DOI:
10.1039/D2RA06573H
(Paper)
RSC Adv., 2023,
13, 2570-2573
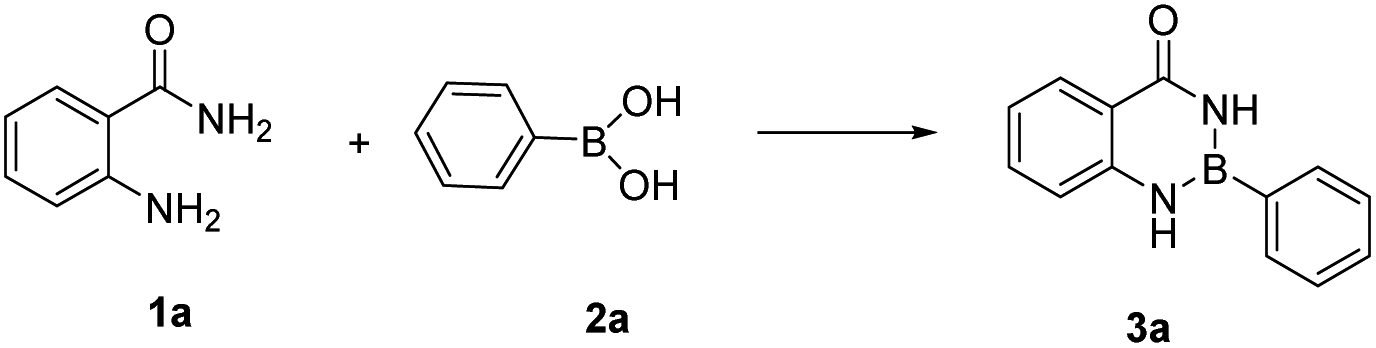
A rapid construction of 1,3,2-benzodiazaborininones [R–B(aam)] from boronic acids and anthranilamides†
Received
18th October 2022
, Accepted 20th December 2022
First published on 18th January 2023
Abstract
A simple, efficient and mild methodology for the synthesis of 1,3,2-benzodiazaborininones [R–B(aam)] from boronic acids and anthranilamides on ethyl acetate is described. A series of 1,3,2-benzodiazaborininones were prepared in moderate to excellent yields at room temperature without dehydrating agents, metal catalysts, corrosive acids or other additives. Meanwhile, a multi-gram scale reaction is also performed to ensure the scalability of the reaction, and the product can be conveniently isolated by simple filtration.
Introduction
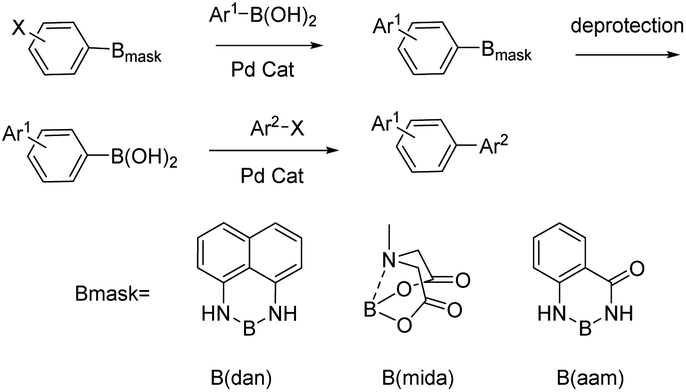
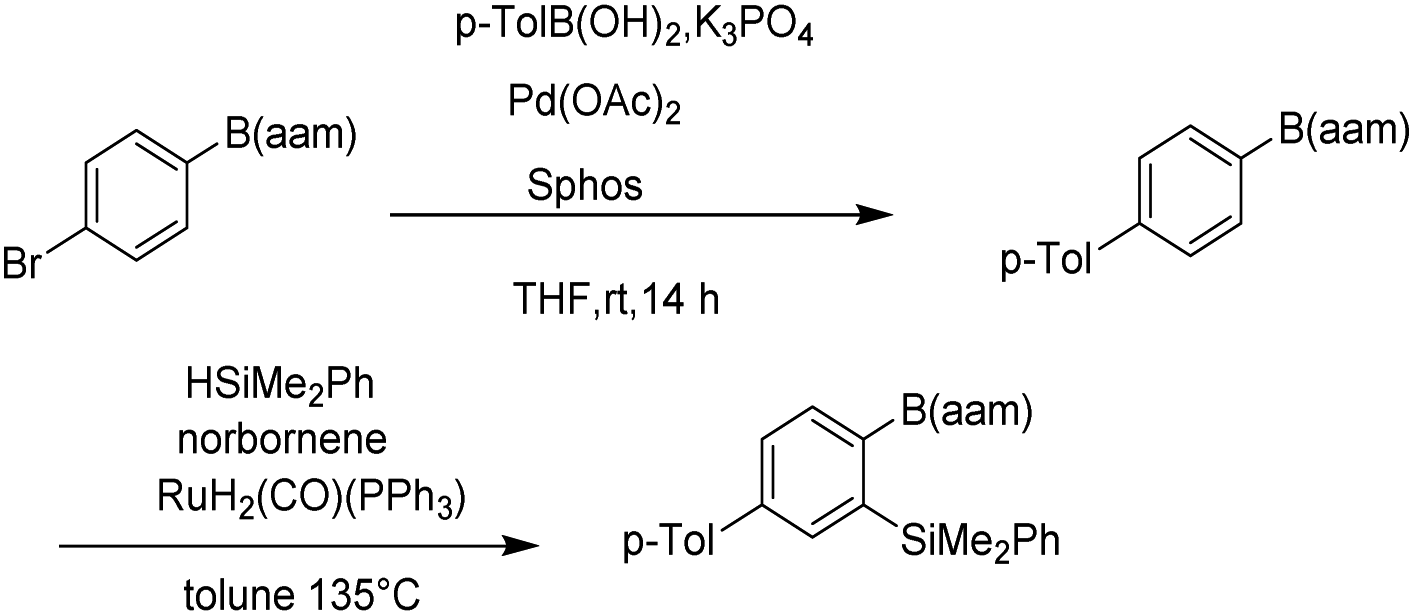
Organoboron compounds play an important role in drug synthesis, medicinal chemistry, and biochemistry.1–6 For example, boronic esters have been developed as triggers for ROS-activated prodrugs for the treatment of cancer patients in recent years.7,8 They also have been regarded as especially popular organometallic reagents9,10 for constructing carbon–carbon and carbon–heteroatom bonds, owing to their broad availability and their compatibility with a multitude of functional groups.11,12 At the same time, Boron(B)–nitrogen(N)-containing heteroaromatic compounds continue to arouse interest in the fields of materials chemistry13–15 and fluorescence imaging.16–20 Since Suginome reported on boron-masking strategy in Suzuki–Miyaura coupling (SMC) using 1,8-diaminonaphthalene (danH2)21–24 and N-methyliminodiacetoxy (mida)25–29 as boron-masking