
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew iron(III) complex of bis-bidentate-anchored diacyl resorcinol on a Fe3O4 nanomagnet: C–H bond oxygenation, oxidative cleavage of alkenes and benzoxazole synthesis †
Mona Majedi a,
Elham Safaei*a and
Sašo Gyergyekb
a,
Elham Safaei*a and
Sašo Gyergyekb
aDepartment of Chemistry, College of Sciences, Shiraz University, Shiraz, 71454, Iran. E-mail: e.safaei@shirazu.ac.ir
bDepartment for Synthesis of Materials, Jožef Stefan Institute, Jamova cesta 39, 1000 Ljubljana, Slovenia
First published on 30th January 2023
Abstract
We have synthesized a novel, bis-bidentate, covalently anchored, 4,6-diacetyl resorcinol (DAR) ligand on silica-coated magnetic Fe3O4 nanoparticles and the corresponding bi-metallic iron(III) complex (Fe3O4@SiO2-APTESFe2LDAR). Both the chemical nature and the structure of the homogeneously heterogenized catalyst were investigated using physico-chemical techniques. The results obtained by XPS, XRD, FT-IR, TGA, VSM, SEM, TEM, EDX, ICP and AAS revealed a magnetic core, a silica layer and the grafting of a binuclear iron complex on the Fe3O4@SiO2-APTES, as well as its thermodynamic stability. Despite many reports of metal complexes on different supports, there are no reports of anchored, bi-metallic complexes. To the best of our knowledge, this is the first report of a bi-active site catalyst covalently attached to a support. This study focuses on the catalytic activity of an as-synthesized, bi-active site catalyst for C–H bond oxygenation, the oxidative cleavage of alkenes, and the multicomponent, one-pot synthesis of benzoxazole derivatives with excellent yields from readily available starting materials. Our results indicated high conversion rates and selectivity under mild reaction conditions and simple separation using a magnetic field. The leaching and recyclability tests of the catalyst were investigated for the above processes, which indicated that all the reactions proceed via a heterogeneous pathway and that the catalyst is recyclable without any tangible loss in catalytic activity for at least 8, 5 and 5 cycles for C–H bond oxygenation, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond cleavage and benzoxazole synthesis, respectively.
C bond cleavage and benzoxazole synthesis, respectively.
Introduction
Schiff base ligands and especially their metal complexes are widely used in materials science.1–8 Nowadays, polynuclear Schiff base-metal complexes are attracting a remarkable level of interest.9,10 Among these materials, the bis-bidentate 4,6-diacetyl resorcinol (DAR) ligand11 and some DAR-based complexes, such as dioxido and oxidomethoxido vanadium(V), have been synthesized and their catalytic activities investigated.12,13Heterogeneous catalysts have emerged as a possible way of overcoming homogeneous catalysts disadvantages.14,15 They were prepared by immobilizing the homogeneous catalysts on the surface of solid carriers.16–21 Among the supports, silica-coated magnetic nanoparticles with thermal stability, simple synthesis and functionalization, easily retrievable with a magnet, and with low toxicity and cost have attracted much attention.22–27
One of the most catalytic oxidation reactions is alkane strong C–H-bond oxidation.28–33 Similar to alkanes, alkenes are some of the most important starting materials for the synthesis of their corresponding carbonyl or carboxylic compounds to epoxides, alcohols, aldehydes, ketones and carboxylic acids34–38 with many applications in agriculture and industry.39–42
Among these carbonyl compounds, adipic acid is a key intermediate in the production of nylon-6,6.43 The production of this class of organic compounds involves efficient CC bond oxidation using stoichiometric oxidants such as O3, KMnO4, RuO2 and OsO4 or second- or third-row transition-metal salts or complexes with NaIO4, TBHP and H2O2 as the oxidant.44–46
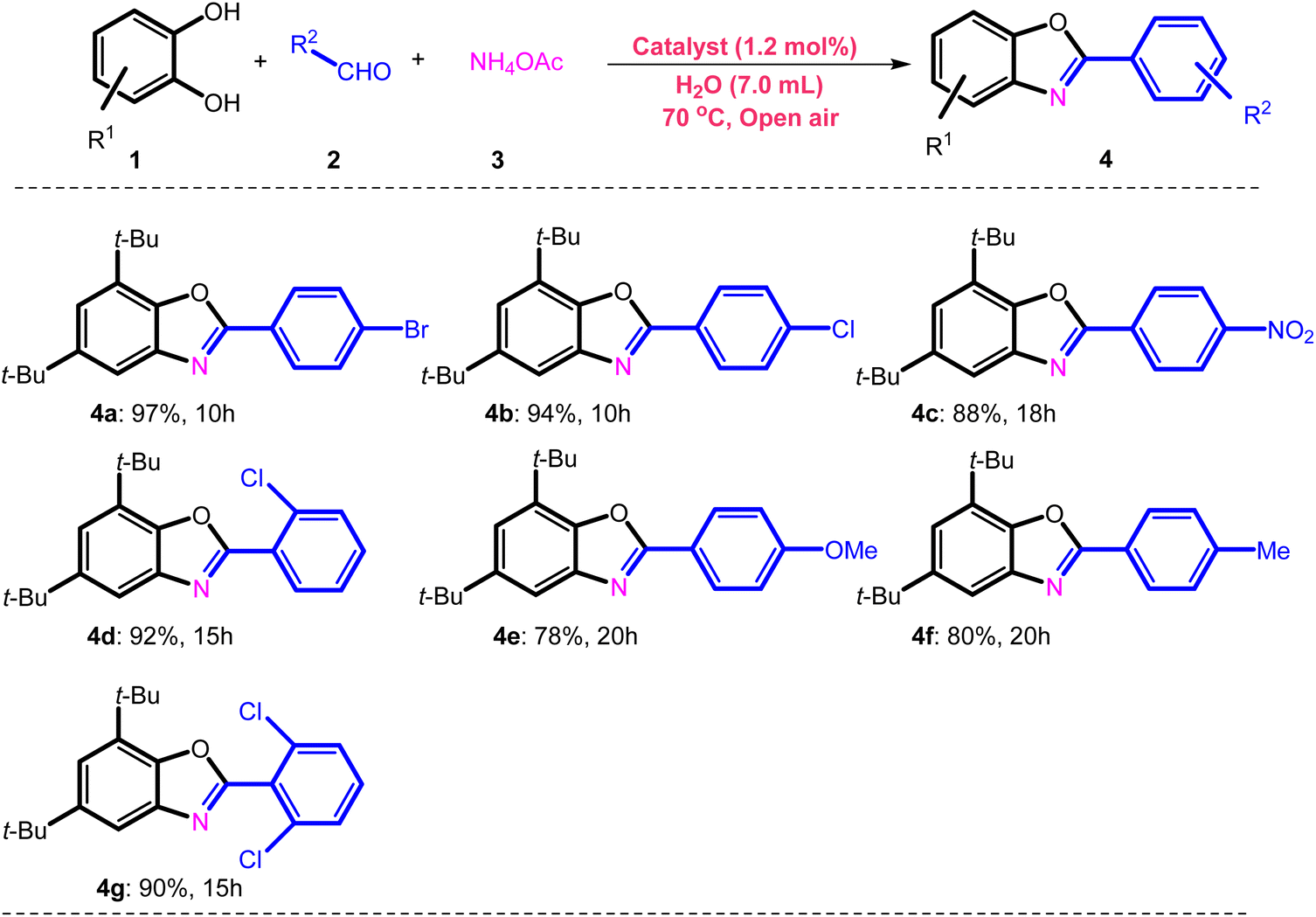
Benzoxazole synthetic reactions (Scheme 1),47 another challenging organic transformation, have generally advanced well, but they are dependent on the consumption of highly poisonous, expensive, corrosive reagents and solvents, non-recyclable catalysts and harsh reaction conditions. These difficulties led to an improvement of catalytic systems, with economical and time-saving one-pot procedures making use of easily available, low-cost materials under mild conditions.48–50
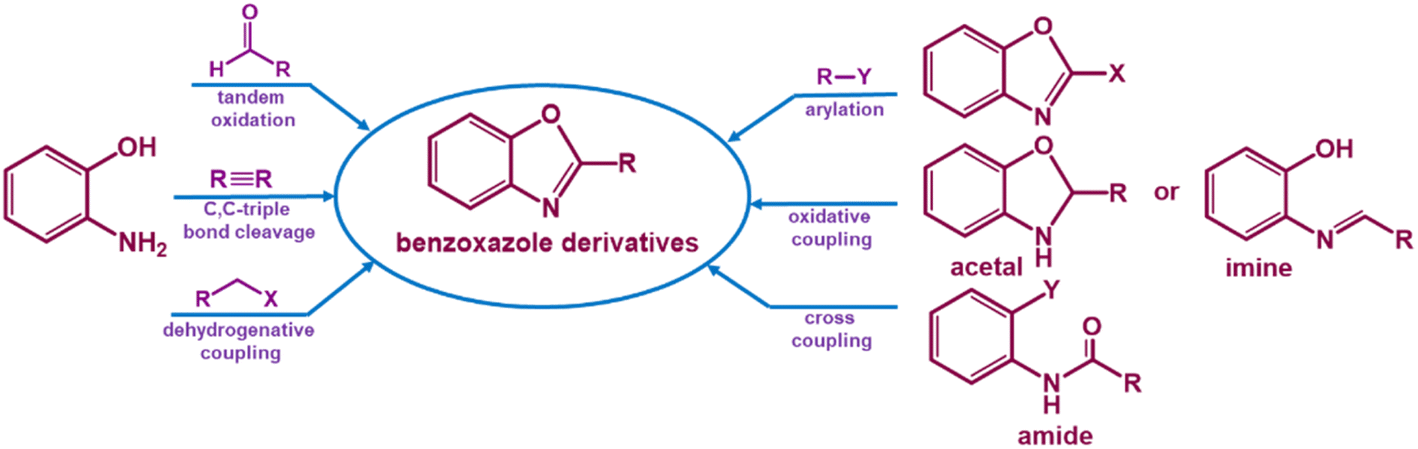 | ||
| Scheme 1 Some reported procedures for synthesizing a benzoxazole ring. | ||
Benzoxazole derivatives were found to have a vast range of pharmacological and biological actions, and several different synthetic procedures were applied to construct benzoxazole derivatives51–57 (Scheme 1).
Here, we applied a 4,6-diacyl resorcinol ligand for covalently anchoring on the silica-coated magnetic nanoparticles to design and fabricate a new bimetallic, homogeneously heterogenized catalyst (Scheme 2) to extend green and tolerable nano-catalysts for the above-mentioned challenging organic transformations under mild conditions.
