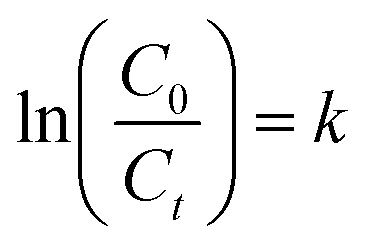DOI:
10.1039/D2RA07509A
(Paper)
RSC Adv., 2023,
13, 2574-2586
Synthesis of cerium oxide embedded perovskite type bismuth ferrite nanocomposites for sonophotocatalysis of aqueous micropollutant ibuprofen†
Received
25th November 2022
, Accepted 7th January 2023
First published on 17th January 2023
Abstract
Ibuprofen is potentially toxic and carcinogenic for freshwater ecosystems and poses a serious threat to human health by affecting kidney function. The present study focused on the sunlight-controlled degradation of ibuprofen from water using a novel magnetically separable cerium oxide-embedded bismuth ferrite heterostructure. Catalysts were synthesized by solvothermal and co-precipitation methods and characterized by X-ray diffractometry, transmission electron microscopy, X-ray photoelectron spectroscopy, UV-vis optical absorption spectroscopy, and nitrogen adsorption. This study investigated the effect of photocatalysis, sonolysis, sonophotolysis, and sonophotocatalysis on the degradation of ibuprofen in water. Pseudo-first-order and second-order kinetics were applied to evaluate the rate of reaction for ibuprofen degradation. The addition of 5% CeO2 to the BiFeO3 significantly increased the surface area and pore volume of bismuth ferrite, which enhanced their photocatalytic degradation efficiency by 2.28 times in terms of ibuprofen mineralization. Sonolysis treatment alone and in combination with photolysis led to the degradation of ibuprofen, but with the formation of intermediate products. Positive synergy was observed when sonolysis was combined with photocatalysis in terms of the mineralization of ibuprofen and the degradation of intermediates along with their parent compound. It was proposed that, compared to photocatalytic mineralization, the ultrasound-assisted advanced oxidation process resulted in the conversion of ibuprofen to its mineralization products.
1. Introduction
Global consumption of pharmaceuticals has increased 2.8 times over the past 15 years.1 The Organization for Economic Co-operation and Development (OECD) countries estimate that US$ 800 billion was spent on prescription medications in 2015.2 In 2013 alone, about 100 new formulations or chemical entities were approved for clinical use by the US Food And Drug Administration (FDA).3 In 2015, the global pharmaceutical market sale was US$ 1.06 trillion with an average annual growth rate of 5.2%. The market size in North America was US$ 363.2 billion, in Europe – US$ 315.1 billion, and Asia – US$ 281.3 billion.4 Thus, the amount of pharmaceuticals in wastewater has increased enormously over the past few years due to their high production and consumption.5 Various persistent pharmaceuticals and personal care products (PPCPs) have been found in aquatic ecosystems, such as wastewater treatment effluents, and ground and surface waters.6 They eventually find their way into the natural environment mainly through wastewater, which is a serious concern to human and aquatic life as it causes toxicity, endocrine disruption, and antibiotic resistance.7
Ibuprofen (IBP) is one of the key pharmaceuticals in the non-steroidal anti-inflammatory drug (NSAIDs) class. IBP is the third most popular drug in the world used to treat fever, pain, inflammation, or minor injuries.8 The annual production of ibuprofen worldwide is more than 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons, and about 70–80% of this amount is excreted by patients in its original form or the form of its metabolites and ends up in wastewater.9 Their detection in the influent and effluent of wastewater treatment plants (WWTPs)10,11 have demonstrated that current systems do not effectively remove this compound before it enters the environment. Hence, the partial removal of ibuprofen by conventional WWTPs suggests the need to develop advanced technologies that can eliminate it. It has been found that advanced oxidation processes (AOPs) based on heterogeneous photocatalysis are the best and most efficient way to degrade toxic organic effluents compared to conventional techniques due to fast oxidation, high efficiency, and cost-effectiveness.12 Recently, a new group of nanomaterials, which exists in the class of perovskite structure, has attracted considerable interest for photocatalytic applications. Among all the ABX3 type photocatalysts,13 a Bi-contained oxide photocatalyst, such as bismuth ferrite (BiFeO3), is a new and promising third-generation photocatalyst sensitive to visible light, having a rhombohedral distorted perovskite for photodegradation of organic pollutants.14 BiFeO3 (BFO) has received a lot of attention due to its remarkable properties, such as the bandgap decreases in the visible light region, exhibiting multiferroic behaviour at room temperature (25 °C), which allows the anions and cations to be efficiently separated, and excellent chemical stability.15 BFO as a multiferroic material exhibits ferroelectric and magnetic ordering, simultaneously, due to the 6 s2 lone pair of electrons in Bi3+ ions and partially filled 3d5 shell of the Fe3+ ions, respectively.16,17 It exhibits multiferroic behaviour due to the simultaneous existence of sites with variable charges and inequivalent magnetic ions.18 So far, BFO has been used to degrade methylene blue,19 methyl orange,20 rhodamine B,21 congo red,22 cefixime,23 oxalic acid and norfloxacin.24
000 tons, and about 70–80% of this amount is excreted by patients in its original form or the form of its metabolites and ends up in wastewater.9 Their detection in the influent and effluent of wastewater treatment plants (WWTPs)10,11 have demonstrated that current systems do not effectively remove this compound before it enters the environment. Hence, the partial removal of ibuprofen by conventional WWTPs suggests the need to develop advanced technologies that can eliminate it. It has been found that advanced oxidation processes (AOPs) based on heterogeneous photocatalysis are the best and most efficient way to degrade toxic organic effluents compared to conventional techniques due to fast oxidation, high efficiency, and cost-effectiveness.12 Recently, a new group of nanomaterials, which exists in the class of perovskite structure, has attracted considerable interest for photocatalytic applications. Among all the ABX3 type photocatalysts,13 a Bi-contained oxide photocatalyst, such as bismuth ferrite (BiFeO3), is a new and promising third-generation photocatalyst sensitive to visible light, having a rhombohedral distorted perovskite for photodegradation of organic pollutants.14 BiFeO3 (BFO) has received a lot of attention due to its remarkable properties, such as the bandgap decreases in the visible light region, exhibiting multiferroic behaviour at room temperature (25 °C), which allows the anions and cations to be efficiently separated, and excellent chemical stability.15 BFO as a multiferroic material exhibits ferroelectric and magnetic ordering, simultaneously, due to the 6 s2 lone pair of electrons in Bi3+ ions and partially filled 3d5 shell of the Fe3+ ions, respectively.16,17 It exhibits multiferroic behaviour due to the simultaneous existence of sites with variable charges and inequivalent magnetic ions.18 So far, BFO has been used to degrade methylene blue,19 methyl orange,20 rhodamine B,21 congo red,22 cefixime,23 oxalic acid and norfloxacin.24
Although BFO has been reported to be one of the most reactive and active photocatalysts in visible light, pure bismuth ferrite has low photodegradation activity for organic pollutants.25 Their photocatalytic efficiency is limited due to the e− – h+ recombination on the surface and in the bulk, the relatively low level of the conduction band to their redox potential, and the limited surface area for the reaction.14 Heterostructure formation is one of the best modification approaches that significantly enhances the performance or photodegradation activity of pristine BFO by improving its stability, dispersion, and recyclability. Because, in heterojunction e− – h+ shuttling from one semiconductor to another inhibits the recombination of charge carriers, as a result, the e− – h+ pair's lifetime increases.26 Over the last few years, advancements in ultrasound-assisted AOP techniques have gained considerable interest in organic pollutants mitigation.27 The increased degradation efficiency of catalysts is due to the synergistic effect of the hybrid system, which produces more reactive species, which promotes the degradation of pollutants.28 Although sonochemical degradation has become effective for mitigation purposes, due to the hydrophilic nature of the degradation intermediates, complete elimination cannot be achieved. To reduce this problem, it is necessary to study the combination of sonochemical processes with advanced oxidation technologies.8 This work involves the fabrication of modified BFO by the formation of a heterostructure with a rare earth oxide, cerium oxide (CeO2). The individual and combined effects of photocatalysis, sonolysis, sonophotolysis, and sonophotocatalysis on ibuprofen degradation were also the focus of the present research. To the best of our knowledge, this is the first study on the formation of CeO2 embedded BFO heterostructure to analyze the effect of CeO2 addition in the BFO lattice. Furthermore, their application with or without the different ultrasonic treatments to examine the photodegradation behaviour of aqueous ibuprofen is another uniqueness of the present study.
2. Materials and methods
2.1. Chemicals
Bismuth nitrate pentahydrate (Bi(NO3)3·5H2O, DAEJUNG), Eisen(iii)-chloride-6-hydrate (FeCl3·6H2O SIGMA-ALDRICH®), ceric ammonium nitrate ((NH4)2Ce (NO3)6, SIGMA-ALDRICH®), nitric acid (HNO3, 65% pure, SIGMA-ALDRICH®), ethylene glycol ((CH2OH)2, AnalaR®), cetrimonium bromide (CTAB) (CH3(CH2)15N(Br)(CH3), SIGMA-ALDRICH®), ammonium hydroxide (NH4OH, 30–33% pure, SIGMA-ALDRICH®), hydrochloric acid (HCl, 37% pure, AnalaR NORMAPUR®) and sodium hydroxide (NaOH, SIGMA-ALDRICH®). All the chemicals were highly pure and used without any further purification.
2.2. Synthesis of photocatalyst
2.2.1. Synthesis of BiFeO3. BiFeO3 was synthesized by dissolving 16 mmol of Bi(NO3)3·5H2O in 15 mL of 3 mol L−1 nitric acid solution and then adding it to ethylene glycol containing 16 mmol FeCl3·6H2O while stirring on a magnetic stirrer at 100 °C for 2 h. Then the synthesized product was decomposed in ambient air at 500 °C for 2 h after drying at 130 °C.