groups, iterative SMC has been widely used in the synthesis of complex oligoaromatic hydrocarbons and natural products (Fig. 1). Besides, other protected boron moieties30,31 were successively reported later such as 1,3,2-benzodiazaborininones [R–B(aam)].32–35 Furthermore, the unique advantage of B(aam) is that it can serve as an ortho-directing group in catalytic Ar–H silylation34 (Fig. 2) and it plays an important role in the stereoselective borohydride reaction of alkynes catalyzed by iridium.36 Therefore, the synthesis of compound R–B(aam) is of great significance.
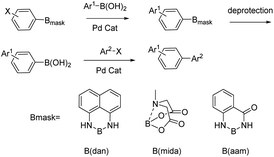 |
| | Fig. 1 Iterative Suzuki–Miyaura coupling using masked haloarylboronic acids for selective synthesis of oligoarenes. | |
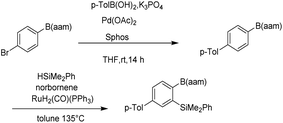 |
| | Fig. 2 Boron-masking strategy with B(aam). | |
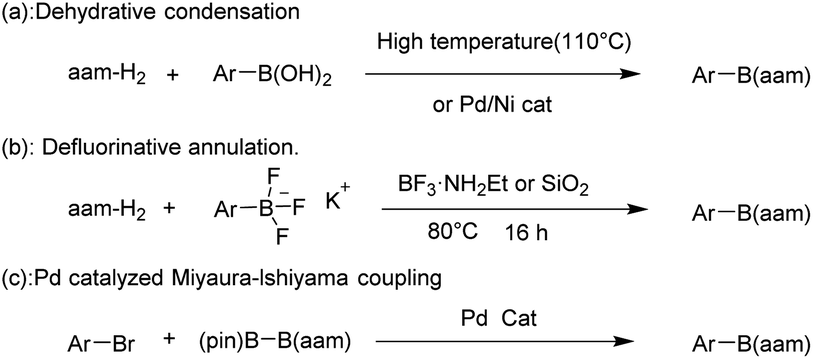
Conventional methods for constructing R–B(aam) reported in the literature include the following synthetic protocols: condensation reaction of boric acids with anthranilamide (aam-H2) in high temperature (110 °C)37 or Ni/Pd-catalyzed.38 Besides, the synthesis of these compounds by the reaction of potassium phenyltrifluoborate with aam-H2 (ref. 39) and the reaction of Ar–X with (pin)B–B(aam) (pin = pinacol ester) catalyzed by Pd40 (Fig. 3). Although high yields can be achieved using these methods, several factors must be considered, including strict reaction conditions that always involve high temperatures, long reaction times, and complicated combinations of reagents. From the green chemistry point of view, the use of green catalysts41,42 or catalyst-free reactions is the mainstream. There is still a great need to develop simple, efficient, clean and high-yield methods to synthesize these compounds.