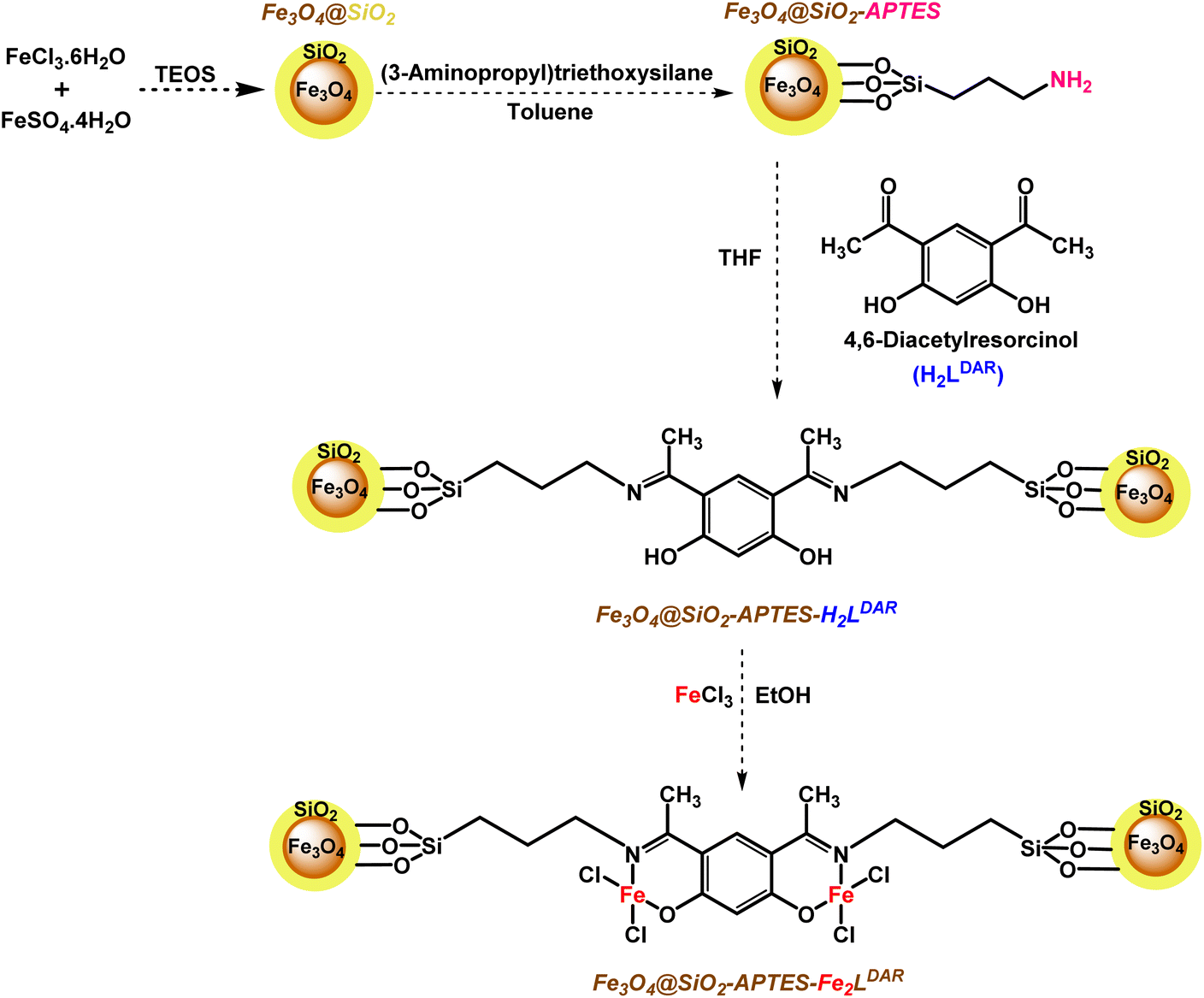 | ||
| Scheme 2 Schematic synthetic pathway for Fe3O4@SiO2-APTES-Fe2LDAR catalyst. | ||
Results and discussion
Synthesis and characterization of Fe3O4@SiO2-APTESFe2LDAR
Our new, bi-active-site catalyst was synthesized by covalently anchoring the 4,6-diacetyl resorcinol ligand on the silica-coated iron oxide nanoparticles (Fe3O4@SiO2-APTESH2LDAR), as illustrated by the pathway in Scheme 2. The reaction was followed by the metalation of Fe3O4@SiO2-APTESH2LDAR by anhydrous FeCl3 (Scheme 2).Fourier-transform infrared spectroscopy was applied to investigate the covalently grafted ligand and its bimetallic complex on the silica-coated magnetic nanoparticles. The peak at 544 cm−1 can be ascribed to the Fe–O stretching vibration, while the peaks at 800–1300 cm−1 are the asymmetric and symmetric Si–O–Si and Si–OH stretching. These suggest the presence of a magnetite core and a silica shell on the MNP surfaces of all the particles. In addition, the signals at 2880, 2930 cm−1 (symmetric and asymmetric stretching, –CH2 group), 3423 cm−1 (stretching vibration, NH bond), and 1480 cm−1 (bending vibration, NH2 and CH2 groups) confirmed that amino propyl groups were effectively anchored on the silica-coated magnetic nanoparticles. After reacting with the FeCl3 salt, the imine peak emerged at 1620 cm−1 by anchoring the ligand on the support, and shifting it to lower frequencies, which confirmed the successful synthesis of the catalyst (Fig. 1).
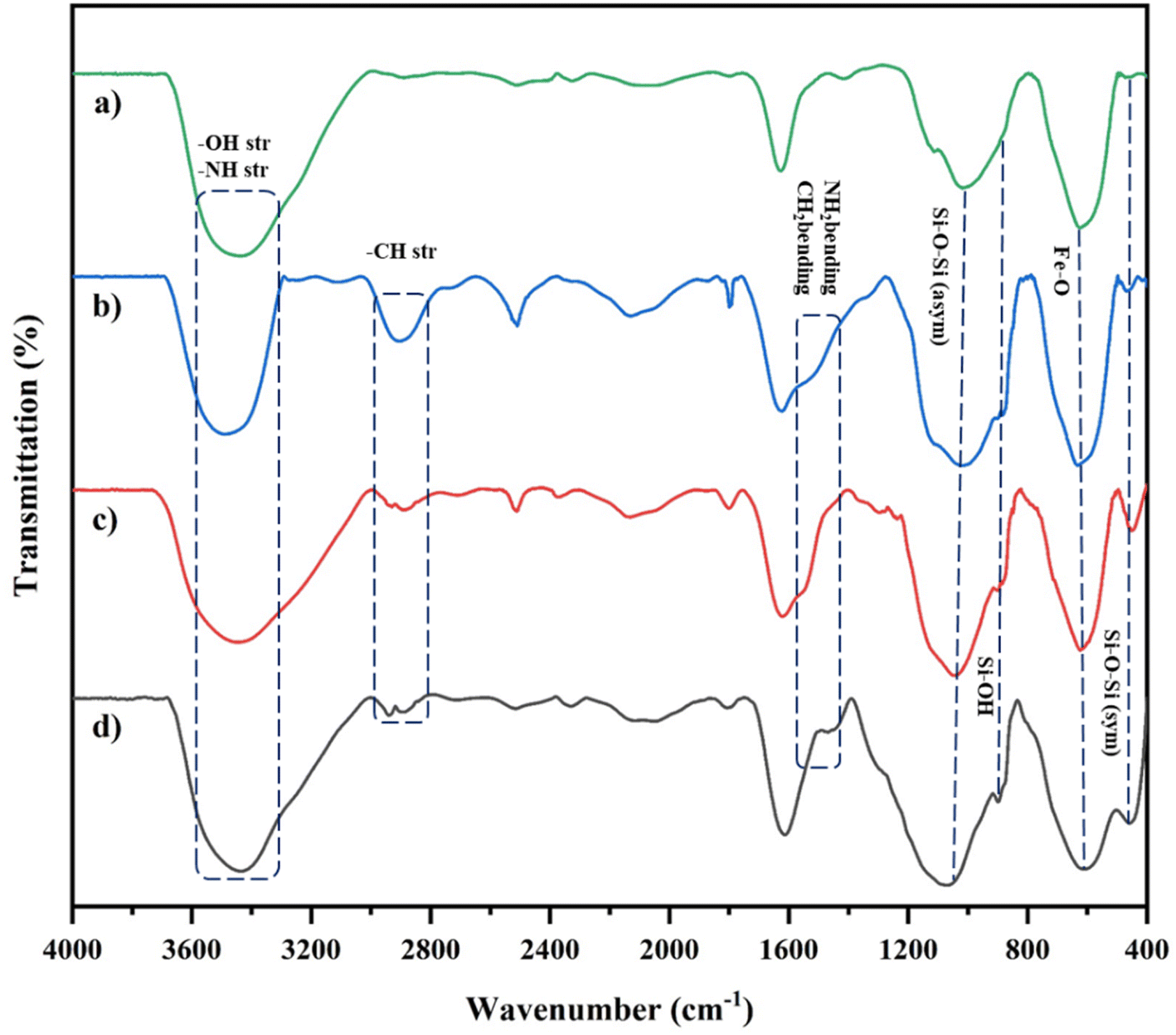 | ||
| Fig. 1 (a) FTIR spectra of Fe3O4@SiO2; (b) Fe3O4@SiO2-APTES; (c) Fe3O4@SiO2-APTES-H2LDAR; (d) Fe3O4@SiO2-APTES-Fe2LDAR. | ||
The morphology, size and structure of the Fe3O4@SiO2-APTESFe2LDAR were investigated by TEM and FE-SEM. As shown in Fig. 2, this catalyst has a smooth surface with a spherical monotonous morphology. According to the FE-SEM data, the average diameter of the synthesized nanoparticles was less than 40 nm (Fig. 2).
 | ||
| Fig. 2 FE-SEM images Fe3O4@SiO2-APTES-Fe2LDAR catalyst. | ||
The energy-dispersive X-ray analysis (EDX) revealed the presence of Fe, Si, C, N and O in the final catalyst. In addition, the amount of loading iron on our catalyst was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) to be 0.3 mmol g−1, which agrees with the EDX result (Fig. S1†). The TEM analysis shows that the iron oxide nanoparticles are well crystallized, and in the range 5–8 nm, coated with a thin, amorphous SiO2 layer (Fig. 3). The EDXS analysis of the amorphous shell (marked with a white arrow in Fig. 3(b)) showed the presence of Si, O and, to a much lesser extent, of Fe. In contrast, the analysis in the area indicated by the black arrow (Fig. 3(b)) had a larger content of Fe (in addition to Si and O).
 | ||
Fig. 3 TEM images of the Fe3O4@SiO2-APTESFe2LDAR catalysts at lower (a) and higher (b) magnifications. The inset in (a) is a selected-area electron-diffraction pattern acquired from the area indicated in (a) and indexed according to the cubic spinel structure (Space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). The black arrow in (b) shows an iron-oxide nanoparticle, while the white arrow shows an amorphous SiO2 layer. m). The black arrow in (b) shows an iron-oxide nanoparticle, while the white arrow shows an amorphous SiO2 layer. | ||
XRD analysis was used to investigate the crystalline structure of the Fe3O4@SiO2-APTESFe2LDAR. Major characteristic peaks were detected at 30.10°, 35.70°, 43.10°, 53.60°, 57.10° and 62.70° corresponding to the (220), (311), (400), (422), (511) and (440) planes, which indicate a cubic inverse spinel structure without impurity phases (Fig. S2†).
The thermal stability of the magnetic nanoparticles (Fe3O4@SiO2-APTESFe2LDAR) was examined using TGA. There was a remarkable mass loss in the three steps shown in the TGA curve of the catalyst (Fig. S3†). The mass loss around 200 °C is probably due to the elimination of organic groups of unbounded aminopropyl part of 3-aminopropyltriethoxysilane or DAR covalently bound to it. An additional mass loss from 250 to 520 °C can be attributed to the decomposition of 3-aminopropyltriethoxysilane. The surface of the silica magnetic nanoparticles was coated with 3-aminopropyltriethoxysilane. The decrease in the mass loss at higher temperature demonstrated the anchoring of the ligand on the support and subsequently the metalation of the nanoparticles. The above result confirmed the thermal stability of the catalyst.
The magnetic properties of Fe3O4@SiO2-APTESFe2LDAR were investigated with a magnetometer. According to Fig. S4,† the hysteresis loop is perfectly reversible, illustrating the superparamagnetic nature of the as-synthesized catalyst. The Fe3O4@SiO2-APTESFe2LDAR catalyst showed a saturation magnetization value of about 13 emu g−1. As a result, the Ms value of the fabricated catalyst (Fe3O4@SiO2-APTESFe2LDAR) is sufficient to ensure its facile retrieval from the solution with a magnet.
To determine the presence of all the elements and evaluate of the iron's valence state in the fabricated catalyst, X-ray photoelectron spectroscopy was employed. The binding-energy levels of 56.37, 717.06 and 730.11 eV are ascribed to Fe 3p, Fe 2p3/2 and Fe 2p1/2, respectively, which match well with the oxidation states +2 and +3 for the Fe ions. The full-range XPS curve illustrated peaks for C 1s, N 1s, O 1s, Si 2s and Si 2p (Fig. 4). Peaks at 108.58 eV and 160.31 eV were also observed, which confirmed the presence of the silica shell in the catalyst (Fig. S5E and F†). Furthermore, peaks related to O 1s, N 1s and C 1s were found at 538.79, 406.85 and 291.21 eV, which showed the successful attachment of the ligand DAR to the silica-coated MNPs (Fig. S5†).