2.2.2. Synthesis of CeO2/BiFeO3. To prepare the CeO2/BiFeO3 nanocomposite, the calcined BiFeO3 particles were dispersed in an aqueous solution containing the desired amount of cerium ammonium nitrate as a cerium precursor. Then, the appropriate amount of CTAB in distilled water was added to the solution. Finally, the contents were stirred for 2 h after adding 10 mL of NH3 solution. Then the solution was filtered and dried at 110 °C for 5 h. Finally, the synthesized powder was calcined at 600 °C for 2 h.
2.3. Characterization of photocatalyst
Various techniques were used to characterize the synthesized photocatalyst. To analyze the crystallinity X-ray diffraction (XRD) patterns were obtained by Bruker D8 Discover XRD instrument. A scan of incident angle from 10° to 70° was obtained using CuKα 1.5418 radiation. Particle size and surface morphology were investigated using transmission electron microscopy (TEM). JEOL (JEM-2100) transmission electron microscope was used to record electron micrographs of synthesized nanomaterials. To analyze the chemical composition of the prepared samples, X-ray photoelectron spectroscopy (XPS) was carried out using the Omicron system (Al Kα 1486.7 eV X-ray source operated at 15 keV) at constant analyzer energy (CAE) of 100 eV for a survey and 20 eV for a detailed scan. UV-visible optical absorption spectra were obtained from 250 to 800 nm using PerkinElmer (Lambda 950) spectrophotometer to study the optical properties of the synthesized photocatalyst. The N2 adsorption–desorption isotherm was obtained using Micromeritics ASAP 2460 2.01 system. All samples were degassed overnight at 150 °C before analysis. The bandgap energy of the synthesized photocatalyst was calculated using eqn (1):where “Eg” is bandgap energy (eV) and “λ” is the wavelength (nm).29
2.4. Sono/photocatalysis
Photocatalytic or sonophotocatalytic treatment of ibuprofen was performed using 20 mg L−1 concentration of ibuprofen in a Pyrex glass beaker, which was stirred on a magnetic stirrer in direct sunlight for 3 h. SW-3H SonoSwiss water bath ultrasonicator (ultrasonic frequency: 50/60 Hz, ultrasonic power: 280 W) was used to provide ultrasound waves for sonophotocatalysis. The experiments were carried out during September and October, 2019 with an average sunlight intensity ranging from 8 × 104 lux to 9 × 104 lux. SMART SENSOR® Intell Instruments™ digital lux meter was used to measure the intensity of sunlight. The pollutant-to-catalyst mass ratio was varied by changing the catalyst dose in the range from 0.25 g L−1 to 1 g L−1. The pH of the solution was adjusted to 3–9 by adding HCl and NaOH. AD8000 Adwa pH meter was used to adjust the pH of the solution. The zero-charge point of the photocatalyst was determined by a simple salt addition method. The stability of the synthesized photocatalyst was determined by reusing it for three consecutive cycles. For this, the photocatalyst was separated from the ibuprofen solution and then thoroughly washed with distilled water and dried at 110 °C before further use.
2.5. Analytical determinations
UV-vis spectrophotometer (T80+ – PG instruments) was used to monitor the ibuprofen salt by measuring the sample absorbance. The quantitative estimation of photocatalytic degradation of ibuprofen was evaluated using the calibration curve. The degradation of the drug was examined by the destruction of ibuprofen's corresponding band at 222 nm. The percentage degradation of ibuprofen was calculated by using the given formula in eqn (2):| | |
% degradation = (((C0 − Ct)/C0) × 100)
| (2) |
here, C0 and Ct are the initial concentration and the final concentration after time t, respectively. The photocatalytic degradation rate of ibuprofen was quantified by applying the following relationship illustrated in eqn (3):| |
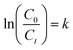 | (3) |
Here, “k” is the rate constant, “C0” is the initial concentration of ibuprofen and “Ct” is the concentration of ibuprofen at the time “t”. The rate constant (k) values were obtained from the slope of their regression plots and the regression coefficient (R2). The formation of degradation intermediates was analyzed by simply examining their corresponding band at 262 nm in a UV-vis spectrophotometer, and this was also confirmed by high-performance liquid chromatography (HPLC). Chromatographic separation was performed on a UFLC SHIMADZU system equipped with an online mobile phase degasser unit (model: DGU-20 A5R), a 20 μL sample loop, a pump (model: LC-20AT), a photodiode array detector (model: SPD-20A), and a column oven (CTO-20AC), all obtained from Shimadzu Corporation (Kyoto, Japan). The mobile phase used consisted of a mixture of acetonitrile and 0.2% formic acid in water in a ratio of 60:40 (v/v) with a flow rate of 0.8 mL min−1. Separation was performed on a LiChrospher C18 HPLC 150 × 4.6 mm × 5 μm column obtained from Merck (Darmstadt, Germany). Photodiode array detection was performed at wavelengths of 200 nm. To prepare samples for HPLC analysis, all liquid samples were filtered using 0.45 μm membrane filters. 20 mL of chloroform was used twice to extract each 10 mL sample with a separating funnel. Then these samples were allowed to evaporate at room temperature. Finally, the obtained residues were dissolved in 10 mL of analytical-grade methanol.
3. Results and discussion
3.1. Photocatalyst characterization
Cerium oxide and bismuth ferrite catalysts were synthesized by solvothermal and co-precipitation methods. The structural characteristics and surface morphology of the synthesized materials were examined using X-ray diffraction (XRD) and transmission electron microscopy (TEM), respectively. Fig. 1a shows the XRD pattern of the synthesized photocatalysts and Fig. 1(b and c) represents the comparison of synthesized photocatalysts with their standard data as XRD stick patterns. The XRD pattern of pristine CeO2 shown in Fig. 1 is in good agreement with the reference card (JCPDS No. 3403940).30 All diffraction peaks in these spectra identified as (111), (200), (220), (311), (222) and (400) are crystal planes of CeO2, which indicates their cubic fluorite phase crystals.31 According to JCPDS No. 86–1518, the diffraction peaks at 2θ values 32.6, 33.5, 39.3, 41, 46.7, 49.8, 57.5, 58.8, corresponding to (104), (110), (006), (202), (024), (116), (018) and (214) crystal planes of pure BiFeO3 phase with rhombohedral perovskite structure32 as shown in Fig. 1. Additional peaks were also observed at 2θ values 24.2, 26, 35.7, 54.2, 63.1 corresponding to the (120), (021), (310), (142) and (004) crystal planes of Bi2Fe4O9 according to the JCPDS No. 25–0090. The appearance33 of Bi2Fe4O9 is consistent with other synthesis reports.34 It may not be possible to avoid these secondary phases due to the crystallization of BiFeO3 in the air during its synthesis. Moreover, it is difficult to synthesize the single-phase BiFeO3 because Bi2Fe4O9 is thermodynamically more stable than BiFeO3.35
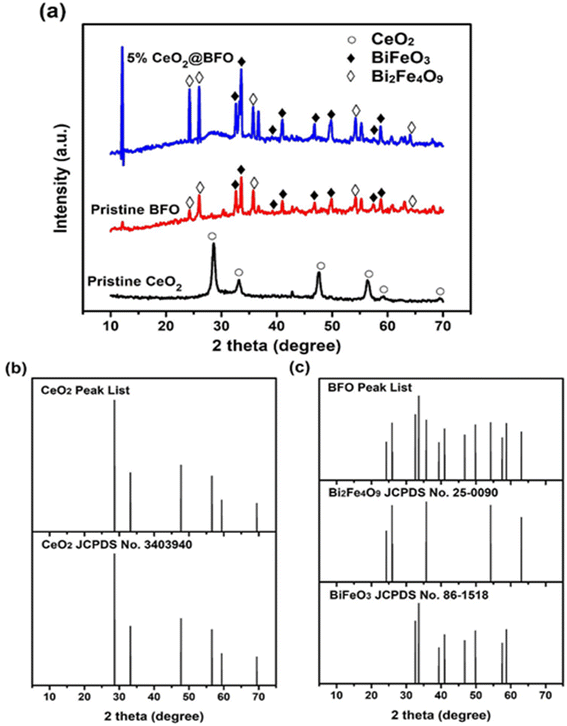 |
| | Fig. 1 (a) XRD patterns of pristine CeO2; pristine BFO; and 5% CeO2@BFO nanocomposite, (b and c) XRD stick pattern comparison of CeO2, BiFeO3 and Bi2Fe4O9. | |
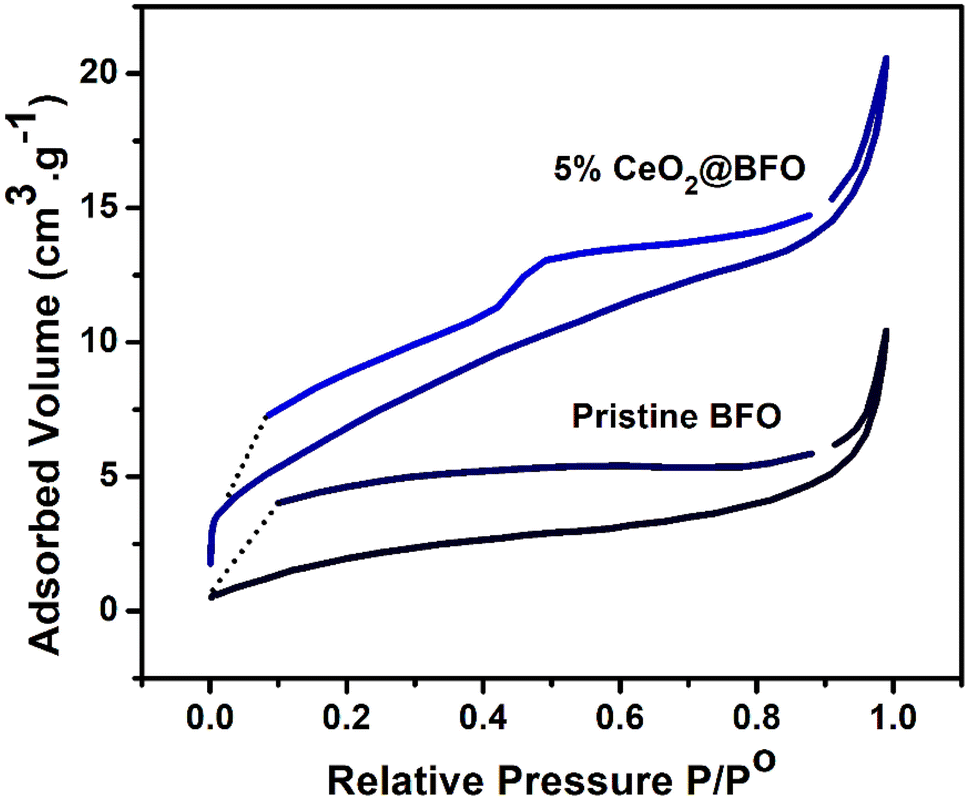
The N2 adsorption–desorption isotherm of the synthesized photocatalyst exhibited a type-IV isotherm with the formation of an H1-type hysteresis loop (Fig. 2), which confirms their mesoporous structure.36 Analysis of the adsorption data by the BET method revealed that the surface area of 5% CeO2@BFO nanocomposite was 26.22 m2 g−1, which was higher than the original BFO (8.45 m2 g−1). The observed inflection intensity due to the capillary condensation37 at relative pressures (p/p0) ranged about 0.1 and 1 indicating the mesoporous characteristics of the synthesized photocatalyst. Furthermore, the pore volumes of the original BFO and 5% CeO2@BFO were obtained to be 0.016 cm3 g−1 and 0.032 cm3 g−1, respectively. It can be assumed that the formation of the nanocomposite led to an increase in the surface area and pore volume of the pristine BFO, which is an important factor in increasing the photocatalytic efficiency of the photocatalyst.
