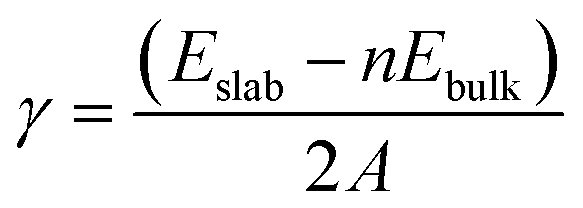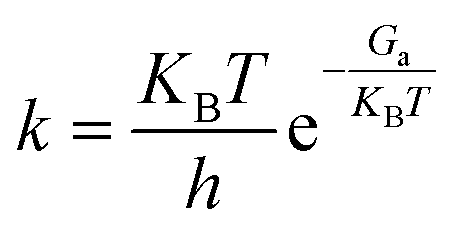DOI:
10.1039/D2RA06744G
(Paper)
RSC Adv., 2023,
13, 4032-4039
Revealing the crystal facet effect on N2O formation during the NH3-SCR over α-MnO2 catalysts†
Received
25th October 2022
, Accepted 16th January 2023
First published on 27th January 2023
Abstract
The detailed atomic-level mechanism of the effect induced by engineering the crystal facet of α-MnO2 catalysts on N2O formation during ammonia-selective catalytic reduction (NH3-SCR) was ascertained by combining density functional theory (DFT) calculations and thermodynamics/kinetic analysis. The surface energies of α-MnO2 with specific (100), (110), and (310) exposed planes were calculated, and the adsorptions of NH3, NO, and O2 on three surfaces were analyzed. The adsorption energies showed that NH3 and NO molecules could be strongly adsorbed on the surface of the α-MnO2 catalyst, while the adsorption of O2 was weak. Moreover, the key steps in the oxidative dehydrogenation of NH3 and the formation of NH2NO as well as dissociation of NH2 were studied to evaluate the catalytic ability of NH3-SCR reaction and N2 selectivity. The results revealed that the α-MnO2 catalyst exposed with the (310) plane exhibited the best NH3-SCR catalytic performance and highest N2 selectivity, mainly due to its low energy barriers in NH3 dehydrogenation and NH2NO generation, and difficulty in NH2 dissociation. This study deepens the comprehension of the facet-engineering of α-MnO2 on inhibiting N2O formation during the NH3-SCR, and points out a strategy to improve their catalytic ability and N2 selectivity for the low-temperature NH3-SCR process.
1 Introduction
Nitrogen oxides (NOx, including N2O, NO, and NO2) emitted from the burning of fossil fuels are one of the main air pollutants in urban areas, and can cause serious environmental problems, such as photochemical smog, acid rain, and ozone depletion.1–5 In general, 95% of NOx pollutants are composed of NO, which is toxic and harmful to the health of humans and animals, such as stimulating the lungs and thus triggering many diseases, like cancer and asthma.6–8 Among various denitrification methods, the selective catalytic reduction process (SCR) using NH3 as the reducing agent (NH3-SCR) has been a highly efficient method for removing NOx emitted from stationary and mobile sources in which NOx is converted into harmless N2 and H2O by the following “Standard SCR” reaction: 4NO + 4NH3 + O2 → 4N2 + 6H2O.9,10 However, the non-ignorable byproduct of N2O generation during the NH3-SCR process seems prominently to be: 4NO + 4NH3 + 3O2 → 4N2O + 6H2O.11 N2O has been included in the list of regulated greenhouse gas (GHG) pollutants because of its strong global warming potential, which is 298 times that of CO2.12 Therefore, it is a major challenge to restrain the N2O formation during the high-activity NH3-SCR process.
A reasonable selection and design of catalysts is an efficient strategy to regulate the NH3-SCR reaction. Compared with the traditional commercial V2O5–WO3/TiO2 catalysts, many studies have focused on the development of low-temperature SCR catalysts, including metal oxide-based catalysts, like Fe,13,14 Mn,15,16 Ce,17 Cr,18 W,19 Cu,20,21 and zeolite-based catalysts, like CuSn/ZSM-5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 and Cu-ZSM-5.23 Among various catalysts, manganese-based catalysts have been widely studied and applied due to their low cost and outstanding low-temperature denitrification performance, whereby their abundant Lewis acid sites, variable valence states of Mn, and strong redox ability contribute to their excellent catalytic activity at low temperature, especially under 200 °C.10,24–27 For instance, Yang et al.28 found that α-MnO2 exhibited the most superior SCR performance for NOx conversion compared with other crystal phases of β, γ, and δ-MnO2 at a lower temperature of 50–120 °C. However, the low N2 selectivity currently extremely restricts the application of Mn-based catalysts because the strong oxidation ability of tetravalent Mn leads to more N2O formation.
22 and Cu-ZSM-5.23 Among various catalysts, manganese-based catalysts have been widely studied and applied due to their low cost and outstanding low-temperature denitrification performance, whereby their abundant Lewis acid sites, variable valence states of Mn, and strong redox ability contribute to their excellent catalytic activity at low temperature, especially under 200 °C.10,24–27 For instance, Yang et al.28 found that α-MnO2 exhibited the most superior SCR performance for NOx conversion compared with other crystal phases of β, γ, and δ-MnO2 at a lower temperature of 50–120 °C. However, the low N2 selectivity currently extremely restricts the application of Mn-based catalysts because the strong oxidation ability of tetravalent Mn leads to more N2O formation.
Manipulating the exposed crystal facets of metal and metal oxide catalysts is an efficient strategy to influence the catalytic activity and selectivity significantly.29–31 This can be attributed to the different exposed atomic arrangement, distorted electronic structure, and synergistic interactions in the surface of exposed facets. Up to now, three exposed crystal facets, namely the (100), (110), and (310), of the α-MnO2 catalyst were designed to investigate the catalytic activity.32 α-MnO2 with the exposed high-index facet (310) exhibited a much better activity in the catalytic oxidation of formaldehyde (HCHO) and in the catalytic ozonation of odorous CH3SH thanks to its higher surface energy than α-MnO2 (100) and α-MnO2 (110).33–35 Whereas, Au/α-MnO2-110 exhibited the highest catalytic activity compared to Au/α-MnO2-100 and Au/α-MnO2-310 in the catalytic combustion of propane because of the lowest oxygen vacancy formation energy of α-MnO2 (110), which benefited the formation of Auδ+ species and therefore promoted the breakage of the C–H bond in propane.36 Generally, the reactivity and activity of facets are proportional to their surface energy, whereby facets with a high surface energy are usually more reactive in heterogeneous reactions.37 Unfortunately, the detailed mechanism of the effect induced by engineering the crystal facet of MnO2 on the catalytic ability of the NH3-SCR reaction and N2O formation, as an important issue, have rarely been deeply understood, and thus deserve further investigation.