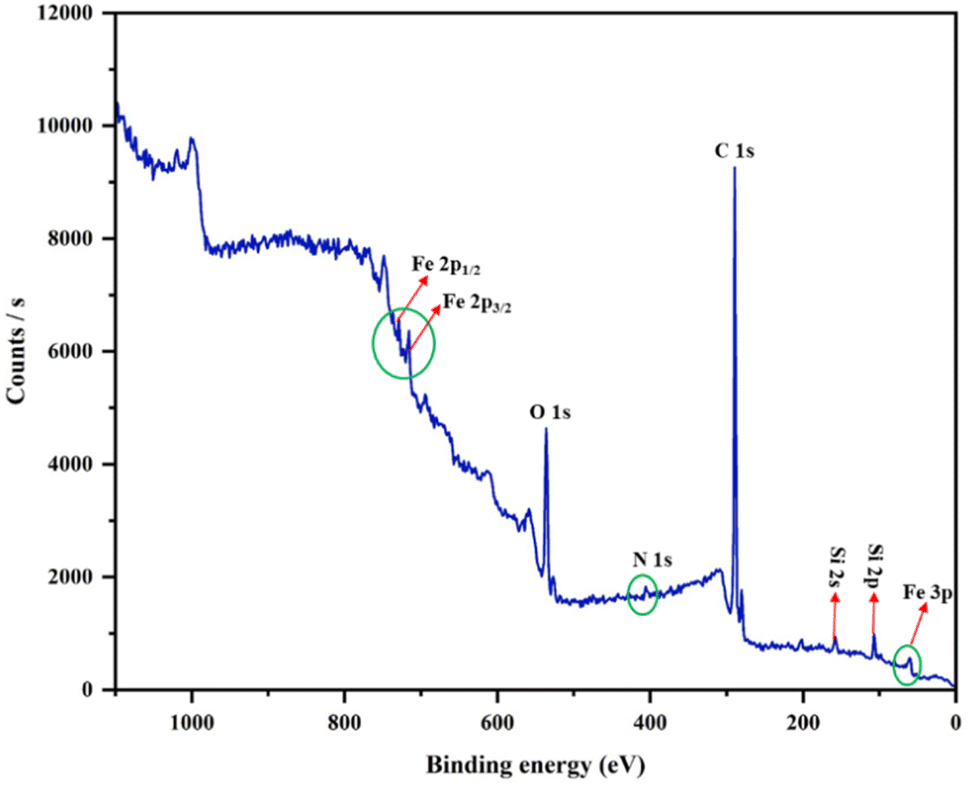 | ||
| Fig. 4 X-ray photoelectron spectroscopy (XPS) spectrum of catalyst (Fe3O4@SiO2-APTES-Fe2LDAR). | ||
Catalytic activities
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | Major product | TBHPb (eq.) | Time (h) | Yieldc (%) | Selectivityd (%) to major product | TONe |
a Reaction conditions: catalyst, substrate (1 mmol), O2 balloon, H2O (1 mL), T = room temperature.b TBHP (70% in H2O).c Conversions were determined by GC using biphenyl as an internal standard (molar ratio of substrate to internal standard is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).d Selectivity% = [(product%)/(products%)] × 100.e TON = [(moles of alkane converted)/(moles of catalyst)].f 40.0 mg ≈ 1.2 mol%.g 30.0 mg ≈ 0.9 mol%. :1).d Selectivity% = [(product%)/(products%)] × 100.e TON = [(moles of alkane converted)/(moles of catalyst)].f 40.0 mg ≈ 1.2 mol%.g 30.0 mg ≈ 0.9 mol%. |
|||||||
| 1f | 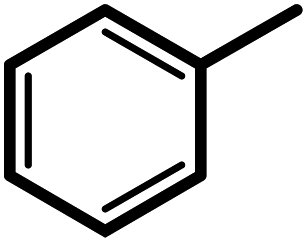 |
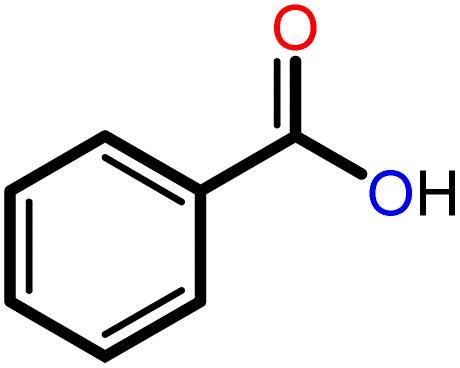 |
3 | 20 | 91 | 100 | 75 |
| 2f | 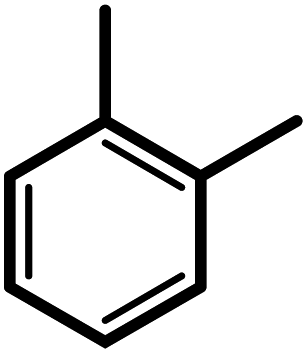 |
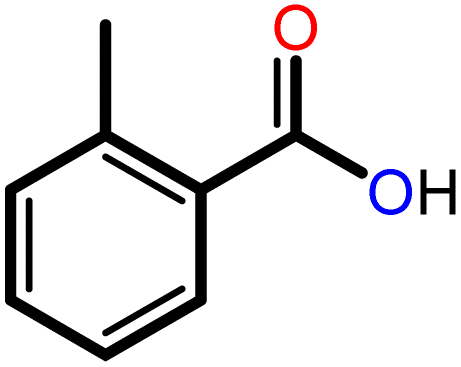 |
3 | 20 | 93 | 100 | 77 |
| 3f | 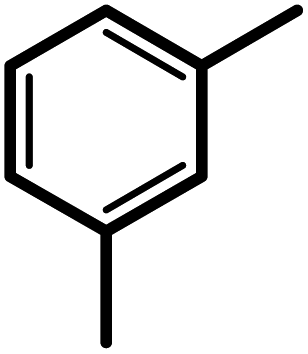 |
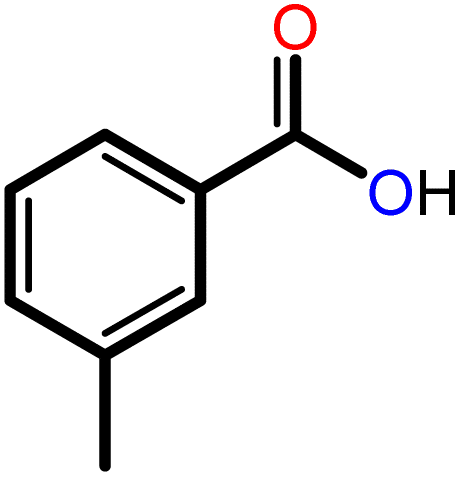 |
3 | 20 | 89 | 100 | 74 |
| 4f | 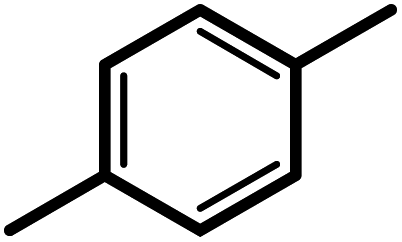 |
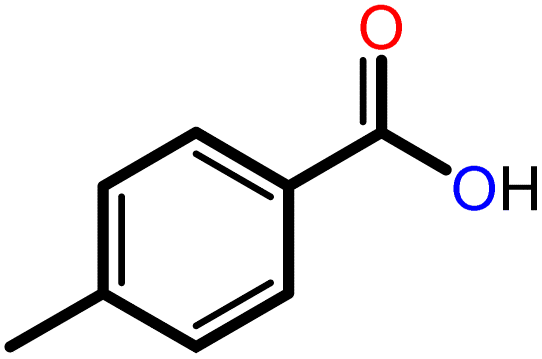 |
3 | 20 | 99 | 100 | 82 |
| 5f |  |
 |
3 | 20 | 100 | 100 | 83 |
| 6f |  |
 |
4 | 20 | 84 | 100 | 70 |
| 7f | 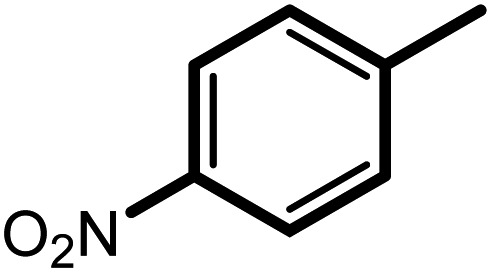 |
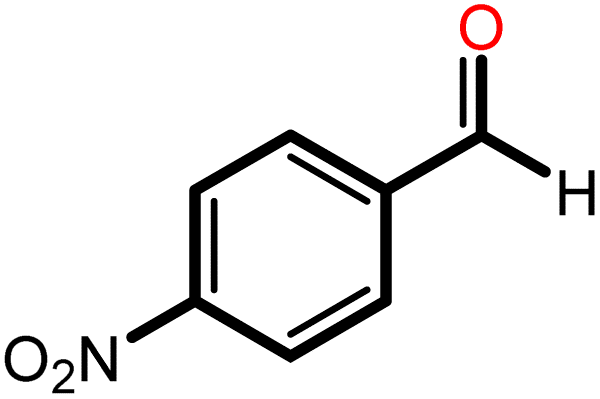 |
4 | 20 | 81 | 100 | 67 |
| 8g | 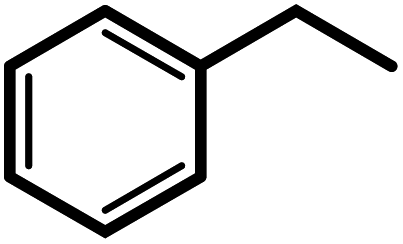 |
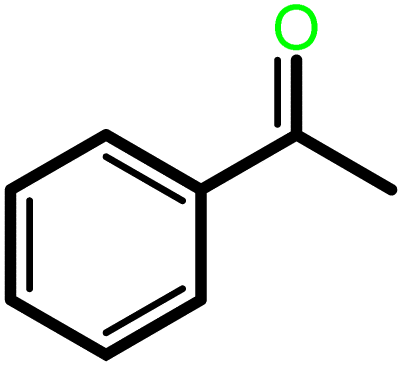 |
— | 15 | 100 | 100 | 111 |
| 9f | 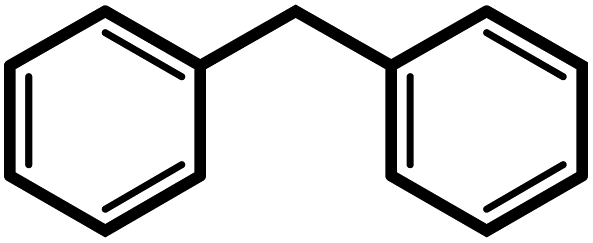 |
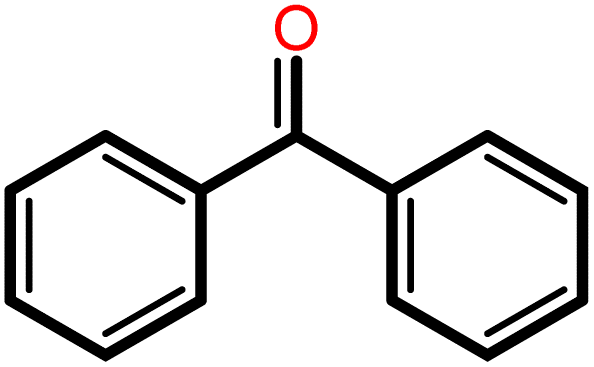 |
3 | 20 | 73 | 100 | 61 |
| 10f | 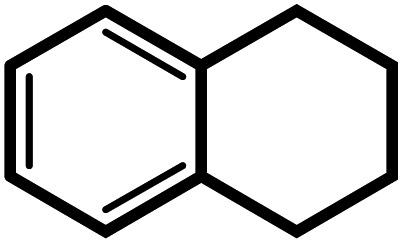 |
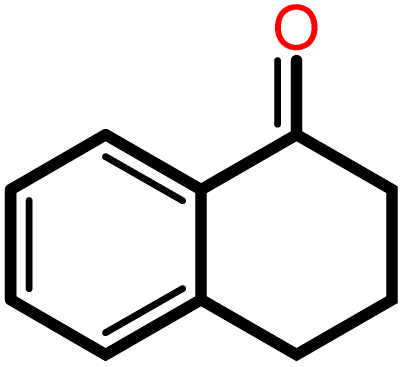 |
3 | 22 | 65 | 82 | 54 |
| 11f |  |
 |
3 | 22 | 87 | 100 | 72 |
We studied the rate vs. [substrate] monitoring of the C–H activation reactions with various concentrations of the substrate. Fig. 5 shows a linear relationship was obtained for the reaction rate versus [S]. Fig. 5 suggests that the reaction includes two steps. In the first step, the rate of reaction is very slow with a small slope (K = 0.001) since O2 as an oxidant reacts with the catalyst to produce a high-valent iron oxo species, which in turn attaches to the substrate with a higher rate (K = 0.01).