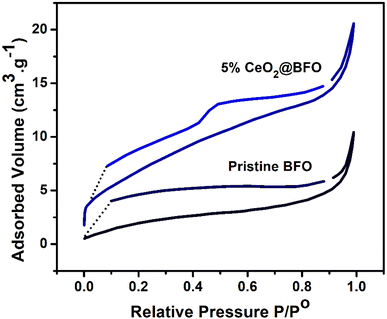 |
| | Fig. 2 N2 adsorption–desorption isotherms of the pristine BFO and 5% CeO2@BFO. | |
The surface morphology of the synthesized photocatalyst was investigated by transmission electron microscopy (TEM) as shown in Fig. 3. Fig. 3(a and b) represents the TEM image of pristine CeO2 appearing as irregularly shaped particles. As shown in Fig. 3(c and d) the pristine BFO nanoparticles were roughly spherical and were randomly distributed with an average particle size of 5–25 nm. The TEM image of the 5% CeO2@BFO composite, shown in Fig. 3(e and f), revealed that CeO2 particles were sparsely deposited on the agglomerated clusters of BFO.
 |
| | Fig. 3 TEM images of (a and b) pristine CeO2; (c and d) pristine BFO and (e and f) 5% CeO2@BFO composite. | |
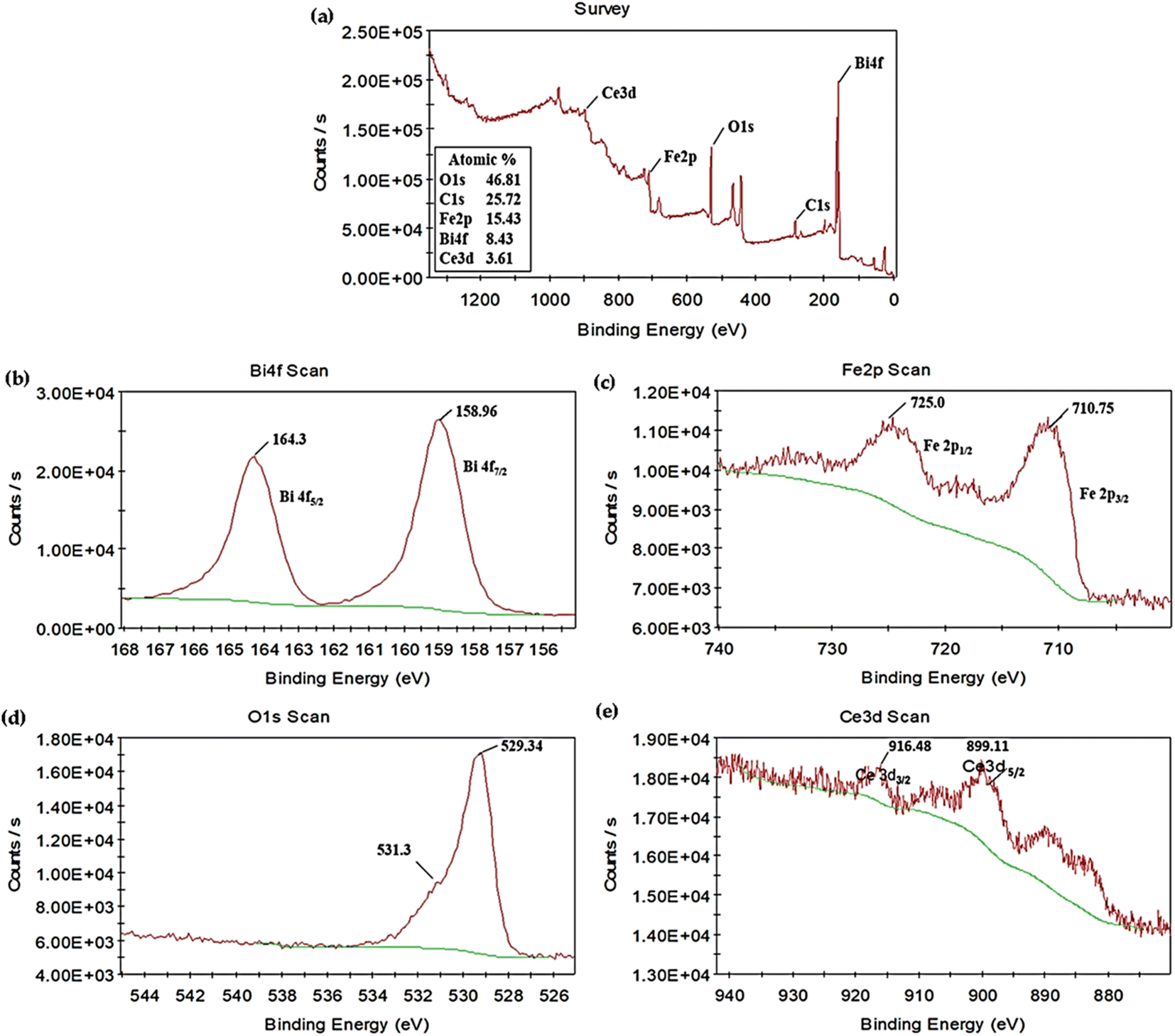
X-ray photoelectron spectroscopy was performed to further analyze the chemical composition of the synthesized photocatalyst. The full-scan XPS spectrum of 5% CeO2@BFO photocatalyst illustrated in Fig. 4a confirmed the coexistence of the constituent elements Bi, Fe, O and Ce.24 The C element was also observed in the spectrum, possibly due to contamination of the surface in the air. The high-resolution XPS core spectra of Bi 4f, Fe 2p, O 1s and Ce 3d are also represented in Fig. 4b–e. As shown in Fig. 4b, two peaks corresponding to Bi 4f5/2 and Bi 4f7/2 were found with binding energies values of 164.3 and 158.96 eV, respectively,14 confirming the Bi3+ characteristics of bismuth.24 Fig. 4c shows the element Fe in the XPS spectrum, which exhibited two peaks around 725.0 and 710.15 eV, corresponding to Fe 2p1/2 and Fe 2p3/2, respectively,21 which confirms the Fe3+ oxidation state of iron.24 Binding energies around 531.3 and 529.34 eV, as shown in Fig. 4d, correspond to oxygen adsorbed on the surface and Fe–O bonds in the lattice. Moreover, in Fig. 4e, two major peaks were observed at binding energies of 916.48 and 899.11 eV, corresponding to Ce 3d3/2 and Ce 3d5/2, indicating the Ce4+ form of cerium.38 Based on the results of the XPS analysis, it was concluded that the element Bi, Fe and Ce existed in the valence state Bi3+, Fe3+ and Ce4+ in as-synthesized 5% CeO2@BFO photocatalyst.
