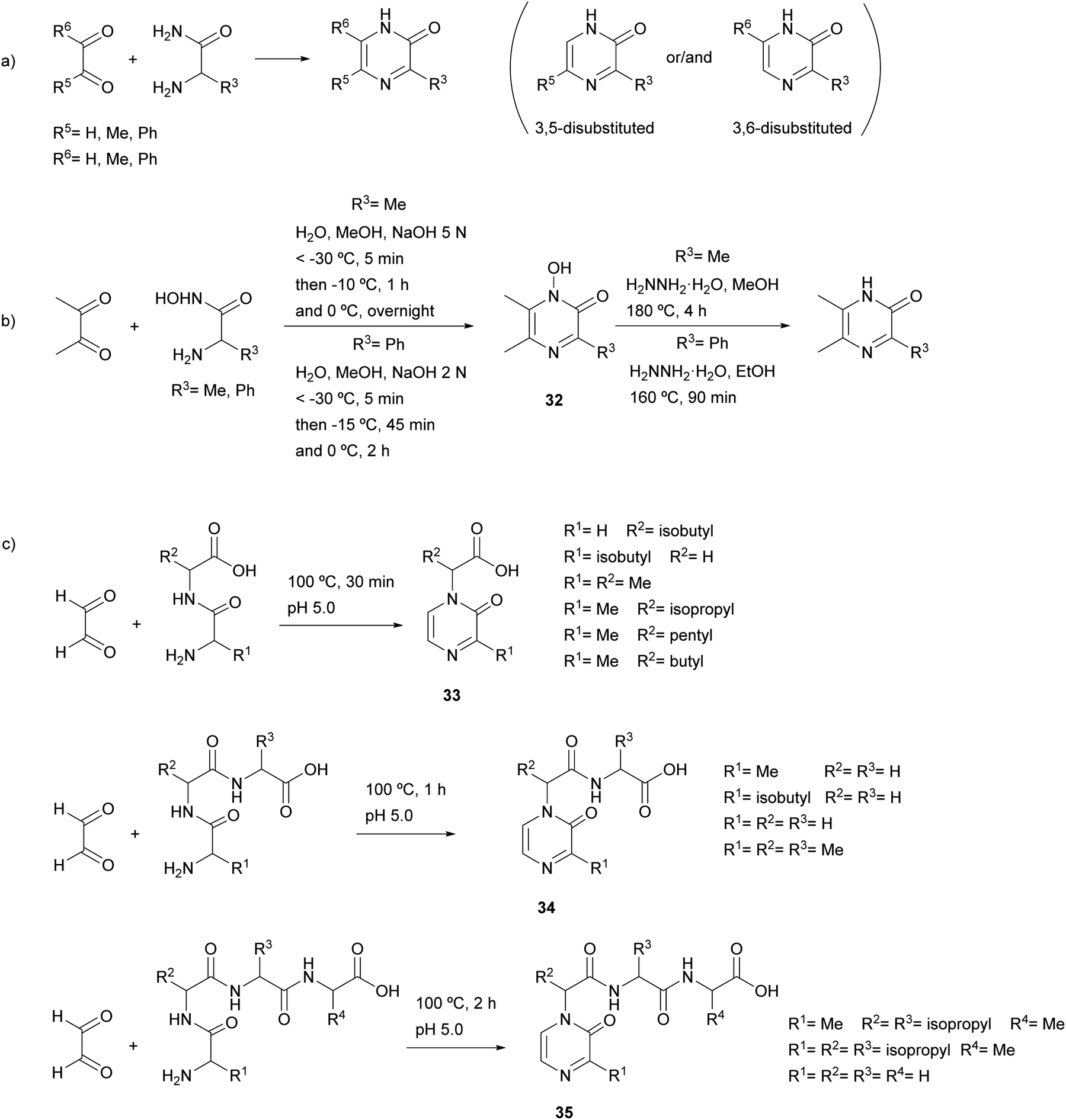
Much more versatile and useful variations of the method were developed, in which the starting components are more readily available and avoid the self-condensation of the starting free α-amino ketone. With the aim of ensuring formation of 3-alkyl-5-aryl-2(1H)-pyrazinones 22a and avoiding the 3,6-disubstituted isomer, Bradbury et al. started from an α-amino ketone incorporating an aryl group, which was reacted with a sodium glyoxylate derivative. Ammonium acetate was used as nitrogen source leading to the cyclization of the resulting diketoamide in refluxing ethanol (Scheme 3a).38 More recently, Johannes et al. extended this method to the synthesis of 3,5-diaryl-2(1H)-pyrazinones 22b by using an arylglyoxylic acid and carrying out the cyclization under microwave irradiation (MWI) (Scheme 3a).39 The versatility of this method allowed the preparation of a variety of derivatives incorporating different heterocyclic substituents with electron-donating and electron-withdrawing groups. In a study focussed on the identification of platelet-derived growth factor receptor (PDGFR) β-inhibitors, Johannes et al. also coupled a glyoxylic acid bearing a 3,4,5-trimethoxyphenyl with tryptamine, though an additional oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) before the cyclization step was necessary in order to obtain the corresponding 3,5-diaryl-2(1H)-pyrazinone 23 (Scheme 3b).40,41 3,5-Bis(3-indolyl)-2(1H)-pyrazinones 24a–b were also prepared by reacting 3-indolylglyoxylic chloride with an α-amino ketone conveniently derivatized with an indolyl group, the cyclization being accomplished in excess of liquid ammonia at 80 °C under 50 psi for three days42 or in aqueous ammonia at 100 °C for two days (Scheme 3c).43 This approach allowed the synthesis of precursors of 6′-debromo- and 6′,6′′-didebromo-cis-3,4-dihydrohamacanthin B.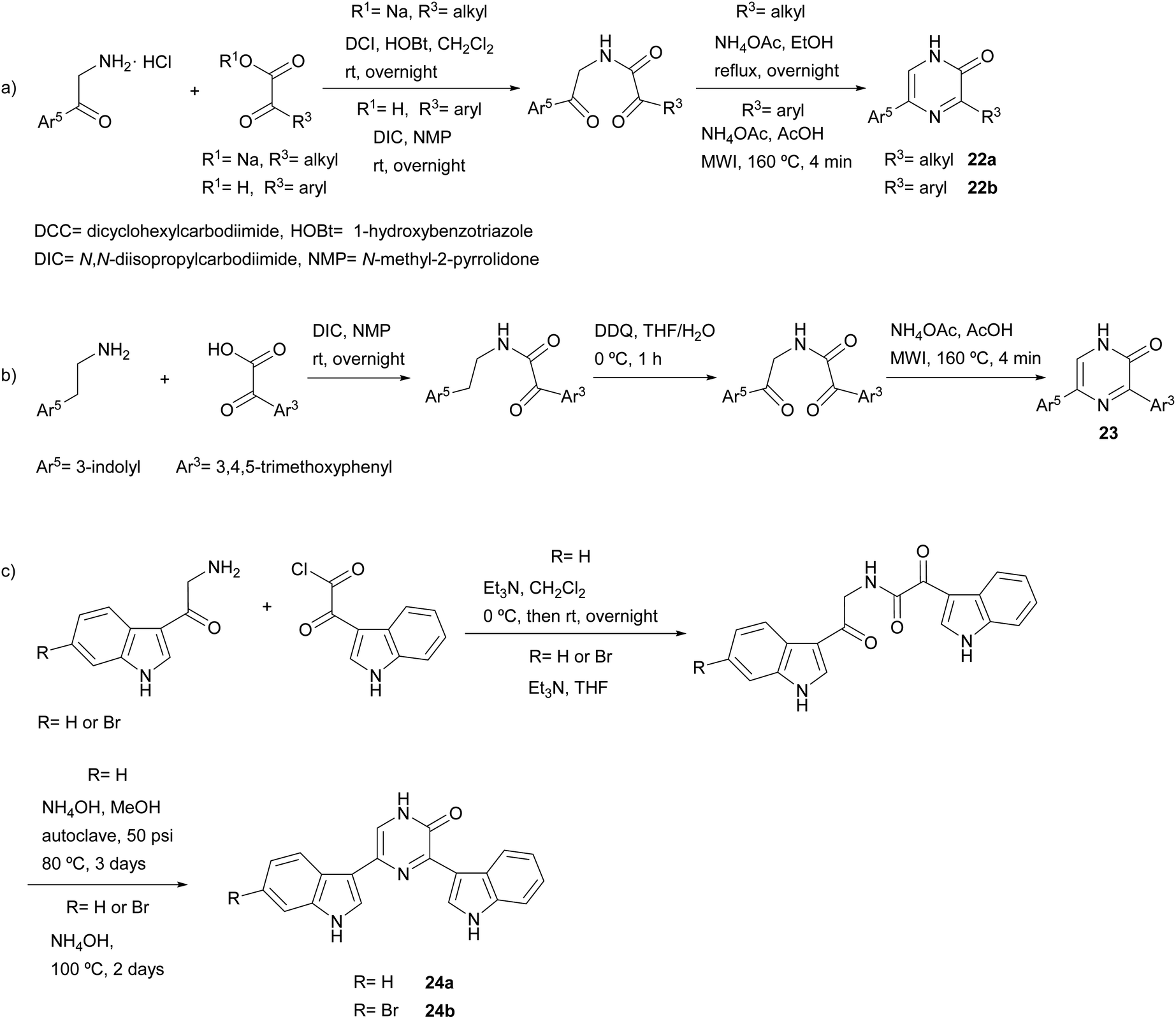 | ||
| Scheme 3 Synthesis of 2(1H)-pyrazinones from an α-amino ketone or tryptamine and a glyoxylic acid derivative. | ||
2.3 Synthesis from diketopiperazines
2(1H)-Pyrazinones can also be synthesized from diketopiperazines (2,5-piperazinediones). Unlike the other approaches included in this review, this method does not start from one or more acyclic products to form the pyrazinone ring. However, diketopiperazines can be prepared very easily through, for example, dehydration of amino acids, and so are readily available pyrazinone precursors.This method, first developed by Baxter and Spring (1947),44 involved the conversion of diketopiperazines into chloro substituted pyrazines, which were then transformed into pyrazinones (Scheme 4). Heating the diketopiperazines with phosphoryl chloride afforded a mixture of mono- and dichloropyrazines. The 2-chloropyrazine yielded a 2(1H)-pyrazinone by treatment with alkali or, alternatively, with sodium ethoxide followed by hydrolysis with hydrochloric acid. The first diketopiperazines used were symmetric, such as DL-alanine anhydride and DL-isoleucine anhydride, which provided 3,6-dimethyl-2(1H)-pyrazinone (25a) and 3,6-(di-sec-butyl)-2(1H)-pyrazinone (25b), respectively (Scheme 4a).44 Two years later, Gallagher et al. reported the first example of direct transformation of a diketopiperazine into a 2(1H)-pyrazinone by exposure to phosphoryl chloride. In particular, DL-phenylglycine anhydride was converted to 3,6-diphenyl-2(1H)-pyrazinone (26a), presumably via loss of hydrogen chloride from a monochloro dihydropyrazine intermediate (Scheme 4b).45 Treatment of DL-leucine anhydride with phosphoryl chloride also resulted in the direct dehydration of the diketopiperazine, affording 3,6-diisobutyl-2(1H)-pyrazinone (2b) together with a mixture of chloropyrazines (Scheme 4b).2 This reaction allowed the synthesis and elucidation of the structure of flavacol (3,6-diisobutyl-2(1H)-pyrazinone) 2b.
 | ||
| Scheme 4 Synthesis of 2(1H)-pyrazinones from diketopiperazines. | ||
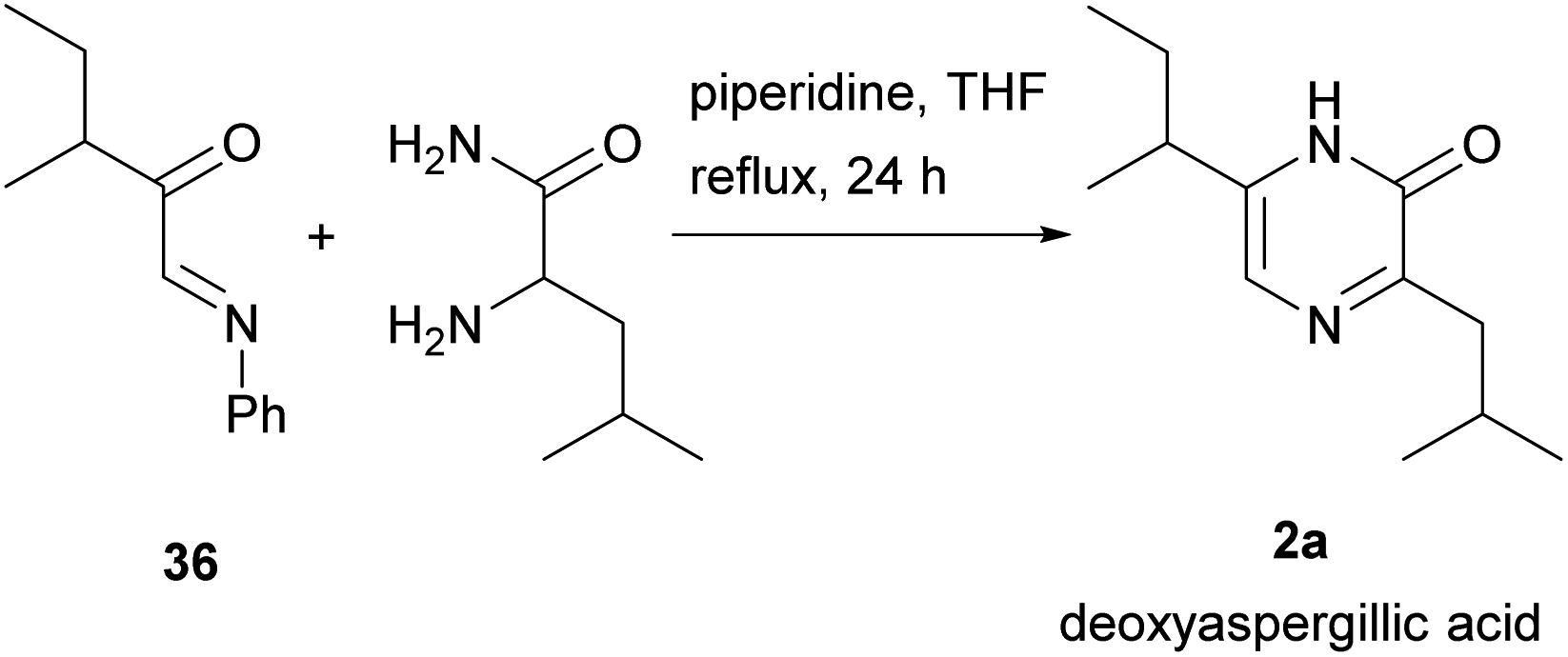
Unsymmetrical diketopiperazines were then used, one of the first being DL-leucyl-isoleucyl anhydride (Scheme 4b).46 Subjecting this anhydride to phosphoryl chloride led to a mixture from which 3-(sec-butyl)-6-isobutyl-2(1H)-pyrazinone (26b) could be isolated. This pyrazinone is an isomer of deoxyaspergillic acid (6-(sec-butyl)-3-isobutyl-2(1H)-pyrazinone) 2a.