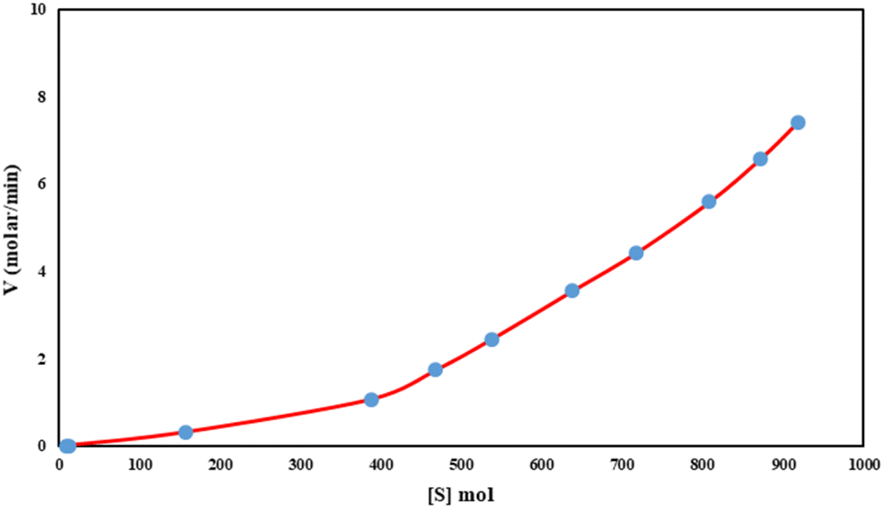 | ||
| Fig. 5 Rate vs. [substrate] monitoring profile of Fe3O4@SiO2-APTES-Fe2LDAR. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) C bond oxidative cleavage. The oxidative cleavage of some alkenes using a bimetallic catalyst has been extensively examined in the literature. First, the oxidative cleavage of 1-hexene as a typical substrate (2 mmol), using O2 as a green oxidant and Na2SO3 as a co-catalyst in the presence of various amounts of the catalyst at ambient temperature, was examined. Of the different amounts of catalyst, 0.050 g had the highest catalytic activity (Table S3,† entry 4). The CC bond oxidation could not be applied without a solvent (entry 9). The conversion could be increased to 94% using CH3CN as a green solvent, which is a better conversion result than with the H2O/acetone mixture (Table S3,† entries 4 and 10). According to Table S3,† different reaction times were also examined. The highest conversion and selectivity were obtained at 15 h (entries 4 and 5). In addition, different amounts of Na2SO3 as a co-catalyst were investigated (Table S3,† entries 4, 6 and 7). The conversion increased from 67% to 94% in the presence of 2 mmol Na2SO3. In the blank test, where there is no catalyst, and in the presence of FeCl3 under the reaction conditions there was no oxidative cleavage of 1-hexene (Table S3,† entry 1 and 14). Also, other nanoparticles such as Fe3O4@SiO2-APTES, Fe3O4@SiO2-APTESH2LDAR under the reaction conditions were much less effective for the CC bond oxidation than the Fe3O4@SiO2-APTESFe2LDAR (Table S3,† entries 11–13). Afterwards, the CC bond oxidation of cyclohexene was examined. This reaction proceeded to result in carbonyl-containing compounds (Table S4†). The use of 0.050 g of catalyst at RT led to the highest activity of cyclohexene to adipic acid with O2 as the oxidant and Na2SO3 as the co-catalyst (Table S4,† entries 2–4). Cyclohexene can be transformed into 87% adipic acid in CH3CN as the best solvent (Table S4,† entries 4 and 8). According to Table S4,† in solvent-free conditions cyclohexene cannot be converted to adipic acid (Table S4,† entries 9–11). As shown in Table S4† (entry 4), 2 mmol of Na2SO3 increased the conversion of adipic acid up to 87% after 24 h. Therefore, the Fe3O4@SiO2-APTESFe2LDAR catalyst can cleave cyclohexene to adipic acid using 0.050 g of catalyst, CH3CN, 2 mmol Na2SO3 in the presence of O2 as oxidant at RT (Table S4,† entry 4). Furthermore, the Fe3O4@SiO2-APTESFe2LDAR catalyst was applicable to the CC bond oxidation of diverse alkenes under these conditions (Table 2). It is worth noting that both epoxide and acid products can be produced after breaking the CC bond in the reaction solution. However, one of these products forms in larger amounts due to the selective performance of the catalyst. The nature of the alkenes is significant in the conversion of the products. Linear terminal alkenes show a remarkably higher activity in this oxidative cleavage than the cyclic alkenes.
C bond oxidative cleavage. The oxidative cleavage of some alkenes using a bimetallic catalyst has been extensively examined in the literature. First, the oxidative cleavage of 1-hexene as a typical substrate (2 mmol), using O2 as a green oxidant and Na2SO3 as a co-catalyst in the presence of various amounts of the catalyst at ambient temperature, was examined. Of the different amounts of catalyst, 0.050 g had the highest catalytic activity (Table S3,† entry 4). The CC bond oxidation could not be applied without a solvent (entry 9). The conversion could be increased to 94% using CH3CN as a green solvent, which is a better conversion result than with the H2O/acetone mixture (Table S3,† entries 4 and 10). According to Table S3,† different reaction times were also examined. The highest conversion and selectivity were obtained at 15 h (entries 4 and 5). In addition, different amounts of Na2SO3 as a co-catalyst were investigated (Table S3,† entries 4, 6 and 7). The conversion increased from 67% to 94% in the presence of 2 mmol Na2SO3. In the blank test, where there is no catalyst, and in the presence of FeCl3 under the reaction conditions there was no oxidative cleavage of 1-hexene (Table S3,† entry 1 and 14). Also, other nanoparticles such as Fe3O4@SiO2-APTES, Fe3O4@SiO2-APTESH2LDAR under the reaction conditions were much less effective for the CC bond oxidation than the Fe3O4@SiO2-APTESFe2LDAR (Table S3,† entries 11–13). Afterwards, the CC bond oxidation of cyclohexene was examined. This reaction proceeded to result in carbonyl-containing compounds (Table S4†). The use of 0.050 g of catalyst at RT led to the highest activity of cyclohexene to adipic acid with O2 as the oxidant and Na2SO3 as the co-catalyst (Table S4,† entries 2–4). Cyclohexene can be transformed into 87% adipic acid in CH3CN as the best solvent (Table S4,† entries 4 and 8). According to Table S4,† in solvent-free conditions cyclohexene cannot be converted to adipic acid (Table S4,† entries 9–11). As shown in Table S4† (entry 4), 2 mmol of Na2SO3 increased the conversion of adipic acid up to 87% after 24 h. Therefore, the Fe3O4@SiO2-APTESFe2LDAR catalyst can cleave cyclohexene to adipic acid using 0.050 g of catalyst, CH3CN, 2 mmol Na2SO3 in the presence of O2 as oxidant at RT (Table S4,† entry 4). Furthermore, the Fe3O4@SiO2-APTESFe2LDAR catalyst was applicable to the CC bond oxidation of diverse alkenes under these conditions (Table 2). It is worth noting that both epoxide and acid products can be produced after breaking the CC bond in the reaction solution. However, one of these products forms in larger amounts due to the selective performance of the catalyst. The nature of the alkenes is significant in the conversion of the products. Linear terminal alkenes show a remarkably higher activity in this oxidative cleavage than the cyclic alkenes.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Major product | Time (h) | Yieldb (%) | Selectivityc (%) to major product | TONd |