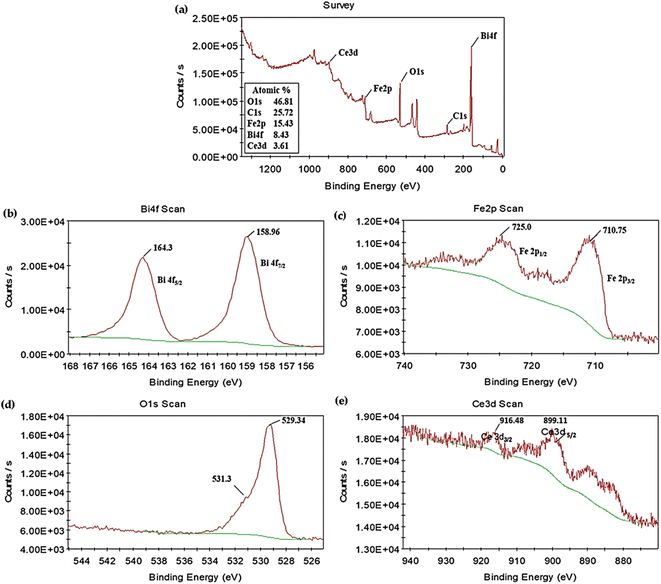 |
| | Fig. 4 (a) XPS survey spectra of 5% CeO2@BFO nanocomposite; high-resolution XPS spectra of (b) Bi 4f (c), Fe 2p (d), O 1s and (e) Ce 3d. | |
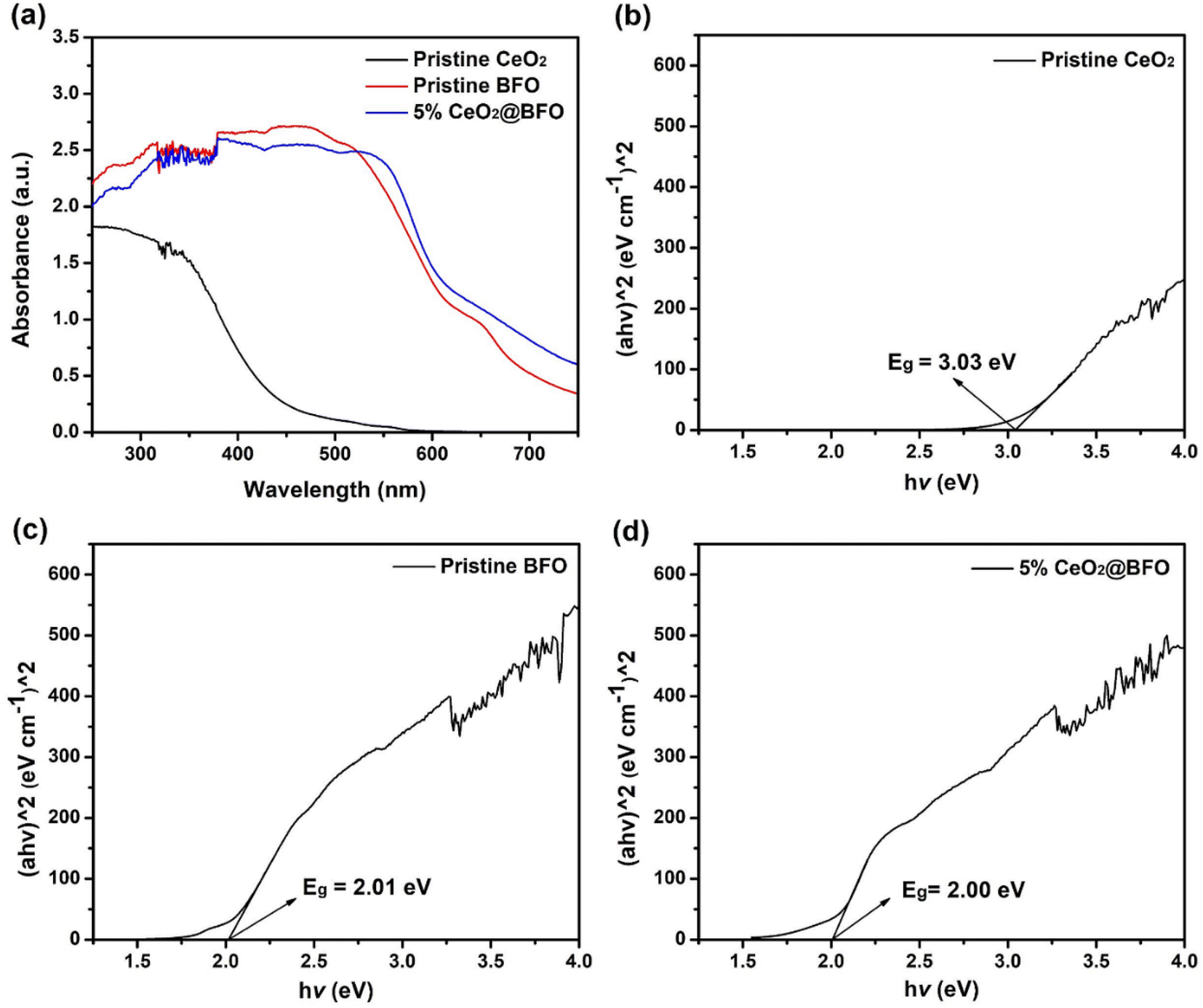
The UV-vis optical absorption spectra as shown in Fig. 5a were used to measure the optical absorption and bandgap energy of the photocatalysts. From the experimental data, the bandgap absorption edge of CeO2 was around 440 nm. While BFO and their composites exhibited absorption around 670 nm, which is clearly in the visible light region, as shown in Fig. 5a. The absorption spectrum was used to create the Tauc's plots ((αhv)^2 vs. hv)21 as shown in Fig. 5 (b–d), from which the bandgap energies of CeO2, BFO and 5% CeO2@BFO were estimated to be 3.03 eV, 2.01 eV and 2.00 eV, respectively. The optical absorption results of pristine BFO are very similar to the previously reported study.14 It was observed that when the composite of bismuth ferrite (BiFeO3) with CeO2 was prepared, there was a slight change in the bandgap energies of BFO and 5% CeO2@BFO, which indicated that their optical absorption is very similar. So, from the above results, it can be concluded that the synthesized photocatalyst may promote the degradation of organic pollutants upon exposure to visible light.
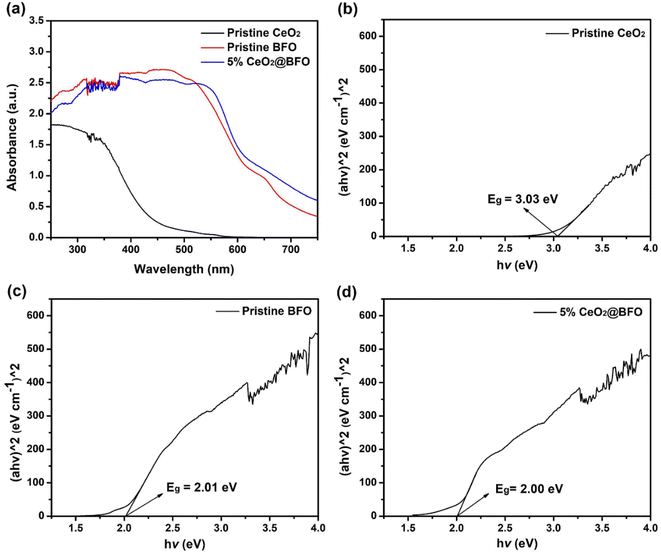 |
| | Fig. 5 (a) UV-vis optical absorption spectra and corresponding Tauc's plots of (b) Pristine CeO2, (c) BFO and (d) CeO2@BFO composite. | |
3.2. Effect of CeO2 loading on IBP photocatalytic degradation
Fig. 6a shows the efficiency of photodegradation of IBP in direct sunlight with different amounts of CeO2 (from 2% to 10% CeO2) in the nanocomposite. 13% degradation of ibuprofen was observed within 3 h when exposed to direct sunlight without the addition of a photocatalyst. Direct photolysis involves photo-oxidation of the ibuprofen molecule due to the production of very few hydroxyl radicals. The proposed reaction mechanism of photolysis is illustrated by the following eqn (4) and (5):
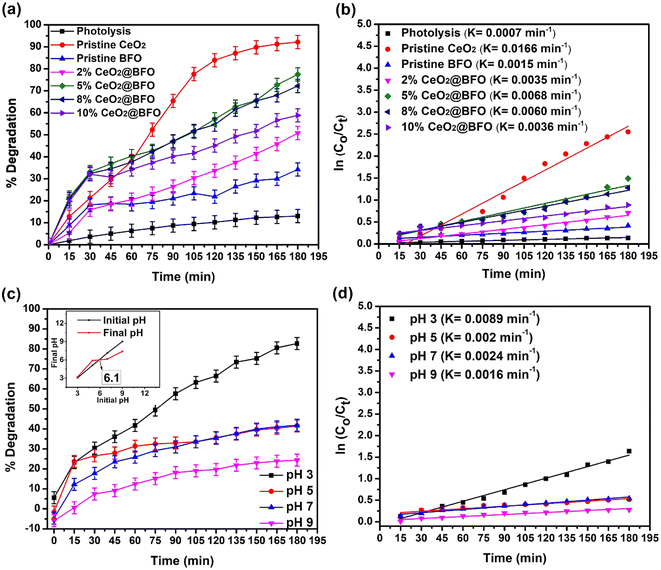 |
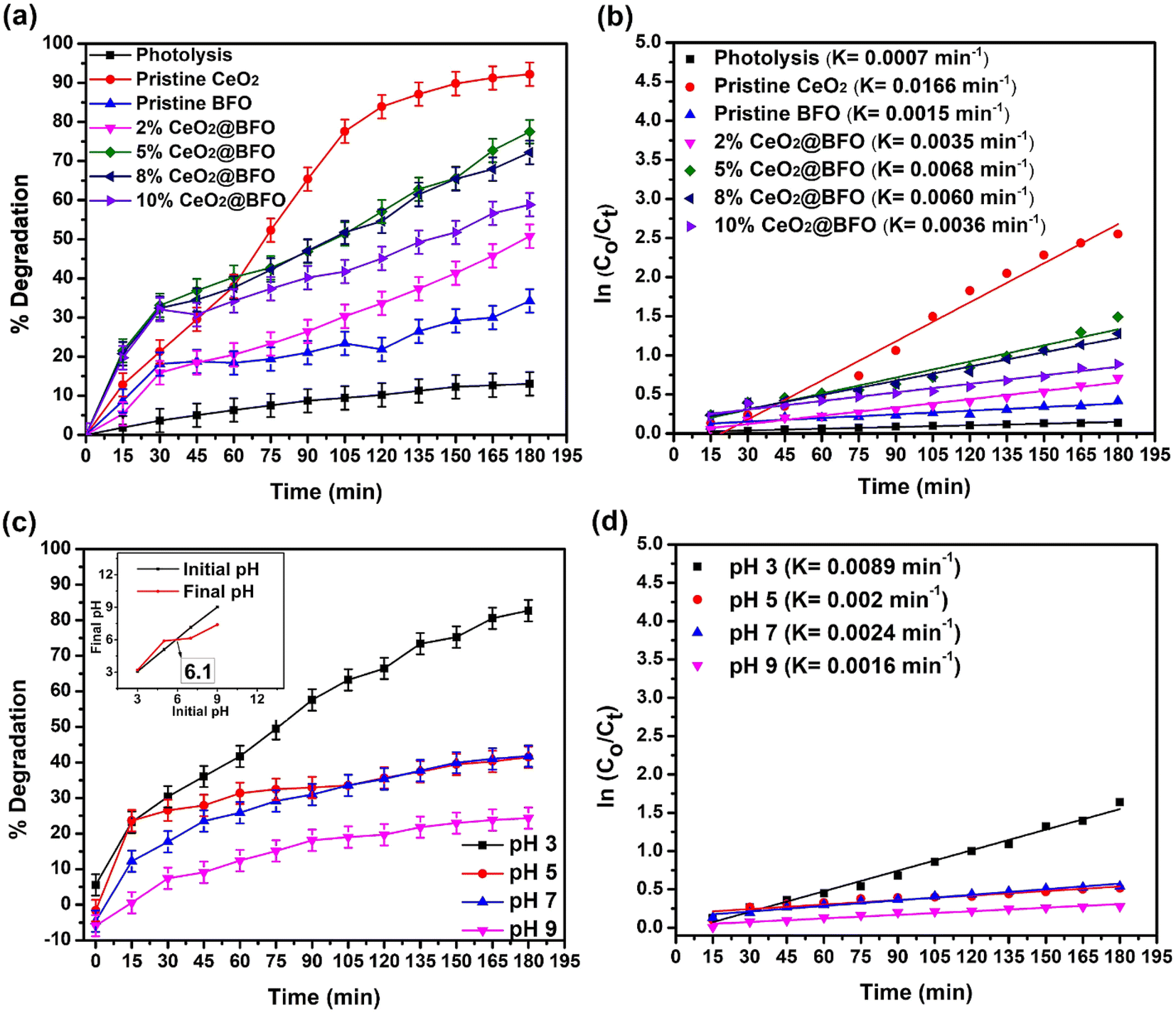
| | Fig. 6 (a) Effect of CeO2 loading on BFO in their nanocomposite for the photocatalytic degradation of IBP, time-dependent degradation percentage (b) reaction kinetics with catalyst choice (c) effect of pH on photodegradation of IBP, time-dependent degradation percentage at various pH; catalyst's point of zero charge (inset) and (d) reaction kinetics at pH change (catalyst: 5%CeO2@BFO, catalyst dose = 0.5 g L−1, IBP concentration = 20 mg L−1, pH = 4.3 (natural), light source = direct sunlight). | |
So, the small change in IBP concentration under direct sunlight without the addition of a photocatalyst is clear evidence that sunlight controls the photocatalysis of IBP degradation. Furthermore, only 34% of IBP were photodegraded over the pristine BFO after 180 min of sunlight irradiation, which is explained by their poor photodegradation activity.25 Pristine CeO2 showed 92.2% degradation of IBP within 180 minutes of exposure. This comparatively higher photodegradation activity of CeO2 can be explained by the efficient redox potential due to its unique 4f electronic configuration and Ce4+/Ce3+ valence states transitions make it an effective photocatalyst.30 Thus, the addition of up to 5%CeO2 in the BFO lattice significantly increased the activity of the pristine BFO against IBP degradation, increasing the photodegradation efficiency from 34% to 77.5%, as shown in Fig. 6a. This can be explained by the fact that the addition of cerium ion in the BFO lattice resulted in the creation of discrete empty energy levels