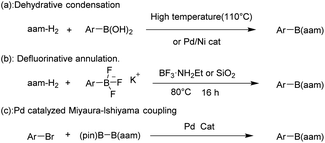 |
| | Fig. 3 Common methods for the synthesis of R–B(aam). | |
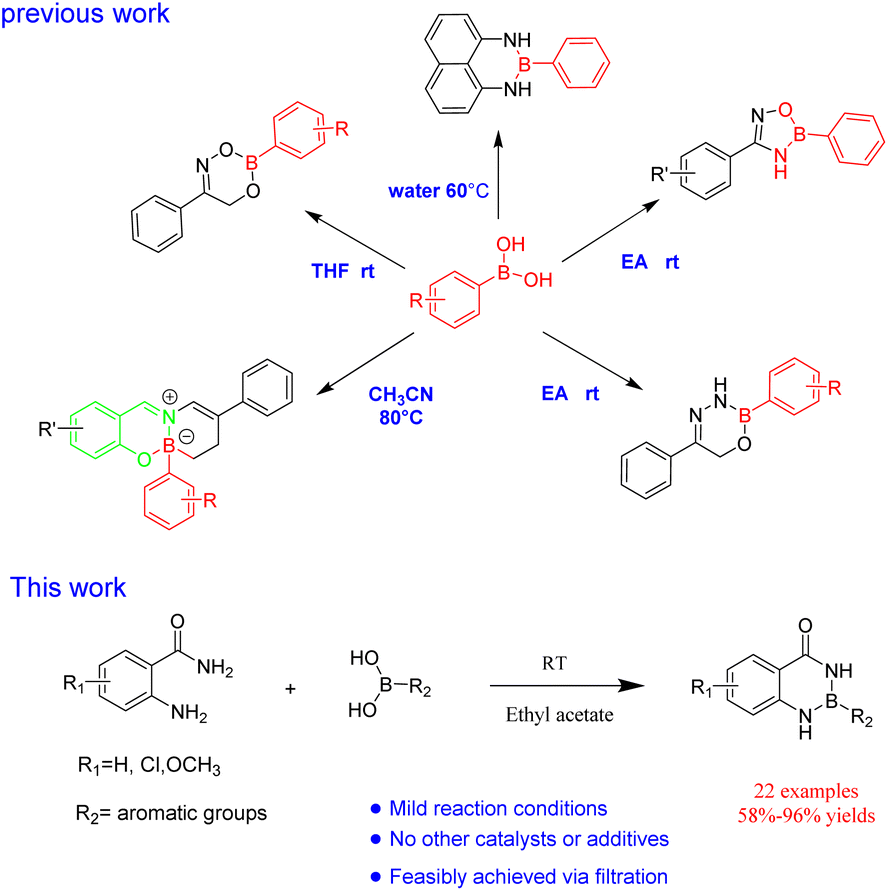
In our previous work, R–B(dan), oxdiazobium compound, dioxazobium compound and oxdiazobium compound were synthesized by a simple method43,44(Fig. 4). At the same time, the green synthesis of borazine heterocycles should be further expanded. Herein, we report an efficient protocol for the synthesis of R–B (aam) in ethyl acetate without any other catalysts or additives (Fig. 4).