| a Reaction conditions: catalyst (50.0 mg, 1.5 mol%), substrate (2 mmol), O2 balloon, H2O/CH3CN (2 mL), Na2SO3 (2 mmol, 0.24 g), T = room temperature.b Isolated yield.c Selectivity% = [(product%)/(products%)] × 100.d TON = [(moles of alkene converted)/(moles of catalyst)]. | ||||||
| 1 |  |
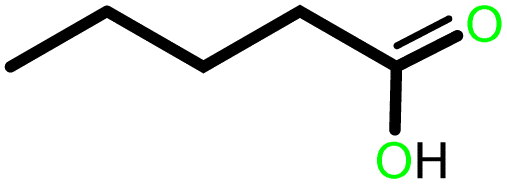 |
15 | 94 | 100 | 62 |
| 2 | 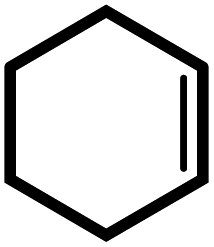 |
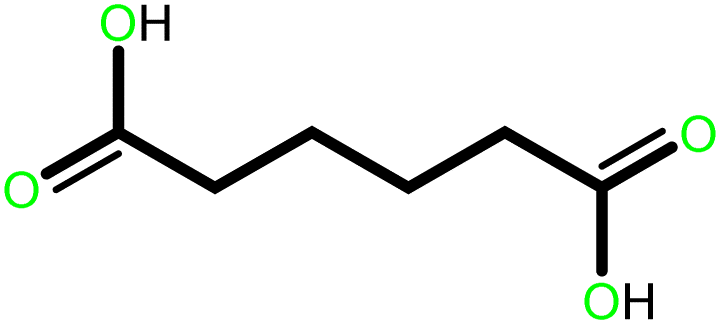 |
24 | 87 | 100 | 58 |
| 3 | 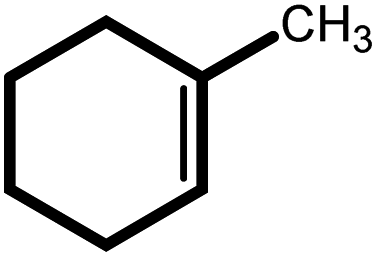 |
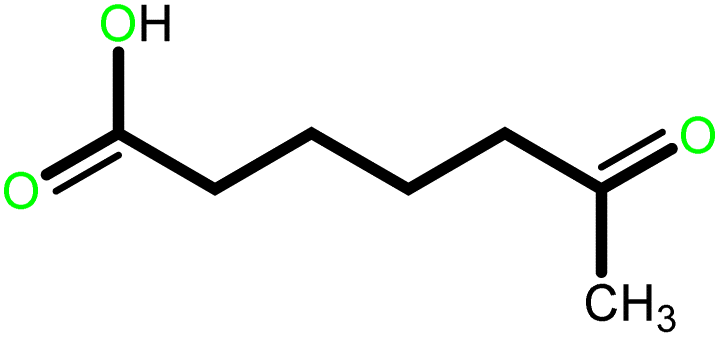 |
24 | 82 | 100 | 54 |
| 4 |  |
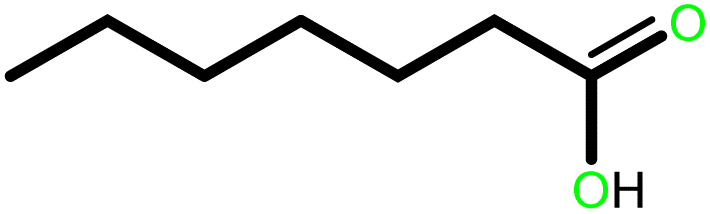 |
15 | 92 | 100 | 61 |
| 5 | 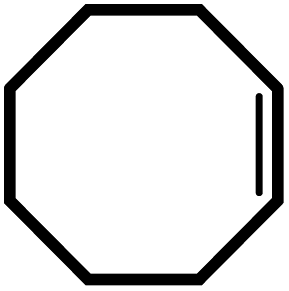 |
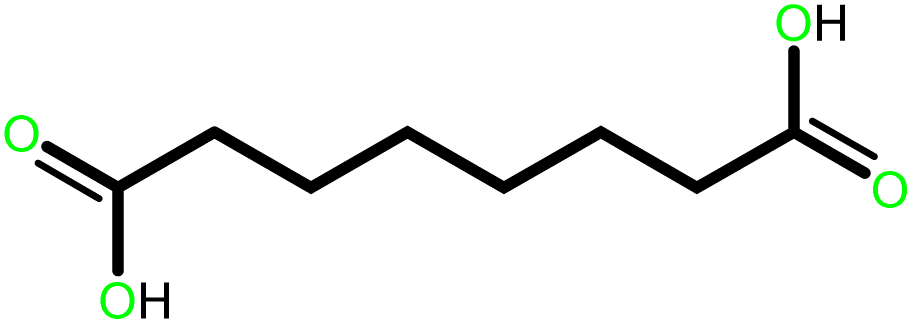 |
20 | 90 | 100 | 60 |
| 6 | 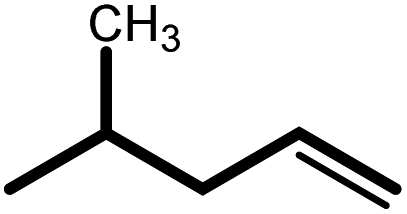 |
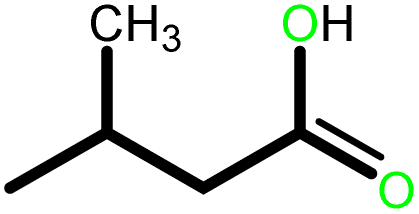 |
18 | 90 | 100 | 60 |
| 7 | 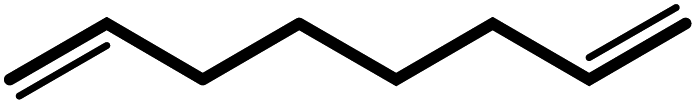 |
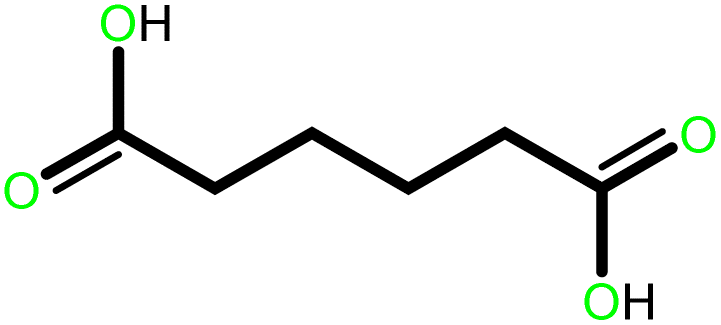 |
24 | 91 | 100 | 60 |
| 8 | 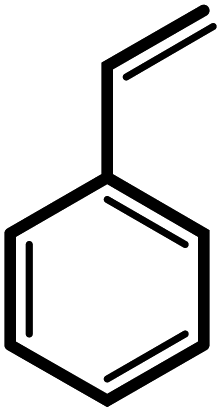 |
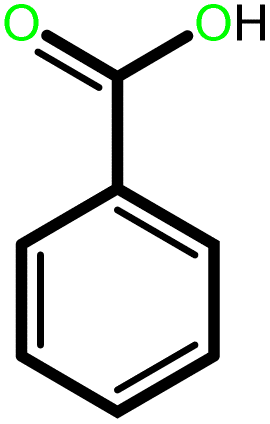 |
24 | 92 | 100 | 61 |
| 9 | 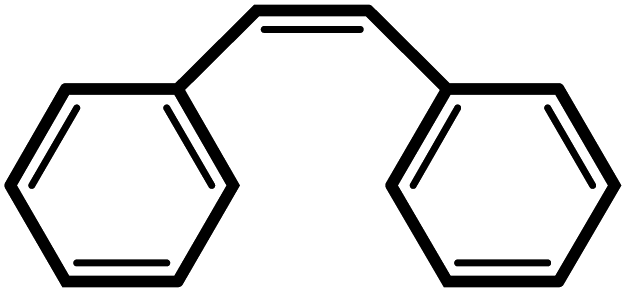 |
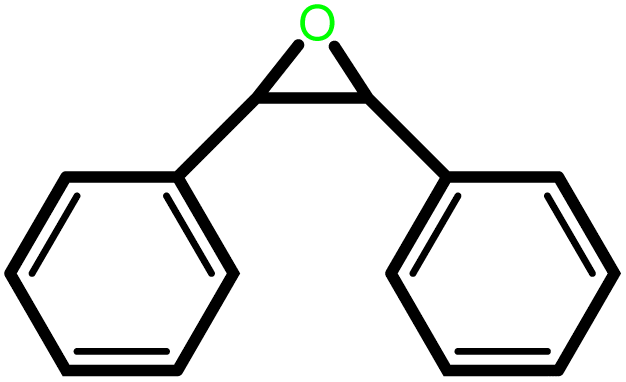 |
24 | 93 | 100 | 62 |
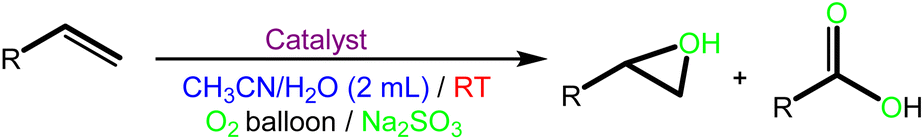
| a Reaction conditions: Fe3O4@SiO2-APTES-Fe2LDAR (40.0 mg, 1.2 mol%), 3,5-di-tert-butylbenzene-1,2-diol (0.5 mmol), NH4OAc (0.5 mmol) and aldehyde (0.5 mmol), H2O (7 mL), 70 °C and reflux. |
|---|
 |
Recyclability of the catalyst and the hot-filtration test
Among the diverse properties of a catalyst, recyclability is the most fundamental one as this relates to the performance and economics of a process from the industrial viewpoint. So, to prove the recyclability of our catalyst, after each cycle the catalyst was collected from the solution using a magnet, rinsed with acetone and dried under vacuum. Fig. 6a shows that the catalyst was used for eight consecutive runs without a tangible decline in the product yield. In addition, to examine the possibility of leaching the catalyst, Sheldon's hot-filtration test was applied for the C–H bond oxidation reaction under optimized conditions. During the C–H bond oxidation reaction of ethylbenzene, the catalyst was magnetically eliminated from the solution after 7.5 h. After this the reaction was continued without the catalyst for 7.5 h (Table 4). Finally, the reaction was stopped and monitored by GC to characterize the product yield, which rejected leaching and proved that the catalyst proceeded the reaction pathway. After completing the CC bond oxidation of 1-hexene under these conditions, the Fe3O4@SiO2-APTESFe2LDAR catalyst could be easily retrieved from the solution with a magnet. No significant leaching of the Fe ions was observed using atomic absorption spectroscopy (AAS). Also, the retrieved Fe3O4@SiO2-APTESFe2LDAR catalyst without a considerable change in the conversion and selectivity could be reused for a fifth run (Fig. 6c). Furthermore, for the benzoxazole synthesis 3,5-di-tert-butylbenzene-1,2-diol (0.5 mmol), 4-bromobenzaldehyde (0.5 mmol) and NH4OAc (0.5 mmol), the data obtained using atomic absorption spectroscopy (AAS) confirmed that the leached amount of Fe after the completion of the reaction was very low and that the nature of the catalyst for these reactions is heterogeneous. It is noteworthy that the retrieved Fe3O4@SiO2-APTESFe2LDAR catalyst had a comparable activity even after the fifth run in the benzoxazoles synthesis (Fig. 6b).
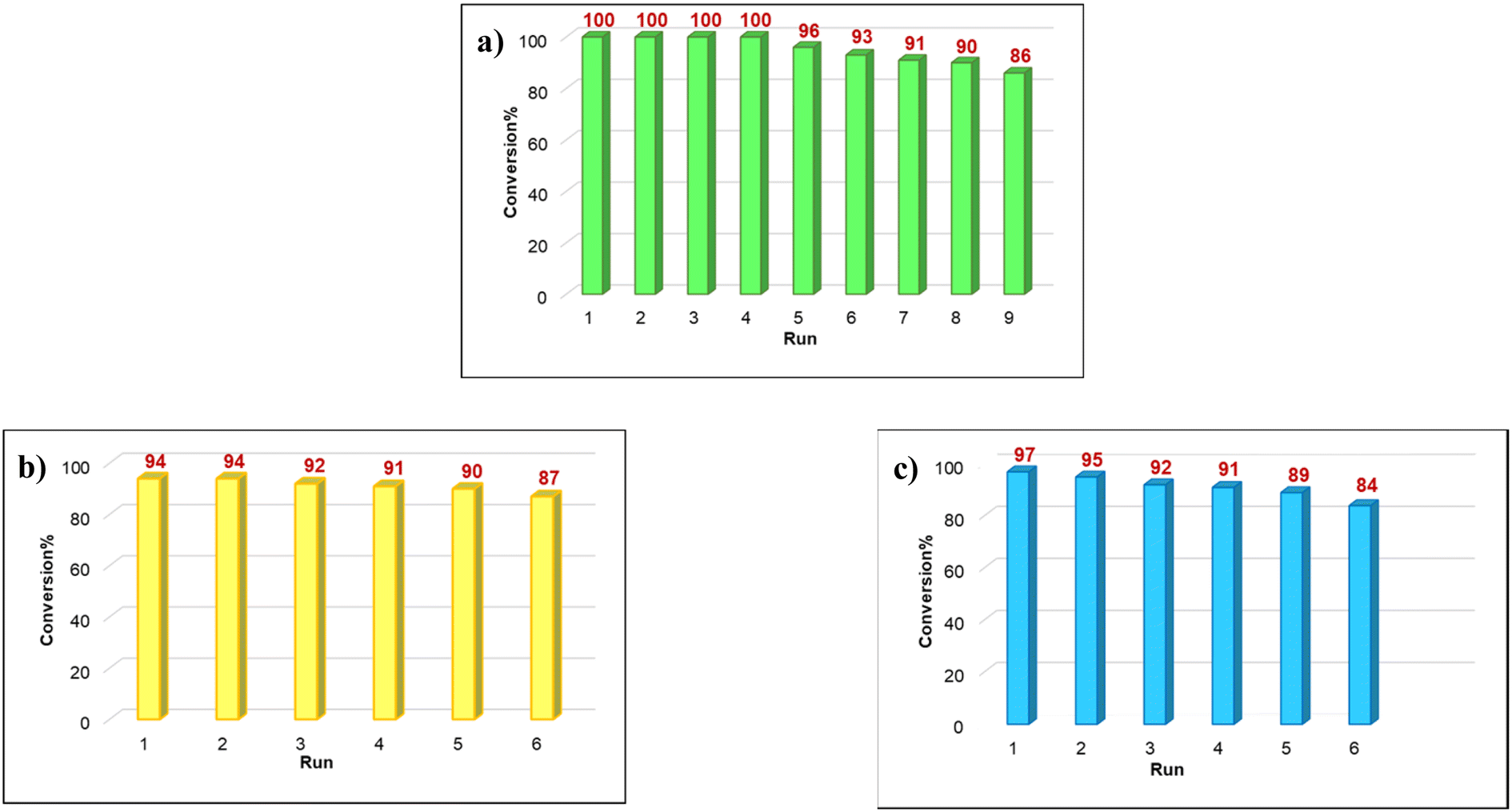 | ||
| Fig. 6 (a) Recycling of catalyst in the oxidation of alkanes; (b) oxidative cleavage of alkenes; (c) reaction synthesis of benzoxazoles. | ||
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst type | Catalyst amount (mg) | Timeb (h) | Temp. (°C) | Conv.c (%) |
| a Reaction conditions: catalyst (30.0 mg ≈ 0.9 mol%), substrate (1 mmol), O2 balloon, H2O (1 mL), T = room temperature.b Hot filtration test.c Conversions were determined by GC using biphenyl as an internal standard (molar ratio of substrate to internal standard is 1:1). |
|||||
| 1 | Fe3O4@SiO2APTESFe2LDAR | 30.0 | 7.5 (before filtering) | 25 | 27 |
| 2 | Fe3O4@SiO2APTESFe2LDAR | 30.0 | 7.5 (after filtering) | 25 | 34 |
| 3 | Fe3O4@SiO2APTESFe2LDAR | 30.0 | 15 | 25 | 100 |
The C–H/CC bond oxidation reactions and the benzoxazoles synthesis using heterogeneous bimetallic catalysts under mild conditions are rare. Therefore, we compared our outcomes with some other catalyst systems (Table 5). This showed that Fe3O4@SiO2-APTESFe2LDAR has excellent applicability in both the C–H/CC bond oxidation reactions and the synthesis of benzoxazoles.