In view of its adverse effect, many studies have been conducted on the N2O formation mechanism in the NH3-SCR process, in order to contrapuntally put forward effective control solutions. In principle, the favored reaction pathways of the NH3-SCR process were proposed as: (i) NH3 adsorbs on the catalyst surface, getting activated and dissociated to NH2 species, (ii) NO molecules in the gaseous or adsorbed state react with NH2 to form the key NH2NO intermediate species, (iii) NH2NO decomposes into N2 and H2O, (iv) removal of the surface H atoms from the catalyst.24,38–40 The kinetic estimation revealed that the dehydrogenation of NH3 to NH2 and the formation of NH2NO were rate-limiting steps in the NH3-SCR process, and the reaction of the adsorbed NO with NH2 to generate NH2NO was more kinetically preferred.24,41 In addition, the strong oxidation ability of Mn-based catalysts can result in the oxidative dehydrogenation of NH2 to NH species, which is inducive to N2O formation.11 On this account, increasing NH2 species and decreasing NH species are favorable for the NH3-SCR activity and N2 selectivity, which is an effective strategy to screen suitable catalysts. Therefore, the reaction pathways for the dehydrogenation of NH3 and formation of NH2NO could be used to evaluate the catalytic ability and the N2 selectivity during the NH3-SCR reaction with different crystal facets of α-MnO2.
In this article, the crystal facet effect on N2O formation during the NH3-SCR over α-MnO2 catalysts was first evaluated through the DFT+U calculation. The α-MnO2 catalysts with different exposed surfaces, including (100), (110), and (310), were constructed to investigate the effect of engineering the crystal facet at an atomic level. Some key steps of NH3-SCR on MnO2 catalysts have been proposed, including the oxidative dehydrogenation of NH3 to NH2 and then to NH species, which determine the selectivity of the NH3-SCR reaction. The catalytic ability and N2 selectivity were primarily studied by the thermodynamics and kinetic analysis. This study revealed the crystal facet effect on N2O formation during the NH3-SCR over Mn-based catalysts, and provides a promising strategy for developing robust Mn-based catalysts with good catalytic ability and a high N2 selectivity for the low-temperature NH3-SCR process.
2 Methods
All of the spin-polarized density functional theory (DFT) calculations were implemented in the Vienna ab initio simulation package (VASP).42,43 The Projector Augmented Wave (PAW) method was used to simulate the interaction between ions and electrons. The Generalized Gradient Approximation (GGA) and Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional were used to deal with the exchange–correlation potential between electrons.44 In addition, the Mn element in α-MnO2 belonged to the transition metal element of the third period, in which existed a strong Coulomb effect between the electrons in the d layer. Therefore, a DFT+U method was used to correct the strong Coulomb interaction between the 3d electrons in the outer layer of Mn atoms with the values of Ueff = 5.5.45,46 According to reports in the literature, α-MnO2 should be calculated under the antiferromagnetic spin-polarized structure.47
For the structural optimization calculations, all the energy of the system converged to 1 × 10−5 eV, and the force on each atom was less than 0.03 eV Å−1. In the static self-consistent field (SCF) calculations, the electronic energies converged to 1 × 10−6 eV. Searches for the transition states along the reaction coordinate were calculated using the climbing image nudged elastic band (CI-NEB) method.48 All the located transition states were confirmed by vibrational frequency analysis to ensure that there was only one imaginary frequency. The cutoff energy was set to 450 eV and a 4 × 2 × 1 k-point mesh using the Monkhorst–Pack method was used to express the integral of the Brillouin zone. All the electronic energies marked with “E” include the ground state energy of the electron (εele) and the contribution of the zero-point vibrational energy (ZPE) calculated by the formula:
where
εele and ZPE were calculated by VASP and VASPKIT code.
49
The surface energy (γ) of each surface model was calculated with
| |
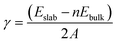 | (2) |
where
Eslab is the energy of the surface model,
Ebulk is the energy of the cell model,
n is the number of unit cells that a surface model contains,
A represents the area of the slab surface, and 2 represents that each slab model had the same upper and lower surfaces.
To understand the strength of the adsorption between the gas molecules and the catalyst surface, the adsorption energy (Eads) was calculated according to the following formula:
| | |
Eads = E(adsorbate/surface) − E(surface) − E(adsorbate)
| (3) |
where
E(adsorbate/surface),
E(surface), and
E(adsorbate) represent the total energy of the adsorbate adsorbed to the catalyst surface, the energy of the surface model, and the energy of the adsorbate (gas molecules), respectively.
The reaction energy (ΔErxn) and energy barrier (Ea) of each elementary reaction were calculated using the following formulas:
where
EIS,
ETS, and
EFS represent the total energy of the reaction initial state, transition state, and final state, respectively.
To understand the effect of temperature on the reaction, the calculated energies (E) were corrected for the Gibbs free energy (G) in the following formulas:27
| | |
Ggas = εele + ZPE + ΔG0→T
| (6) |
| | |
Gsolid = εele + ZPE + ΔU0→T − TS
| (7) |
where
εele is the electron energy of the system in the ground state, ZPE is the zero-point correction energy,
T is the temperature,
S is the entropy of the system, and Δ
U0→T and Δ
G0→T are the contribution of the temperature to enthalpy and the Gibbs free energy. Also,
Ggas represents the energy of gas molecules and
Gsolid represents the energy of catalysts and adsorbed species. All the thermodynamic quantities were processed with VASPKIT code.
49
In order to better measure the speed of each elementary reaction, the reaction rate constant k was calculated according to the transition state theory (TST)50 as per the following formula:
| |
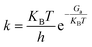 | (8) |
where
KB is the Boltzmann constant (1.380649 × 10
−23 J K
−1),
h is Planck's constant (6.62607015 × 10
−34 J s),
T is the temperature (K), and
Ga is the energy barrier with the correction for the Gibbs free energy from
Ea through formulas
(5)–(7).
3 Results and discussion
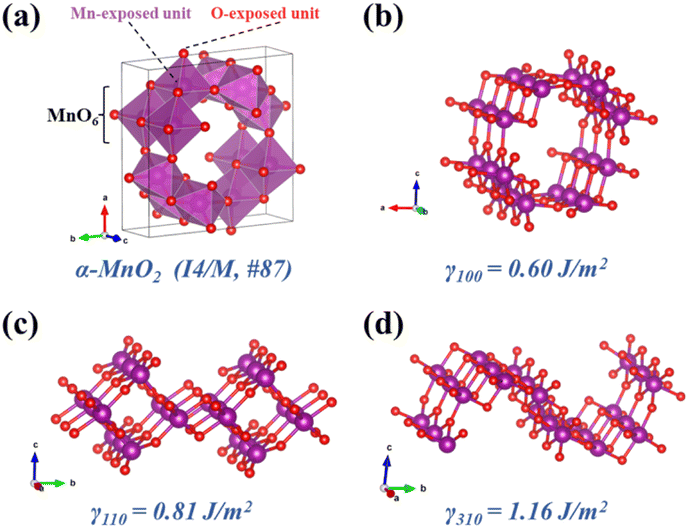
3.1 Models and surface energies
The cell model structure of α-MnO2 was established first (Fig. 1a), in which it has a (2 × 2) tunnel structure and the space group I4/m.47,51 with the relaxation lattice constants obtained by geometric optimization as a = b = 9.850 Å, c = 2.925 Å. As shown in Fig. 1b–d, the surface models of α-MnO2 with three crystal facets of (100), (110), and (310) were constructed to further understand the difference in the exposed facets of α-MnO2 and its effect on N2O formation. All the models maintained stoichiometric MnO2 ratios and proper surface sites for the adsorption of gas molecules. A vacuum layer of 15 Å was added to the three models to avoid the interaction between the slabs in the z-direction. The calculated surface energies of the slab models of α-MnO2 (100), (110), and (310) were 0.60, 0.81, and 1.16 J m−2 respectively, which indicated that α-MnO2 (100) had thermodynamically stable facets with the lowest surface energy.