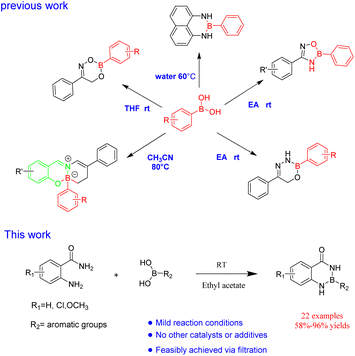 |
| | Fig. 4 The synthesis of R–B(aam). | |
Results and discussion
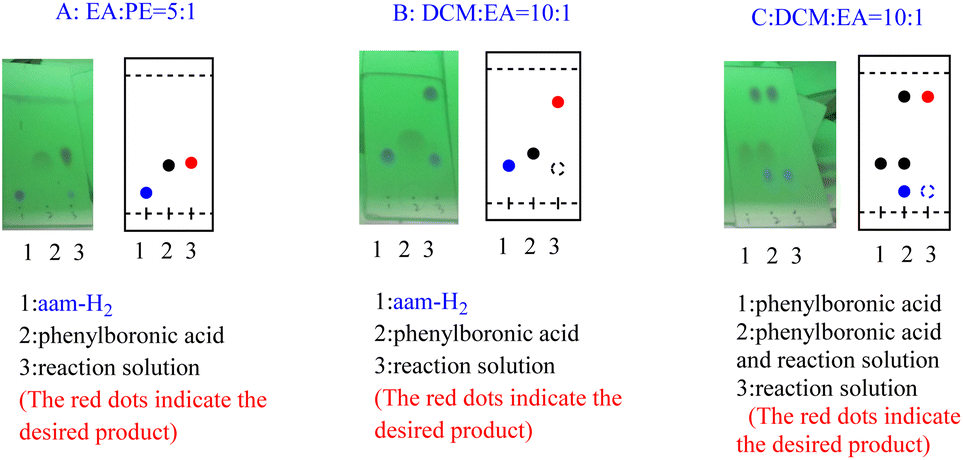
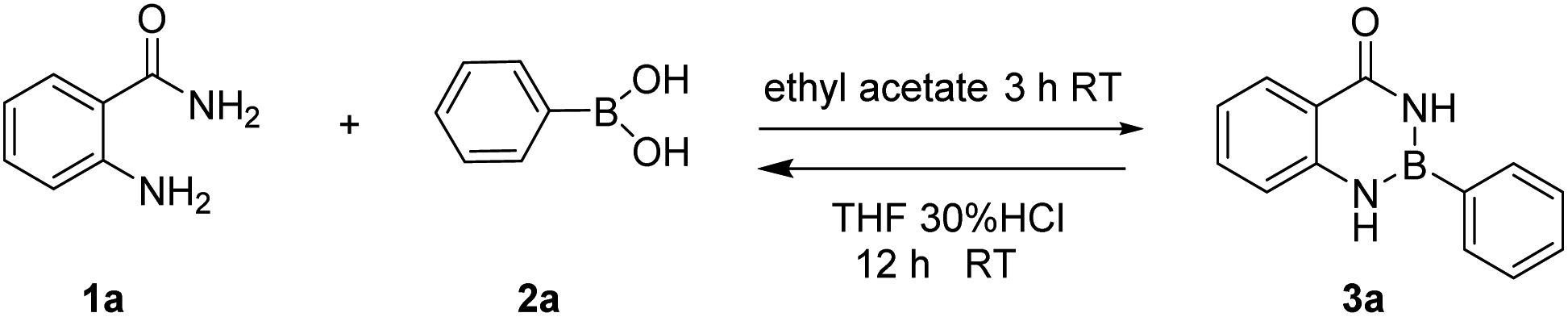
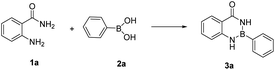
In our initial trials, we first carried out the reaction of amm-H2 with phenylboronic using TiCl4 as a dehydrating agent in the tetrahydrofuran (THF) at room temperature for 4 h. The product 3a was obtained in 93% yield (Table 1, entry 1), The structure of the product was established by NMR analysis. To our delight, the yield of the product was 90% in the absence of TiCl4 at room temperature (Table 1, entry 2). Then the reaction was carried out in different solvents (including ethanol, methanol, N,N-Dimethylformamide (DMF), ethyl acetate, etc.) for 0.5 h (Table 1, entries 3–12) revealed that the highest yield (70%) was obtained in ethyl acetate (Table 1, entry 7). The yields increased gradually with extended reaction times (Table 1, entries 13–15), and an excellent yield of Ar–B(aam) was generated in 93% (Table 1, entry 15) as the reaction times was increased to 3 hours. Interestingly, the desired product 3a was precipitated from the ethyl acetate (Fig. 5) and could be isolated by simple filtration in 75% yield (93% yield for column chromatography). But with the increase of reaction time and temperature continuously, the reaction failed to give the expected yield (Table 1, entries 16–19). The optimum conditions were chosen for 3 h at room temperature (RT) in this reaction system. It was worth nothing that the progress of the reaction was monitored by TLC (DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EA = 10:1) in all the cases (Fig. 6).
:EA = 10:1) in all the cases (Fig. 6).