| Entry | Catalyst | Reaction conditions | Conv. (%) | Major product |
|---|---|---|---|---|
| C–H bond oxygenation | ||||
| 1 (ref. 60) | Fe3O4MNPs (10 mol%) | Ethylbenzene (1 mmol), TBHP (70% aq., 0.5 mmol), T = 120 °C, time = 3 h | 93 | Acetophenone |
| 2 (ref. 61) | HKUST-1@Cu–CuFe2O4 (15.0 mg) | Ethylbenzene (1 mmol), CH3CN (5 mL), N-hydroxyphthalimide (10 mol%), T = 60 °C, 1 atm O2, time = 9 h | 93 | Acetophenone |
| 3 (ref. 62) | Mn(OH)x/γ-Al2O3 (0.03 mmol) | Ethylbenzene (40 mmol), O2 flow (5 mL min−1), benzoic acid (0.4 mmol), T = 135 °C, time = 24 h | 74 | Acetophenone |
| 4 (ref. 63) | FeVO4/g-C3N4 (50.0 mg) nanocomposites | Ethylbenzene (10 mmol), CH3CN (6 mL), H2O2 (25 mmol), T = 60 °C time = 4 h | 91 | Acetophenone |
| 5 (ref. 64) | Mn–N–C@SiO2, (30 mg) | Ethylbenzene (10 mL), O2 (0.8 MPa), T = 120 °C time = 5 h | 12 | Acetophenone |
| 6 (ref. 65) | Ruthenium complex (0.004 mmol) | Ethylbenzene (4 mmol), TBHP (12 mmol), T = 40 °C time = 26 h | 82 | Acetophenone |
| 7 (ref. 66) | Mn@SiO2/Al2O3, mixed oxide (50.0 mg) | Ethylbenzene (1.14 mL, 2 mmol), TBHP 80% (1.34 mL, 2 mmol), T = 80 °C time = 24 h | 67 | Acetophenone |
| 8 this work | Fe3O4@SiO2-APTES-Fe2LDAR (30.0 mg, 0.9 mol%) | Ethylbenzene (1 mmol), H2O (1 mL), RT, time = 15 h, O2 | 100 | Acetophenone |
|
||||
| CC bond oxidative cleavage |
||||
| 9 (ref. 67) | SiRu(H2O)W11O39 ((C6H13)4N)5 (0.2 mol%) | Cyclohexene (1 mmol), 5 equiv. KHSO5, 1,2-dichloroethane/H2O 1:1, time = 4 h, T = 60 °C |
67 | Adipic acid |
| 10 (ref. 68) | WO(O2)2L22− (L = oxalic acid), (1 mol%) | Cyclohexene (0.3 mmol), 30% H2O2 (134 mL), time = 24 h, T = 94 °C | 96 | Adipic acid |
| 11 (ref. 69) | Cu-salen-PPh3, (8 mmol) | Cyclohexene (1 mmol), 2.6 equiv. 30% H2O2, H3PO4, [Emim]PF6 CH2Cl2 (1:1), time = 3 h |
96 | Cyclohexen oxide |
| 12 (ref. 70) | C-PMAn-V, (27–38 wt%) (30.0 mg) | Cyclohexene (1 mmol), 8 equiv. TBHP, H2O, time = 32 h, T = 105 °C | 67 | Adipic acid |
| 13 (ref. 71) | POM on oxide supports (50 wt%) | Cyclohexene (1 mmol), 3–4 equiv. 30% H2O2, 2-methylpropan-2-ol, time = 24 h, T = 60 °C | 90 | Adipic acid |
| 14 (ref. 72) | Ru on charcoal, (1 mol%) | Cyclohexene (1 mmol), 1.1 equiv. NaIO4, MeCN:EtOAc:H2O 1:1:1 (v/v), time = 7.5 h |
42 | Adipic acid |
| 15 this work | Fe3O4@SiO2-APTES-Fe2LDAR (50.0 mg, 1.5 mol%) | Cyclohexene (2 mmol), H2O (2 mL), NaSO3 (2 mmol), RT, time = 24 h, O2 | 87 | Adipic acid |
|
||||
| Benzoxazole synthesis | ||||
| 16 (ref. 73) | (FeLAPIP)2O (1.2 mol%) | Bromo aminophenol (1 mmol), TBHP (70%, 0.4 mL), H2O (5 mL), time = 8 h | 83 | 4,6-Di-tert-butyl-2-(4-boromophenyl)-1H-benzo[d]oxazole |
| 17 (ref. 74) | Fe(III)-salen (3 mol%) | 3-Bromobenzaldehyde (1 mmol), EtOH (5 mL), T = 50 °C, time = 5 h | 80 | 4,6-Di-tert-butyl-2-(3-boromophenyl)-1H-benzo[d]oxazole |
| 18 (ref. 75) | RuCl3 | 4-Bromobenzaldehyde (1 mmol), [bmim]BF4 1 mL, T = 80 °C, time = 10 h | 78 | 4,6-Di-tert-butyl-2-(4-boromophenyl)-1H-benzo[d]oxazole |
| 19 this work | Fe3O4@SiO2-APTES-Fe2LDAR (40.0 mg, 1.2 mol%) | 4-Bromobenzaldehyde (1 mmol), H2O (7 mL), T = 70 °C, time = 10 h | 97 | 4,6-Di-tert-butyl-2-(4-boromophenyl)-1H-benzo[d]oxazole |
Proposed mechanisms
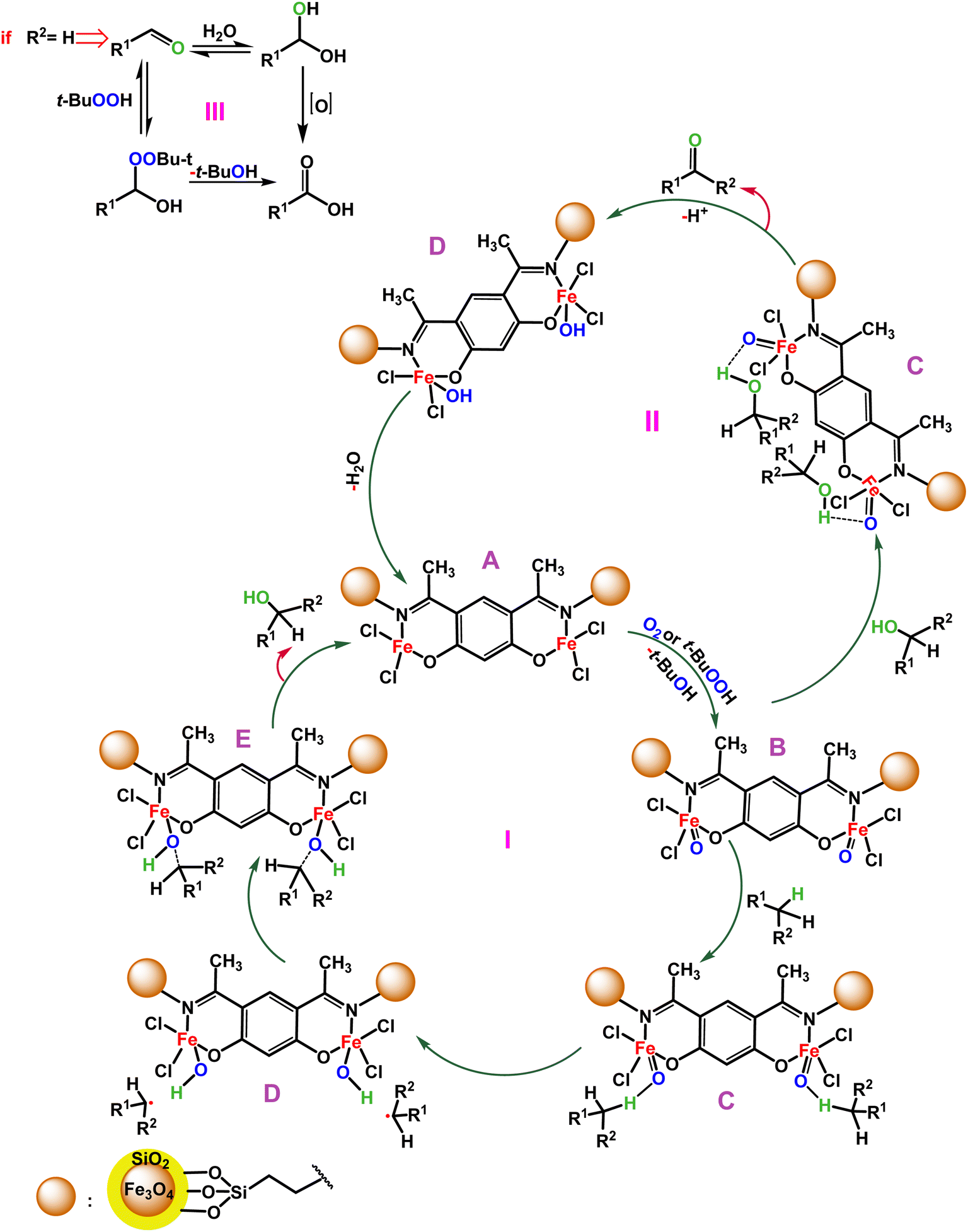 | ||
| Scheme 3 Proposed mechanism for C–H bond oxygenation of the Fe3O4@SiO2-APTES-Fe2LDAR catalyst to different products (I) alcohol, (II) ketone, (III) acid. | ||
C bond oxidative cleavage. The possible oxidation mechanism of the CC bond in the presence of the Fe3O4@SiO2-APTES-Fe2LDAR catalyst involves forming a high-valent iron-oxo species too. According to Scheme 4, SO32− reduces the Fe(III) to Fe(II), which is very stimulated to react with the alkene. In the following, we postulate that the oxidation proceeds by an initial coordination of alkene to the Fe(II) centre of the Fe3O4@SiO2-APTESFe2LDAR catalyst (B). Then the reaction is followed by an interaction with molecular O2 to produce the superoxo radical (C). Afterwards, the intermediate (C) undergoes cyclization with the alkene (D). Then dioxetane (E) was produced during the resulting peroxo metallacycle decomposition. It disintegrated to produce the benzaldehyde,80 the further oxidation of which produces benzoic acid (Scheme 4).
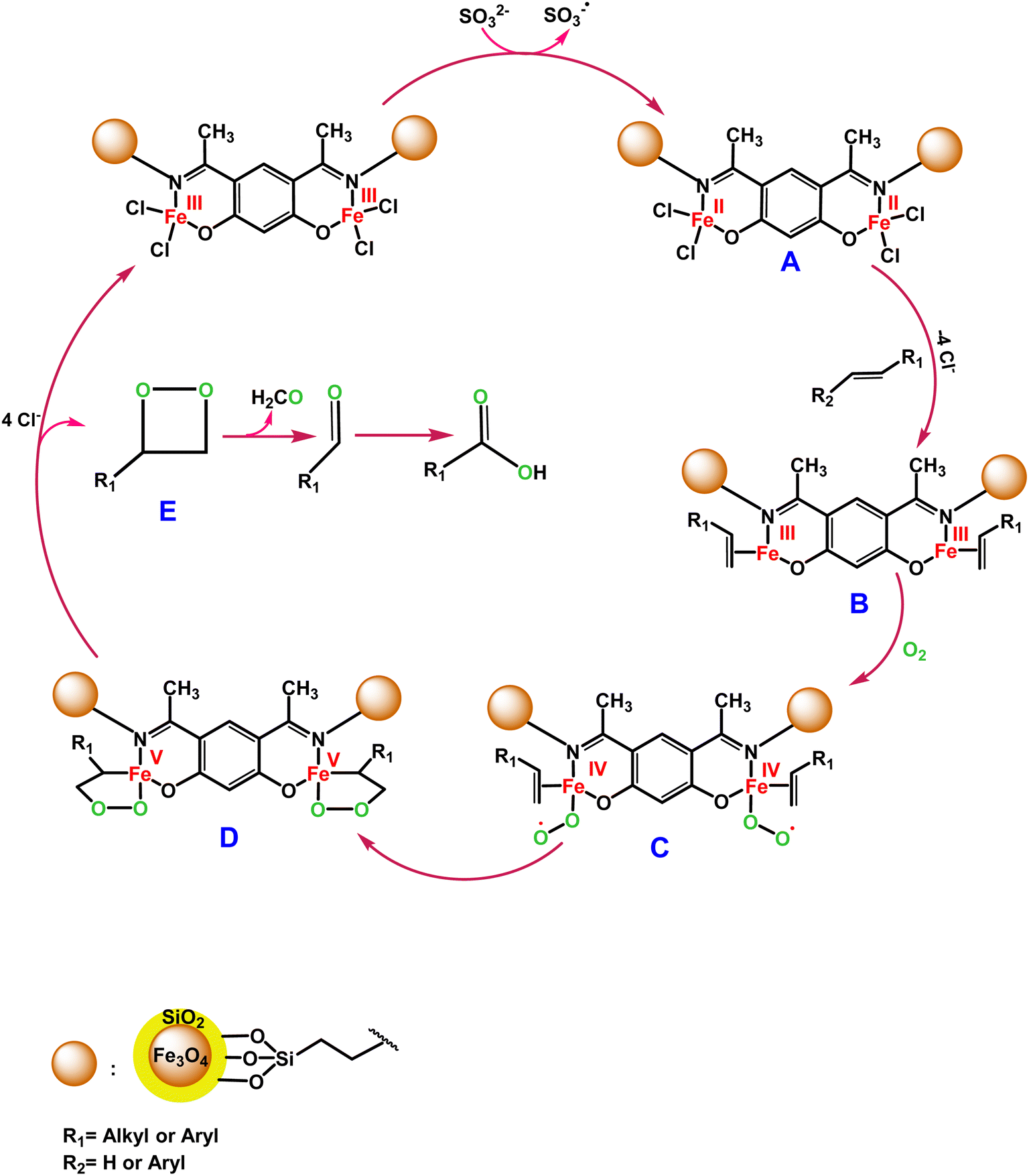 | ||
| Scheme 4 Proposed mechanism for CC bond oxidative cleavage of the Fe3O4@SiO2-APTES-Fe2LDAR catalyst. | ||
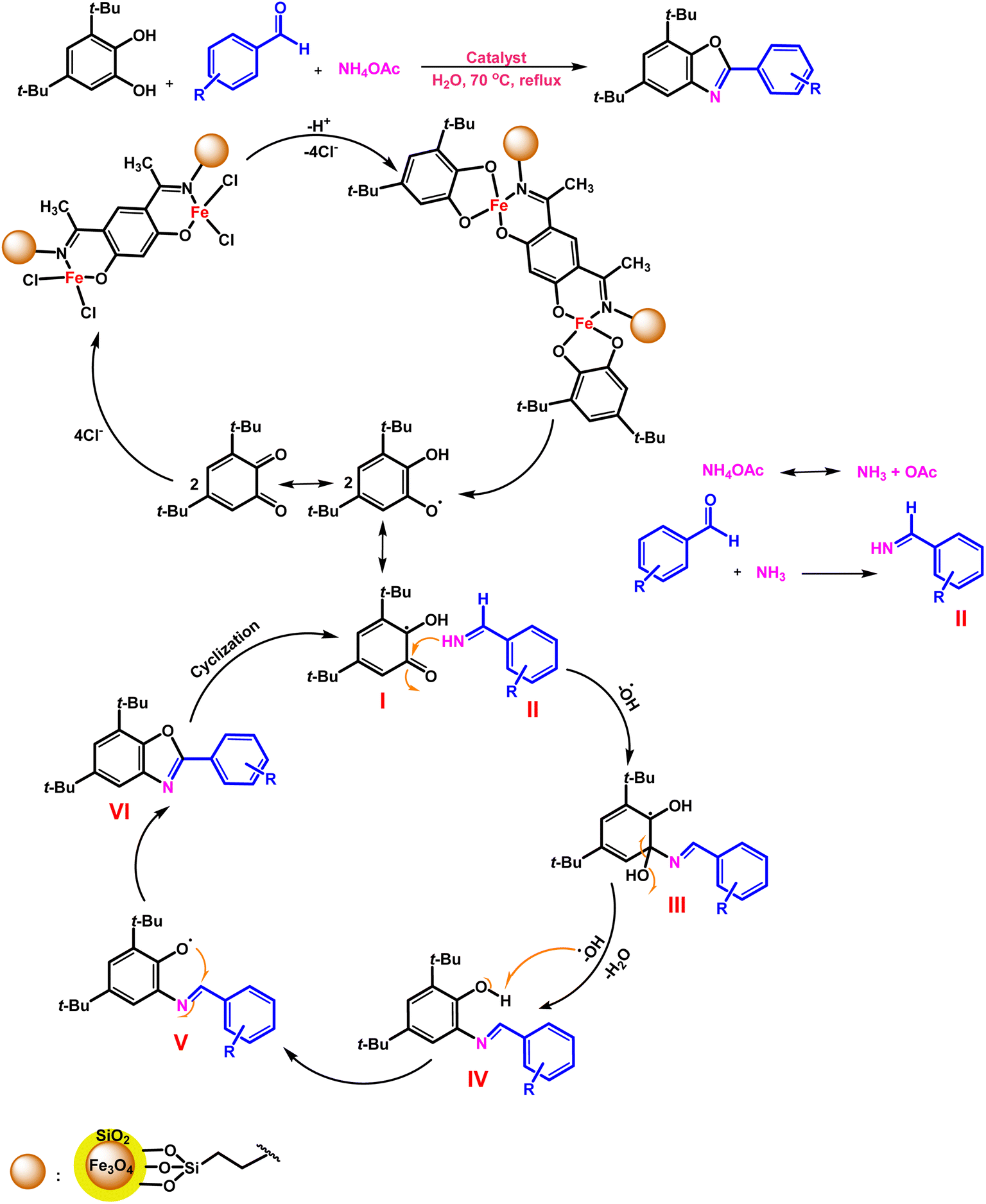 | ||
| Scheme 5 Proposed mechanism for benzoxazoles synthesis reaction of the Fe3O4@SiO2-APTES-Fe2LDAR catalyst. | ||
Conclusion
For the first time the synthesis and characterization of a homogeneously heterogenized binuclear iron(III) complex of covalently anchored 4,6-diacetyl resorcinol on silica-coated magnetic nanoparticles (Fe3O4@SiO2-APTESFe2LDAR) is reported. Moreover, the catalytic application of this catalyst for C–H and CC bond oxidation of a wide range of alkanes and alkenes was investigated. In addition, a one-pot multicomponent reaction for the synthesis of benzoxazole derivatives from 3,5-di-tert-butylbenzene-1,2-diol, diverse aldehydes and ammonium acetate as the nitrogen source was performed. The as-synthesized bimetallic (or better as the bi-active catalyst) displays remarkable catalytic activities under mild reaction conditions and in water. Based on our knowledge, the C–H oxidation of alkanes is mostly performed under the harsh reactions of high temperature and organic solvents, and iron complexes usually do not show good reactivity for this class of organic reactions unless harsh conditions are applied. Furthermore, excellent conversion and selectivity in CC bond oxidation is observed with the addition of an appropriate amount of Na2SO3 as the co-catalyst in comparison to the mentioned bimetallic catalyst. An analysis of the products shows that except in one case, some oxidative cleavages of the cyclic and terminal alkenes was observed. Among them, producing adipic acid and some other bifunctional carboxylic acids are important in terms of polymerization. Importantly, the recyclability and reusability of this bimetallic catalyst remained after the eighth, fifth and fifth consecutive runs for C–H, CC bond oxidation and in the benzoxazole synthesis, respectively, without any tangible loss in catalytic activities. Moreover, the leaching tests in the C–H/CC bond oxidation and the benzoxazole synthesis process confirmed that the binuclear complex was immobilized tightly to the magnetite nanoparticles and the reactions proceed via a heterogeneous pathway. The designing of a bi-ferric catalyst and the dramatic effect of having two active sites in one structure on the catalyst function makes this a credible candidate for green chemistry.