within the band gap of the BFO due to the unique 4f levels of cerium and creation of oxygen vacancies to trap the photogenerated electrons, thereby decreasing the electron–hole recombination. So the higher CeO2 load on BFO provides larger interfaces for effective photogenerated e−–h+ pairs separation. However, a further increase in the mass ratio of CeO2 to BFO decreased the photodegradation activity, since an excessive load of CeO2 results in the agglomeration of particles, which negatively affects the charge transfer by occupying more active sites on the surface of BFO. The increase in photodegradation efficiency of the pristine BFO can also be evident from the study of the reaction kinetics illustrated in Fig. 6b. A comparison between pseudo-first-order and second-order kinetics applied on different operational parameters is provided in Table S1.† The addition of CeO2 significantly increased the reaction rate constant from 0.0015 min−1 to 0.0068 min−1. The photocatalytic degradation rate of ibuprofen is consistent with the pseudo-first-order kinetics model. The apparent rate constant (k) values obtained from the slope of their regression plots and the regression coefficient (R2) revealed that the photocatalytic degradation of ibuprofen is fully consistent with pseudo-first-order kinetics.
3.3. Effect of initial pH
The effect of pH on the ability of any drug to photodegrade is very significant. It affects the surface charge of the photocatalyst and affects the ionic species present in the solution. Lowering the pH increases the adsorption of pharmaceuticals due to the electrostatic force of attraction between the positively charged surface of the photocatalyst and the non-ionized form of the pharmaceutical species, hence increasing the rate of degradation.23 The effect of initial pH on the photodegradation efficiency of IBP and their reaction rate are shown in Fig. 6(c and d). An increase in the photodegradation of ibuprofen was observed with a decrease in the pH of the solution from 9 to 3. The point of zero charge of the catalyst was about pH 6.1 (Fig. 6c(inset)). Therefore, at lower pH values, the surface of BFO is likely to be positively charged and the pharmaceuticals are in non-ionized form, as indicated by their pKa value (4.9 in the case of IBP). While, at high pH, BFO is negatively charged and pharmaceuticals species are in their ionized form, which also has a negative charge, resulting in electrostatic repulsion. This minimizes the pollutant's adsorption on the surface of the photocatalyst, hence, reducing the photodegradation efficiency. The adsorption of pollutant molecules on the surface of the photocatalyst is very important during photodegradation.21 Moreover, the pKa value of ibuprofen is 4.9 and at a pH higher than its pKa, ibuprofen is deprotonated. While at a pH lower than its pKa value, ibuprofen remains protonated and is not in its molecular form that is less polar and can be easily removed from the water. Therefore, a pH of 2–4 is most favorable for the mineralization of ibuprofen from water.39
3.4. Effect of catalyst dose
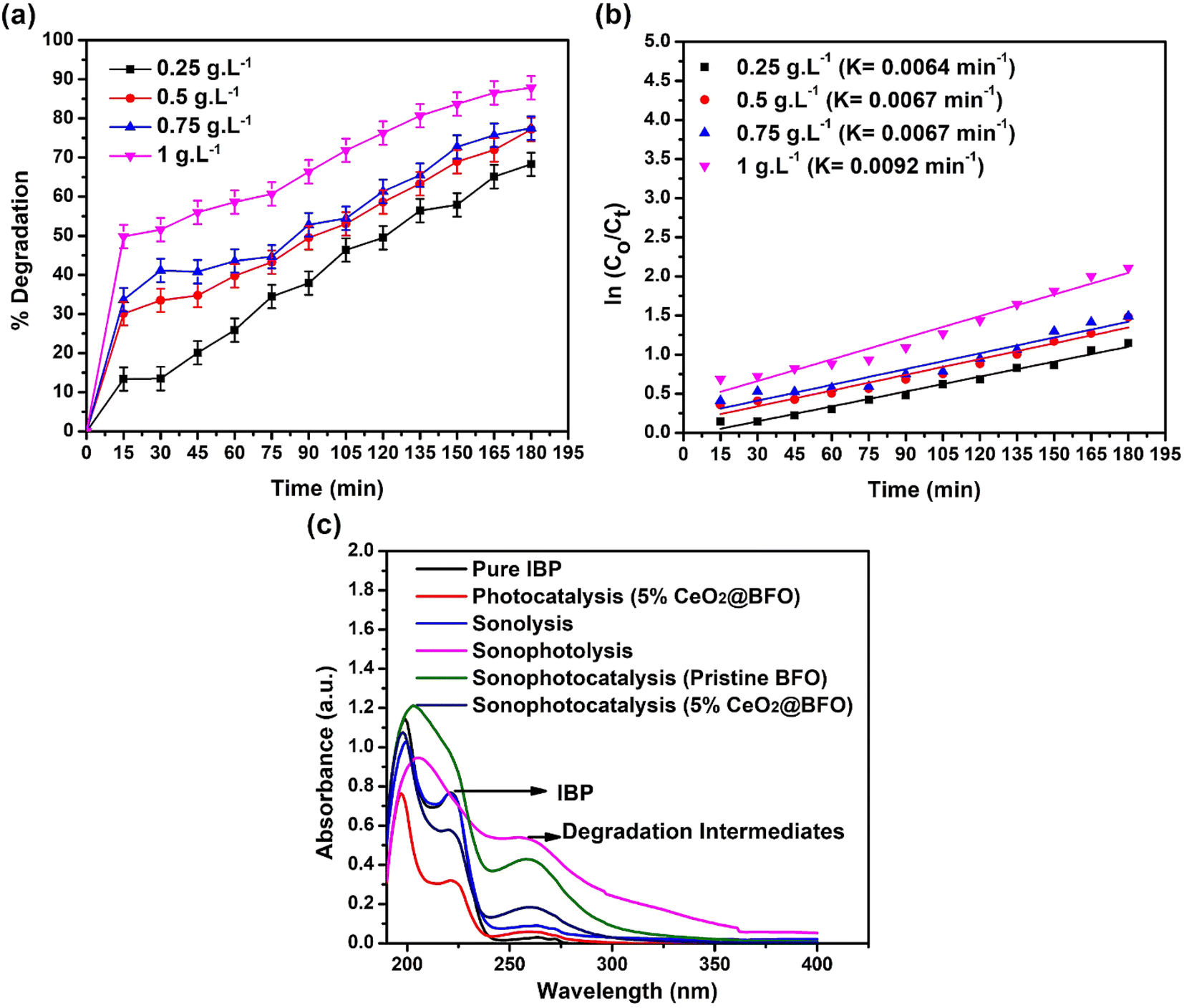
Increasing the catalyst dose significantly increased the rate of IBP photodegradation reaction from 0.0064 min−1 to 0.0092 min−1, when the catalyst dose was increased from 0.25 g L−1 to 1 g L−1. The overall 68.25% to 87.85% degradation was achieved at these catalyst concentrations as shown in Fig. 7(a and b). This is simply explained by the fact that increasing the catalyst dose increases the number of available active sites on the catalyst surface, which enhances the formation of radical species that promote photodegradation.