 |
| | Fig. 1 Crystal structure of (a) bulk α-MnO2, (b) α-MnO2 (100), (c) α-MnO2 (110), and (d) α-MnO2 (310). Purple and red circles denote Mn and O atoms, respectively. | |
3.2 Adsorption of gas molecules
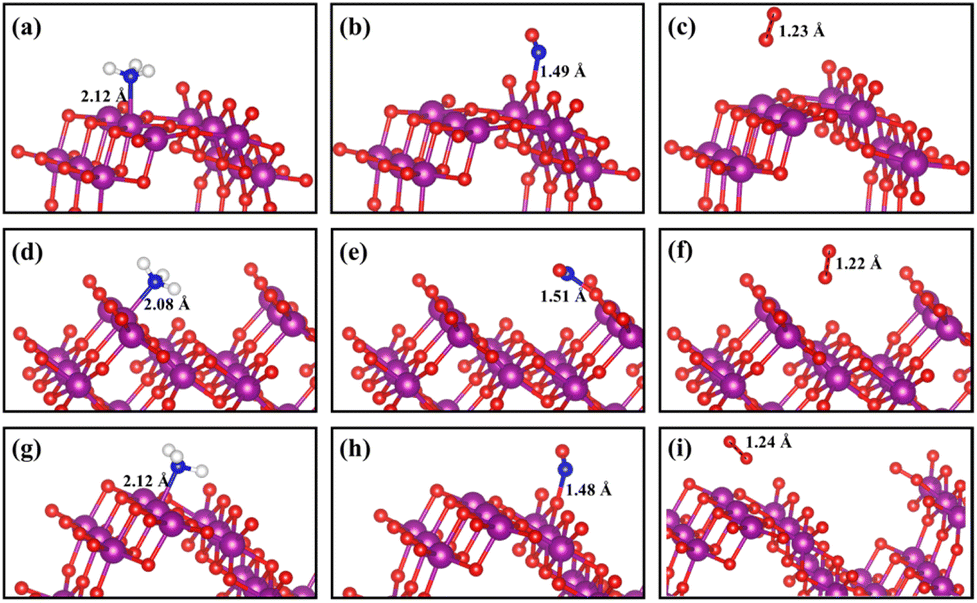
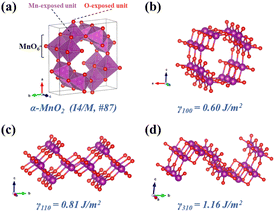
The adsorption and activation of reactant molecules on the active sites of catalysts are regarded as important steps in the NH3-SCR process. Three key gas molecules (NH3, NO, and O2)52 in the NH3-SCR process on α-MnO2 catalysts with three crystal facets were tested first to obtain their adsorption energy and the most favorable adsorption site. The test results are shown in Fig. S1† and the optimum configurations are shown in Fig. 2. It can be concluded the NH3 molecules were easily adsorbed on the α-MnO2 surface, with the N atom bonding to the surface Mn atom, where the Mn cations acted as a Lewis acid site to have more empty orbitals to accept electron pairs.53,54 The calculated adsorption energies of NH3 molecules on the surfaces of α-MnO2 (100), (110), and (310) were −1.12, −1.82, and −1.08 eV, respectively, as shown in Table 1, which all had strong chemical adsorption with the catalysts and showed a table-shaped exothermic process.
 |
| | Fig. 2 Adsorption configurations of NH3, NO, and O2 molecules on α-MnO2 surfaces with different crystal facets: (a–c) (100); (d–f) (110); (g–i) (310). Purple, red, dark blue, and white circles denote Mn, O, N, and H atoms, respectively. | |
Table 1 Adsorption energies (eV) of three gas molecules on α-MnO2 (100), (110), and (310) facets with zero-point vibrational energy (ZPE) contribution
| |
NH3 |
NO |
O2 |
| (100) |
−1.12 |
−2.23 |
−0.05 |
| (110) |
−1.82 |
−2.64 |
−0.27 |
| (310) |
−1.08 |
−1.96 |
−0.09 |
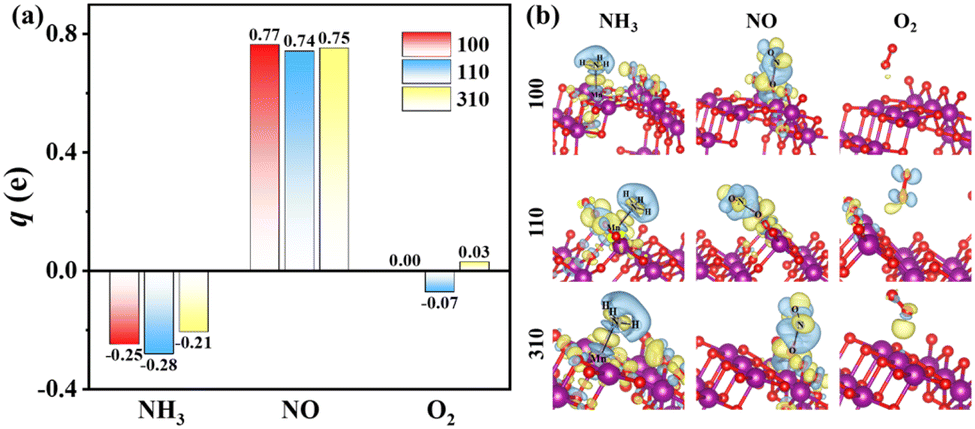
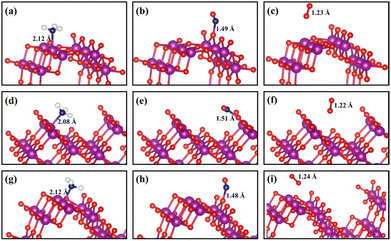
The charge-density difference of NH3 molecule adsorbed on α-MnO2 surfaces, as shown in Fig. 3a, demonstrated that there was more electron consumption around the H atom and more electron accumulation around the N atom, which showed that part of the electrons were transferred from the H atom to N atom. In the adsorption site, especially around the Mn atom, there was more electron accumulation, which illustrated that electrons were transferred from NH3 to α-MnO2 catalysts. The NH3 molecules acted as electron donors in the adsorption on α-MnO2 (100), (110), and (310) surfaces, which lost 0.25e, 0.28e, and 0.21e, respectively, by Bader charge analysis, as shown in Fig. 3b. The calculated results verified the role of the Lewis acid site that the Mn atom behaved as, which may facilitate the activation of NH3 in the NH3-SCR process.55
 |
| | Fig. 3 (a) Bader charges variations of NH3, NO, and O2 molecules adsorbed on α-MnO2 (100), (110), and (310) facets. (b) Charge-density differences of the adsorption structures. Blue and yellow density regions represent electron density depletion and accumulation respectively (isosurface levels set as 0.0025 e Å−3). | |
The adsorption of NO molecules on α-MnO2 (100) (110), and (310) facets are also shown in Fig. 2. Different from the results of the NH3 molecules, NO molecules preferred the O site, forming an O–N–O structure, which indicated that no competitive adsorption occurred between NH3 and NO molecules over α-MnO2. The calculated adsorption energies on three facets were −2.23, −2.64, and −1.96 eV, as listed in Table 1, which all presented a strong process of chemical adsorption. The Bader charges analysis displayed that NO molecules acted as electron acceptors after adsorption, which obtained 0.77e, 0.74e, and 0.75e on α-MnO2 (100) (110), and (310), respectively.