This procedure was also the first step in syntheses of arglecin 3a and argvalin 3b from DL-alanyl-leucyl anhydride and DL-alanyl-valyl anhydride, respectively, and of their analogues 27 and 28 (Scheme 5).47,48 As an example, the synthesis of arglecin involved heating DL-alanyl-leucyl anhydride in phosphoryl chloride at 140 °C. A mixture of chloropyrazines was obtained from which 2-chloro-3-isobutyl-6-methylpyrazine (29) was isolated. Refluxing 29 with sodium methoxide in methanol for 6 h yielded the 2-methoxyderivative 30. The methyl group at position 6 was subsequently derivatized as a nitroguanidino substituent in five steps, giving rise to 31. Finally, hydrogenation of 31 followed by hydrolysis with 10% HCl afforded arglecin.47
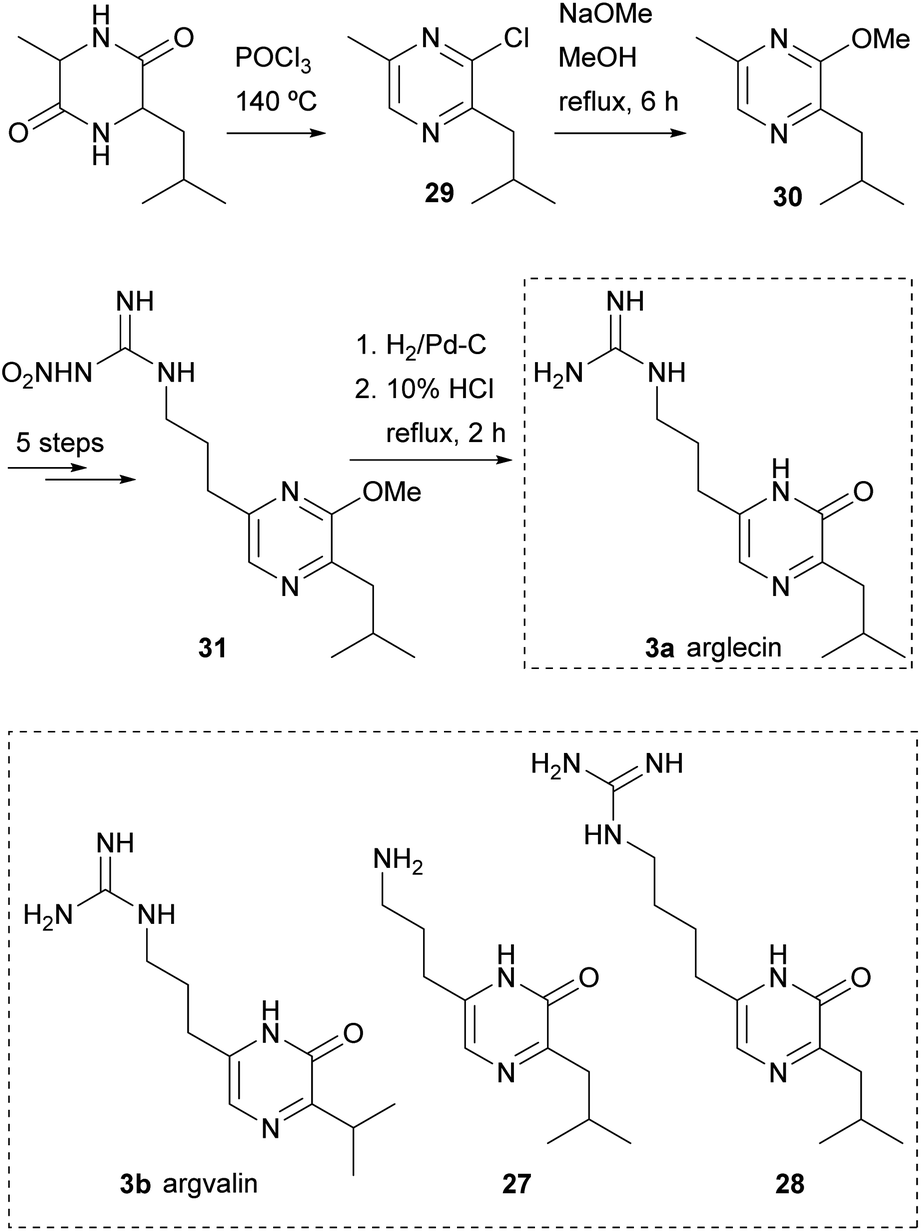 | ||
| Scheme 5 Structure and synthesis of arglecin; structures of argvalin, and analogues 27 and 28. | ||
2.4 Synthesis from α-amino acid amides and 1,2-dicarbonyl compounds or analogues
 | ||
| Scheme 6 Synthesis of 2(1H)-pyrazinones from α-amino acid amides using the method of Jones and Karmas and Spoerri. | ||
In the original report by Jones, the reaction was performed using free α-amino acid amides and in the presence of a base (sodium or potassium hydroxide or piperidine) (general Scheme 6a).49 The main drawback of this method was the preparation of the free α-amino acid amides, especially those with low molecular weight. Later, Karmas and Spoerri showed that the condensation also proceeds starting from the hydrohalides of the amino acid amides, which are easier to synthesise and, therefore, more readily accessible.50 The condensation was also carried out in alkaline medium or, in case of decomposition of the amino acid amide, with a prior neutralization with silver oxide. Moreover, it was shown that the efficiency of the reaction depended on the temperature and the rate of the addition of the base, the best conditions for obtaining 2(1H)-pyrazinone being around −50 °C and a rate of addition of a NaOH solution between 8.0 and 8.6 mmol min−1.51
The scope of this reaction has been shown to be wide, having been conducted with many 1,2-diketones and 1,2-ketoaldehydes (glyoxal, methylglyoxal, diacetyl, phenylglyoxal, benzil) as well as α-amino acid amides (aminomalonamide, glycine amide, alanine amide, methionine amide, tyrosine amide, α-aminophenylacetamide, leucine amide, α-aminobutyramide, norvaline amide and valine amide) (Scheme 6a). Moreover, the use of α-aminohydroxamic acids, instead of α-amino acid amides, allows the preparation of pyrazinones through the formation of the corresponding 1-hydroxy-2(1H)-pyrazinones 32 and a subsequent reduction step with hydrazine (Scheme 6b).52
Condensation of an α-amino acid amide with an unsymmetrical 1,2-dicarbonyl compound can occur in one or both of two directions, providing one of the two possible isomers or a mixture of both. In particular, when methylglyoxal reacted with a variety of α-amino acid amides the 5-methyl-2(1H)-pyrazinone was formed as a single or major isomer (Scheme 6a),49,50,53 whereas if the reaction mixture contained sodium bisulfite, the formation of the 6-methyl isomer was favoured.54 In another study, the structure of the product resulting from methylglyoxal and alanine amide was demonstrated by X-ray crystallography to be 3,5-dimethyl-2(1H)-pyrazinone.55
Glyoxal also reacted with dipeptides, tripeptides and tetrapeptides leading to 2(1H)-pyrazinones 33, 34 and 35 bearing a carboxylic acid, an acylamino acid or an acylpeptide at position 1, respectively (Scheme 6c).56,57 In these examples, the reaction took place between the two carbonyl groups of glyoxal and the N-terminal amino group and the amide nitrogen of the amino acid at position 1 in the peptide sequences.
 | ||
| Scheme 7 Synthesis of 2(1H)-pyrazinones from α-aminoamides and 1,2-diketone mono Schiff bases. | ||
 | ||
| Scheme 8 2(1H)-Pyrazinones with potential applications prepared following Jones and Karmas and Spoerri procedure. | ||
In the medicinal field, a collection of potential allosteric Akt kinase inhibitors bearing a 2(1H)-pyrazinone ring was synthesized (Scheme 8b).61 The two regioisomers 38 resulting from the reaction of a benzil derivative and different amino acid amides were screened for their Akt inhibitory activity. Derivatives selective for Akt1, Akt2 or both Akt1/Akt2 were identified. Moreover, 2(1H)-pyrazinones prepared following the Jones' method were employed for the synthesis of nucleosides. In a study concerning this topic, Benjahad et al. reported piperazin-2-one nucleosides 39 resulting from the reduction of pyrazinones linked to a ribofuranose (Scheme 8c).62 The synthesis of the corresponding pyrazinones was performed by treating glyoxal with glycine amide, alanine amide or 2-aminododecanamide. Two of these piperazin-2-one nucleosides bearing a decyl chain showed moderate activity against the Visna virus related to the human immunodeficiency virus. In another work, Weber et al. focussed their attention on the prebiotic synthesis of nucleosides (Scheme 8d). First, the synthesis of 2(1H)-pyrazinones from sugars (glyceraldehyde or erythrose) through reaction with an amino acid amide (glycine amide, alanine amide or leucine amide) was explored.63 The corresponding 3,5- and 3,6-disubstituted pyrazinone isomers 40a and 40b were obtained. A few years later, aldopentoses, such as ribose, arabinose, xylose and lyxose, were subjected to similar conditions.64 Although the results were promising, pyrazinones were not obtained in sufficient quantity to study their usefulness in pyrazine nucleic acid synthesis. Therefore, pyrazinones were prepared through traditional organic synthesis. In particular, D-ribose was reacted with alanine amide, which afforded the two possible regioisomers 41a and 41b. In another study, in an attempt to find new antiviral nucleoside analogues, 3-oxo-3,4-dihydropyrazine-2-carboxamide (42) was prepared from glyoxal and 2-aminopropanediamide, and then glycosylated with a ribofuranose derivative (Scheme 8e).65
2.5 Synthesis from 2-chloro ketone oximes and α-amino acid esters
3,6-Disubstituted 2(1H)-pyrazinones were also synthesised from 2-chloroketone oximes and α-amino acid esters through formation of bonds between N-1 and C-2 and between N-4 and C-5.66 This method afforded pyrazinones 42 and it was also successfully applied to the preparation of deoxyaspergillic acid (2a) (Scheme 9).67 In the case of 2a, reaction of 1-chloro-3-methyl-2-pentanone oxime and L-leucine ethyl ester led to the corresponding N-(2-oximinoalkyl)-amino acid ester 43, which was then subjected to saponification of the ester, conversion of the oxime into a ketone, esterification of the carboxylic acid and treatment with ammonia with subsequent oxidation of the ring.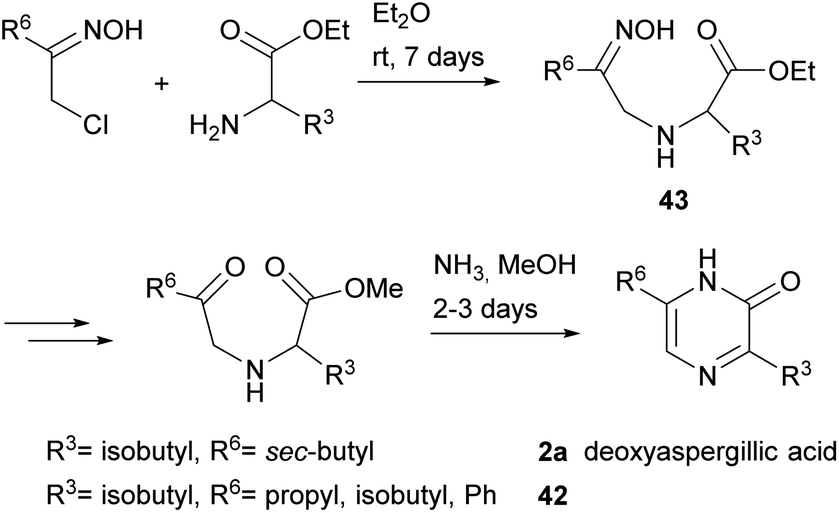 | ||
| Scheme 9 Synthesis of 2(1H)-pyrazinones from 2-chloroketone oximes and α-amino acid esters. | ||
2.6 Synthesis from an amino acetal or amino alcohol, an oxalate and an amine
In 1975 Cheeseman et al. described a sequence for the preparation of the pyrazinone core as an alternative of the Strecker reaction, avoiding the high toxicity associated with trimethylsilyl cyanide (TMSCN). The key steps of this strategy involve the formation of the C-3–N-4 bond and a cyclization between N-1 and C-6.68This synthetic route starts with diethyl oxalate reacting with an equimolar quantity of 2,2-dimethoxyethylamine to afford oxamate 44 that is converted with benzylamine into the corresponding N-benzyl-N′-(2,2-dimethoxyethyl)oxamide 45 (Scheme 10a). This N-1-protected oxamide undergoes cyclization under reflux in acetic acid with a catalytic amount of HCl. The resulting pyrazinedione 46 is then treated with hexanemethyldisilazane (HDMS) to afford 1-benzyl-3-trimethylsilyloxypyrazin-2(1H)-one (47), that can be used as a precursor of further functionalized pyrazinone scaffolds. Specifically, Cheeseman and co-workers used this strategy for the preparation of pyrazine nucleosides as analogues of the natural pyrimidine nucleosides with potential anti-tumoral activity. These compounds were obtained by the condensation of 47 with the corresponding acylglycosyl chloride, using silver perchlorate.68
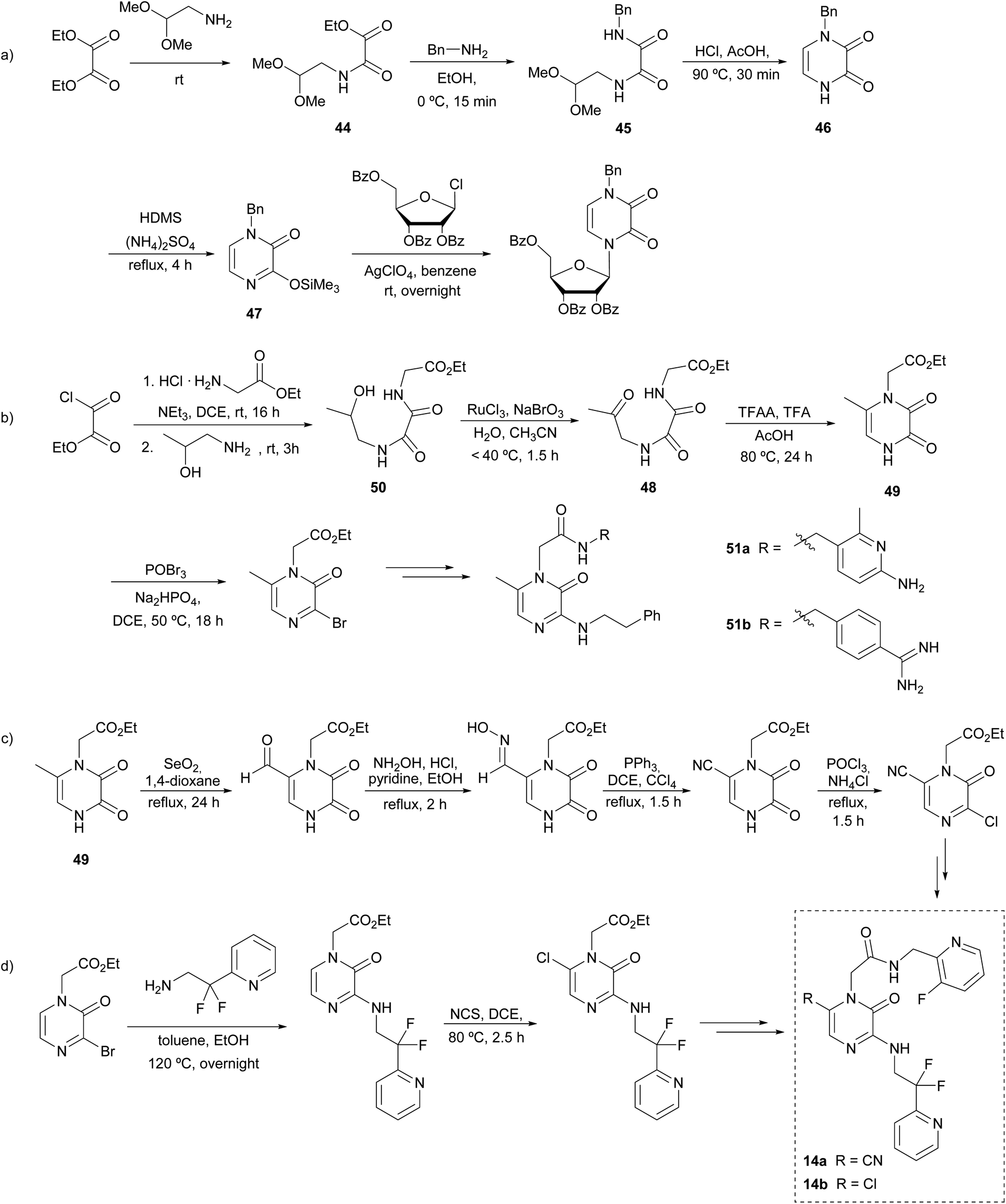 | ||
| Scheme 10 Synthesis of 2(1H)-pyrazinones from an aminoacetal or amino alcohol, an oxalate, and an amine, and further derivatizations. | ||
From this method, Fleitz et al. considered a similar approach for the construction of the pyrazinone core in a multi-kilogram scale synthesis (Scheme 10b).69 In their approach, the cyclization precursor 48 contained an acetyl group instead of the dimethylacetal-protected aldehyde initially reported by Cheeseman, which after an acidic treatment afforded the 6-methyl-pyrazine-2,3-dione 49. The oxamide 48 was synthesised from the oxidation of the corresponding alcohol 50, which was previously obtained from a two-step condensation starting from ethyl oxalyl chloride. Moreover, the presence of an acetate group at N-1 in 49 allowed further functionalization after the selective hydrolysis of this ester. Likewise, the position 3 of the pyrazinedione 49 can be easily brominated using POBr3. This halogenation allows nucleophilic substitution of amines to take place at this position. All in all, based on this strategy, the synthesis of the thrombin inhibitor 51a was described.
Later, the incorporation of monocyclic70 and bicyclic71 arginine derivatives at position 1 was also assessed to improve the selectivity and thrombin inhibitory activity of the previously reported 51a, leading to the description of 51b as the analogue with the best biological profile (Scheme 10b). This derivatization was achieved following a similar strategy: the cyclic arginine surrogates were bound to N-1 through the hydrolysis of the acetate group affording the carboxylic acid and subsequent coupling of the corresponding amine precursors mediated by 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), 1-hydroxybenzotriazole (HOBt) and Et3N at room temperature.
The identification of the metabolic activation pathways of previously reported thrombin inhibitors led to the optimization of 51a in order to increase the oral bioavailability and pharmacokinetics.72,73 Burgey et al. found that compounds with methyl groups at position 6 had low half-life in plasma because of oxidation of this position during metabolism.22 In contrast, when it was replaced by a chlorine or a nitrile group (14a,b), the inhibitory activity was enhanced and half-lives considerably increased. The synthetic pathway to introduce a nitrile group at position 6 is represented in Scheme 10c: starting from a 6-methylpyrazinedione 49 an oxidation of the 6-methyl with SeO2 afforded the aldehyde, which was converted into the nitrile after forming the oxime and its subsequent reduction. On the other hand, the introduction of a chlorine at C-6 is achieved by reaction with N-chlorosuccinimide (NCS) in 1,2-dichloroethane (DCE) (Scheme 10d). From these intermediates, target molecules 14a and 14b are easily achieved following the strategy discussed previously.
2.7 Synthesis of 3,5-dihalo-2(1H)-pyrazinones from oxalic halides and α-aminonitriles: Hoornaert's method
Vekemans et al. reported in 1983 the most general method for the preparation of 3,5-dichloro- and 3,5-dibromo-2(1H)-pyrazinones.74 This method involves the treatment of an α-aminonitrile 52 with an excess of the corresponding oxalyl halide in toluene or o-dichlorobenzene at 70–100 °C for 4–6 h or at room temperature for several days (Scheme 11). The α-aminonitrile provides the substituents at N-1 and C-6 of the pyrazinone core and it is usually obtained from a primary amine, an aldehyde and a cyanide source via a Strecker type reaction. If the primary amine is poorly nucleophilic, the α-aminonitrile 52 can also be prepared via a two-step procedure involving the synthesis of an imine followed by reaction with TMSCN (Scheme 11).20 To form the pyrazinone, the salt of the α-aminonitrile must be used to avoid the formation of symmetrical oxamides. Free-base aminonitriles are employed only when they have poor nucleophilicity (e.g. R1 = Ph). | ||
| Scheme 11 Synthesis of 3,5-dihalo-2(1H)-pyrazinones using Hoornaert's method. | ||
The mechanism for the formation of the pyrazinone core involves first the acylation of the α-aminonitrile to form an oxamoyl halide 53 (bond between N-1 and C-2) (Scheme 12). Next, addition of HX to the nitrile, tautomerization to the enamine 54 and subsequent cyclization affords a cyclic pyrazine-2,3-dione 55 (bond between N-4 and C-3). This intermediate then reacts with an excess of the oxalyl halide allowing the introduction of the halogen atom at position 3 with release of CO2, CO and X−. This step can be favoured by adding a tetraalkylammonium halide, acting as an additional halide source.