Experimental
Materials and methods
All chemical reagents and solvents were supplied by Acros Organics, Sigma Aldrich, Fluka and Merck and used as received. The FT-IR spectra of nanoparticles were recorded in the range 400–4000 cm−1 in the solid state on a Shimadzu FT-IR 8300 spectrophotometer. Scanning electron microscopy (SEM) (JEOL, JSM-5600) was used to observe the morphology of the catalyst. Iron oxide nanoparticles and silica coating were observed by transmission electron microscopy (TEM) (JEOL JEM 2100), while elemental composition was determined using energy-dispersive X-ray spectroscopy (EDXS) (JEOL Jed 2300). The X-ray diffraction (XRD) pattern was captured by a PHILLIPS PW1730 (using step size of 0.05 and time per step of 1 s). Using a PerkinElmer 240-B microanalyzer, the elemental analysis (C, H and N) was accomplished. The hysteresis curve was measured with a vibrating-sample magnetometer at RT from −15000 to +15000 Oe. Thermal gravimetric analyses (TGA) were recorded under nitrogen on Netzsch STA 409 PC/PG instrument with a heating rate of 20 °C min−1 up to 800 °C. To measure the electronic state of the catalyst elements, an XPS multilab 2000 with Al-Kα radiation was used. The EDX analysis was obtained with a Tescan Mira (II) at an acceleration voltage of 15 kV. The content of Fe in the catalyst was calculated using an inductively coupled plasma-optical emission spectrometer (ICP-OES). The mixture reactions were monitored by thin-layer chromatography (TLC) and gas chromatography (GC, Agilent gas chromatograph model 7890 A) using a flame-ionization detector. The 1H NMR spectra were recorded with a 250 MHz Bruker Avance instrument in CDCl3 or DMSO-d6 as solvents and tetramethyl silane (TMS) as internal standard.Synthesis of Fe3O4@SiO2-APTES
The magnetic Fe3O4 MNPs, silica-coated nanoparticles (Fe3O4·SiO2) and the functionalized (Fe3O4@SiO2-APTES) form were acquired via the reported method.84Synthesis of 4,6-diacetylresorcinol covalently anchored on nanomagnet (Fe3O4@SiO2-APTESH2LDAR) and its iron(III) complex (Fe3O4@SiO2-APTESFe2LDAR)
Fe3O4@SiO2-APTESH2LDAR was prepared by adding 4,6-diacetylresorcinol (4.37 mmol, 0.85 g) to a suspension containing Fe3O4@SiO2-APTES and dry THF (25 mL) with vigorous stirring for 4 days at room temperature. Afterwards the solid product was gathered with a magnet and rinsed with ethanol, CH2Cl2, acetone and dried under vacuum at 70 °C for 24 h.The modified MNPs (Fe3O4@SiO2-APTES-H2LDAR) were scattered in absolute ethanol (20 mL), then triethyl amine (0.5 mmol, 0.050 g) was slowly added to the solution after 45 min stirring at room temperature, FeCl3 (0.5 mmol, 0.082 g) was added to the resulting mixture, then was again stirred for 4 days at room temperature. Finally, the brown product was gathered using a magnetic field and rinsed with EtOH, CH2Cl2, acetone and dried in a vacuum oven at 80 °C for 24 hours (Fe3O4@SiO2-APTESFe2LDAR).
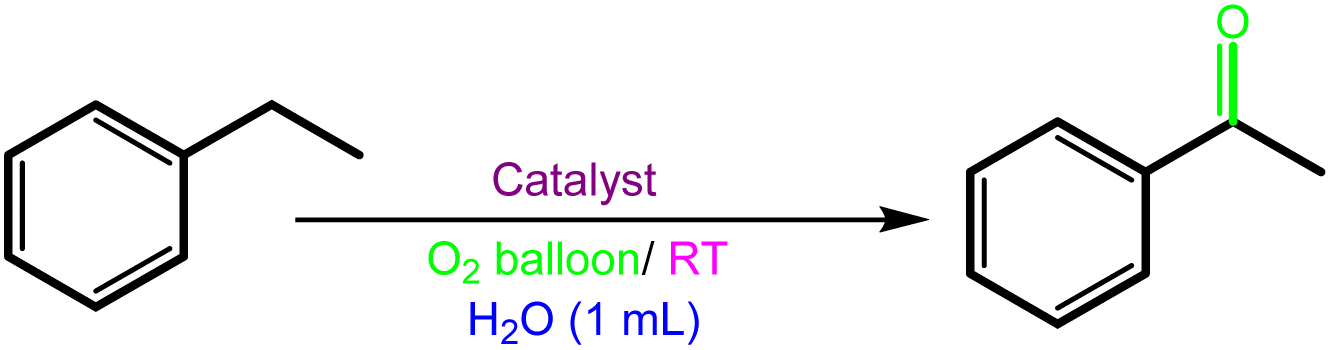
The C–H bond oxidation procedure
To a 10 mL flask containing a mixture of H2O (1 mL, saturated with O2 for 15 minutes), TBHP (2–4 eq) as the oxidant and substrate (1 mmol), Fe3O4@SiO2-APTESFe2LDAR (0.040 g, 1.2 mol%) was added and the mixture was stirred in the presence of O2 (from a balloon) at RT. The reaction's progress was followed by TLC. At the end, the mixture was elicited with ethyl acetate then the catalyst recycled with a magnetic field. Finally, the products were characterized via gas chromatography and 1H NMR spectroscopy.The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C bond oxidative cleavage procedure
C bond oxidative cleavage procedure
To a 10 mL flask containing a mixture of CH3CN (2 mL, saturated with O2 for 15 minutes), Na2SO3 (2 mmol) as the co-catalyst and substrate (2 mmol) was added Fe3O4@SiO2-APTESFe2LDAR (0.050 g, 1.5 mol%) and the mixture was stirred in the presence of O2 (from a balloon) at RT. Finally, the aforementioned mixture was elicited with ethyl acetate and the catalyst recycled with a magnetic field. At the end, the products were characterized by 1H NMR spectroscopy.
The synthesis of benzoxazoles procedure
3,5-di-tert-butylbenzene-1,2-diol (0.5 mmol), aldehyde (0.5 mmol), NH4OAc (0.5 mmol, 0.05 g) in H2O (7 mL) and catalyst (0.040 g, 1.2 mol%) were added to a 15 mL round-bottom flask. Then, the mixture was stirred at 70 °C magnetically under refluxing. The reaction's progress was followed by TLC. Finally, the mixture was elicited with ethyl acetate then the catalyst recycled using a magnetic field. Utilizing a mixture of n-hexane/EtOAc as an eluent, the residue was purified by column chromatography on silica to arrive at the pure product. Finally, the products were characterized by 1H NMR spectroscopy.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
The authors are grateful to the Department of Chemistry, Shiraz University for support. The authors disclosed receipt of the following support for the research of this article: this work was supported by the Iran High-Tech Laboratory Network [grant number: 29810 LabsNet]. Sašo Gyergyek acknowledge the financial support from the Slovenian Research Agency (research core funding No. P2–0089). The authors also acknowledge the Centre of Excellence in Nanoscience and Nanotechnology – Nanocenter for the use of the Jeol JEM-2100 transmission electron microscope.References
- W. Al Zoubi and Y. G. Ko, J. Organomet. Chem., 2016, 822, 173–188 CrossRef CAS.
- A. Erxleben, Inorg. Chim. Acta, 2018, 472, 40–57 CrossRef CAS.
- M. T. Kaczmarek, M. Zabiszak, M. Nowak and R. Jastrzab, Coord. Chem. Rev., 2018, 370, 42–54 CrossRef CAS.