For O2 molecules, the optimized adsorption configurations showed that they did not tend to be bonded on the surface. The adsorption energies of O2 on the (100), (110), and (310) surfaces were −0.05, −0.27, and −0.09 eV, respectively. All of them were less than 0.5 eV, coinciding with the results from the adsorption of O2 on the surface of β-MnO2.56 Only weak physical adsorption of O2 molecule could happen on pure α-MnO2 surfaces, which indicated that O2 could not directly be chemisorbed on the catalysts surface. Instead, physically adsorbed O2 molecule could react with the surface-active species, such as intermediate H atom to form *OH and H2O, to participate in the reaction in the NH3-SCR process.24,56 Besides, O2 could also be activated by the defected slab containing an oxygen vacancy.24 It is reported that α-MnO2 (310) has the lowest formation energy of an oxygen vacancy of 0.33 eV than that of (100) and (110),33 which indicates that the O2 molecule is beneficial to be adsorbed and dissociated on defected the α-MnO2 (310) surface to participate in the removal of surface H atom and the generation of H2O.
3.3 Key reaction pathways
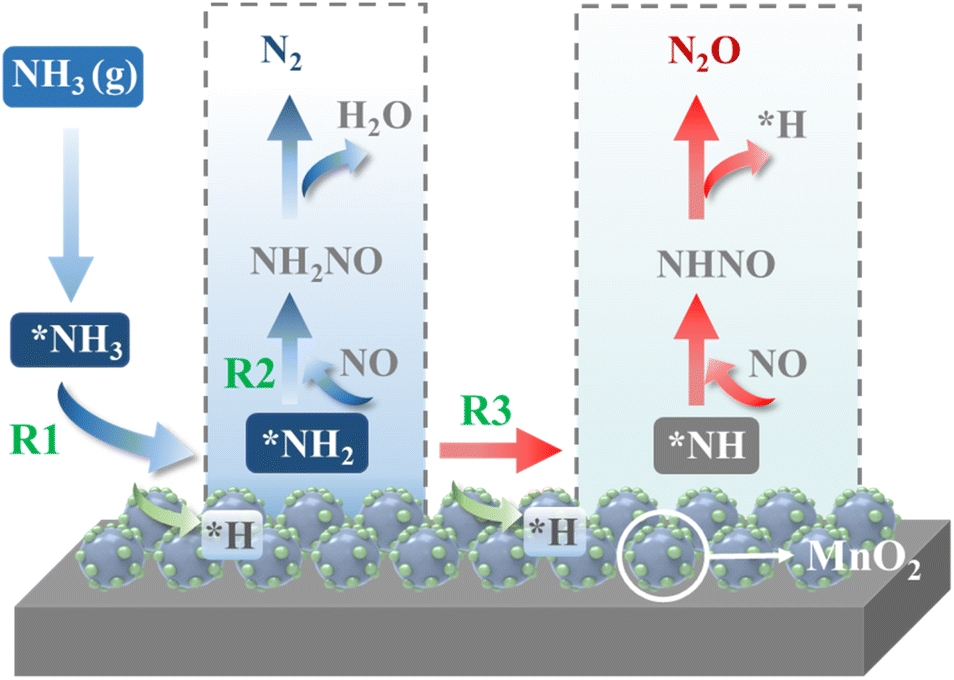
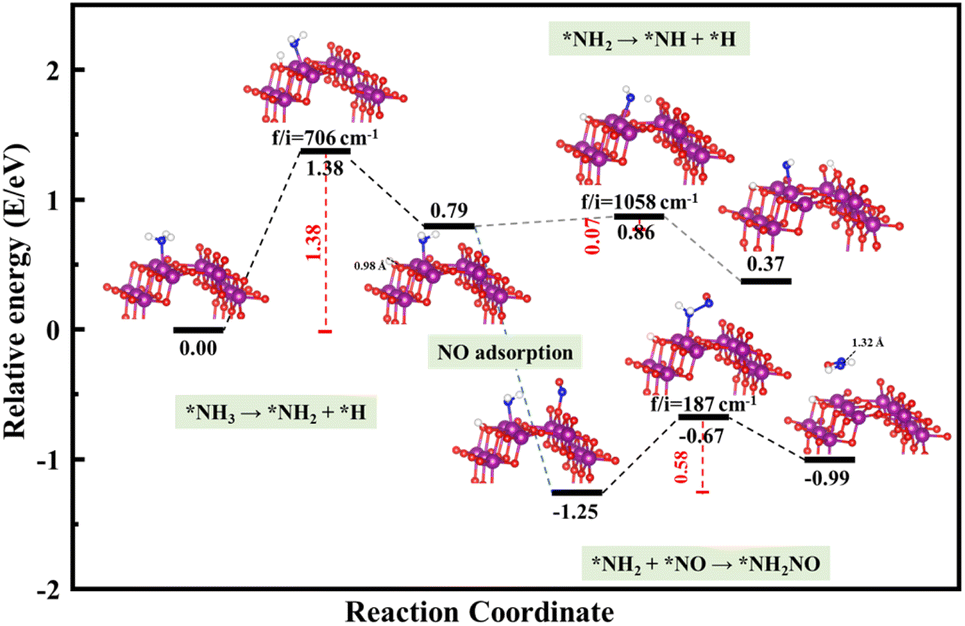
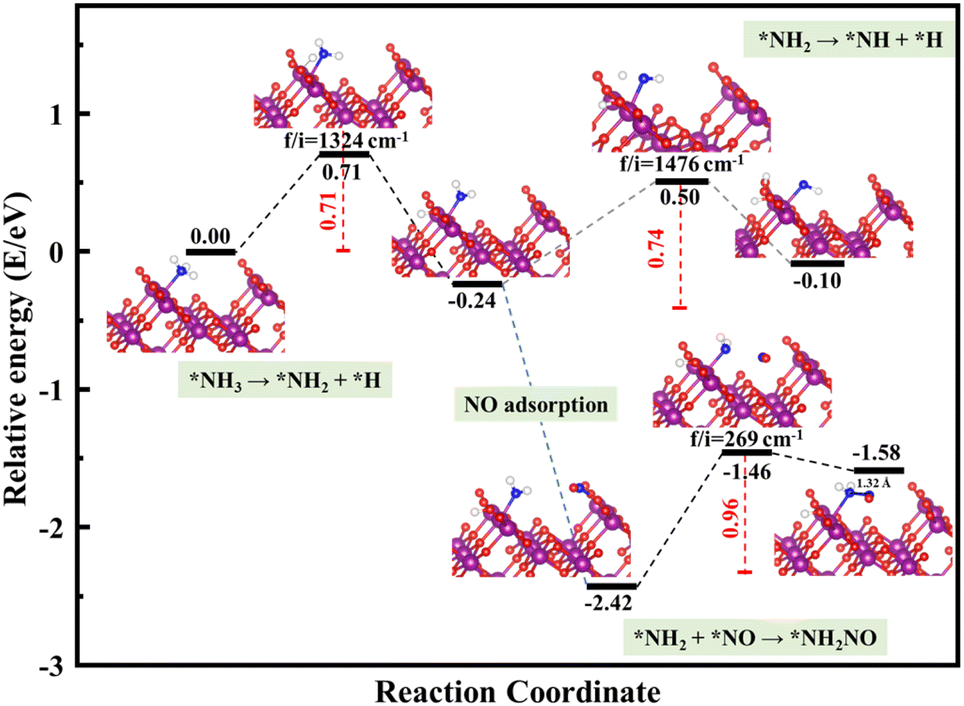
The schemed reaction pathways depicted in Fig. 4 have been widely employed to illuminate the mechanism of N2O formation during the NH3-SCR reaction. Here, the three key reaction steps of the dehydrogenation of NH3 and formation of NH2NO were used to evaluate the NH3-SCR catalytic ability and the N2 selectivity discussed in the introduction of this paper: step R1: NH3 → NH2 + H, where the activated NH3 dissociated to NH2 species; step R2: NH2 + NO → NH2NO, where the NO reacts with NH2 to form the NH2NO following the L–H mechanism; step R3: NH2 → NH + H, where the NH2 species are oxidized to NH. The energy profiles of R1, R2, and R3 over the three surfaces of α-MnO2 (100) (110), and (310) and the corresponding geometries of all the intermediates and transition states are depicted in Fig. 5–7. The values of the energy barriers and reaction energies for the R1–3 reactions over the three crystal facets of α-MnO2 are summarized in Table 2.