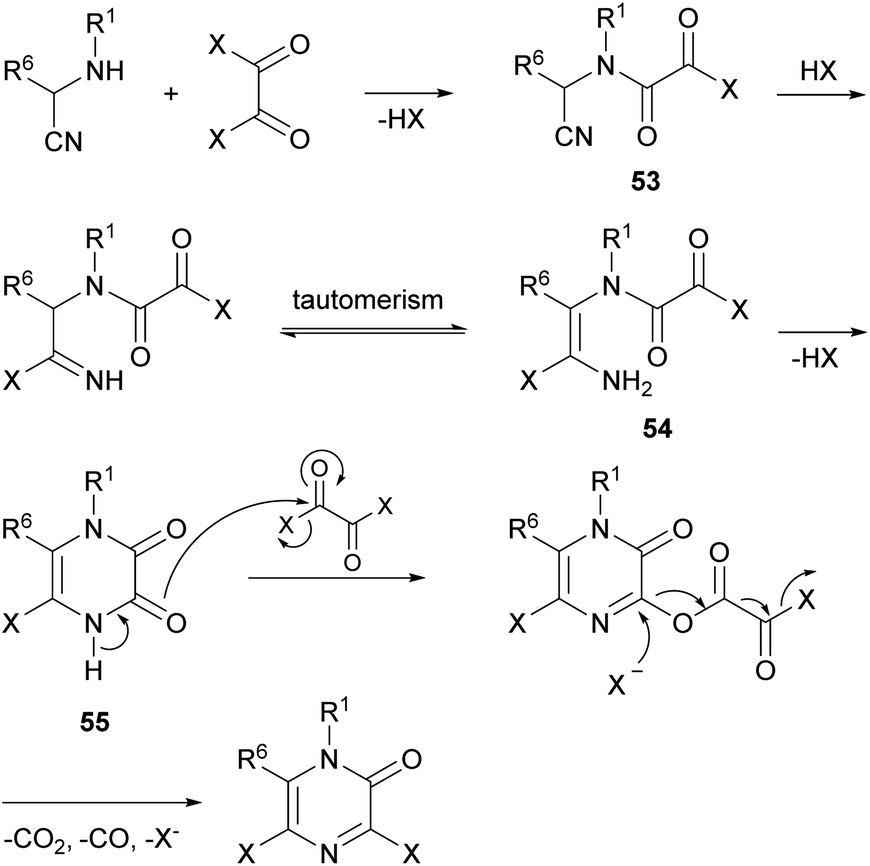 | ||
| Scheme 12 Mechanism for the formation of 3,5-dihalo-2(1H)-pyrazinones. | ||
This method has been employed to obtain 3,5-dihalo-2(1H)-pyrazinones with a range of substituents at positions 1 and 6. However, it fails when R1 is an acyl group and is difficult when R6 is an electron-withdrawing group. In addition, when R1 and/or R6 are bulky groups, the yields are low because the conversion of the pyrazine-2,3-dione into the dihalopyrazinone is not efficient.75 It has been reported that this halogenation step can be accelerated by adding a catalytic amount of DMF after the reaction of the α-aminonitrile with the oxalyl halide.20,75
Applying this procedure, Tulinsky et al. described the asymmetric synthesis of atropisomeric 6-aryl-3,5-dichloro-2(1H)-pyrazinones (Scheme 13).76,77 They observed that the cyclization of the diastereomerically pure (S,S)- and (R,R)-aminonitriles 56a and 56b, derived from o-iodobenzaldehyde, (R)- or (S)-α-methylbenzylamine and NaCN, provided the diastereomerically pure pyrazinones (aS,S) 57a and (aR,R) 57b, respectively. They concluded that the configuration at the nitrile-bearing carbon determined the configuration of the asymmetric axis and that the steric bulk of the ortho-halo substituent influenced the selectivity.
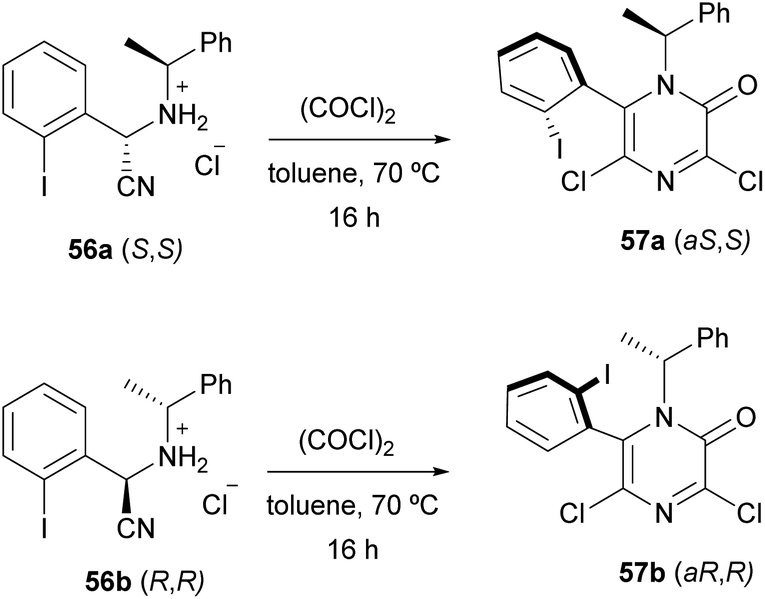 | ||
| Scheme 13 Synthesis of atropisomeric 6-aryl-3,5-dichloro-2(1H)-pyrazinones. | ||
With the aim of taking advantage of combinatorial principles and to pave the way for the generation of libraries of biologically interesting structures, Kaval et al. transferred the Hoonaert's method to solid phase (Scheme 14).78 A Wang amide resin was used as solid support which was treated with the corresponding aldehydes and TMSCN to yield the α-aminonitriles. Following acidification, the resulting α-aminonitrile salts were cyclized with an excess of oxalyl chloride in toluene providing the resin-bound 3,5-dichloro-2(1H)-pyrazinones bearing a range of substituents at position 6.
 | ||
| Scheme 14 Solid-phase synthesis of 3,5-dichloro-2(1H)-pyrazinones. | ||
Gising et al. demonstrated that the speed of Hoonaert's method can be improved using MWI.79–81 They developed a rapid one-pot, two-step protocol that involved: (i) a microwave-assisted Strecker reaction to provide the α-aminonitrile and (ii) addition of HCl gas, then oxalyl chloride and cyclization under MWI (Scheme 15).
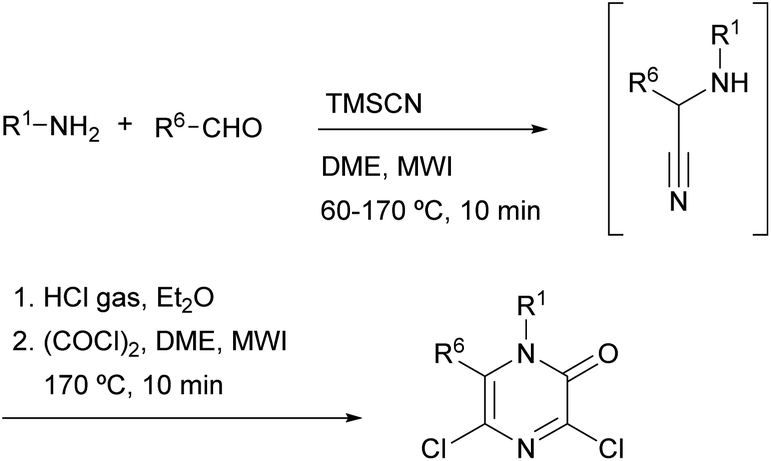 | ||
| Scheme 15 Synthesis of 3,5-dichloro-2(1H)-pyrazinones under MWI. | ||
Researchers from Bristol-Myers Squibb described an optimized procedure that allowed the synthesis of 3,5-dihalo-2(1H)-pyrazinones in multigram quantities, involving lower reaction temperatures and the use of lower boiling point solvents.82,83 This procedure consists of the preparation of the α-aminonitrile via the nucleophilic substitution of chloroacetonitrile with a primary amine in the presence of KI and K2CO3 in MeCN followed by acidification and final treatment with oxalyl chloride or oxalyl bromide in toluene (Scheme 16).
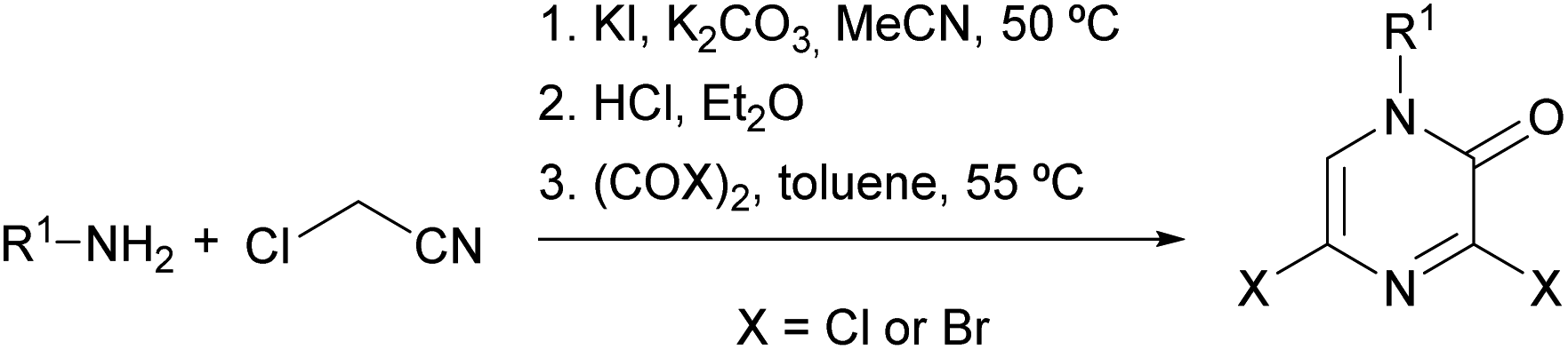 | ||
| Scheme 16 Synthesis of 3,5-dihalo-2(1H)-pyrazinones starting from a primary amine and chloroacetonitrile. | ||
Starting from 3,5-dihalo-2(1H)-pyrazinones, Bristol-Myers Squibb researchers developed a family of N-3-(hetero)aryl-2(1H)-pyrazinones as corticotropin-releasing factor-1 receptor (CRF1R) antagonists that could be useful for the treatment of psychiatric disorders.18,82,83,88–90 The general structure 58 of this family of 2(1H)-pyrazinones and two of the most representative members, BMS-665053 (59) and BMS-764459 (11), are depicted in Fig. 3. Both compounds are highly potent (IC50 ≤ 1.0 nM) and selective CRF1R antagonists with good pharmacokinetic properties and efficacy in rats, but BMS-764459 shows an improved metabolic profile.88,89 Based on this activity as CRF1R antagonists, later they prepared fluorinated analogues and explored their potential as Positron Emission Tomography (PET) radioligands.91,92
 | ||
| Fig. 3 2(1H)-Pyrazinones developed as CRF1R antagonists. | ||
The synthesis of this family of compounds, with general structure 58, was achieved via a base-mediated reaction or a palladium-catalyzed site-selective coupling of the corresponding (hetero)aryl amine with the 3,5-dihalo-2(1H)-pyrazinone 60a or 60b (Scheme 17),18,82,83,88–90 which were prepared as shown in Scheme 16. 5-Bromo-2(1H)-pyrazinone 58b served as a starting material for the incorporation of substituents at C-5 of the pyrazinone via palladium-catalyzed cross-coupling reactions (Scheme 18).18 The des-halo analogue was obtained by hydrogenation in presence of Pd/C.18 Analogues bearing a methyl group at position 5 were also prepared following an alternative method that involved the synthesis of a 3-chloro-5-methyl-2(1H)-pyrazinone intermediate by the condensation of an α-amino ketone and ethyl chlorooxacetate using a similar approach to that discussed in Section 2.2.18
 | ||
| Scheme 17 Synthesis of 5-halo-2(1H)-pyrazinones 58. | ||
 | ||
| Scheme 18 Derivatization of 5-bromo-2(1H)-pyrazinone 58b. | ||
Sandström's group developed a family of achiral inhibitors of the hepatitis C virus NS3 protease based on 2(1H)-pyrazinones with general structure 61 (Fig. 4). They chose this scaffold for its ability to retain the H-bonding pattern of the peptide backbone and to act as a β-strand inducer. The starting point of their design was tripeptide 62, and their first study led to the identification of compounds 61a and 61b (Fig. 4).80 The former was taken as a lead for further optimization.81 This second study provided 61c and 61d as two of the best 2(1H)-pyrazinone-based inhibitors and showed the important role of elongated substituents at C-6 for the activity.
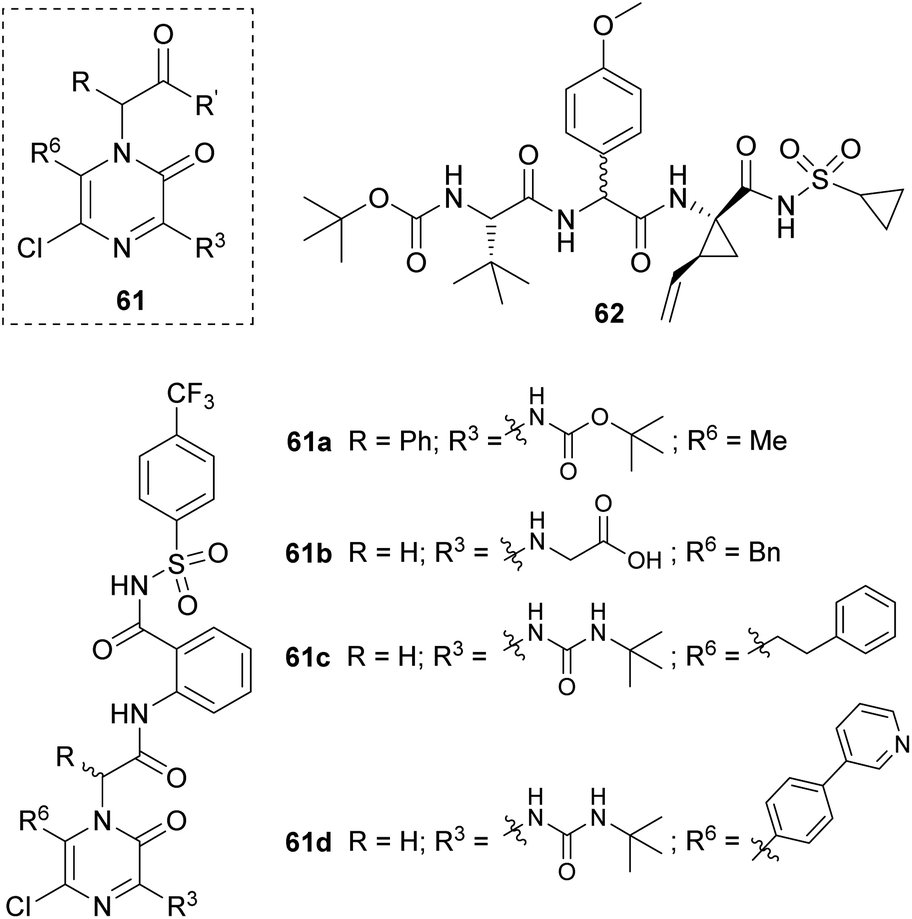 | ||
| Fig. 4 2(1H)-Pyrazinones developed as inhibitors of the hepatitis C virus NS3 protease. | ||
The synthesis of the pyrazinone scaffold was performed via Hoornaert's method using MWI (Scheme 15).79 The resulting 3,5-dichloro-2(1H)-pyrazinone, incorporating the required substituents at positions 1 and 6, was selectively derivatized at position 3 with tert-butyl carbamate or tert-butylurea via a palladium-catalyzed Buchwald N-arylation coupling using Pd(OAc)2, Xantphos and Cs2CO3 in 1,2-dimethoxyethane (DME) under MWI. Alternatively, this position could be derivatized with an amino group via a nucleophilic aromatic substitution with the corresponding amine in presence of DIEA in CH3CN.80,81
In another study, Harker et al. described the synthesis of 2-pyridone boronic acid derivatives in multigram quantities via a [4 + 2] cycloaddition between a 3,5-dichloro-2(1H)-pyrazinone and a trimethylsilyl alkynylboronate (Scheme 19).93 The corresponding pyrazinones were prepared on multigram scale following the procedure described by Bristol Myers Squibb researchers (Scheme 16).82,83 The cycloaddition was carried out at 180 °C in the absence of solvent for <2 h and provided the expected 2-pyridone boronic acid derivatives as a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of regioisomers. These could be separated using high performance counter-current chromatography (HPCC) and were further derivatized to provide a range of functionalized 2-pyridones.
:1 mixture of regioisomers. These could be separated using high performance counter-current chromatography (HPCC) and were further derivatized to provide a range of functionalized 2-pyridones.