Table 1 Optimization of reaction conditionsa
|
| Entry |
Catalyst |
Solvent |
Temp |
Time (h) |
Yieldb (%) |
| Reaction condition: 1 (1 mmol), 2 (0.83 mmol), TiCl4 (16 mol%), solvent (3 mL). Isolated yield for column chromatography. RT: room temperature. |
| 1 |
TiCl4 |
THF |
RTc |
4 |
93 |
| 2 |
— |
THF |
RTc |
4 |
90 |
| 3 |
— |
THF |
RTc |
1 |
50 |
| 4 |
— |
Ethanol |
RTc |
1 |
62 |
| 5 |
— |
Methanol |
RTc |
1 |
61 |
| 6 |
— |
DMF |
RTc |
1 |
30 |
| 7 |
— |
Ethyl acetate |
RTc |
1 |
70 |
| 8 |
— |
DMSO |
RTc |
1 |
20 |
| 9 |
— |
Water |
RTc |
1 |
15 |
| 10 |
— |
Dichloromethane |
RT |
1 |
68 |
| 11 |
— |
Chloroform |
RT |
1 |
61 |
| 12 |
— |
Ether |
RT |
1 |
60 |
| 13 |
— |
Ethyl acetate |
RTc |
1.5 |
78 |
| 14 |
— |
Ethyl acetate |
RTc |
2 |
82 |
| 15 |
— |
Ethyl acetate |
RTc |
3 |
93 |
| 16 |
— |
Ethyl acetate |
RTc |
3.5 |
93 |
| 17 |
— |
Ethyl acetate |
RTc |
4 |
93 |
| 18 |
— |
Ethyl acetate |
40 °C |
3 |
93 |
| 19 |
— |
Ethyl acetate |
60 °C |
3.5 |
93 |
 |
| | Fig. 5 Reaction phenomenons and results. | |
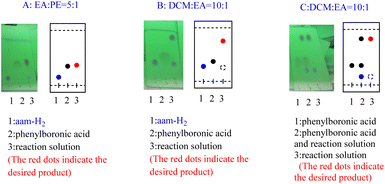 |
| | Fig. 6 The progress of the reaction was monitored by TLC. | |
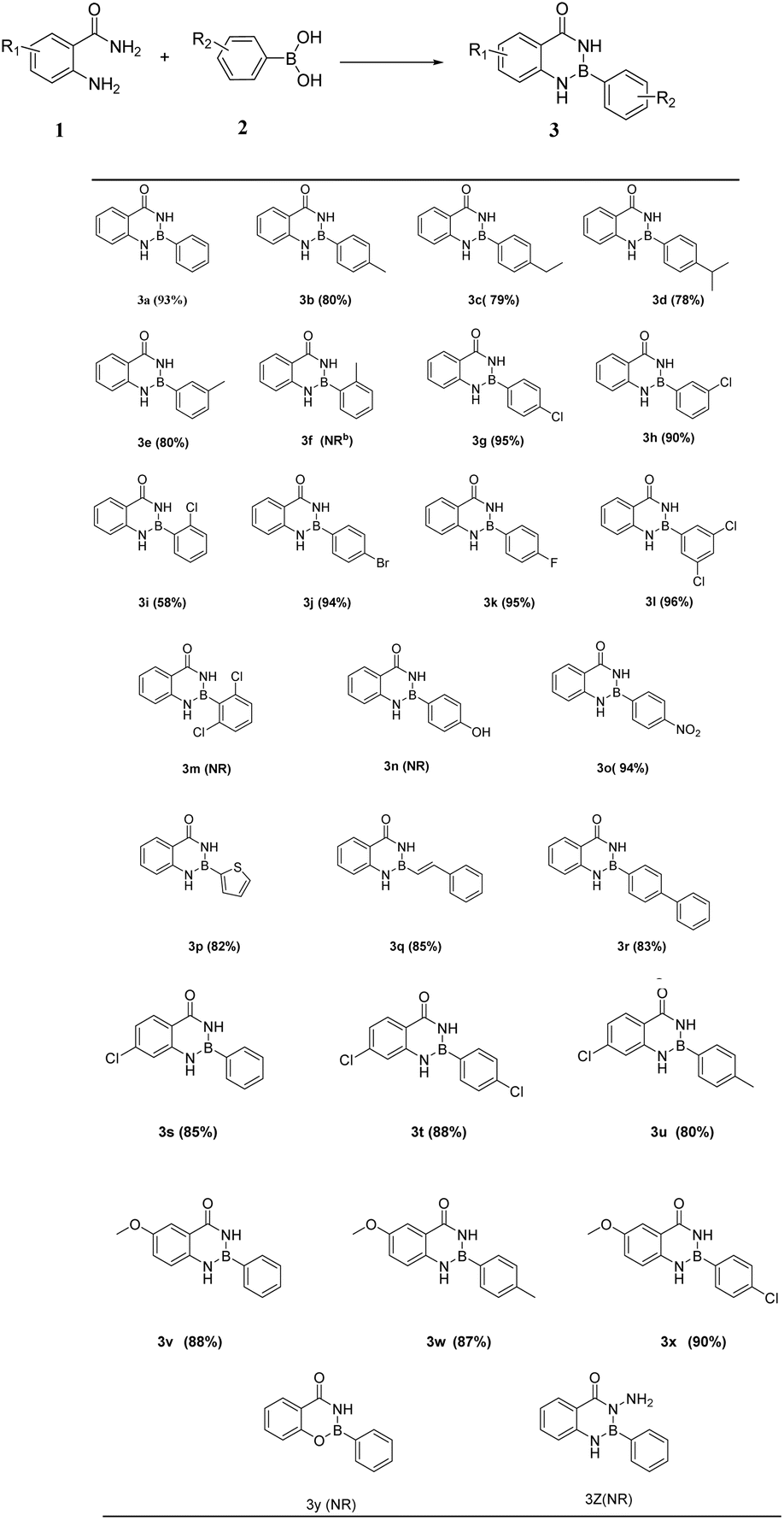
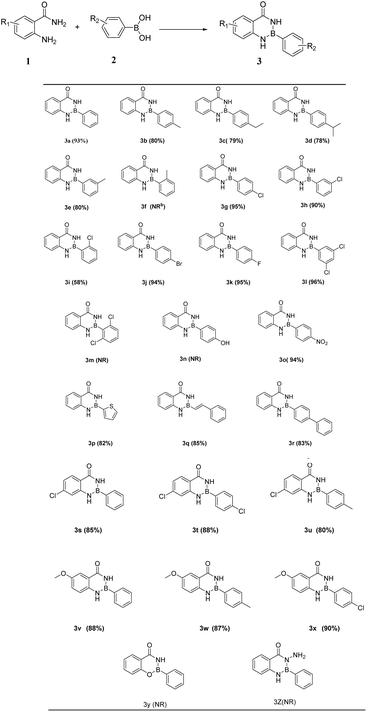
With the optimized reaction conditions in hand, substituted anthranilamides 1 with boronic acids 2 were used to investigate the scope of the reaction. Results were displayed in Table 2. Various phenylboronic acids containing different substituent on the para-position, such as methyl, ethyl, isopropyl and halogen