- L. Lecarme, L. Chiang, J. Moutet, N. Leconte, C. Philouze, O. Jarjayes, T. Storr and F. Thomas, Dalton Trans., 2016, 45, 16325–16334 RSC.
- X. Ran, L. Wang, Y. Lin, J. Hao and D. Cao, Appl. Organomet. Chem., 2010, 24, 741–747 CrossRef CAS.
- S. Rana, J. P. Biswas, A. Sen, M. Clémancey, G. Blondin, J.-M. Latour, G. Rajaraman and D. Maiti, Chem. Sci., 2018, 9, 7843–7858 RSC.
- H. L. Singh, Res. Chem. Intermed., 2011, 37, 1087–1101 CrossRef CAS.
- M. More, P. Joshi, Y. Mishra and P. Khanna, Mater. Today Chem., 2019, 14, 100195 CrossRef CAS PubMed.
- A. Nishinaga, H. Ohara, H. Tomita and T. Matsuura, Tetrahedron Lett., 1983, 24, 213–216 CrossRef CAS.
- A. P. Vieira, C. A. Wegermann and A. M. D. C. Ferreira, New J. Chem., 2018, 42, 13169–13179 RSC.
- A. A. Emara and A. A. Abou-Hussen, Spectrochim. Acta, Part A, 2006, 64, 1010–1024 CrossRef PubMed.
- M. R. Maurya, N. Jangra, F. Avecilla and I. Correia, Eur. J. Inorg. Chem., 2019, 2019, 314–329 CrossRef CAS.
- M. Sutradhar, M. A. Andrade, S. A. Carabineiro, L. M. Martins, M. d. F. C. Guedes da Silva and A. J. Pombeiro, Nanomater., 2021, 11, 1456 CrossRef CAS PubMed.
- P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410–421 RSC.
- E. M. McGarrigle and D. G. Gilheany, Chem. Rev., 2005, 105, 1563–1602 CrossRef CAS PubMed.
- M. Lashanizadegan and Z. Zareian, Catal. Lett., 2011, 141, 1698–1702 CrossRef CAS.
- S. K. Maiti, S. Dinda, M. Nandi, A. Bhaumik and R. Bhattacharyya, J. Mol. Catal. A: Chem., 2008, 287, 135–141 CrossRef CAS.
- M. R. Maurya, A. K. Chandrakar and S. Chand, J. Mol. Catal. A: Chem., 2007, 274, 192–201 CrossRef CAS.
- S. More, S. Jadhav, R. Salunkhe and A. Kumbhar, Mol. Catal., 2017, 442, 126–132 CrossRef CAS.
- R. Ratti, S. Kaur, M. Vaultier and V. Singh, Catal. Commun., 2010, 11, 503–507 CrossRef CAS.
- K. B. Sidhpuria, A. L. Daniel-da-Silva, T. Trindade and J. A. Coutinho, Green Chem., 2011, 13, 340–349 RSC.
- G. Chouhan, D. Wang and H. Alper, Chem. Commun., 2007, 4809–4811 RSC.
- T. Hara, T. Kaneta, K. Mori, T. Mitsudome, T. Mizugaki, K. Ebitani and K. Kaneda, Green Chem., 2007, 9, 1246–1251 RSC.
- V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J.-M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS PubMed.
- V. Polshettiwar and R. S. Varma, Green Chem., 2010, 12, 743–754 RSC.
- E. Rafiee and S. Eavani, Green Chem., 2011, 13, 2116–2122 RSC.
- M. A. Zolfigol, V. Khakyzadeh, A. R. Moosavi-Zare, A. Rostami, A. Zare, N. Iranpoor, M. H. Beyzavi and R. Luque, Green Chem., 2013, 15, 2132–2140 RSC.
- W. A. Duetz, J. B. Van Beilen and B. Witholt, Curr. Opin. Biotechnol., 2001, 12, 419–425 CrossRef CAS PubMed.
- N. A. M. Fadzil, M. H. A. Rahim and G. P. Maniam, Chin. J. Catal., 2014, 35, 1641–1652 CrossRef CAS.
- F. Löbermann, P. Mayer and D. Trauner, Angew. Chem., Int. Ed., 2010, 49, 6199–6202 CrossRef PubMed.
- N. Viswanadham, S. K. Saxena and H. Ala'a, Mater. Today Chem., 2017, 3, 37–48 CrossRef.
- C. Zhu, J.-X. Liang, Y. Meng, J. Lin and Z. Cao, Chin. J. Catal., 2021, 42, 1030–1039 CrossRef CAS.
- P. Mohammadpour and E. Safaei, RSC Adv., 2020, 10, 23543–23553 RSC Synfacts, 2020, 16(09), 1088..
- M. Sankaralingam, M. Balamurugan and M. Palaniandavar, Coord. Chem. Rev., 2020, 403, 213085 CrossRef.
- M. Mirzaee, B. Bahramian and M. Mirebrahimi, Chin. J. Catal., 2016, 37, 1263–1274 CrossRef CAS.
- P. Spannring, P. C. Bruijnincx, B. M. Weckhuysen and R. J. K. Gebbink, Catal. Sci. Technol., 2014, 4, 2182–2209 RSC.
- Y. Tao, O. De Luca, B. Singh, A. J. Kamphuis, J. Chen, P. Rudolf and P. P. Pescarmona, Mater. Today Chem., 2020, 18, 100373 CrossRef CAS.
- G. Urgoitia, R. SanMartin, M. T. Herrero and E. Domínguez, ACS Catal., 2017, 7, 3050–3060 CrossRef CAS.
- C. Lin and L. Shen, ChemCatChem, 2019, 11, 961–968 CrossRef CAS.
- D. B. Miklos, C. Remy, M. Jekel, K. G. Linden, J. E. Drewes and U. Hübner, Water Res., 2018, 139, 118–131 CrossRef CAS PubMed.
- D. Saini, R. Aggarwal, S. Anand, N. Satrawala, R. Joshi and S. Sonkar, Mater. Today Chem., 2020, 16, 100256 CrossRef CAS.
- D. Wang, J. R. Gardinier and S. V. Lindeman, Dalton Trans., 2019, 48, 14478–14489 RSC.
- H. Jiang, H. Gong, Z. Yang, X. Zhang and Z. Sun, React. Kinet. Catal. Lett., 2002, 75, 315–321 CrossRef CAS.
- P. Daw, R. Petakamsetty, A. Sarbajna, S. Laha, R. Ramapanicker and J. K. Bera, J. Am. Chem. Soc., 2014, 136, 13987–13990 CrossRef CAS PubMed.
- R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977–1986 RSC.
- R. Willand-Charnley, T. J. Fisher, B. M. Johnson and P. H. Dussault, Org. Lett., 2012, 14, 2242–2245 CrossRef CAS PubMed.
- R. Sattar, R. Mukhtar, M. Atif, M. Hasnain and A. Irfan, J. Heterocycl. Chem., 2020, 57, 2079–2107 CrossRef CAS.
- B. Jiang, T. Rajale, W. Wever, S. J. Tu and G. Li, Chem.–Asian J., 2010, 5, 2318–2335 CrossRef CAS PubMed.
- F. Bahrami, F. Panahi, F. Daneshgar, R. Yousefi, M. B. Shahsavani and A. Khalafi-Nezhad, RSC Adv., 2016, 6, 5915–5924 RSC.
- M. M. Heravi, E. Hashemi, Y. S. Beheshtiha, K. Kamjou, M. Toolabi and N. Hosseintash, J. Mol. Catal. A: Chem., 2014, 392, 173–180 CrossRef CAS.
- S. K. Alla, P. Sadhu and T. Punniyamurthy, J. Org. Chem., 2014, 79, 7502–7511 CrossRef CAS PubMed.
- N. Khatun, S. Guin, S. K. Rout and B. K. Patel, RSC Adv., 2014, 4, 10770–10778 RSC.
- A. R. Tiwari and B. M. Bhanage, Org. Biomol. Chem., 2016, 14, 7920–7926 RSC.
- L. Wang, Z.-G. Ma, X.-J. Wei, Q.-Y. Meng, D.-T. Yang, S.-F. Du, Z.-F. Chen, L.-Z. Wu and Q. Liu, Green Chem., 2014, 16, 3752–3757 RSC.
- H.-Z. Xie, Q. Gao, Y. Liang, H.-S. Wang and Y.-M. Pan, Green Chem., 2014, 16, 2132–2135 RSC.
- D. Yang, X. Zhu, W. Wei, N. Sun, L. Yuan, M. Jiang, J. You and H. Wang, RSC Adv., 2014, 4, 17832–17839 RSC.
- F. Zhu, J.-L. Tao and Z.-X. Wang, Org. Lett., 2015, 17, 4926–4929 CrossRef CAS PubMed.
- S. Ojala, N. Koivikko, T. Laitinen, A. Mouammine, P. K. Seelam, S. Laassiri, K. Ainassaari, R. Brahmi and R. L. Keiski, Catalysts, 2015, 5, 1092–1151 CrossRef CAS.
- S. Scirè and L. F. Liotta, Appl. Catal., B, 2012, 125, 222–246 CrossRef.
- M. Zarghani and B. Akhlaghinia, RSC Adv., 2016, 6, 38592–38601 RSC.
- S. Fan, W. Dong, X. Huang, H. Gao, J. Wang, Z. Jin, J. Tang and G. Wang, ACS Catal., 2017, 7, 243–249 CrossRef CAS.
- M. Arshadi and M. Ghiaci, Appl. Catal., A, 2011, 399, 75–86 CrossRef CAS.
- Y. Kuwahara, Y. Yoshimura and H. Yamashita, Catal. Sci. Technol., 2016, 6, 442–448 RSC.
- S. Samanta and R. Srivastava, Appl. Catal., B, 2017, 218, 621–636 CrossRef CAS.
- Y. Zhang, L. Zhao, H. Zhang, R. Huang and J. Zhao, Appl. Organomet. Chem., 2017, 31, e3709 CrossRef.
- L. Fu, S. Zhao, Y. Chen and Z. Liu, Chem. Commun., 2016, 52, 5577–5580 RSC.
- R. Neumann and C. Abu-Gnim, J. Am. Chem. Soc., 1990, 112, 6025–6031 CrossRef CAS.
- Y. Deng, Z. Ma, K. Wang and J. Chen, Green Chem., 1999, 1, 275–276 RSC.
- R. Dileep and B. Rudresha, RSC Adv., 2015, 5, 65870–65873 RSC.
- Z. Saedi, S. Tangestaninejad, M. Moghadam, V. Mirkhani and I. Mohammadpoor-Baltork, Catal. Commun., 2012, 17, 18–22 CrossRef CAS.
- S. Jarupinthusophon, U. Thong-In and W. Chavasiri, J. Mol. Catal. A: Chem., 2007, 270, 289–294 CrossRef CAS.
- V. Kumar, V. P. Reddy, R. Sridhar, B. Srinivas and K. R. Rao, Synlett, 2009, 2009, 739–742 CrossRef.
- E. Safaei, Z. Alaji, F. Panahi, A. Wojtczak and J. Z. Jagličić, New J. Chem., 2018, 42, 7230–7236 RSC.
- H. Sharghi, J. Aboonajmi and M. Aberi, J. Org. Chem., 2020, 85, 6567–6577 CrossRef CAS PubMed.
- X. Fan, Y. He, Y. Wang, X. Zhang and J. Wang, Chin. J. Chem., 2011, 29, 773–777 CrossRef CAS.
- R. D. Jana, A. Das and T. K. Paine, Inorg. Chem., 2021, 60, 5969–5979 CrossRef CAS PubMed.
- J. L. Lee, D. L. Ross, S. K. Barman, J. W. Ziller and A. S. Borovik, Inorg. Chem., 2021, 60, 13759–13783 CrossRef CAS PubMed.
- M. Guo, T. Corona, K. Ray and W. Nam, ACS Cent. Sci., 2018, 5, 13–28 CrossRef PubMed.
- T. Karimpour, E. Safaei and B. Karimi, RSC Adv., 2019, 9, 14343–14351 (Synfacts, 15, 0918) RSC.
- A. Gonzalez-de-Castro and J. Xiao, J. Am. Chem. Soc., 2015, 137, 8206–8218 CrossRef CAS PubMed.
- H. Yuan, W.-J. Yoo, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2012, 134, 13970–13973 CrossRef CAS PubMed.
- D. Xu, W. Wang, C. Miao, Q. Zhang, C. Xia and W. Sun, Green Chem., 2013, 15, 2975–2980 RSC.
- H. Sharghi, M. Aberi and M. M. Doroodmand, Adv. Synth. Catal., 2008, 350, 2380–2390 CrossRef CAS.
- J. Javidi, M. Esmaeilpour and M. R. Khansari, RSC Adv., 2015, 5, 73268–73278 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06818d |
| This journal is © The Royal Society of Chemistry 2023 |