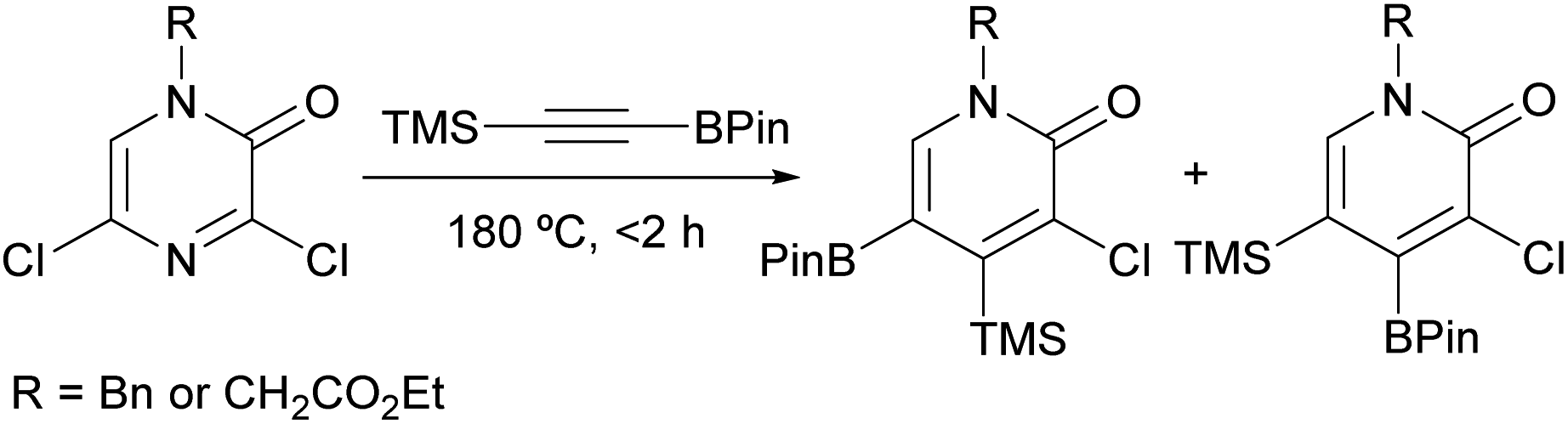 | ||
| Scheme 19 Synthesis of 2-pyridones from 2(1H)-pyrazinones. | ||
In 2006, DuPont patented the tubulin-modulating fungicide 12, containing a pyrazinone core (Fig. 5).19 Later, they performed a structure–activity relationship analysis incorporating a range of modifications at positions N-1, C-3, C-5 and C-6 of the 2(1H)-pyrazinone ring.20 Best derivatives, highlighting 63a–d and 64a–b included in Fig. 5, generally contained a branched alkyl group at N-1, a N-linked pyrazole at position 3, a chloro group at position 5 and a 2,4,6-trisubstituted phenyl ring at position 6. These compounds showed fungicidal activity against economically important plant pathogenic fungi and also exhibited activity similar to that of placlitaxel against rhabdomyosarcoma cells.
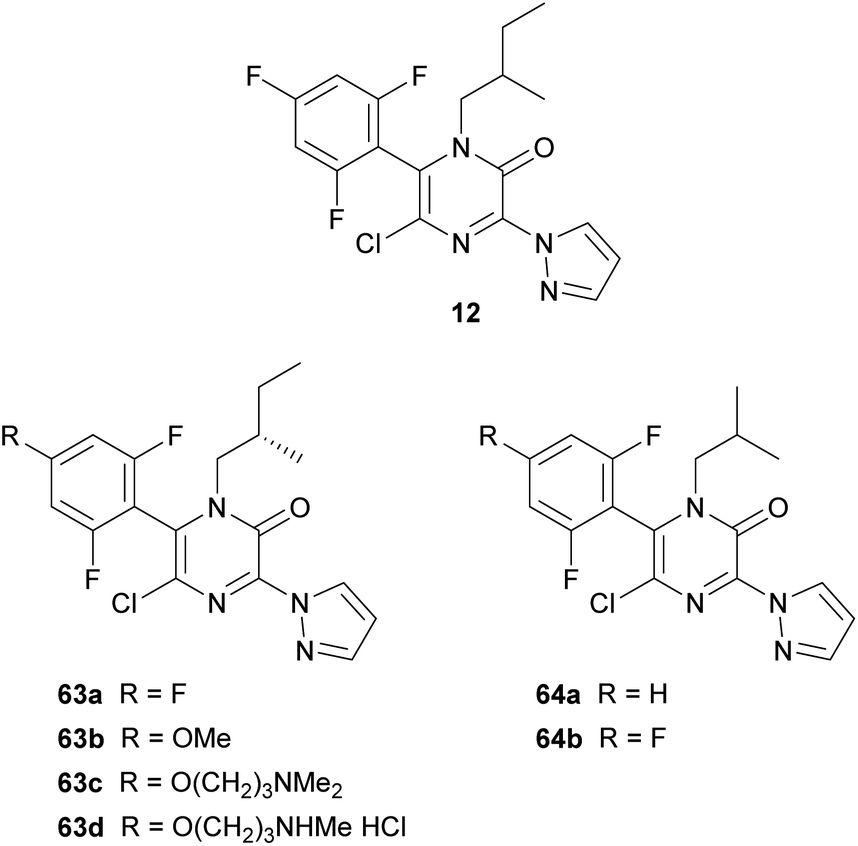 | ||
| Fig. 5 2(1H)-Pyrazinones developed as tubulin-modulating compounds. | ||
The substituent present at position 3 was introduced by treating the corresponding 3,5-dichloro-2(1H)-pyrazinone, obtained as described in Scheme 11, with a variety of nucleophiles or by subjecting it to a metal-catalyzed cross-coupling reaction, yielding compounds containing an N- or a C-linked heterocycle.20 Amide groups were also introduced by treatment of the 3,5-dichloro-2(1H)-pyrazinone with N-cyanomethylbenzotriazoles in the presence of a base and subsequent oxidation with peracetic acid.
Yang et al. synthesized a collection of 1,3,5-trisubstituted-2(1H)-pyrazinones 65 and studied their activity as PB2 inhibitors (Fig. 6).94 PB2 is a subunit of influenza RNA-dependent RNA polymerase and it is an important target for the design of anti-influenza drugs. The structure of these new 2(1H)-pyrazinones was based on VX787 (66), a PB2 inhibitor that has entered phase III clinical trials, by replacing the pyrimidine ring with a pyrazinone moiety. The best analogue 65a is depicted in Fig. 6.
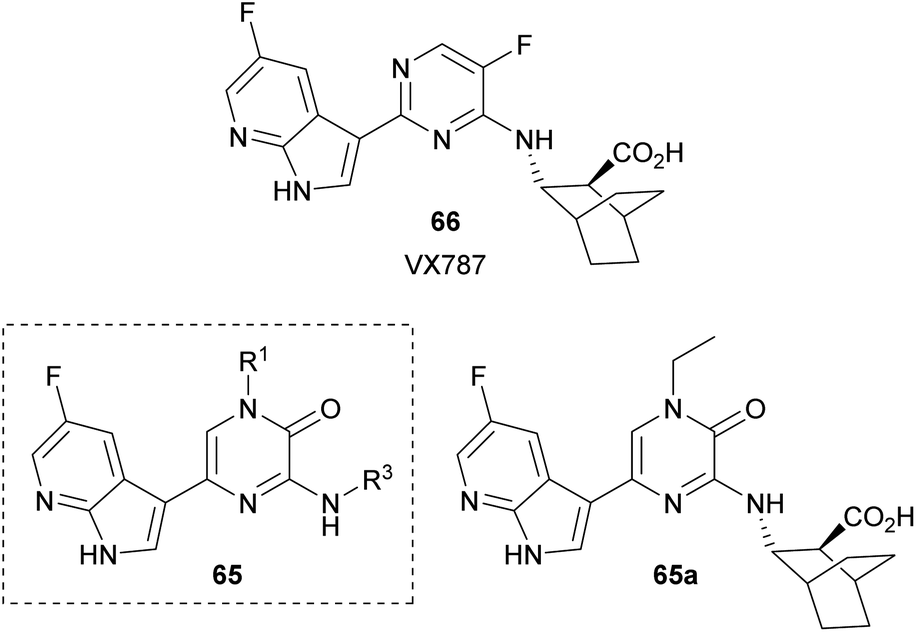 | ||
| Fig. 6 2(1H)-Pyrazinones developed as PB2 inhibitors. | ||
This family of compounds was prepared either from the commercially available 3,5-dibromo-1-methyl-2(1H)-pyrazinone or from a 3,5-dibromo-2(1H)-pyrazinone prepared following the procedure described in Scheme 16. This step provided the group at C-1. The substituent at C-3 was incorporated by reaction with the corresponding amine in presence of DIEA whereas the substituent at C-5 was introduced via a Suzuki–Miyaura cross-coupling using the required 7-azaindole-3-boronic acid pinacol ester.
Researchers from AstraZeneca, starting from their lead compound AZD6703 (67), designed analogues 68 containing a 2(1H)-pyrazinone core instead of the quinazolinone scaffold (Fig. 7).23 Their aim was the development of selective p38α mitogen-activated protein kinase (MAPK) inhibitors. p38α plays a relevant role in the modulation of the pro-inflammatory signalling. They performed a structure–activity relationship analysis by incorporating a variety of aminoalkyl groups at position 3 of the pyrazinone ring and identified compound 15 with in vitro and in vivo biological properties for an inhaled drug candidate.
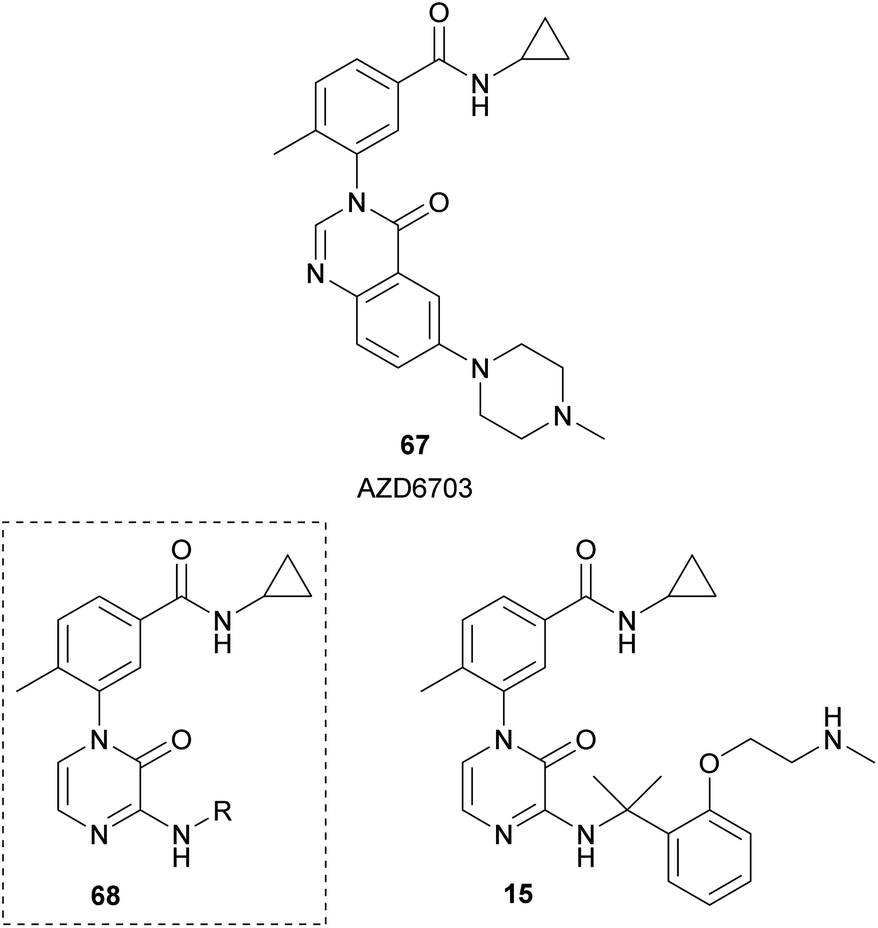 | ||
| Fig. 7 2(1H)-Pyrazinones developed as p38α MAP kinase inhibitors. | ||
The synthesis of this family of compounds was carried out from the required 1-aryl-3,5-dibromo-2(1H)-pyrazinone, which was obtained as depicted in Scheme 16. The aminoalkyl group at position 3 was incorporated via a nucleophilic aromatic substitution with the corresponding amine in presence of Et3N. The bromine at position 5 was removed through hydrogen transfer hydrogenation using ammonium formate and Pd/C in EtOH under MWI.
2.8 Synthesis from ethylenediamine and methyl pyruvate
Masuda et al. described in 1981 another strategy in which the 1,2- and 4,3-bonds of the final pyrazinone are made (Scheme 20).95 Methyl pyruvate was condensed with ethylenediamine to afford 3-methyl-5,6-dihydro-2(1H)-pyrazinone 69 via imine and amide formation. In the next step, a strange alkylation was achieved by reaction of 69 with an aldehyde or ketone under alkaline conditions. Although the authors first reported that reaction had taken place at position 5, it was later demonstrated by NOE experiments and X-ray crystallography that 6-alkyl-3-methyl-2(1H)-pyrazinones 70 were obtained.96 The process probably involves deprotonation, prototropy and 6-alkylation at 5,6-enamine. Compounds 70 were further converted into pyrazines bearing an alkoxy, a phenoxy, an alkylthio or a phenylthio group with interesting odour properties.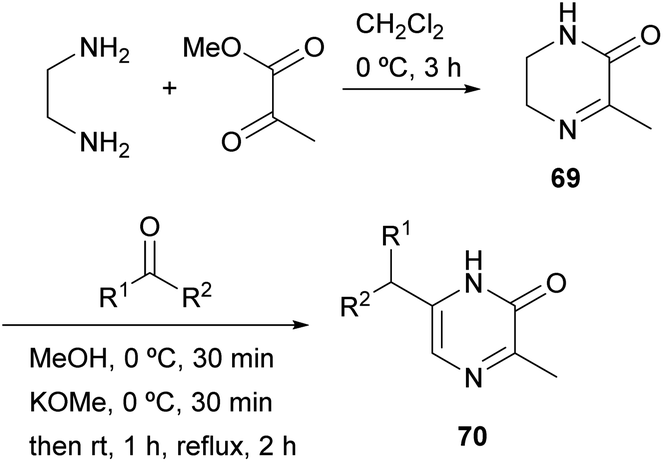 | ||
| Scheme 20 Synthesis of 2(1H)-pyrazinones from ethylendiamine and methyl pyruvate. | ||
2.9 Synthesis from dipeptides
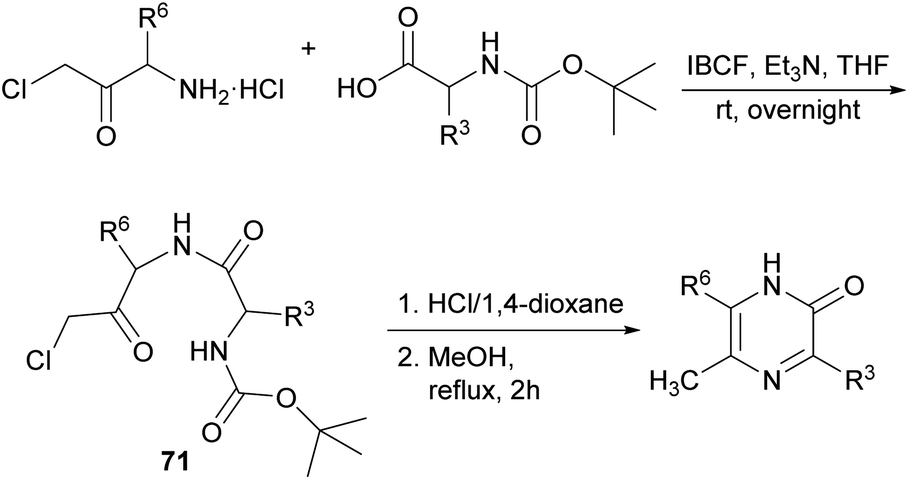 | ||
| Scheme 21 Synthesis of 2(1H)-pyrazinones from Boc-protected amino acids and a chloromethyl amino ketone. | ||
For the last step, the presence of the chlorine atom in the dipeptidyl derivative plays a key role as it notably increases the electrophilicity of the carbonyl group, and it is easily released as a good leaving group favouring the formation of the pyrazinone. Moreover, the reaction can occur independently of the stereoisomerism of the starting reagent.99 From this route, 2-acetoxypyrazines can be easily accessed, which have been proposed as useful acetylating reagents, reacting selectively with amino groups of amino acids in the presence of hydroxy groups.100
This method constitutes an efficient strategy to provide highly substituted rings in high yields under mild conditions, depending on the amino acids employed that determine R3 and R6 in the final pyrazinone core: when aspartate or glutamate are used as components of the starting dipeptidyl chloromethyl ketone, pyrazinone carboxylates can be obtained. Nevertheless, in contrast to that observed for position 6, carboxymethyl groups at R3 rapidly decarboxylate due to the low electron density of this position.101
In addition, amino groups can be easily introduced as substituents at positions 3 and/or 6, protected with a benzyloxycarbonyl (Z) group prior to the cyclization. Although the catalytic hydrogenation of the Z group of aminomethyl attached to position 6 causes instantaneous and selective deamination of this position,102 when larger aminoalkyl chains are introduced, the Z group can be removed and the amine further functionalized by coupling to various amino acids.
Based on this strategy, the development of opioid mimetics constitutes an interesting application for pyrazinone-containing compounds. Thus, Okada et al. synthesised a series of analogues of the pentapeptide enkephalin, a natural ligand of opioid receptors.103–105 Specifically, they incorporated a pyrazinone ring with aminoalkyls of different lengths as R3 or R6, at which a tyrosine was attached. Based on the results which pointed to a strong binding to the μ-opioid receptor and good lipophilicity given by the pyrazinone core, several modifications were prepared. Among them, the inclusion of 2′,6′-dimethyl-L-tyrosine (Dmt) as in 72 (Fig. 8) has been reported to enhance receptor affinity, and to exhibit higher analgesic and antinociceptive activities.106–112 Moreover, Li et al. reported that N-allylation of Dmt in these scaffolds allows the conversion of μ-opioid agonists into potent μ-opioid antagonists.113
 | ||
| Fig. 8 General structures of pyrazinone-containing opioid derivatives with Dmt and somatostatin analogues. | ||
An additional example of an application related to pyrazinone derivatives synthesised following this strategy was reported by Miyazaki et al. whose work was aimed at the development of analogues of the somatostatin, a cyclic tetradecapeptide involved in the inhibition of growth hormone secretion.114,115 Particularly, they designed novel cyclic analogues which included a pyrazinone ring instead of the disulfide bond. The resulting compounds, with a general structure 73, exhibited antiproliferative activity on tumour cells, but also presented interesting properties: pyrazinone-containing peptides are able to cross the blood–brain barrier, as well as the epithelial membrane of gastrointestinal tracts. Also, the presence of the heterocycle increases the lipophilicity, enhancing the interaction with cell membranes, and offers higher resistance to enzymatic degradation.