groups, and the product (Table 2 3b-3d, 3g, 3j, 3k) was obtained in high yield of 80–95%. Moreover, one or two different groups were introduced into the meta-position of phenylboronic acid, and the yield of the product (Table 2 3e, 3h, 3l) also was 80–96%. Similarly, the reaction was also applicable to boric acid containing different aromatic groups, such as 4-vinylphenylboronic acid, 2-thiopheneboronic acid and 4-biphenylboronic acid, and could still proceed smoothly in 82–85% yield (Table 2 3p-3r). However, it should be noted that a halogen in the ortho-position of phenylboronic acid (Table 2 3i) gave the target product only in 58%. Especially, phenylboronic acid couldn't react with aam-H2 when it contains a methyl group at the ortho-position (Table 2 3f), which indicates that steric hindrance has a great influence. And boronic acids bearing strong electron-withdrawing substituents such as nitro underwent the reaction smoothly to afford target product 3o. Nevertheless, product formation was not observed when the strong electron-donating substituents such as hydroxyl group were attached (Table 2 3n). Thus, the electronic effects of substituents on the phenylboronic acid showed obvious influence on the reaction outcome. To further examine the substrate scope of the reaction, anthranilamides with substituents on benzene ring were employed. All products (Table 2 3s-3x) were afforded in good yields followed by the same procedure. In addition, anthranilic acid hydrazide and salicylamide do not react with phenylboronic acid.