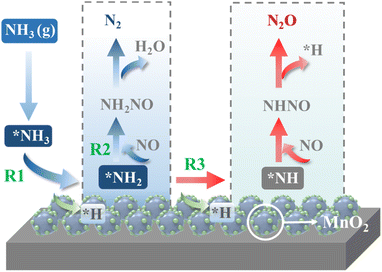 |
| | Fig. 4 Schematic of the reaction pathways for N2 and N2O formation during the NH3-SCR reaction over MnO2. | |
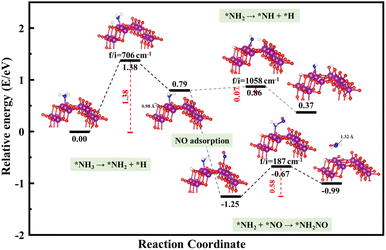 |
| | Fig. 5 Reaction pathway for R1–3 over α-MnO2 (100) coupled with the calculated structures. Purple, red, dark blue, and white circles denote Mn, O, N, and H atoms, respectively. | |
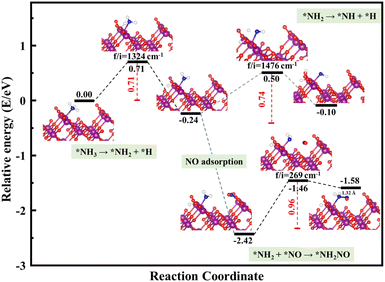 |
| | Fig. 6 Reaction pathway for R1–3 over α-MnO2 (110) coupled with the calculated structures. Purple, red, dark blue, and white circles denote Mn, O, N, and H atoms, respectively. | |
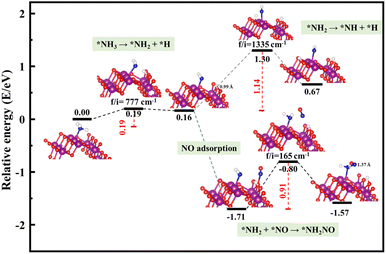 |
| | Fig. 7 Reaction pathway for R1–3 over α-MnO2 (310) coupled with the calculated structures. Purple, red, dark blue, and white circles denote Mn, O, N, and H atoms, respectively. | |
Table 2 Energy barriers (Ea, eV) and reaction energies (ΔErxn, eV) of three elementary reactions on α-MnO2 (100), (110), and (310) facets
| Step |
Elementary reaction |
(100) |
(110) |
(310) |
| Ea |
ΔErxn |
Ea |
ΔErxn |
Ea |
ΔErxn |
| R1 |
NH3 → NH2 + H |
1.38 |
0.79 |
0.71 |
−0.24 |
0.19 |
0.16 |
| R2 |
NH2 + NO → NH2NO |
0.58 |
0.26 |
0.96 |
0.84 |
0.91 |
0.14 |
| R3 |
NH2 → NH + H |
0.07 |
−0.42 |
0.74 |
0.14 |
1.14 |
0.51 |
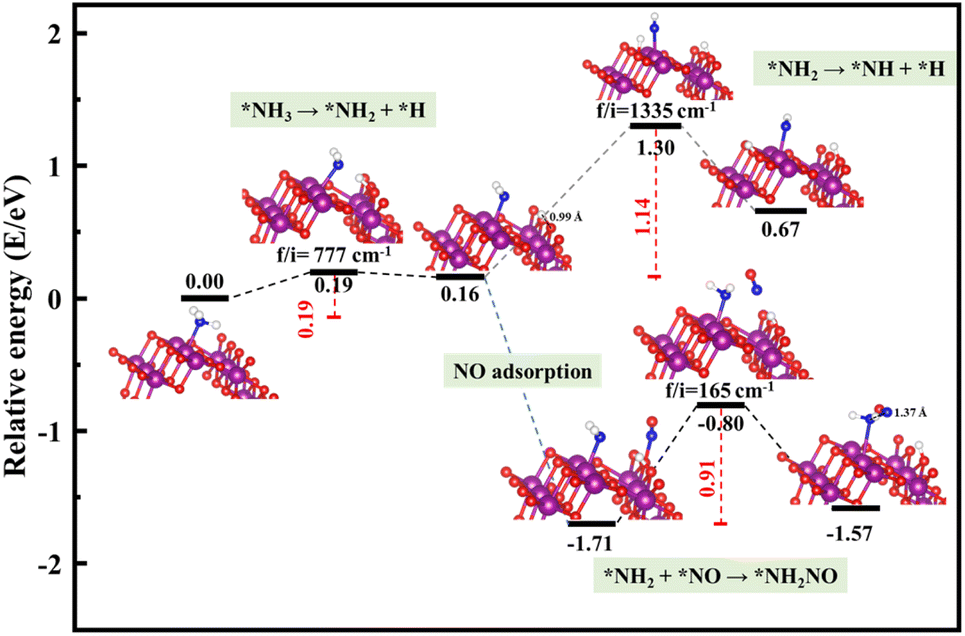
Starting from the reaction for R1, an NH3 molecule was adsorbed and activated by the catalysts while the adjacent O site on the catalyst surface acted as a fitted acceptor for H atoms in the NH3 molecule, enlarging the distance between H and NH2. At the end of this reaction, a H atom dissociated from the NH3 molecule combined with surface oxygen atoms to form a hydroxyl group, and the final O–H bond lengths on the three crystal facets were 0.98, 0.98, and 0.99 Å, respectively. The calculated results showed that the process of NH3 dissociation on the α-MnO2 (100) and (310) surfaces was endothermic for both, with reaction energies of 0.79 and 0.16 eV. However, it was slightly exoergic for NH3 dissociating on the surface of α-MnO2 (110), with a reaction energy of −0.24 eV. Moreover, α-MnO2 (100) had the highest energy barrier (1.38 eV) for NH3 dissociation compared to α-MnO2 (110) and (310), indicating that the difficulty in dissociation of NH3 may be the key factor to reduce its SCR activity. The (310) facet had the lowest energy barrier of 0.19 eV for NH3 to dissociate, which would be more favorable to activate the NH3 molecule and facilitate the SCR process.