In fact, in 1996, the research group that first developed the cyclization of dipeptidyl chloromethyl ketones, devised a route to pyrazinones without a substituent at position 5 starting from a dipeptidyl aldehyde 74 (Scheme 22).116 This aldehyde was obtained from a Boc protected α-amino Weinreb amide 75, which was subjected to Boc removal followed by coupling with the second Boc amino acid in the sequence and final selective reduction of the Weinreb amide to an aldehyde. After Boc removal of 74, cyclization was promoted at room temperature in MeCN. Following this approach, besides the 2(1H)-pyrazinones 76a–b, the natural products deoxymuta-aspergillic acid 76c, deoxyaspergillic acid 2a and flavacol 2b were prepared. Moreover, the stereochemistry of the chiral carbon atom of deoxyaspergillic acid 2a was determined to be S. Taking advantage of this method, Jiang and Gu prepared 3,6-bis(3-indolyl)-2(1H)-pyrazinones (77), corresponding to the core of dragmacidin D, the key starting amino acids in this case being N-Boc-indolylglycine and the Weinreb amide 78 also derived from indolylglycine (Scheme 23).42
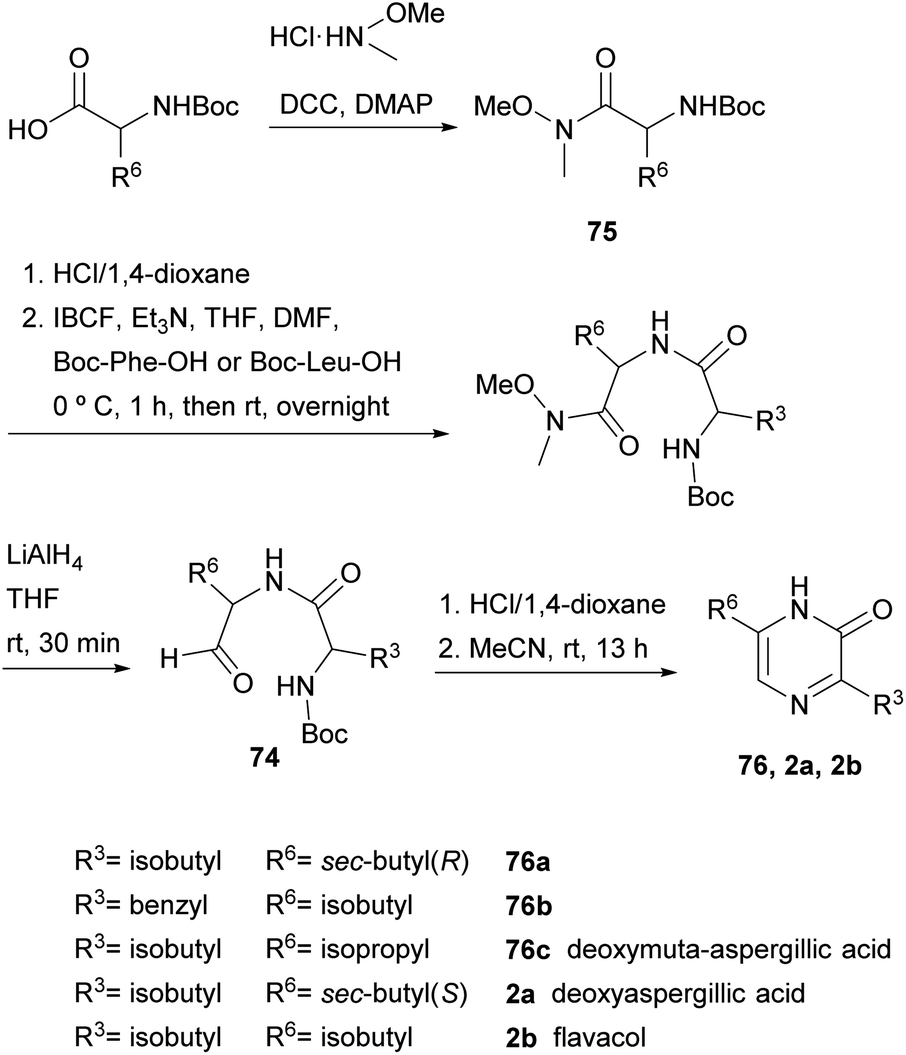 | ||
| Scheme 22 Synthesis of 2(1H)-pyrazinones from dipeptidyl aldehydes. | ||
 | ||
| Scheme 23 Synthesis of 3,6-bis(3-indolyl)-2(1H)-pyrazinone. | ||
Alternative approaches to obtain the dipeptidyl aldehydes were reported, thus Aubé et al. used a dipeptidyl ester 79 that was reduced to aldehyde with DIBAL or a dipeptidyl alcohol 80 that was oxidized to aldehyde with Dess-Martin periodinane (DMP) (Scheme 24). Phevalin (2c) was obtained following both approaches.117,118 Tyrvalin (2c), leuvalin (2d), phileucin (2e) and the six synthetic analogs 81a–f were also prepared using an amino alcohol as starting material for the corresponding dipeptidyl aldehydes.118 Formation of the pyrazinones was achieved in trifluoroacetic acid (TFA) via a one-pot Boc removal, ring closure and spontaneous oxidation.
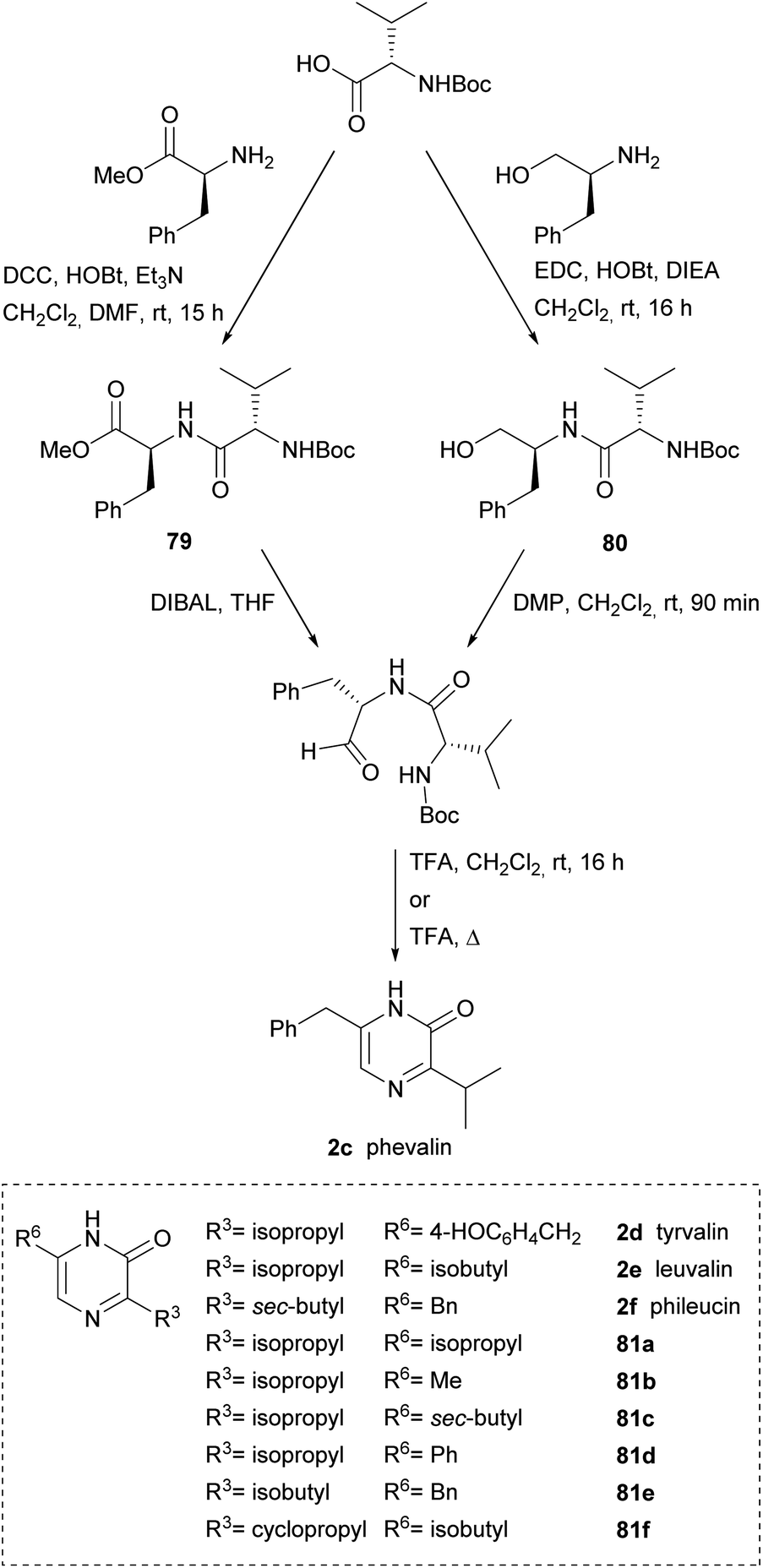 | ||
| Scheme 24 Two routes for the preparation of dipeptidyl aldehydes as precursors of 3,6-disubstituted pyrazinones. | ||
In 2004, Adam et al. and Matsushita et al. took this method a step further. On the one hand, the former authors prepared 2(1H)-pyrazinones again substituted at position 5, this time by cyclising dipeptidyl ketones 82 (Scheme 25).119 These intermediates were readily obtained through coupling Boc-protected amino acids with α-amino ketones or with α-amino secondary alcohols, the latter followed by an additional oxidation with DMP. After Boc removal of 82 with HCl, stirring the resulting dipeptidyl ketones in pyridine at 80 °C afforded the expected 5-phenyl or 5-methyl-2(1H)-pyrazinones 83 in high yields.
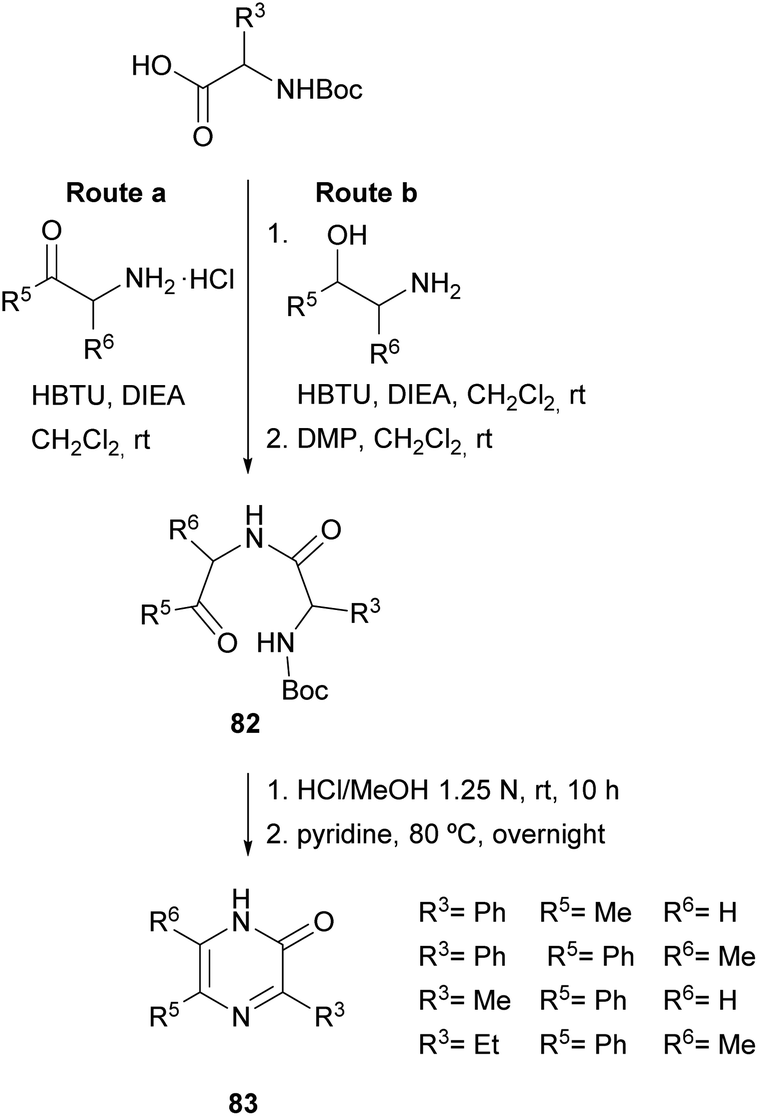 | ||
| Scheme 25 Synthesis of 2(1H)-pyrazinones from dipeptidyl ketones. | ||
Matsushita et al. carried out N–H insertion reactions of α-diazo-β-ketoesters with Boc amino acid amides in the presence of a rhodium octanoate catalyst to afford α-acylamino-β-ketoesters 84 as precursors of pyrazinones (Scheme 26).120 Their subsequent treatment with TFA at 100 °C led to Boc removal and cyclisation, yielding 2(1H)-pyrazinones 85 with a methyl or phenyl at position 5 and with a carboxylate substituent at position 6. To gain more insight into this method, the authors extended it to the solid phase. Accordingly, α-diazo-β-ketoesters linked to a JandaJel polymer were used and converted to the resin-bound pyrazinones 86. The procedure as described in solution was followed, except for the cyclization which was performed in acetic acid after Boc removal with TFA. Before cleavage from the resin, these pyrazinones 86 were then alkylated at N-1 or converted into pyrazines through reaction with POBr3 and subsequent solid-phase Suzuki coupling with aryl boronic acids.
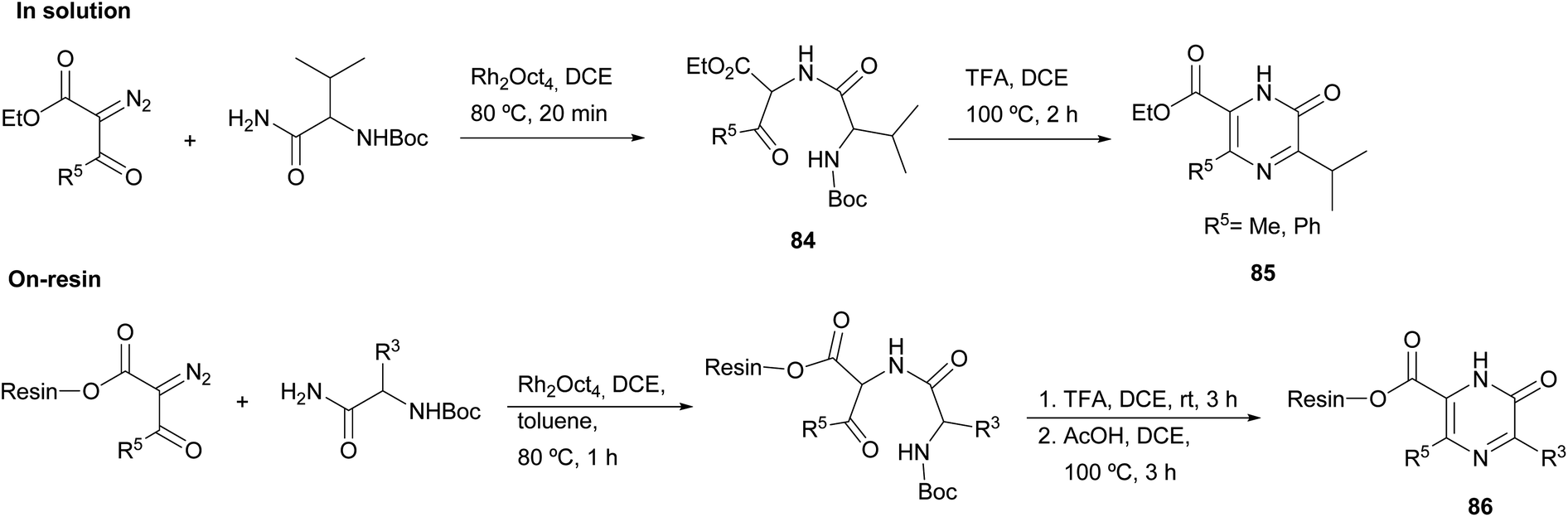 | ||
| Scheme 26 The use of N–H insertion reactions for the preparation of 2(1H)-pyrazinones either in solution or on solid phase. | ||
Dipeptides bearing a carboxamide group at the C-terminus have also been converted into 3,5,6-trisubstituted-2(1H)-pyrazinones, the substituent at position 5 being a carboxamide. With the aim of obtaining dipeptidyl carboxamides 87, Basso et al. used the PADAM strategy, based on a Passerini multicomponent reaction, amine deprotection and acyl migration (Scheme 27).121 In particular, they reacted an N-Fmoc α-amino aldehyde with N-Boc α-amino acids and an isocyanide, and then removed the Fmoc group with Et2NH, which also led to spontaneous intramolecular acyl migration affording α-hydroxy amides 88. The hydroxyl group of 88 was then oxidized with 2-iodoxybenzoic acid (IBX) to reach the desired dipeptidyl carboxamides 87. Final treatment of 87 with TFA not only allowed Boc removal and in situ cyclization, but also the unexpected aromatisation of the ring, giving rise to the corresponding 3,5,6-trisubstituted-2(1H)-pyrazinones.