Table 2 Substrate scope for the synthesis of Oxadiazaborole 3a,b
| Reaction conditions: 1 (1 mmol),2 (0.83 mmol), ethyl acetate (3 mL), 3 h, RT (room temperature), yields refer to isolated yield for column chromatography. NR: no reaction. |
 |
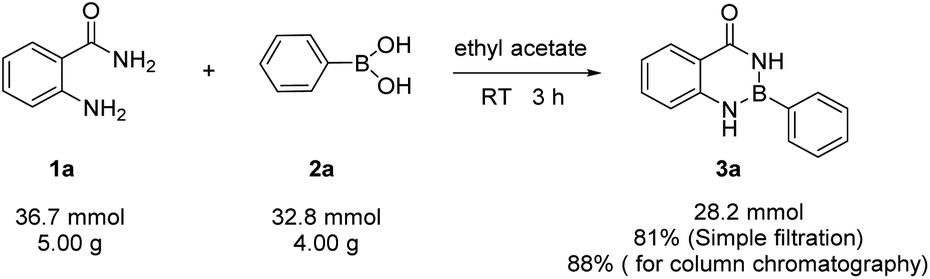
The gram-scale reaction was also carried out to demonstrate the practicability of the methodology (Scheme 1). The aam-H2 (5.00 g, 36.7 mmol) was treated with phenylboronic (4.00 g, 32.8 mmol) in ethyl acetate (100 mL). As expected, the product Ar–B(aam) 3a was obtained in 88% yield (6.42 g, by column chromatography purification) which indicted the scalability of the reaction. Furthermore, the desired product could be isolated by simple filtration in 81% yield (5.96 g). Therefore, with merit of simplicity, convenience and quickness, this method can be applied in industrial production. Moreover, Hydrolysis reaction of Ar–B(aam) were performed as follows (Scheme 2) from the reaction conditions found in the literature.21,45
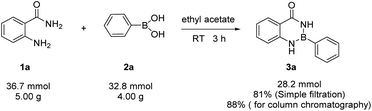 |
| | Scheme 1 Multi-gram scale reaction of aam-H2 with phenylboronic acid. | |
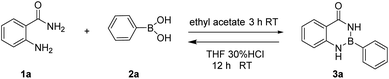 |
| | Scheme 2 Hydrolysis reaction of Ar–B(aam). | |
Conclusions
We have developed a mild and efficient protocol for the synthesis of R–B(aam) through the reaction of boric acids with anthranilamides. Moderate to excellent yields of the products were obtained for 3 h at room temperature without any catalysts or other additives. The multi-gram scale reaction was also found that the product in was afforded excellent yields successfully, and the product can be conveniently isolated by simple filtration. Considering the potential importance of the R–B(aam) in SMC reactions and materials science, the newly developed green methodology will be efficacious to chemists and industrialists.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- S. J. Baker, J. W. Tomsho and S. J. Benkovic, Chem. Soc. Rev., 2011, 40, 4279–4285 RSC.
- S. Kuwano, Y. Hosaka and T. Arai, Org. Biomol. Chem., 2019, 17, 4475–4482 RSC.
- F. Meng, K. P. McGrath and A. H. Hoveyda, Nature, 2014, 513, 367–374 CrossRef CAS PubMed.
- G. R. Mereddy, A. Chakradhar, R. M. Rutkoski and S. C. Jonnalagadda, J. Organomet. Chem., 2018, 865, 12–22 CrossRef CAS.
- T. Ogiyama, M. Yamaguchi, N. Kurikawa, S. Honzumi, Y. Yamamoto, D. Sugiyama and S. Inoue, Bioorg. Med. Chem., 2016, 24, 3801–3807 CrossRef CAS PubMed.
- C. Xu, H. Li, Z. Wang, X. Lou and W. Fu, Monatsh. Chem., 2014, 145, 767–773 CrossRef CAS.
- Y. Ai, O. N. Obianom, M. Kuser, Y. Li, Y. Shu and F. Xue, ACS Med. Chem. Lett., 2019, 10, 127–131 CrossRef CAS PubMed.
- X.-M. Jiang, X.-R. Liu, A. Chen, X.-Z. Zou, J.-F. Ge and D.-W. Gao, Eur. J. Org. Chem., 2022, e202101463 CAS.
- A. Yuasa, K. Nagao and H. Ohmiya, Beilstein J. Org. Chem., 2020, 16, 185–189 CrossRef CAS PubMed.
- T. Wakamatsu, K. Nagao, H. Ohmiya and M. Sawamura, Beilstein J. Org. Chem., 2015, 11, 2444–2450 CrossRef CAS PubMed.
- T. Ohe, N. Miyaura and A. Suzuki, Synlett, 1990, 221–223 CrossRef CAS.
- W. J. Teo, X. X. Yang, Y. Y. Poon and S. Z. Ge, Nat. Commun., 2020, 11, 8 CrossRef PubMed.
- R. N. Dhital and H. Sakurai, Asian J. Org. Chem., 2014, 3, 668–684 CrossRef CAS.
- X. Y. Lu, C. T. Yang, J. H. Liu, Z. Q. Zhang, X. Lu, X. Lou, B. Xiao and Y. Fu, Chem. Commun., 2015, 51, 2388–2391 RSC.
- L.-J. Xiao, L. Cheng, W.-M. Feng, M.-L. Li, J.-H. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2018, 57, 461–464 CrossRef CAS PubMed.
- K. Boknevitz, J. S. Italia, B. Li, A. Chatterjee and S. Y. Liu, Chem. Sci., 2019, 10, 4994–4998 RSC.
- P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed.
- F. Lindl, S. Lin, I. Krummenacher, C. Lenczyk, A. Stoy, M. Mueller, Z. Lin and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 338–342 CrossRef CAS PubMed.
- W. M. Wan, D. Tian, Y. N. Jing, X. Y. Zhang, W. Wu, H. Ren and H. L. Bao, Angew. Chem., Int. Ed., 2018, 57, 15510–15516 CrossRef CAS PubMed.
- P. Zhao, D. O. Nettleton, R. G. Karki, F. J. Zecri and S. Y. Liu, ChemMedChem, 2017, 12, 358–361 CrossRef CAS PubMed.
- N. Iwadate and M. Suginome, J. Organomet. Chem., 2009, 694, 1713–1717 CrossRef CAS.
- N. Iwadate and M. Suginome, Org. Lett., 2009, 11, 1899–1902 CrossRef CAS PubMed.
- N. Iwadate and M. Suginome, J. Am. Chem. Soc., 2010, 132, 2548 CrossRef CAS PubMed.
- H. Noguchi, K. Hojo and M. Suginome, J. Am. Chem. Soc., 2007, 129, 758–759 CrossRef CAS PubMed.
- G. R. Dick, E. M. Woerly and M. D. Burke, Angew. Chem., Int. Ed., 2012, 51, 2667–2672 CrossRef CAS PubMed.
- E. P. Gullis and M. D. Burke, J. Am. Chem. Soc., 2007, 129, 6716–6717 CrossRef PubMed.
- C. He and X. Pan, Macromolecules, 2020, 53, 3700–3708 CrossRef CAS.
- D. M. Knapp, E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2009, 131, 6961 CrossRef CAS PubMed.
- J. Q. Li, A. S. Grillo and M. D. Burke, Acc. Chem. Res., 2015, 48, 2297–2307 CrossRef CAS PubMed.
- S. Darses and J.-P. Genet, Chem. Rev., 2008, 108, 288–325 CrossRef CAS PubMed.
- G. A. Molander and N. Ellis, Acc. Chem. Res., 2007, 40, 275–286 CrossRef CAS PubMed.
- H. Ihara, M. Koyanagi and M. Suginome, Org. Lett., 2011, 13, 2662–2665 CrossRef CAS PubMed.
- S. Kamio, I. Kageyuki, I. Osaka and H. Yoshida, Chem. Commun., 2019, 55, 2624–2627 RSC.
- M. Koyanagi, N. Eichenauer, H. Ihara, T. Yamamoto and M. Suginome, Chem. Lett., 2013, 42, 541–543 CrossRef CAS.
- S. Kamio, I. Kageyuki, I. Osaka and H. Yoshida, Chem. Commun., 2019, 55, 2624–2627 RSC.
- H. Yoshida, M. Kimura, H. Tanaka, Y. Murashige, I. Kageyuki and I. Osaka, Chem. Commun., 2019, 55, 5420–5422 RSC.
- S. S. Chissick, M. J. S. Dewar and P. M. Maitlis, J. Am. Chem. Soc., 1961, 83, 2708–2711 CrossRef CAS.
- H.-J. Wang, M. Zhang, W.-J. Li, Y. Ni, J. Lin and Z.-H. Zhang, Adv. Synth. Catal., 2019, 361, 5018–5024 CrossRef CAS.
- G. H. M. Davies, A. Mukhtar, B. Saeednia, F. Sherafat, C. B. Kelly and G. A. Molander, J. Org. Chem., 2017, 82, 5380–5390 CrossRef CAS PubMed.
- S. Kamio, I. Kageyuki, I. Osaka, S. Hatano, M. Abe and H. Yoshida, Chem. Commun., 2018, 54, 9290–9293 RSC.
- S. P. Ding, M. J. Hulsey, H. An, Q. He, H. Asakura, M. Gao, J. Y. Hasegawa, T. Tanaka and N. Yan, CCS Chem., 2021, 3, 1814–1822 CrossRef CAS.
- M. J. Hülsey, B. Zhang, Z. Ma, H. Asakura, D. A. Do, W. Chen, T. Tanaka, P. Zhang, Z. Wu and N. Yan, Nat. Commun., 2019, 10, 1330 CrossRef PubMed.
- Q. Kuang, X. Hu, Y. Li, S. Shang, X. Huang, S. Liao, Y. Song, W. Ma, S. Li, A. Liu, X. Liu, X. Zhang and J. Yuan, Org. Biomol. Chem., 2022, 20, 6413–6417 RSC.
- S. Liao, X. Hu, Y. Li, X. Wang, D. Li, Q. Wang, Y. Wang, X. Huang, P. Xu, H. Wu, X. Li and J. Yuan, Tetrahedron, 2021, 90, 132205 CrossRef CAS.
- H. Noguchi, K. Hojo and M. Suginome, J. Am. Chem. Soc., 2007, 129, 758–759 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and copies of 1H, 13C, 11B NMR and HRMS. See DOI: https://doi.org/10.1039/d2ra06573h |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a