Subsequently, the NO molecule desorbed from the O site approached the NH2 species and bonded to the N atom to form the NH2NO intermediate (the process for R2), with N–N bond lengths of 1.32, 1.32, and 1.37 Å on the (100), (110), and (310) facets, respectively. The calculated energy barriers were 0.58, 0.96, and 0.91 eV, respectively. Concerning the formation of N2O, the further oxidative dehydrogenation of NH2 to NH (the process for R3) was also investigated. After one H atom dissociated from the NH3 molecule to the surface O atom on the α-MnO2 surface, another H atom dissociated from NH2 species to the adjacent oxygen site of the catalyst surface. Our results showed that all the processes on the three catalysts were endothermic with positive reaction energy. The reaction energies of this reaction on α-MnO2 (100), (110), and (310) were 0.07, 0.74, and 0.91 eV, respectively. It is worth noting that the NH2 intermediate could be easily converted into NH species over the α-MnO2 (100) with a low energy barrier of 0.07 eV, resulting in poor reactivity and N2 selectivity.
Overall, α-MnO2 (100) had high NH3 dissociation (1.38 eV) and low NH2 dissociation (0.07 eV) energy barriers, which means it faces the problem of poor activity and nitrogen selectivity. The energy barriers of NH3 dissociation and NH2NO generation on α-MnO2 (110) were 0.71 and 0.96 eV, which were similar to the reported results of 1.10 eV on α-MnO2 (110) and lower than the results of 1.15 eV on γ-Fe2O3 (110) and 1.27 eV on CeO2 (111).38 However, the high-index facet of α-MnO2 (310) had a low energy barrier for NH3 dissociation (0.19 eV) and a high energy barrier for NH formation (1.14 eV), which exhibited the excellent reaction activity and good N2 selectivity compared with the (100) and (110) facets.
3.4 Thermodynamic and kinetic analysis
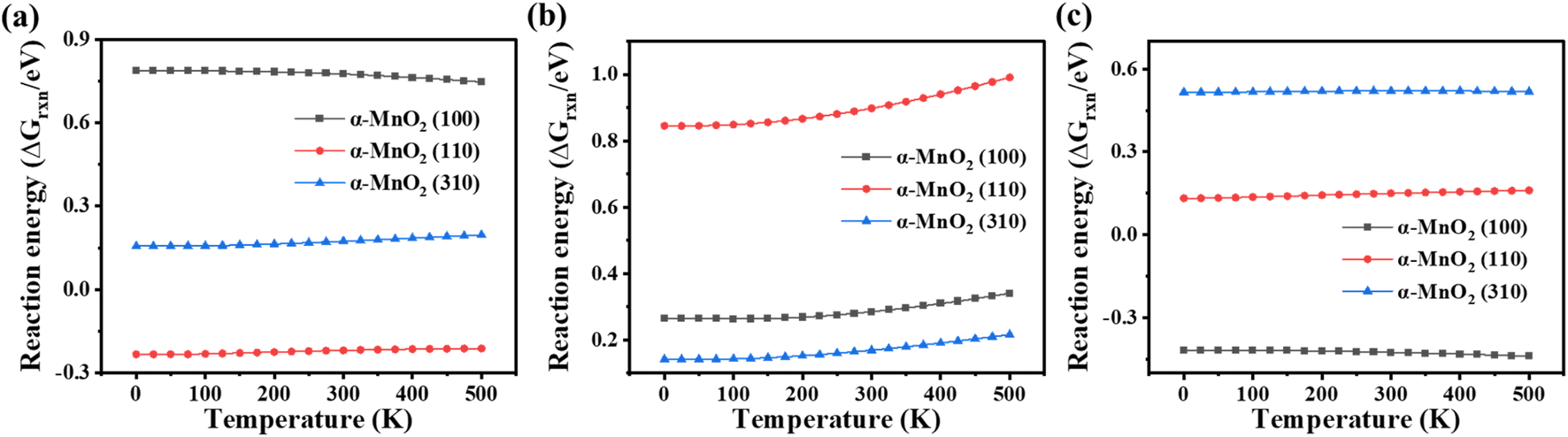
Temperature plays an important role in the NH3-SCR process. Therefore, the variations of the reaction energy of each elementary reaction were analyzed by the correction of the Gibbs free energy (formulas (4)–(7)) to obtain the thermodynamic properties. The reaction energies with the correction for the Gibbs free energy of processes R1–3 on α-MnO2 exposing different crystal facets at temperatures below 500 K were plotted, and are shown in Fig. 8. The results indicated that for α-MnO2 (100), the high ΔGrxn of R1 restricted the dissociation of NH3. Besides, as the temperature decreased, the reaction free energy value of R1 on α-MnO2 (100) became more positive, which became more limited. The reaction for NH2 dissociation was exergonic on α-MnO2 (100), and spontaneous as well, which could lead to poor N2 selectivity. For α-MnO2 (110), the reaction of NH3 dissociation was exergonic, and had a certain spontaneity. As the temperature decreased, the ΔGrxn of NH2NO generation on α-MnO2 (110) became less exergonic, but its higher reaction free energy value than that of α-MnO2 (100) and (310) still restricted this reaction and led to a poor catalytic activity. The reaction free energy values of R1 and R2 on α-MnO2 (310) both decreased with the decrease in temperature, keeping at a low endergonic value. Moreover, the ΔGrxn value of R3 on α-MnO2 (310) was higher than that of R1 and R2, as well as on α-MnO2 (100) and (110), showing it was the thermodynamically rate-limiting step, indicating that α-MnO2 (310) had good catalytic activity and N2 selectivity.