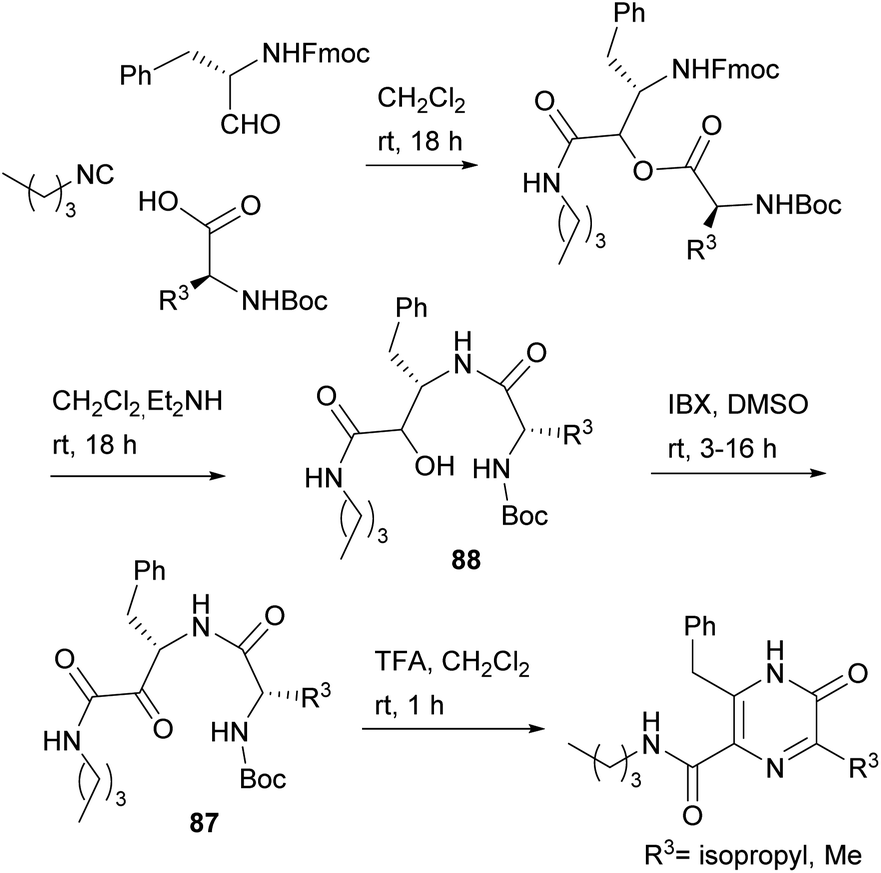 | ||
| Scheme 27 Synthesis of 2(1H)-pyrazinones from dipeptidyl carboxamides. | ||
2.10 Synthesis through an Ugi condensation
Faggi et al. proposed a novel strategy for the synthesis of pyrazinones, with a carboxamide at position 6, through an Ugi four-component condensation between an arylglyoxal (R5 = Ar), a glyoxylic acid, an isocyanide, and a primary amine (Scheme 28a).122 The condensation affords the intermediate 89, which in the presence of ammonia undergoes a Davidson cyclization providing polysubstituted pyrazinone-6-carboxamides 90.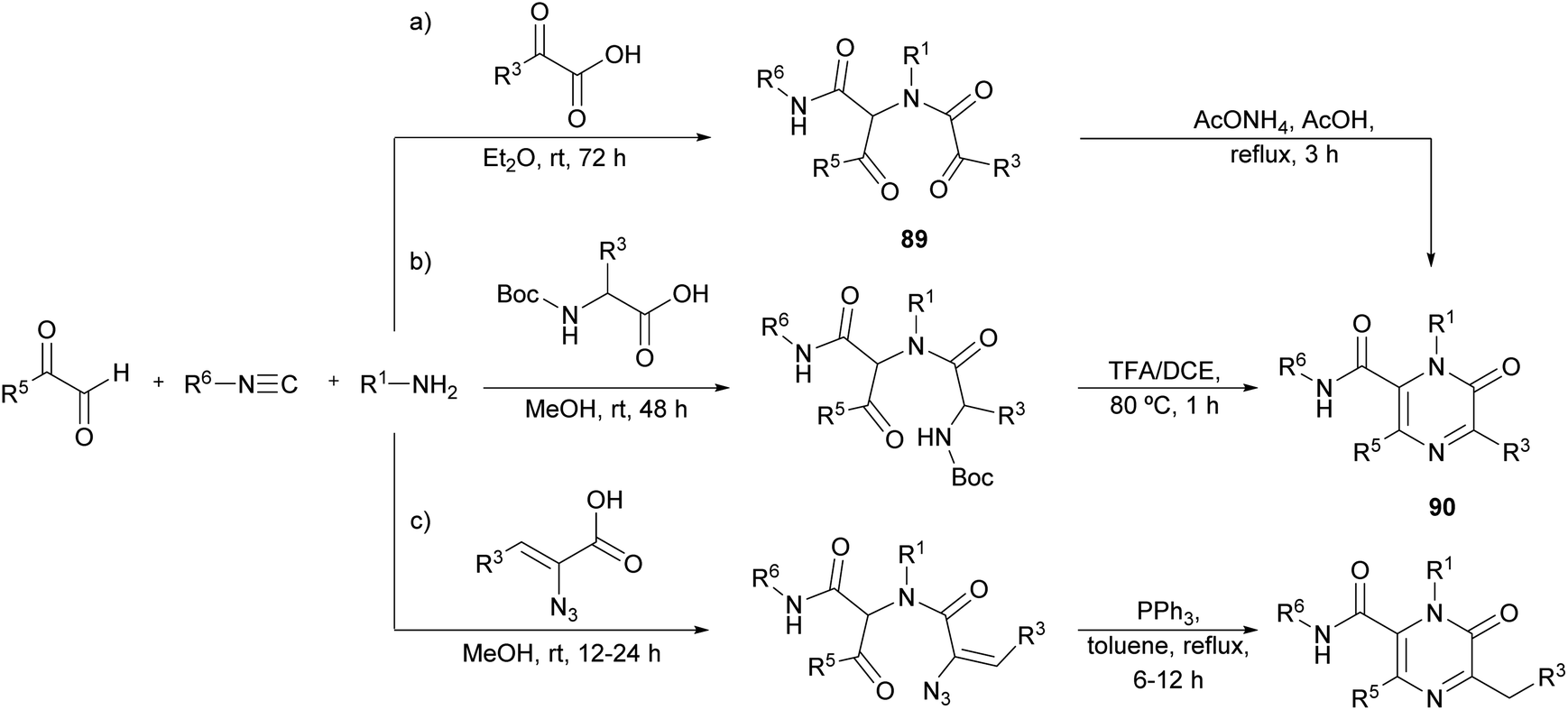 | ||
| Scheme 28 Synthesis of 2(1H)-pyrazinones by means of Ugi reactions. | ||
Later, Azuaje et al. further developed this Ugi-based strategy employing a N-Boc protected amino acid instead of the glyoxylic acid (Scheme 28b).123 In their one-pot approach, after the four-component condensation, a subsequent step involving a deprotection/cyclization strategy with TFA/DCE provides pyrazinone 90. Interestingly, they reported that when 2 N HCl/ether at 0 °C was employed to cleave the Boc group, a 3,4-dihydropyrazin-2(1H)-one was isolated instead of the target pyrazinone. The resulting pyrazinones have been proven to be efficient adenosine receptor antagonists.124
More recently, Wu et al. reported a similar approach employing α-azidovinyl acids (Scheme 28c). After the Ugi condensation, the resulting intermediate was treated with PPh3 leading to a one-pot Staudinger/Aza-Wittig/isomerization cascade giving 2(1H)-pyrazinones in an efficient way.125
These strategies based on four-component reactions allow the assembly of pyrazinones with a wide variety of substituents depending on the readily available substrates involved.
2.11 Synthesis from α-azido-N-allylamides
Zhang et al. reported a synthesis of 5-formyl-2(1H)-pyrazinones starting from α-azido-N-allylamides via a copper(II)-catalyzed reaction under an O2 atmosphere, that involves the formation of the bond between N-4 and C-5 (Scheme 29).126 This procedure provided, in moderate to good yields, 5-formyl-2(1H)-pyrazinones 91 bearing an electron-rich aryl, a phenyl, a 4-chlorophenyl or a 4-bromophenyl at position 5 and a methyl, a benzyl or a 2,6-dimethylphenyl at N-1.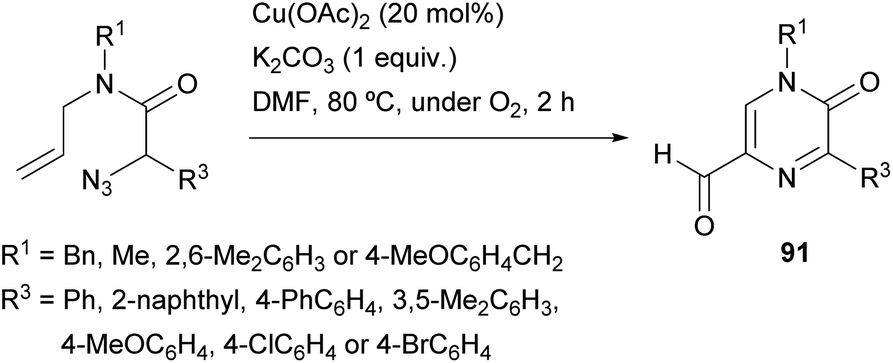 | ||
| Scheme 29 Synthesis of 2(1H)-pyrazinones from α-azido-N-allylamides. | ||
The proposed pathway for this transformation involves first a 1,3-dipolar cycloaddition between the azido group and the alkene moiety to yield a bicyclic triazoline 92 which could undergo denitrogenative fragmentation giving a bicyclic aziridine 93 (Scheme 30). Next, single-electron oxidation of this intermediate with Cu(OAc)2 and deprotonation would result in ring opening of the aziridine yielding a primary alkyl radical, which would be transformed into a copper peroxy species 94 by reaction with O2. Finally, elimination of copper hydroxide and oxidation of the resulting 5-formyl dihydropyrazinone 95 would give the corresponding 5-formyl-2(1H)-pyrazinone 91.
 | ||
| Scheme 30 Proposed mechanism for the synthesis of 2(1H)-pyrazinones from α-azido-N-allylamides. | ||
2.12 Synthesis from 3,4-disubstituted 4-amino-5(4H)-isoxazolones
Becalli and Marchesini described a synthesis of 3,5,6-trisubstituted 2(1H)-pyrazinones from 3,4-disubstituted 4-amino-5(4H)-isoxazolones (Scheme 31).127 This method is an alternative to the protocol first described by Jones for the preparation of 2(1H)-pyrazinones starting from α-amino acid amides and 1,2-dicarbonyl compounds, discussed in Section 2.4. First,49 the corresponding 3,4-disubstituted 4-amino-5(4H)-isoxazolone 96 is treated with a 2-oxoacyl chloride in the presence of Et3N and 4-(dimethylamino)pyridine (DMAP). Next, hydrogenation of the resulting intermediate, using the Lindlar catalyst, results in the reduction of the N–O bond, decarboxylation and intramolecular cyclization, yielding the 3,5,6-trisubstituted 2(1H)-pyrazinone.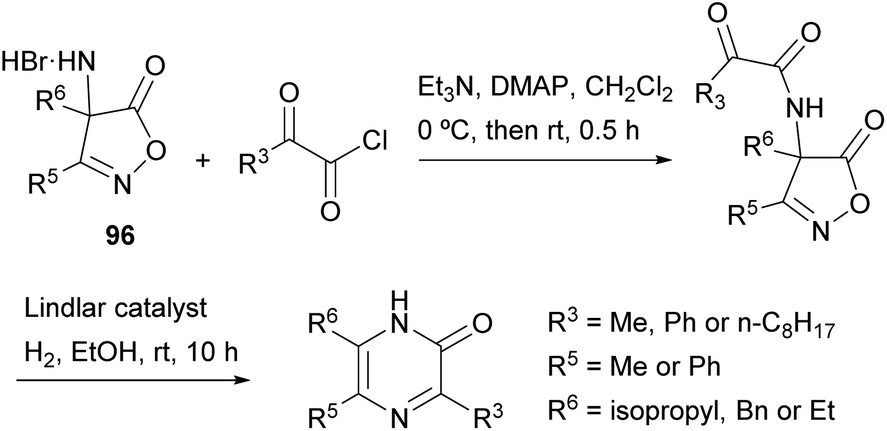 | ||
| Scheme 31 Synthesis of 2(1H)-pyrazinones from 4-amino-5(4H)-isoxazolones. | ||
2.13 Synthesis from mesoionic oxazoles and an isocyanide
Saijo et al. described a novel strategy to afford 2(1H)-pyrazinones by the ring transformation of mesoionic 1,3-oxazolium-5-olates by a base-mediated reaction with p-toluenesulfonylmethyl isocyanide (TosMIC) (Scheme 32).128 Later, they found that this methodology could also be applied to ethyl isocyanoacetate as a reagent bearing an active methylene group.129 In both cases, the 4-trifluoroacetyl-1,3-oxazolium-5-olate 97 is obtained by the cyclodehydration of a N-acyl-N-alkylglycine with trifluoroacetic anhydride. Moreover, if an excess of TosMIC is used, an imidazo[1,4-a]pyrazin-8(7H)-one derivative 98 is produced, due to the high electrophilic character of C-3 of the 2(1H)-pyrazinone core.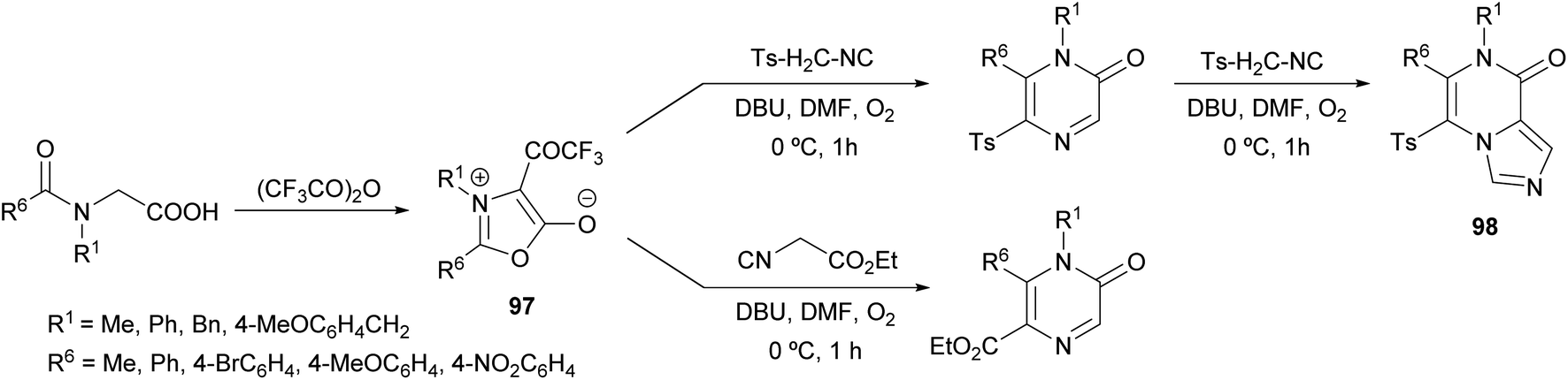 | ||
| Scheme 32 Preparation of mesoionic oxazoles and their use in the synthesis of 2(1H)-pyrazinones. | ||
The mechanism of this reaction would involve the initial deprotonation of the active methylene group of TosMIC by 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) and a nucleophilic attack of the resulting carbanion to the C-2 position of the oxazolium affording the intermediate 99, which is converted into 100 by proton transfer (Scheme 33).128 This intermediate is in equilibrium with 101, which after a decarboxylation promotes a nucleophilic attack causing the ring closure. After a proton transfer, intermediate 102 would react with O2 forming a superoxide complex, which suffers a recombination affording the pyrazinone after the elimination of the trifluoroacetate anion.