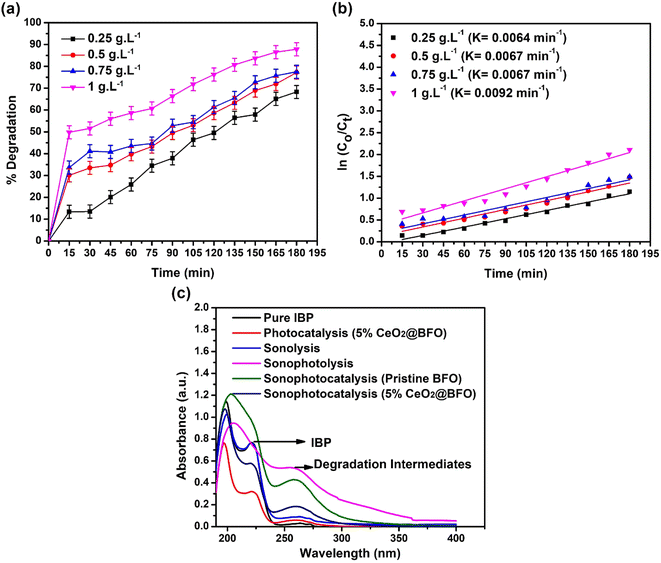 |
| | Fig. 7 (a) Effect of catalyst dose for the degradation of IBP as time-dependent degradation percentage; (b) reaction kinetics; and (c) sonophotocatalytic degradation UV spectrum of IBP (catalyst: 5% CeO2@BFO, IBP concentration: 20 mg L−1, pH = 4.3 (natural), light source = direct sunlight, ultrasound frequency = 50/60 Hz, ultrasound temperature = 30 °C, ultrasound power = 0 W).28 | |
3.5. Sonophotocatalytic degradation of IBP
Ultrasonication of IBP promotes the generation of several transformation intermediates with the degradation of the parent compound by the sequential attack of hydroxyl radicals. It increases the production of hydroxyl radicals in the reaction, which leads to the formation of intermediates because hydroxyl radicals do not show selectivity towards various functional groups, while other reactive species (for example O2˙−, h+, e−) participate in other oxido-reductive pathways. Ultrasonication degrades the IBP and results in the formation of intermediates such as substituted phenols, aromatic carboxylic acids, and others. When OH˙ radicals attack the propanoic acid and isobutyl chain in the ibuprofen structure, a variety of transformation by-products are formed.40 To evaluate the feasibility of the sonophotocatalytic degradation process sonolysis, sonophotolysis, sonophotocatalysis with pristine BFO and sonophotocatalysis with 5% CeO2@BFO, was conducted and the results are shown in Fig. 7c. The results demonstrated that the ultrasound-assisted photodegradation process led to the formation of IBP degradation intermediates. Sonophotolysis of IBP resulted in mineralization, which was observed from the disappearance of the IBP corresponding peak at 222 nm. At the same time, the peak of IBP degradation intermediates was also observed at 262 nm, which indicates the formation of intermediate products as a result of sonophotolysis.41
The disappearance of the IBP corresponding peak (at 222 nm) indicates that a relatively higher degree of degradation of the IBP parent compound was achieved, but with the formation of intermediates. Sonophotolytic degradation of IBP can be explained by the fact that sono/photo-oxidation of water molecules generate hydroxyl radicals which help break down ibuprofen molecules. Briefly, sonolysis generates H2O2, which can be photolyzed by light radiation to produce known reactive oxygen species (ROS) or hydroxyl radicals. These hydroxyl radicals degrade ibuprofen into their intermediates. The proposed mechanism42 is illustrated by the following eqn (6)–(8), where “)))” refers to ultrasound waves:
| | |
H2O + ))) → ½H2 + ½H2O2 → H2O2 + hv → 2˙OH
| (7) |
Sonophotocatalysis of IBP using pristine BFO efficiently mineralized the parent compound, as seen by the disappearance of the corresponding peak at 222 nm, while the intermediates (peak at 262 nm) still existed. Whereas, the 5% CeO2@BFO composite effectively degraded intermediates as well as IBP, showing much better photodegradation efficiency than the pristine BFO. The intensities of both peaks (parent compound; 222 nm and intermediates; 262 nm) were much lower than the intensities of the peaks formed by sonophotocatalysis with pristine BFO and sonophotolysis alone. It has been observed that the combination of sonolysis with photocatalysis resulted in a positive synergy against the mineralization of IBP.
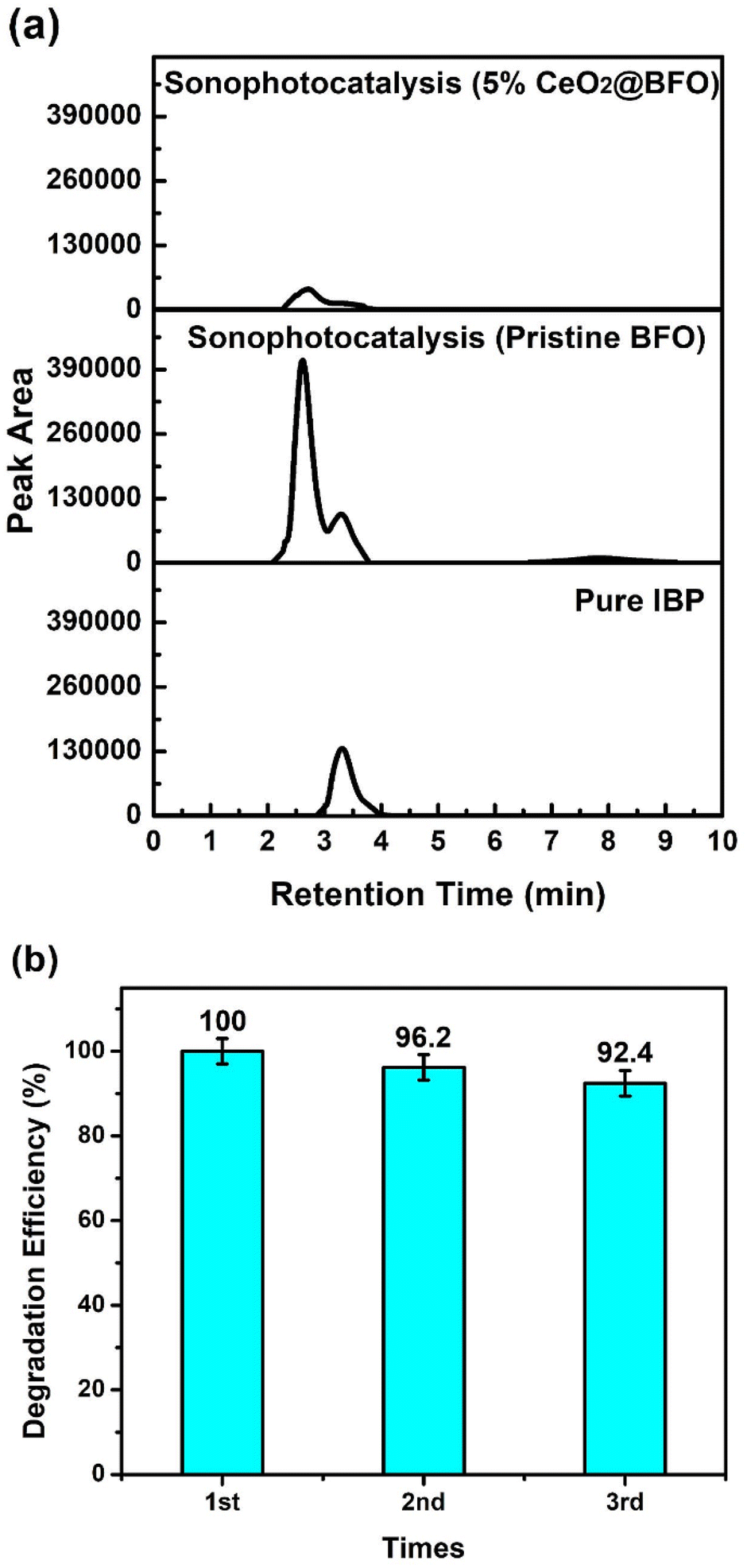
The degradation of IBP together with their intermediates was also confirmed by the high-performance liquid chromatographic analysis presented in Fig. 8a. HPLC analysis showed better agreement with the previously described UV-vis spectrophotometric analysis, as the intermediates specifically formed by the ultrasonication were efficiently mineralized together with the parent compound with 5% CeO2@BFO as compared to pristine BFO. The peak observed after a retention time of three minutes (3.29 min) corresponds to the pure ibuprofen compound with the elementary composition of C13H18O2. After 2.7 min, a significant peak of ibuprofen degradation intermediates was analyzed, which related to hydroxylated and decarboxylated derivatives of ibuprofen with the proposed identification 1-(4-isobutylphenyl) ethan-1-ol, 1-(4-ethyl phenyl)-2-methyl propane-1-ol and 1-(4-ethyl phenyl)-2-methyl propane-2-ol of having an elementary composition of C12H18O, as reported by M. Jiménez-Salcedo and their colleagues.43 These intermediates have also been reported in previous studies.44,45 The possible structures of ibuprofen degradation intermediates were inferred by the reported literature as the photodegradation mechanism of ibuprofen is common which is hydroxylation, decarboxylation or demethylation mainly due to the attack of h+, ˙OH and O2.46–50 As the benzylic carbon in the ibuprofen molecule is known to be the most reactive site in the presence of radical specie (OH),44 the proposed degradation mechanism suggested the attack of OH radicals at 1–2 positions of the iso-butyl group and on the aryl-carboxylic constituents of the ibuprofen molecule is reported to be the main process.43 Moreover, after a retention time of seven minutes (7.8 min), a less pronounced peak was observed after the ultrasonication treatment of ibuprofen, which decreased after the sonophotocatalytic treatment with 5% CeO2@BFO. Possibly, this is due to the ring-opened structure of the ibuprofen derivative, which has an elemental composition of C12H18O3.51
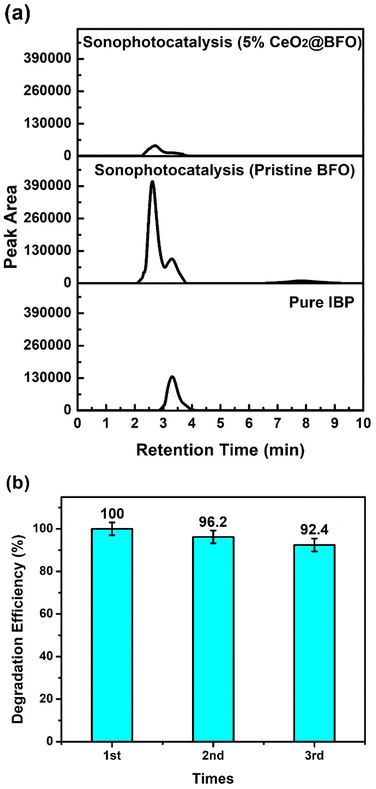 |
| | Fig. 8 (a) HPLC chromatogram of sonophotocatalytic treatments of aqueous ibuprofen and (b) reusability of 5% CeO2@BFO for three consecutive cycles. | |
3.6. Reusability of photocatalyst
Catalyst recovery and reusability are one of crucial aspects when considering their large-scale application which directly impacts the treatment process by reducing the cost. There are some commonly used techniques for catalyst recovery such as centrifugation, filtration and fixation of photocatalysts which usually enhance their capital cost and fixation causes a reduction in photocatalytic efficiency due to decreasing the active surface area.52 Besides these techniques, magnetic separation is known to be a cost-effective and more convenient technique to separate the catalyst for reuse. It is a highly efficient method to isolate the solid catalyst from the solution phase which ensures the continuous and automatic control of wastewater treatment operations.53 Fig. 8b represents the stability of synthesized photocatalyst over three consecutive cycles of ibuprofen photocatalytic treatment, demonstrating the reusability of the catalyst in the following reactions after simple separation with the magnet. The degradation efficiency of the photocatalyst decreased by only about 7.5% after the 3rd cycle of reuse, probably due to a decrease in the available active sites on the catalyst surface. The above results indicated the good stability of as-synthesized photocatalyst which ensure their feasibility to use for continuous treatment operations.
3.7. Comparison study
To analyze the effectiveness of the synthesized photocatalyst, its photodegradation activity was compared to the reported literature. Table 1 shows the work carried out on the photocatalytic activities of bismuth ferrite (BiFeO3) containing photocatalysts for IBP degradation compared to the present study.
Table 1 Photocatalytic activities of bismuth ferrite (BiFeO3) containing photocatalysts, comparison of the present study with reported literature
| S. no. |
Composites |
Pollutants |
Experimental conditions |
% Degradation |
References |
| 1 |
CeO2/BiFeO3 |
Ibuprofen (IBP) |
0.5 g L−1 of PC, 20 mg L−1 of ibuprofen, Direct sunlight |
77.5% in 180 min |
Present study |
| 2 |
BiFeO3/h-BN |
Ibuprofen (IBP) |
0.2 g L−1 of PC, 15 mg L−1 of ibuprofen, two 150 W visible light metal halide lamps |
48% in 120 min |
56 |
| 3 |
ZnAl2O4 supported on lychee-biochar |
Ibuprofen (IBP) |
1 g L−1 of PC, 20 mg L−1 of ibuprofen, UV radiation (Hg, 125 W) |
100% in 120 min |
57 |
| 4 |
g-C3N4/Bi2WO6/rGO |
Ibuprofen (IBP) |
1 g L−1 of PC, 5 mg L−1 of ibuprofen, 300 W Xe lamp |
93.5% in 240 min |
58 |
| 5 |
CoFe2O4/TiO2 |
Ibuprofen (IBP) |
0.5 g L−1 of PC, 100 μM of ibuprofen, simulated solar light (SSL) irradiation (Xe lamp) |
100% in 120 min |
59 |
| 6 |
S-doped BiOBr |
Ibuprofen (IBP) |
0.4 g L−1 of PC, 60 μM of ibuprofen, 300 W Xe lamp |
78% in 60 min |
60 |
| 7 |
Boron nitride nanosheets decorated MIL-53(Fe) |
Ibuprofen (IBP) |
0.1 g L−1 of PC, 10 mg L−1 of ibuprofen, 500 W Xe lamp |
99% in 60 min |
61 |
| 8 |
Co3O4/BiOI |
Ibuprofen (IBP) |
0.8 g L−1 of PC, 10 mg L−1 of ibuprofen, 60 W LED lamp |
99.97% in 60 min |
62 |
It can be seen that comparatively efficient photodegradation activity was achieved for the highest initial concentration (20 mg L−1) of ibuprofen under natural sunlight. Other higher degradation efficiencies may be attributed to variations in operational conditions, mainly UV light, which is known to be more effective than visible light.54,55
4. Conclusion
A novel and magnetically separable heterostructure of cerium oxide and bismuth ferrite composite, sensitive to sunlight, was successfully synthesized by simple co-precipitation and solvothermal method. It was found that the addition of 5% cerium oxide to the composite heterostructure significantly increased the surface area of bismuth ferrite from 8.45 m2 g−1 to 26.22 m2 g−1 without affecting their optical absorption properties and increased the photodegradation efficiency of bismuth ferrite from 34% to 77.5% in terms of ibuprofen mineralization. Catalytic photodegradation of ibuprofen followed a pseudo-first-order kinetics model and the reaction rate of bismuth ferrite was increased from 0.0015 min−1 to 0.0068 min−1 by adding 5% of CeO2 in the heterostructure. On the other hand, analysis of individual and synergistic effects of photocatalysis, sonolysis and sonophotocatalysis on the degradation behaviour of ibuprofen demonstrated that ultrasonication treatment transformed ibuprofen into their degradation intermediates. It was observed that photolysis in combination with sonolysis increased the rate of transformation due to the formation of hydroxyl radicals produced during the photolytic breakdown of H2O2 formed by ultrasonication. The addition of the 5% CeO2@BFO photocatalyst to the sonophotolytic system showed positive synergy for the degradation of ibuprofen with the formation of intermediate products in relatively lower amounts compared to the sonophotolytic system. The catalyst was stable and retained 92% catalytic activity after 3 cycles, so it can be successfully reused without post-operation treatments.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†
Author contributions
Jamshaid Rashid: conceptualization, supervision, material characterization, original draft preparation, manuscript revision and editing. Ali Ahsan: data curation, methodology, experimentation, writing – original draft preparation. Ming Xu: characterization support, writing – reviewing and editing. Irina Savina: writing – reviewing and language editing. Faisal Rehman: data curation, writing – reviewing and language editing.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work presented in this paper.
Acknowledgements
The authors acknowledge Quaid-i-Azam University, Pakistan for funding this research and Professor Ming Xu for extending his courtesy in performing material characterizations.
References
- A. Majumder, B. Gupta and A. K. Gupta, Environ. Res., 2019, 176, 108542 CrossRef CAS PubMed.
- OECD, Health at a Glance 2017, 2017, DOI:10.1787/health_glance-2017-en.
- C. F. Couto, L. C. Lange and M. C. S. Amaral, J. Water Process. Eng., 2019, 32, 100927 CrossRef.
- S. H. Lee, K. H. Kim, M. Lee and B. D. Lee, J. Water Process. Eng., 2019, 31, 100828 CrossRef.
- S. N. Malik, S. M. Khan, P. C. Ghosh, A. N. Vaidya, G. Kanade and S. N. Mudliar, Sci. Total Environ., 2019, 678, 114–122 CrossRef CAS PubMed.
- J. M. Monteagudo, H. El-taliawy, A. Durán, G. Caro and K. Bester, J. Hazard. Mater., 2018, 357, 457–465 CrossRef CAS PubMed.
- M. Khan, C. S. L. Fung, A. Kumar and I. M. C. Lo, J. Hazard. Mater., 2019, 365, 733–743 CrossRef CAS PubMed.
- J. Madhavan, F. Grieser and M. Ashokkumar, J. Hazard. Mater., 2010, 178(1–3), 202–208 CrossRef CAS PubMed.
- D. Liu, H. Zhang, Y. Wei, B. Liu, Y. Lin, G. Li and F. Zhang, Chemosphere, 2018, 209, 998–1006 CrossRef CAS PubMed.
- K. K. Barnes, D. W. Kolpin, E. T. Furlong, S. D. Zaugg, M. T. Meyer and L. B. Barber, Sci. Total Environ., 2008, 402(2–3), 192–200 CrossRef CAS PubMed.
- P. H. Roberts and K. V Thomas, Sci. Total Environ., 2006, 356(1–3), 143–153 CrossRef CAS PubMed.
- L. Chen, J. Tang, L. Song, P. Chen, J. He, C. Au and S. Yin, Appl. Catal., B, 2019, 242, 379–388 CrossRef CAS.
- P. Kanhere and Z. Chen, Molecules, 2014, 19(12), 19995–20022 CrossRef PubMed.
- M. Humayun, A. Zada, Z. Li, M. Xie, X. Zhang, Y. Qu, F. Raziq and L. Jing, Appl. Catal., B, 2016, 180, 219–226 CrossRef CAS.
- S. Irfan, Z. Zhuanghao, F. Li, Y. Chen, G. Liang, J. Luo and F. Ping, J. Mater. Res. Technol., 2019, 8(6), 6375–6389 CrossRef CAS.
- Č. Maria, D. Zagorac, K. Batalovi, J. Radakovi, B. Stojadinovi, V. Spasojevi and R. Hercigonja, Ceram. Int., 2017, 43, 1256–1264 CrossRef.
- E. Gil-González, A. Perejón, P. E. Sánchez-Jiménez, J. M. Criado, & L. A. Pérez-Maqueda, Handbook of Thermal Analysis and Calorimetry, 2018, ch. 16, vol. 6 Search PubMed.
- J. Van Den Brink and D. I. Khomskii, J. Phys.: Condens. Matter, 2008, 20, 434217 CrossRef.
- S. Bharathkumar, M. Sakar and S. Balakumar, J. Phys. Chem. C, 2016, 120(33), 18811–18821 CrossRef CAS.
- X. Wang, W. Mao, Q. Zhang, Q. Wang, Y. Zhu, J. Zhang, T. Yang, J. Yang, X. Li and W. Huang, J. Alloys Compd., 2016, 677, 288–293 CrossRef CAS.
- X. Wang, Y. Lin, X. Ding and J. Jiang, J. Alloys Compd., 2011, 509(23), 6585–6588 CrossRef CAS.
- S. Li, Y.-H. Lin, B.-P. Zhang, C.-W. Nan and Y. Wang, J. Appl. Phys., 2009, 105(5), 056105 CrossRef.
- R. Mostafaloo, M. H. Mahmoudian and M. Asadi-Ghalhari, J. Mater. Res. Technol., 2019, 382, 111926 CAS.
- J. Yin, G. Liao, J. Zhou, C. Huang, Y. Ling, P. Lu and L. Li, Sep. Purif. Technol., 2016, 168, 134–140 CrossRef CAS.
- F. Niu, D. Chen, L. Qin, N. Zhang, J. Wang, Z. Chen and Y. Huang, ChemCatChem, 2015, 7(20), 3279–3289 CrossRef CAS.
- S. Lam, J. Sin and A. Rahman, Mater. Res. Bull., 2017, 90, 15–30 CrossRef CAS.
- N. H. Ince, Ultrason. Sonochem., 2018, 40, 97–103 CrossRef CAS PubMed.
- D. Kanakaraju, B. D. Glass and M. Oelgemöller, J. Environ. Manage., 2018, 219, 189–207 CrossRef CAS PubMed.
- B. Samran, S. Iunput, S. Tonnonchiang and S. Chaiwichian, Phys. B, 2019, 561, 23–28 CrossRef CAS.
- M. Ebadi, O. Amiri and M. Sabet, Sep. Purif. Technol., 2018, 190, 117–122 CrossRef CAS.
- M. Sun, Z. Li, H. Li, Z. Wu, W. Shen and Y. Qing, Electrochim. Acta, 2020, 331, 135366 CrossRef CAS.
- N. Sheoran, V. Kumar and A. Kumar, J. Magn. Magn. Mater., 2018, 475, 30–37 CrossRef.
- S. A. N. H. Lavasani, O. Mirzaee, H. Shokrollahi, A. K. Moghadam and M. Salami, Ceram. Int., 2017, 43(15), 12120–12125 CrossRef CAS.
- E. M. M. Ibrahim, G. Farghal, M. M. Khalaf and H. M. A. El-lateef, J. Nano. Adv. Mat., 2017, 5(1), 33–39 Search PubMed.
- N. A. Lomanova and V. V Gusarov, Nanosyst.: Phys., Chem., Math., 2013, 4(5), 696–705 CAS.
- I. Papadas, J. A. Christodoulides, G. Kioseoglou and G. S. Armatas, J. Mater. Chem. A, 2015, 3(4), 1587–1593 RSC.
- M. W. Kadi, R. M. Mohamed and A. A. Ismail, Opt. Mater., 2020, 104, 109842 CrossRef CAS.
- K. Saravanakumar, M. M. Ramjan, P. Suresh and V. Muthuraj, J. Alloys Compd., 2016, 664, 149–160 CrossRef CAS.
- S. Adityosulindro, L. Barthe, K. González-Labrada, U. J. Jáuregui Haza, H. Delmas and C. Julcour, Ultrason. Sonochem., 2017, 39, 889–896 CrossRef CAS PubMed.
- I. Michael, A. Achilleos, D. Lambropoulou, V. Osorio Torrens, S. Pérez, M. Petrović, D. Barceló and D. Fatta-Kassinos, Appl. Catal., B, 2014, 147, 1015–1027 CrossRef CAS.
- J. Choina, H. Kosslick, Ch. Fischer, G. U. Flechsig, L. Frunza and A. Schulz, Appl. Catal., B, 2013, 129, 589–598 CrossRef CAS.
- C. G. Joseph, G. L. Puma, A. Bono and Y. H. Taufiq-yap, Chem. Eng. Commun., 2015, 202(8), 1061–1068 CrossRef CAS.
- M. Jiménez-Salcedo, M. Monge and M. T. Tena, Chemosphere, 2019, 215, 605–618 CrossRef PubMed.
- L. Lin, W. Jiang, M. Bechelany, M. Nasr, J. Jarvis, T. Schaub, R. R. Sapkota, P. Miele, H. Wang and P. Xu, Chemosphere, 2019, 220, 921–929 CrossRef CAS PubMed.
- N. Jallouli, L. M. Pastrana-Martínez, A. R. Ribeiro, N. F. F. Moreira, J. L. Faria, O. Hentati, A. M. T. Silva and M. Ksibi, Chem. Eng. J., 2018, 334, 976–984 CrossRef CAS.
- N. Liu, J. Wang, J. Wu, Z. Li, W. Huang, Y. Zheng, J. Lei, X. Zhang and L. Tang, Mater. Res. Bull., 2020, 132, 111000 CrossRef CAS.
- N. Liu, W. Dai, F. Fei, H. Xu, J. Lei, G. Quan and Y. Zheng, Sep. Purif. Technol., 2022, 297, 121545 CrossRef CAS.
- N. Liu, F. Fei, W. Dai, J. Lei, F. Bi, B. Wang, G. Quan, X. Zhang and L. Tang, J. Colloid Interface Sci., 2022, 625, 965–977 CrossRef CAS PubMed.
- L. Sruthi, B. Janani and S. S. Khan, Sep. Purif. Technol., 2021, 279, 119709 CrossRef CAS.
- S. Sun, Y. Hu, M. Xu, F. Cheng, H. Zhang and Z. Li, J. Hazard. Mater., 2022, 424, 127698 CrossRef CAS PubMed.
- R. B. Arthur, J. L. Bonin, L. P. Ardill, E. J. Rourk, H. H. Patterson and E. A. Stemmler, J. Hazard. Mater., 2018, 358, 1–9 CrossRef CAS PubMed.
- D. Wang, D. Han, Z. Shi, J. Wang, J. Yang, X. Li and H. Song, Appl. Catal., B, 2018, 227, 61–69 CrossRef CAS.
- G. Su, L. Liu, Q. Kuang, X. Liu, W. Dong, M. Niu, A. Tang and J. Xue, J. Mol. Liq., 2021, 335, 116566 CrossRef CAS.
- F. El-Sayed, V. Ganesh, M. S. A. Hussien, T. H. AlAbdulaal, H. Y. Zahran, I. S. Yahia, M. Sh. Abdel-wahab, M. Shakir and Y. Bitla, J. Mater. Res. Technol., 2022, 19, 4867–4880 CrossRef CAS.
- Z. Wang, V. Srivastava, I. Ambat, Z. Safaei and M. Sillanpää, J. Water Process Eng., 2019, 31, 100808 CrossRef.
- Z. Balta and E. B. Simsek, J. Alloys Compd., 2022, 898, 162897 CrossRef CAS.
- S. Siara, C. Elvis, R. Harishkumar and P. V. Chellam, Mater. Res. Bull., 2022, 145, 111530 CrossRef CAS.
- S. H. Liu and W. T. Tang, Sci. Total Environ., 2020, 731, 139172 CrossRef CAS PubMed.
- H. Gong, W. Chu, Y. Huang, L. Xu, M. Chen and M. Yan, Environ. Pollut., 2021, 276, 116691 CrossRef CAS PubMed.
- Y. Liu, Z. Hu and J. C. Yu, Chemosphere, 2021, 278, 130376 CrossRef CAS PubMed.
- N. Liu, J. Wang, M. Tian, J. Lei, J. Wang, W. Shi, X. Zhang and L. Tang, J. Colloid Interface Sci., 2021, 603, 270–281 CrossRef CAS PubMed.
- M. E. Malefane, U. Feleni, P. J. Mafa and A. T. Kuvarega, Appl. Surf. Sci., 2020, 514, 145940 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Ali Ahsan‡b,
Ming Xua,
Irina Savina
*ab,
Ali Ahsan‡b,
Ming Xua,
Irina Savina