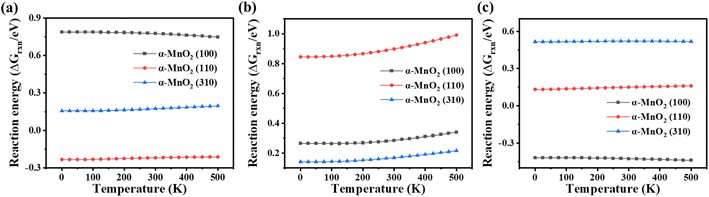 |
| | Fig. 8 Variation of the reaction free energy value (ΔGrxn) under different temperatures for R1–3. (a) NH3 dissociation, (b) NH2NO generation, and (c) NH2 dissociation. | |
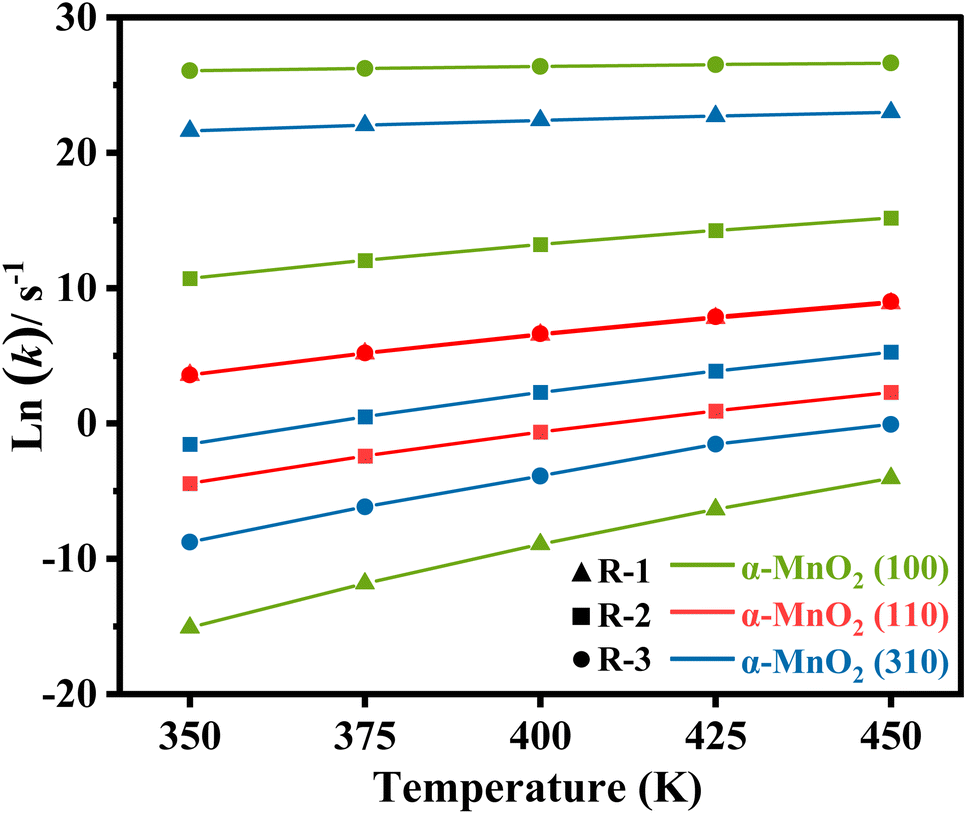
According to the reaction pathway for R1–3, the rate-determining steps could be obtained by formula (8) to perform the kinetic analysis. The reaction rate constants (k) for R1–3 below 500 K are listed in Table S1† and the natural logarithm data ln(k) are plotted in Fig. 9, respectively. The results showed that the rate of reactions R1–3 decreased with the reaction temperature decreasing, especially for the dehydrogenation of NH3 on α-MnO2 (100) and dissociation of NH2 on α-MnO2 (310). Moreover, the (100) had a slightly higher rate constant for the N2O generation than those of the other facets. In particular, the reaction rate constants of R1 and R2 were much larger than that of R3 on α-MnO2 (310) at the research temperatures, indicating that the (310) facet showed remarkable activity and N2 selectivity, in line with the results from the reaction pathway analysis.
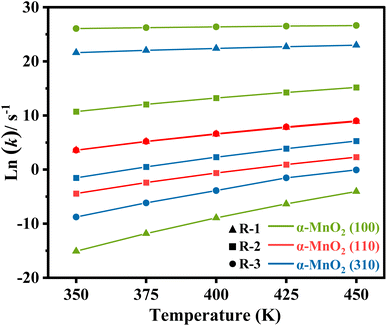 |
| | Fig. 9 Natural logarithm of the reaction rate constant (k) of R1–3 under different temperatures. | |
4 Conclusions
In summary, the effect induced by engineering the crystal facet of α-MnO2 catalysts on N2O formation during the NH3-SCR were systematically investigated by using DFT+U calculations. The adsorption behavior of the key gas molecules in the NH3-SCR process was described by configurations of the gas adsorption on three surfaces of α-MnO2 (100), (110), and (310). It could be found that the NH3 molecule prefers the Lewis acid site (the Mn site) on the α-MnO2 surface, but the NO molecule tends to adsorb on the O site, with both showing strong chemical adsorption. The adsorption energy of the NH3 and NO molecules on the α-MnO2 (110) surface was higher than that of O2. Three key reaction pathways for the dehydrogenation of NH3, formation of NH2NO, and dissociation of NH2 were considered to construct the reaction activity and N2 selectivity. The detailed path for N2O formation was explored by the transition state theory. The effects of different reaction temperatures on the reaction equilibrium and reaction rate were revealed in depth through thermodynamics and kinetics analyses. The results suggested that the high-index facet of α-MnO2 (310) exhibited excellent reaction activity and good N2 selectivity compared with the (100) and (110) facets in the researched temperature range below 500 K. The findings of this study help improve the atomic-level knowledge of the mechanism of N2O formation, and advance the understanding of the roles of the crystal facets of α-MnO2 on the denitration activity and selectivity, which is good reference for the suppression of N2O emission during the low-temperature NH3-SCR.
Author contributions
Jundong Guo: conceptualization, methodology, formal analysis, resources, data curation, writing – original draft, visualization; Fengli Gan: resources, investigation, visualization; Yifan Zhao: resources, investigation, data curation; Jinglin He: resources, investigation, visualization; Tao Gao: formal analysis, software; Xia Jiang: writing – review & editing, supervision, project administration, funding acquisition; Shenggui Ma: methodology, formal analysis, writing – review & editing, supervision, funding acquisition.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
Financial support was provided by the National Natural Science Foundation of China (No. 51978427), Sichuan Science and Technology Program (No. 2021YJ0378, No. 2020YFH0109), and Fundamental Research Funds for the Central Universities.
Notes and references
- G. Jiang, X. Li, M. Lan, T. Shen, X. Lv, F. Dong and S. Zhang, Appl. Catal., B, 2017, 205, 532–540 CrossRef CAS.
- Q. Zhang, Y. Huang, L. Xu, J.-j. Cao, W. Ho and S. C. Lee, ACS Appl. Mater. Interfaces, 2016, 8, 4165–4174 CrossRef CAS PubMed.
- H. Schneider, S. Tschudin, M. Schneider, A. Wokaun and A. Baiker, J. Catal., 1994, 147, 5–14 CrossRef CAS.
- R. Tsekov and P. G. Smirniotis, J. Phys. Chem. B, 1998, 102, 9525–9531 CrossRef CAS.
- H.-F. Wang, Y.-L. Guo, G. Lu and P. Hu, J. Phys. Chem. C, 2009, 113, 18746–18752 CrossRef CAS.
- L. Zhao, G. Dong, L. Zhang, Y. Lu and Y. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 10042–10051 CrossRef CAS PubMed.
- D. P. Chock, A. M. Dunker, S. Kumar and C. S. Sloane, Environ. Sci. Technol., 1981, 15, 933–939 CrossRef CAS PubMed.
- I. M. Orme, R. T. Robinson and A. M. Cooper, Nat. Immunol., 2015, 16, 57–63 CrossRef CAS PubMed.
- J.-K. Lai and I. E. Wachs, ACS Catal., 2018, 8, 6537–6551 CrossRef CAS.
- L. Han, S. Cai, M. Gao, J.-y. Hasegawa, P. Wang, J. Zhang, L. Shi and D. Zhang, Chem. Rev., 2019, 119, 10916–10976 CrossRef CAS PubMed.
- S. Yang, S. Xiong, Y. Liao, X. Xiao, F. Qi, Y. Peng, Y. Fu, W. Shan and J. Li, Environ. Sci. Technol., 2014, 48, 10354–10362 CrossRef CAS PubMed.
- A. Ravishankara, J. S. Daniel and R. W. Portmann, Science, 2009, 326, 123–125 CrossRef CAS PubMed.
- M. Gao, G. He, W. Zhang, J. Du and H. He, Environ. Sci. Technol., 2021, 55, 10967–10974 CrossRef CAS PubMed.
- W. Yang, J. Ren, H. Zhang, J. Li, C. Wu, I. D. Gates and Z. Gao, Fuel, 2021, 302, 121041 CrossRef CAS.
- Z. Fu, G. Zhang, W. Han and Z. Tang, Chem. Eng. J., 2021, 426, 131334 CrossRef CAS.
- Y. Xin, H. Li, N. Zhang, Q. Li, Z. Zhang, X. Cao, P. Hu, L. Zheng and J. A. Anderson, ACS Catal., 2018, 8, 4937–4949 CrossRef CAS.
- X. Sun, R.-t. Guo, S.-w. Liu, J. Liu, W.-g. Pan, X. Shi, H. Qin, Z.-y. Wang, Z.-z. Qiu and X.-y. Liu, Appl. Surf. Sci., 2018, 462, 187–193 CrossRef CAS.
- P. G. Smirniotis, D. A. Peña and B. S. Uphade, Angew. Chem., Int. Ed., 2001, 40, 2479–2482 CrossRef CAS PubMed.
- Y. Inomata, H. Kubota, S. Hata, E. Kiyonaga, K. Morita, K. Yoshida, N. Sakaguchi, T. Toyao, K.-i. Shimizu and S. Ishikawa, Nat. Commun., 2021, 12, 1–11 CrossRef PubMed.
- C. Chen, Y. Cao, S. Liu and W. Jia, Appl. Surf. Sci., 2020, 507, 145153 CrossRef CAS.
- S. Hao, Y. L. Liu and J. Yang, Sci. Total Environ., 2022, 820, 152984 CrossRef CAS PubMed.
- K. Lee, B. Choi, C. Kim, C. Lee and K. Oh, J. Ind. Eng. Chem., 2021, 93, 461–475 CrossRef CAS.
- J. Cheng, R. Xu, N. Liu, C. Dai, G. Yu, N. Wang and B. Chen, Catal. Sci. Technol., 2022, 12, 823–833 RSC.
- H. Yuan, N. Sun, J. Chen, J. Jin, H. Wang and P. Hu, ACS Catal., 2018, 8, 9269–9279 CrossRef CAS.
- X. Shi, J. Guo, T. Shen, A. Fan, Y. Liu and S. Yuan, J. Taiwan Inst. Chem. Eng., 2021, 126, 102–111 CrossRef CAS.
- L.-g. Wei, R.-t. Guo, J. Zhou, B. Qin, X. Chen, Z.-x. Bi and W.-g. Pan, Fuel, 2022, 316, 123438 CrossRef CAS.
- B. Tian, S. Ma, J. Guo, Y. Zhao, T. Gao and X. Jiang, Appl. Surf. Sci., 2022, 601, 154162 CrossRef CAS.
- J. Yang, S. Ren, B. Su, G. Hu, L. Jiang, J. Cao, W. Liu, L. Yao, M. Kong and J. Yang, Catal. Lett., 2021, 151, 2964–2971 CrossRef CAS.
- R. Yang, S. Peng, B. Lan, M. Sun, Z. Zhou, C. Sun, Z. Gao, G. Xing and L. Yu, Small, 2021, 17, 2102408 CrossRef CAS PubMed.
- B. Hu, K. Sun, Z. Zhuang, Z. Chen, S. Liu, W. C. Cheong, C. Chen, M. Hu, X. Cao and J. Ma, Adv. Mater., 2022, 2107721 CrossRef CAS PubMed.
- B. Jiang, Y. Qiu, D. Tian, Y. Zhang, X. Song, C. Zhao, M. Wang, X. Sun, H. Huang and C. Zhao, Adv. Energy Mater., 2021, 2102995 CrossRef CAS.
- R. Yang, Y. Fan, R. Ye, Y. Tang, X. Cao, Z. Yin and Z. Zeng, Adv. Mater., 2021, 33, 2004862 CrossRef CAS PubMed.
- S. Rong, P. Zhang, F. Liu and Y. Yang, ACS Catal., 2018, 8, 3435–3446 CrossRef CAS.
- D. Ding, Y. Zhou, T. He and S. Rong, Chem. Eng. J., 2022, 431, 133737 CrossRef CAS.
- C. He, Y. Wang, Z. Li, Y. Huang, Y. Liao, D. Xia and S. Lee, Environ. Sci. Technol., 2020, 54, 12771–12783 CrossRef CAS PubMed.
- Y. Jian, Z. Jiang, C. He, M. Tian, W. Song, G. Gao and S. Chai, Catal. Sci. Technol., 2021, 11, 1089–1097 RSC.
- Q. Kuang, X. Wang, Z. Jiang, Z. Xie and L. Zheng, Acc. Chem. Res., 2014, 47, 308–318 CrossRef CAS PubMed.
- G. He, M. Gao, Y. Peng, Y. Yu, W. Shan and H. He, Environ. Sci. Technol., 2021, 55, 6995–7003 CrossRef CAS PubMed.
- S. Yang, Y. Liao, S. Xiong, F. Qi, H. Dang, X. Xiao and J. Li, J. Phys. Chem. C, 2014, 118, 21500–21508 CrossRef CAS.
- Z. Chen, F. Wang, H. Li, Q. Yang, L. Wang and X. Li, Ind. Eng. Chem. Res., 2012, 51, 202–212 CrossRef CAS.
- M. Zhang, W. Wang and Y. Chen, Phys. Chem. Chem. Phys., 2018, 20, 2211–2219 RSC.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- D. A. Tompsett, D. S. Middlemiss and M. S. Islam, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 205126 CrossRef.
- X. Ye, X. Jiang, L. Chen, W. Jiang, H. Wang, W. Cen and S. Ma, Appl. Surf. Sci., 2020, 521, 146477 CrossRef CAS.
- E. Cockayne and L. Li, Chem. Phys. Lett., 2012, 544, 53–58 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- S. Canneaux, F. Bohr and E. Henon, J. Comput. Chem., 2014, 35, 82–93 CrossRef CAS PubMed.
- L. Espinal, W. Wong-Ng, J. A. Kaduk, A. J. Allen, C. R. Snyder, C. Chiu, D. W. Siderius, L. Li, E. Cockayne and A. E. Espinal, J. Am. Chem. Soc., 2012, 134, 7944–7951 CrossRef CAS PubMed.
- D. Wang, Q. Chen, X. Zhang, C. Gao, B. Wang, X. Huang, Y. Peng, J. Li, C. Lu and J. Crittenden, Environ. Sci. Technol., 2021, 55, 2743–2766 CrossRef CAS PubMed.
- Y. Peng, W. Yu, W. Su, X. Huang and J. Li, Catal. Today, 2015, 242, 300–307 CrossRef CAS.
- Y. Wang, L. Chen, H. Cao, Z. Chi, C. Chen, X. Duan, Y. Xie, F. Qi, W. Song and J. Liu, Appl. Catal., B, 2019, 245, 546–554 CrossRef CAS.
- F. Lin, Q. Wang, J. Zhang, J. Jin, S. Lu and J. Yan, Ind. Eng. Chem. Res., 2019, 58, 22763–22770 CrossRef CAS.
- X. J. Zhou, Y. Sun, B. Zhu, J. Chen, J. Xu, H. Yu and M. Xu, Fuel, 2022, 318, 123470 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence abc and
Shenggui Ma
abc and
Shenggui Ma