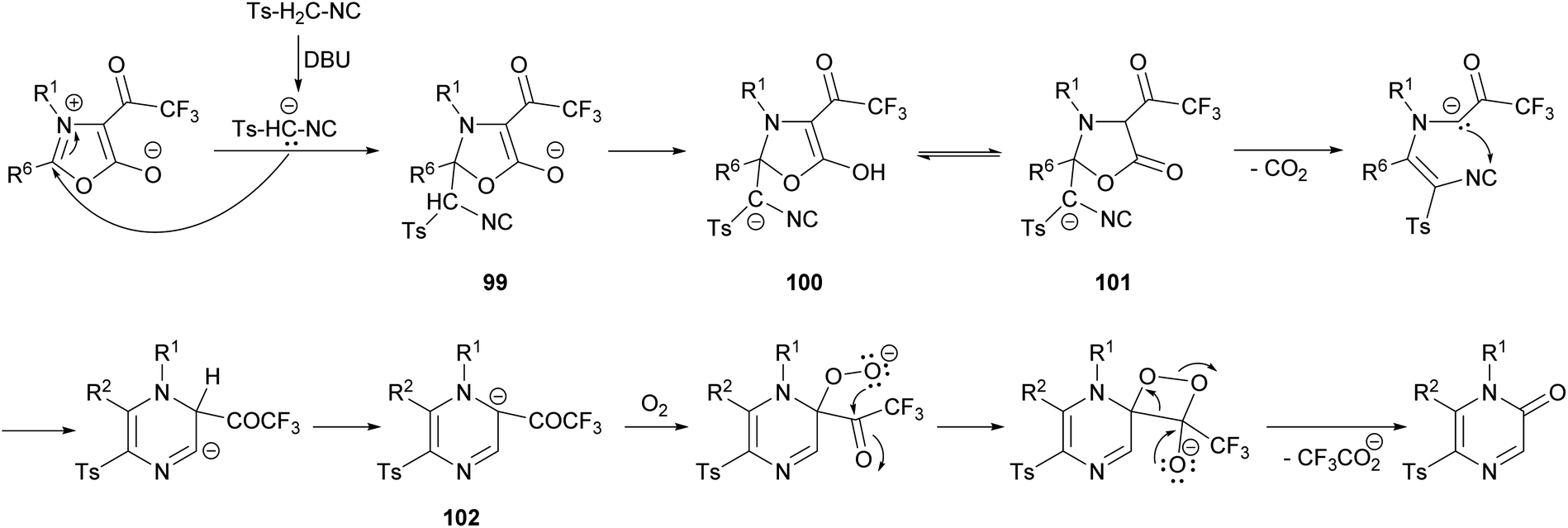 | ||
| Scheme 33 Proposed mechanism for the reaction of mesoionic oxazoles and an isocyanide. | ||
2.14 Synthesis from 2-chloro ketone oximes and an amine in a deep eutectic solvent
In 2017, Perrone et al. reported the first synthesis of 1-alkyl-3,5-diaryl trisubstituted 2(1H)-pyrazinones (103) in deep eutectic solvents (DESs) by means of the coupling between aromatic α-chloro oximes with aliphatic primary amines (Scheme 34).130 α-Chloro ketone oximes, which are reported to be highly reactive, were prepared by a straightforward condensation of α-chloro ketones with hydroxylamine hydrochloride.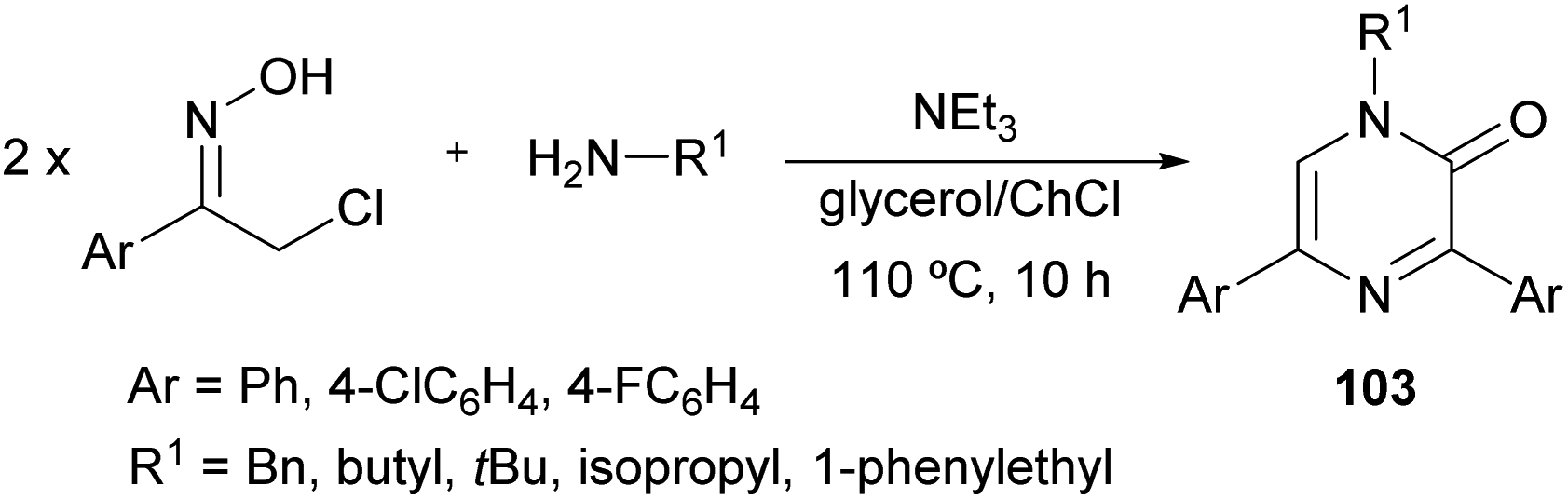 | ||
| Scheme 34 Synthesis of 2(1H)-pyrazinones from α-chloro oximes and an amine. | ||
Regarding the optimization of the reaction conditions, they found that the use of choline chloride (ChCl) based DESs, such as the combination glycerol/ChCl or urea/ChCl, resulted in better yields than using organic solvents like THF or DMF. In all cases, reactions were carried out at 110 °C for 10 h with an excess of the primary amine, and after a simple work-up, pyrazinones were obtained and the DES mixtures could be recovered.
This route constitutes a green and simple way to obtain 2(1H)-pyrazinones. However, this methodology cannot be applied using aliphatic α-chlorinated oximes or aromatic amines as starting reagents. The mechanism of formation could involve an initial nucleophilic attack of the primary amine to the α-chloro oxime, a subsequent base-catalysed tautomerization and a water elimination via N–O bond breaking, affording the 1,4-diaza-1,3-diene 104 (Scheme 35).130 This intermediate reacts with the enolic form of another α-chlorinated oxime through a [4 + 2] cycloaddition. The release of hydroxylamine and chloride favours the aromatization of the ring and a nucleophilic attack of water. A final oxidation step provides the pyrazinone 103.
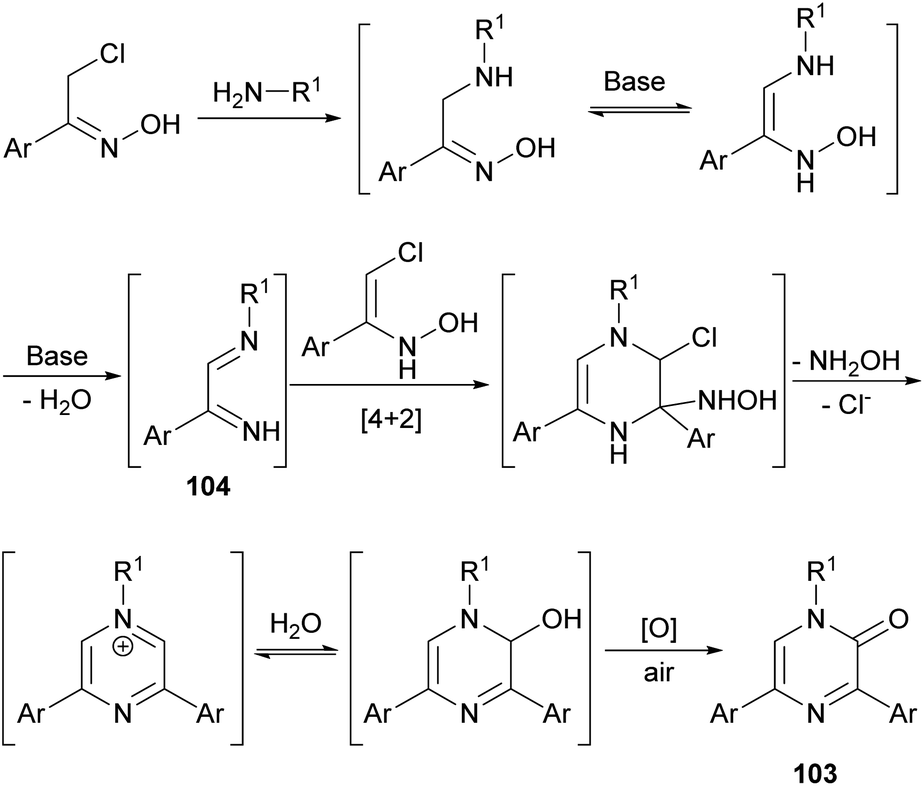 | ||
| Scheme 35 Proposed mechanism for the formation of pyrazinones from α-chloro oximes and an amine. | ||
3 Concluding remarks
The synthesis of the 2(1H)-pyrazinone scaffold can be achieved through the combination of a wide variety of acyclic components as key building blocks, especially α-amino acids (and esters), α-amino ketones (and aldehydes), α-amino acid amides, glyoxylic acid derivatives, α-haloacyl halides, 1,2-dicarbonyl compounds, 2-chloro ketone oximes, α-aminonitriles, dipeptidyl chloromethyl ketones, dipeptidyl aldehydes, ketones and carboxamides, isocyanides, primary amines, and mesoionic oxazoles. The review deals with the various synthetic strategies, starting from the earliest, published at the turn of the 20th century, up to those described in recent years. The ways in which the components are assembled to construct the heterocyclic ring, as well as the details of reaction conditions, are reviewed for each of these. All these approaches pave the way for the preparation of highly functionalized 2(1H)-pyrazinones with interesting biological applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. R.-L. is recipient of a predoctoral fellowship from the University of Girona (IFUdG2020).References
- J. D. Dutcher, J. Biol. Chem., 1947, 171, 321–339 CrossRef CAS.
- G. Dunn, G. T. Newbold and F. S. Spring, J. Chem. Soc., 1949, 2586–2587 RSC.
- M. E. Álvarez, C. B. White, J. Gregory, G. C. Kydd, A. Harris, H. H. Sun, A. M. Gillum and R. Cooper, J. Antibiot., 1995, 48, 1165–1167 CrossRef PubMed.
- M. Zimmermann and M. A. Fischbach, Chem. Biol., 2010, 17, 925–930 CrossRef CAS PubMed.
- N. Böhringer, M. Götschow, G. M. König and T. F. Schäberle, Nat. Prod. Commun., 2017, 12, 107–109 CrossRef.
- S. Umezawa, K. Tatsuta, T. Tsuchiya, H. Umezawa and H. Naganawa, Tetrahedron Lett., 1971, 12, 259–262 CrossRef.
- K. Tatsuta, K. Fujimoto, M. Yamashita, T. Tsuchiya, S. Umezawa and H. Umezawa, J. Antibiot., 1973, 26, 606–608 CrossRef CAS PubMed.
- Y.-Q. Tang, I. Sattler, R. Thiericke, S. Grabley and X.-Z. Feng, Eur. J. Org. Chem., 2001, 2, 261–267 CrossRef.
- R. Jansen, S. Sood, K. I. Mohr, B. Kunze, H. Irschik, M. Stadler and R. Müller, J. Nat. Prod., 2014, 77, 2545–2552 CrossRef CAS PubMed.
- F. Zhang, D. R. Braun, S. R. Rajski, D. DeMaria and T. S. Bugni, Mar. Drugs, 2019, 17, 698 CrossRef CAS PubMed.
- K. Hirano, T. Kubota, M. Tsuda, K. Watanabe, J. Fromont and J. Kobayashi, Tetrahedron, 2000, 56, 8107–8110 CrossRef CAS.
- A. E. Wright, S. A. Pomponi, S. S. Cross and P. McCarthy, J. Org. Chem., 1992, 57, 4772–4775 CrossRef CAS.
- R. J. Capon, F. Rooney, L. M. Murray, E. Collins, A. T. R. Sim, J. A. P. Rostas, M. S. Butler and A. R. Carroll, J. Nat. Prod., 1998, 61, 660–662 CrossRef CAS PubMed.
- A. Cutignano, G. Bifulco, I. Bruno, A. Casapullo, L. Gomez-Paloma and R. Riccio, Tetrahedron, 2000, 56, 3743–3748 CrossRef CAS.
- K. Motohashi, K. Inaba, S. Fuse, T. Doi, M. Izumikawa, S. T. Khan, M. Takagi, T. Takahashi and K. Shin-Ya, J. Nat. Prod., 2011, 74, 1630–1635 CrossRef CAS PubMed.
- Y. Furuta, K. Takahashi, Y. Fukuda, M. Kuno, T. Kamiyama, K. Kozaki, N. Nomura, H. Egawa, S. Minami, Y. Watanabe, H. Narita and K. Shiraki, Antimicrob. Agents Chemother., 2002, 46, 977–981 CrossRef CAS PubMed.
- S. Joshi, J. Parkar, A. Ansari, A. Vora, D. Talwar, M. Tiwaskar, S. Patil and H. Barkate, Int. J. Infect. Dis., 2021, 102, 501–508 CrossRef CAS PubMed.
- R. A. Hartz, V. T. Ahuja, A. G. Arvanitis, M. Rafalski, E. W. Yue, D. J. Denhart, W. D. Schmitz, J. L. Ditta, J. A. Deskus, A. B. Brenner, F. W. Hobbs, J. Payne, S. Lelas, Y.-W. Li, T. F. Molski, G. K. Mattson, Y. Peng, H. Wong, J. E. Grace, K. A. Lentz, J. Qian-Cutrone, X. Zhuo, Y.-Z. Shu, N. J. Lodge, R. Zaczek, A. P. Combs, R. E. Olson, J. J. Bronson, R. J. Mattson and J. E. Macor, J. Med. Chem., 2009, 52, 4173–4191 CrossRef CAS PubMed.
- J. F. Bereznak, P. L. Sharpe, R. B. Sheth, T. M. Stevenson, A. E. Taggi, C.-P. Tseng and W. Zhang, PCT Application WO 2006089060, E. I. DuPont De Nemours, 2006 Search PubMed.
- A. E. Taggi, T. M. Stevenson, J. F. Bereznak, P. L. Sharpe, S. Gutteridge, R. Forman, J. J. Bisaha, D. Cordova, M. Crompton, L. Geist, P. Kovacs, E. Marshall, R. Sheth, C. Stavis and C.-P. Tseng, Bioorg. Med. Chem., 2016, 24, 435–443 CrossRef CAS PubMed.
- J. Heeres, M. R. de Jonge, L. M. H. Koymans, F. F. D. Daeyaert, M. Vinkers, K. J. A. Van Aken, E. Arnold, K. Das, A. Kilonda, G. J. Hoornaert, F. Compernolle, M. Cegla, R. A. Azzam, K. Andries, M.-P. de Béthune, H. Azijn, R. Pauwels, P. J. Lewi and P. A. J. Janssen, J. Med. Chem., 2005, 48, 1910–1918 CrossRef CAS PubMed.
- C. S. Burgey, K. A. Robinson, T. A. Lyle, P. E. J. Sanderson, S. D. Lewis, B. J. Lucas, J. A. Krueger, R. Singh, C. Miller-Stein, R. B. White, B. Wong, E. A. Lyle, P. D. Williams, C. A. Coburn, B. D. Dorsey, J. C. Barrow, M. T. Stranieri, M. A. Holahan, G. R. Sitko, J. J. Cook, D. R. McMasters, C. M. McDonough, W. M. Sanders, A. A. Wallace, F. C. Clayton, D. Bohn, Y. M. Leonard, T. J. Detwiler, J. J. Lynch, Y. Yan, Z. Chen, L. Kuo, S. J. Gardell, J. A. Shafer and J. P. Vacca, J. Med. Chem., 2003, 46, 461–473 CrossRef CAS PubMed.
- P. Raubo, R. Evans and P. Willis, Bioorg. Med. Chem. Lett., 2020, 30, 127412 CrossRef CAS PubMed.
- P. A. Allway, J. K. Sutherland and J. A. Joule, Tetrahedron Lett., 1990, 31, 4781–4782 CrossRef CAS.
- N. D. Yates, D. A. Peters, P. A. Allway, R. L. Beddoes, D. I. C. Scopes and J. A. Joule, Heterocycles, 1995, 40, 331–347 CrossRef CAS.
- D. A. Peters, N. D. Yates, D. I. C. Scopes and J. A. Joule, Heterocycles, 1995, 40, 983–991 CrossRef CAS.
- M. Helliwell, Y. You and J. A. Joule, Heterocycles, 2006, 70, 87–91 CrossRef CAS PubMed.
- E. R. Ashley, E. G. Cruz and B. M. Stoltz, J. Am. Chem. Soc., 2003, 125, 15000–15001 CrossRef CAS PubMed.
- K. M. Allan and B. M. Stoltz, J. Am. Chem. Soc., 2008, 130, 17270–17271 CrossRef CAS PubMed.
- F. R. Japp and J. Knox, J. Chem. Soc., Trans., 1905, 87, 701–707 RSC.
- H. McCombie and E. Parry, J. Chem. Soc., Trans., 1909, 95, 584–590 RSC.
- C. Gastaldi, Gazz. Chim. Ital., 1921, 51, 233–255 Search PubMed.
- B. H. Ingham, J. Chem. Soc., 1927, 692–700 RSC.
- M. Busch and W. Foerst, J. Prakt. Chem., 1928, 119, 287 CrossRef CAS.
- Y. A. Tota and R. C. Elderfield, J. Org. Chem., 1942, 313–319 CrossRef CAS.
- R. A. Baxter, G. T. Newbold and F. S. Spring, J. Chem. Soc., 1947, 370–372 RSC.
- N. Sato, J. Heterocycl. Chem., 1978, 15, 665–670 CrossRef CAS.
- R. H. Bradbury, D. Griffiths and J. E. Rivett, Heterocycles, 1990, 31, 1647–1653 CrossRef CAS.
- E. Johannes, R. Horbert, J. Schlosser, D. Schmidt and C. Peifer, Tetrahedron Lett., 2013, 54, 4067–4072 CrossRef CAS.
- B. Pinchuk, E. Johannes, S. Gul, J. Schlosser, C. Schaechtele, F. Totzke and C. Peifer, Mar. Drugs, 2013, 11, 3209–3223 CrossRef PubMed.
- R. Horbert, B. Pinchuk, E. Johannes, J. Schlosser, D. Schmidt, D. Cappel, F. Totzke, C. Schächtele and C. Peifer, J. Med. Chem., 2015, 58, 170–182 CrossRef CAS PubMed.
- B. Jiang and X. H. Gu, Heterocycles, 2000, 7, 1559–1568 CrossRef.
- F. Y. Miyake, K. Yakushijin and D. A. Horne, Org. Lett., 2002, 4, 941–943 CrossRef CAS PubMed.
- R. A. Baxter and F. S. Spring, J. Chem. Soc., 1947, 25, 1179–1183 RSC.
- J. J. Gallagher, G. T. Newbold, F. S. Spring and J. C. Woods, J. Chem. Soc., 1949, 910–912 RSC.
- J. J. Gallagher, G. T. Newbold, W. Sharp and F. S. Spring, J. Chem. Soc., 1952, 4870–4874 RSC.
- A. Ohta, Y. Aoyagi, Y. Kurihara, K. Yuasa, M. Shimazaki, T. Kurihara and H. Miyamae, Heterocycles, 1987, 26, 3181–3191 CrossRef CAS.
- A. Ohta, Y. Aoyagi, Y. Kurihara, A. Kojima, K. Yuasa and M. Shimazaki, Heterocycles, 1988, 27, 437–444 CrossRef CAS.
- R. G. Jones, J. Am. Chem. Soc., 1949, 71, 78–81 CrossRef CAS PubMed.
- G. Karmas and P. E. Spoerri, J. Am. Chem. Soc., 1952, 74, 1580–1584 CrossRef CAS.
- T. Konakahara and Y. Takagi, Heterocycles, 1978, 9, 1733–1739 CrossRef CAS.
- G. Dunn, J. A. Elvidge, G. T. Newbold, D. W. C. Ramsay, F. S. Spring and W. Sweeny, J. Chem. Soc., 1949, 2707–2712 RSC.
- J. Maguire, D. Paton and F. L. Rose, J. Chem. Soc. C, 1969, 1593–1597 RSC.
- F. L. Muehlmann and A. R. Day, J. Am. Chem. Soc., 1956, 78, 242–244 CrossRef CAS.
- M. Helliwell, Y. Yun and J. A. Joule, Acta Crystallogr., 2006, 62, o955–o956 CAS.
- N. van Chuyen, T. Kurata and M. Fujimaki, Agric. Biol. Chem., 1972, 36, 1257–1258 CrossRef.
- N. van Chuyen, T. Kurata and M. Fujimaki, Agric. Biol. Chem., 1973, 37, 1613–1618 CrossRef.
- R. J. Bergeron and P. G. Hoffman, J. Org. Chem., 1980, 45, 163–165 CrossRef CAS.
- R. M. Seifert, R. G. Buttery, D. G. Guadagni, D. R. Black and J. G. Harris, J. Agric. Food Chem., 1970, 18, 246–249 CrossRef CAS.
- R. M. Seifert, R. G. Buttery, D. G. Guadagni, D. R. Black and J. G. Harris, J. Agric. Food Chem., 1972, 20, 135–137 CrossRef CAS.
- C. W. Lindsley, Z. Zhao, W. H. Leister, R. G. Robinson, S. F. Barnett, D. Defeo-Jones, R. E. Jones, G. D. Hartman, J. R. Huff, H. E. Huber and M. E. Duggan, Bioorg. Med. Chem. Lett., 2005, 15, 761–764 CrossRef CAS PubMed.
- A. Benjahad, R. Granet, P. Krausz, C. Bosgiraud and S. Delebassée, Nucleosides Nucleotides, 1996, 15, 1849–1861 CrossRef CAS.
- A. L. Weber, Orig. Life Evol. Biosph., 2008, 38, 279–292 CrossRef CAS PubMed.
- M. Gately, S. Wong, J. Peoples, D. Galamay, G. Delgado, A. L. Weber and T. Campbell, Nucleosides, Nucleotides Nucleic Acids, 2020, 39, 866–891 CrossRef CAS PubMed.
- C. Pierra, C. Counor, R. Storer and G. Gosselin, Collect. Czech. Chem. Commun., 2011, 76, 1327–1333 CrossRef CAS.
- M. Masaki and M. Ohta, Bull. Chem. Soc. Jpn., 1963, 36, 1177–1179 CrossRef CAS.
- Y. Chigira, M. Masaki and M. Ohta, Bull. Chem. Soc. Jpn., 1966, 39, 1014–1016 CrossRef CAS.
- G. W. H. Cheeseman, A. J. Freestone, R. A. Godwin and T. L. Hough, J. Chem. Soc. Perkin Trans., 1975, 1, 1888–1891 RSC.
- F. J. Fleitz, T. A. Lyle, N. Zheng, J. D. Armstrong III and R. P. Volante, Synth. Commun., 2000, 30, 3171–3180 CrossRef CAS.
- J. E. Reiner, D. V. Siev, G. L. Araldi, J. J. Cui, J. Z. Ho, K. M. Reddy, L. Mamedova, P. H. Vu, K.-S. S. Lee, N. K. Minami, T. S. Gibson, S. M. Anderson, A. E. Bradbury, T. G. Nolan and J. E. Semple, Bioorg. Med. Chem. Lett., 2002, 12, 1203–1208 CrossRef CAS PubMed.
- J. J. Cui, G.-L. Araldi, J. E. Reiner, K. M. Reddy, S. J. Kemp, J. Z. Ho, D. V. Siev, L. Mamedova, T. S. Gibson, J. A. Gaudette, N. K. Minami, S. M. Anderson, A. E. Bradbury, T. G. Nolan and J. E. Semple, Bioorg. Med. Chem. Lett., 2002, 12, 2925–2930 CrossRef CAS PubMed.
- R. Singh, M. V. Silva Elipe, P. G. Pearson, B. H. Arison, B. K. Wong, R. White, X. Yu, C. S. Burgey, J. H. Lin and T. A. Baillie, Chem. Res. Toxicol., 2003, 16, 198–207 Search PubMed.
- P. E. J. Sanderson, K. J. Cutrona, D. L. Dyer, J. A. Krueger, L. C. Kuo, S. D. Lewis, B. J. Lucas and Y. Yan, Bioorg. Med. Chem. Lett., 2003, 13, 161–164 CrossRef CAS PubMed.
- J. Vekemans, C. Pollers-Wieers and G. Hoornaert, J. Heterocycl. Chem., 1983, 20, 919–923 CrossRef CAS.
- W. M. De Borggraeve, B. M. P. Verbist, F. J. R. Rombouts, V. G. Pawar, W. J. Smets, L. Kamoune, J. Alen, E. V. Van der Eycken, F. Compernolle and G. J. Hoornaert, Tetrahedron, 2004, 60, 11597–11612 CrossRef CAS.
- J. Tulinsky, S. A. Mizsak, W. Watt, L. A. Dolak, T. Judge and R. B. Gammill, Tetrahedron Lett., 1995, 12, 2017–2020 CrossRef.
- J. Tulinsky, B. V. Cheney, S. A. Mizsak, W. Watt, F. Han, L. A. Dolak, T. Judge and R. B. Gammill, J. Org. Chem., 1999, 64, 93–100 CrossRef CAS PubMed.
- N. Kaval, W. Dehaen and E. Van der Eycken, J. Comb. Chem., 2005, 7, 90–95 CrossRef CAS PubMed.
- J. Gising, P. Örtqvist, A. Sandström and M. Larhed, Org. Biomol. Chem., 2009, 7, 2809–2815 RSC.
- P. Örtqvist, J. Gising, A. E. Ehrenberg, A. Vema, A. Borg, A. Karlén, M. Larhed, U. H. Danielson and A. Sandström, Bioorg. Med. Chem., 2010, 6512–6525 CrossRef PubMed.
- J. Gising, A. K. Belfrage, H. Alogheli, A. Ehrenberg, E. Åkerblom, R. Svensson, P. Artursson, A. Karlén, U. H. Danielson, M. Larhed and A. Sandström, J. Med. Chem., 2014, 57, 1790–1801 CrossRef CAS PubMed.
- R. A. Hartz, V. T. Ahuja, M. Rafalski, W. D. Schmitz, A. B. Brenner, D. J. Denhart, J. L. Ditta, J. A. Deskus, E. W. Yue, A. G. Arvanitis, S. Lelas, Y.-W. Li, T. F. Molski, H. Wong, J. E. Grace, K. A. Lentz, J. Li, N. J. Lodge, R. Zaczek, A. P. Combs, R. E. Olson, R. J. Mattson, J. J. Bronson and J. E. Macor, J. Med. Chem., 2009, 52, 4161–4172 CrossRef CAS PubMed.
- J. Li, D. Smith, S. Krishnananthan, R. A. Hartz, B. Dasgupta, V. Ahuja, W. D. Schmitz, J. J. Bronson, A. Mathur, J. C. Barrish and B.-C. Chen, Org. Process Res. Dev., 2012, 16, 156–159 CrossRef CAS.
- N. Kaval, K. Bisztray, W. Dehaen, C. O. Kappe and E. Van der Eycken, Mol. Divers., 2003, 7, 125–133 CrossRef CAS PubMed.
- V. G. Pawar and W. M. de Borggraeve, Synthesis, 2006, 2799–2814 CAS.
- N. Kaval, P. Appukkuttan and E. Van der Eycken, Top Heterocycl. Chem., 2006, 1, 267–304 CAS.
- V. P. Mehta, P. Appukkuttan and E. Van der Eycken, Curr. Org. Chem., 2011, 15, 265–283 CrossRef CAS.
- R. A. Hartz, V. T. Ahuja, X. Zhuo, R. J. Mattson, D. J. Denhart, J. A. Deskus, V. M. Vrudhula, S. Pan, J. L. Ditta, Y.-Z. Shu, J. E. Grace, K. A. Lentz, S. Lelas, Y.-W. Li, T. F. Molski, S. Krishnananthan, H. Wong, J. Qian-Cutrone, R. Schartman, R. Denton, N. J. Lodge, R. Zaczek, J. E. Macor and J. J. Bronson, J. Med. Chem., 2009, 52, 7653–7668 CrossRef CAS PubMed.
- X. Zhuo, R. A. Hartz, J. J. Bronson, H. Wong, V. T. Ahuja, V. M. Vrudhula, J. E. Leet, S. Huang, J. E. Macor and Y.-Z. Shu, Drug Metab. Dispos., 2010, 38, 5–15 CrossRef CAS PubMed.
- R. A. Hartz, V. T. Ahuja, W. D. Schmitz, T. F. Molski, G. K. Mattson, N. J. Lodge, J. J. Bronson and J. E. Macor, Bioorg. Med. Chem. Lett., 2010, 1890–1894 CrossRef CAS PubMed.
- J. A. Deskus, D. D. Dischino, R. J. Mattson, J. L. Ditta, M. F. Parker, D. J. Denhart, D. Zuev, H. Huang, R. A. Hartz, V. T. Ahuja, H. Wong, G. K. Mattson, T. F. Molski, J. E. Grace Jr., L. Zueva, J. M. Nielsen, H. Dulac, Y.-W. Li, M. Guaraldi, M. Azure, D. Onthank, M. Hayes, E. Wexler, J. McDonald, N. J. Lodge, J. J. Bronson and J. E. Macor, Bioorg. Med. Chem. Lett., 2012, 22, 6651–6655 CrossRef CAS PubMed.
- D. J. Denhart, D. Zuev, J. L. Ditta, R. A. Hartz, V. T. Ahuja, R. J. Mattson, H. Huang, G. K. Mattson, L. Zueva, J. M. Nielsen, E. S. Kozlowski, N. J. Lodge, J. J. Bronson and J. E. Macor, Bioorg. Med. Chem. Lett., 2013, 23, 2052–2055 CrossRef CAS PubMed.
- W. R. R. Harker, P. M. Delaney, M. Simms, M. J. Tozer and J. P. A. Harrity, Tetrahedron, 2013, 69, 1546–1552 CrossRef CAS.
- J. Yang, J. Du, C. Huang, T. Wang, L. Huang, S. Yang and L. Li, Bioorg. Med. Chem. Lett., 2019, 29, 1609–1613 CrossRef CAS PubMed.
- H. Masuda, M. Yoshida and T. Shibamoto, J. Agric. Food Chem., 1981, 29, 944–947 CrossRef CAS.
- S. Mihara and H. Masuda, J. Agric. Food Chem., 1990, 38, 1032–1034 CrossRef CAS.
- H. Taguchi, T. Yokoi, F. Kasuya, Y. Nishiyama, M. Fukui and Y. Okada, J. Chem. Soc. Chem. Commun., 1994, 247 RSC.
- Y. Okada, H. Taguchi, Y. Nishiyama and T. Yokoi, Tetrahedron Lett., 1994, 35, 1231–1234 CrossRef CAS.
- H. Taguchi, T. Yokoi, M. Tsukatani and Y. Okada, Tetrahedron, 1995, 51, 7361–7372 CrossRef CAS.
- H. Taguchi, K. Hirano, T. Yokoi, K. Asada and Y. Okada, Chem. Pharm. Bull., 1995, 43, 1336–1339 CrossRef CAS PubMed.
- H. Taguchi, T. Yokoi and Y. Okada, Chem. Pharm. Bull., 1996, 44, 2037–2041 CrossRef CAS.
- Y. Okada, Y. Fujisawa, A. Morishita, K. Shiotani, A. Miyazaki, Y. Fujita, T. Li, Y. Tsuda, T. Yokoi, S. D. Bryant and L. H. Lazarus, Tetrahedron Lett., 2002, 43, 8137–8139 CrossRef CAS.
- Y. Okada, M. Tsukatani, H. Taguchi, T. Yokoi, S. D. Bryant and L. H. Lazarus, Chem. Pharm. Bull., 1998, 46, 1374–1382 CrossRef CAS PubMed.
- Y. Okada, A. Fukumizu, M. Takahashi, T. Yokoi, Y. Tsuda, S. D. Bryant and L. H. Lazarus, Chem. Pharm. Bull., 1999, 47, 1193–1195 CrossRef CAS PubMed.
- Y. Okada, A. Fukumizu, M. Takahashi, J. Yamazaki, T. Yokoi, Y. Tsuda, S. D. Bryant and L. H. Lazarus, Tetrahedron, 1999, 55, 14391–14406 CrossRef CAS.
- Y. Jinsmaa, A. Miyazaki, Y. Fujita, T. Li, Y. Fujisawa, K. Shiotani, Y. Tsuda, T. Yokoi, A. Ambo, Y. Sasaki, S. D. Bryant, L. H. Lazarus and Y. Okada, J. Med. Chem., 2004, 47, 2599–2610 CrossRef CAS PubMed.
- Y. Jinsmaa, Y. Okada, Y. Tsuda, K. Shiotani, Y. Sasaki, A. Ambo, S. D. Bryant and L. H. Lazarus, J. Pharmacol. Exp. Ther., 2004, 309, 432–438 CrossRef CAS.
- T. Li, Y. Fujita, K. Shiotani, A. Miyazaki, Y. Tsuda, A. Ambo, Y. Sasaki, Y. Jinsmaa, E. Marczak, S. D. Bryant, S. Salvadori, L. H. Lazarus and Y. Okada, J. Med. Chem., 2005, 48, 8035–8044 CrossRef CAS PubMed.
- K. Shiotani, T. Li, A. Miyazaki, Y. Tsuda, S. D. Bryant, A. Ambo, Y. Sasaki, L. H. Lazarus and Y. Okada, Bioorg. Med. Chem. Lett., 2006, 16, 5793–5796 CrossRef CAS PubMed.
- Y. Koda, K. Shiotani, I. Toth, Y. Tsuda, Y. Okada and J. T. Blanchfield, Bioorg. Med. Chem. Lett., 2007, 17, 2043–2046 CrossRef CAS PubMed.
- K. Shiotani, A. Miyazaki, T. Li, Y. Tsuda, T. Yokoi, A. Ambo, Y. Sasaki, S. Bryant, Y. Jinsmaa, L. Lazarus and Y. Okada, Med. Chem., 2007, 3, 583–598 CrossRef CAS PubMed.
- K. Shiotani, T. Li, A. Miyazaki, Y. Tsuda, T. Yokoi, A. Ambo, Y. Sasaki, S. D. Bryant, L. H. Lazarus and Y. Okada, Bioorg. Med. Chem. Lett., 2007, 17, 5768–5771 CrossRef CAS PubMed.
- T. Li, Y. Jinsmaa, M. Nedachi, A. Miyazaki, Y. Tsuda, A. Ambo, Y. Sasaki, S. D. Bryant, E. Marczak, Q. Li, H. S. Swartzwelder, L. H. Lazarus and Y. Okada, Bioorg. Med. Chem., 2007, 15, 1237–1251 CrossRef CAS PubMed.
- A. Miyazaki, T. Yokoi, Y. Tachibana, R. Enomoto, E. Lee, G. Bokonyi, G. Keri, Y. Tsuda and Y. Okada, Tetrahedron Lett., 2004, 45, 6323–6327 CrossRef CAS.
- A. Miyazaki, Y. Tsuda, S. Fukushima, T. Yokoi, T. Vántus, G. Bökönyi, E. Szabó, A. Horváth, G. Kéri and Y. Okada, Bioorg. Med. Chem. Lett., 2008, 18, 6199–6201 CrossRef CAS PubMed.
- Y. Okada, H. Taguchi and T. Yokoi, Tetrahedron Lett., 1996, 37, 2249–2252 CrossRef CAS.
- Y. Zeng, Q. Li, R. P. Hanzlik and J. Aubé, Bioorg. Med. Chem. Lett., 2005, 15, 3034–3038 CrossRef CAS PubMed.
- R. Ramesh, M. T. Bovino, Y. Zeng and J. Aubé, J. Org. Chem., 2019, 84, 3647–3651 CrossRef CAS PubMed.
- I. Adam, D. Orain and P. Meier, Synlett, 2004, 2031–2033 CAS.
- H. Matsushita, S. H. Lee, K. Yoshida, B. Clapham, G. Koch, J. Zimmermann and K. D. Janda, Org. Lett., 2004, 6, 4627–4629 CrossRef CAS PubMed.
- A. Basso, L. Banfi, G. Guanti, R. Riva and P. Tosatti, Synlett, 2011, 2009–2012 CrossRef CAS.
- C. Faggi, M. García-Valverde, S. Marcaccini, R. Pepino and M. C. Pozo, Synthesis, 2003, 1553–1558 CAS.
- J. Azuaje, A. El Maatougui, J. M. Pérez-Rubio, A. Coelho, F. Fernández and E. Sotelo, J. Org. Chem., 2013, 78, 4402–4409 CrossRef CAS PubMed.
- J. Azuaje, C. Carbajales, M. González-Gómez, A. Coelho, O. Caamaño, H. Gutiérrez-De-Terán and E. Sotelo, Future Med. Chem., 2015, 7, 1373–1380 CrossRef CAS PubMed.
- J. Wu, L. Zhao, M.-L. Yang and M.-W. Ding, J. Org. Chem., 2021, 86, 10755–10761 CrossRef CAS PubMed.
- L. Zhang, J.-Y. Lee, N. Yamazaki and S. Chiba, Synlett, 2011, 2167–2170 CAS.
- E. M. Becalli and A. Marchesini, Synthesis, 1991, 861–862 CrossRef.
- R. Saijo, K.-I. Kurihara, K. Akira, H. Uno and M. Kawase, Tetrahedron Lett., 2013, 54, 4418–4421 CrossRef CAS.
- R. Saijo, H. Sekiya, E. Tamai, K.-I. Kurihara, J. Maki, H. Sakagami and M. Kawase, Chem. Pharm. Bull., 2017, 65, 365–372 CrossRef CAS PubMed.
- S. Perrone, M. Capua, F. Messa, A. Salomone and L. Troisi, Tetrahedron, 2017, 73, 6193–6198 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |