
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetermination of nine prohibited N-nitrosamines in cosmetic products by vortex-assisted dispersive liquid–liquid microextraction prior to gas chromatography-mass spectrometry†
Lorenza Schettino,
Juan L. Benedé and
Alberto Chisvert *
*
GICAPC Research Group, Department of Analytical Chemistry, University of Valencia, 46100 Burjassot, Valencia, Spain. E-mail: alberto.chisvert@uv.es
First published on 19th January 2023
Abstract
An analytical method for the simultaneous determination of nine prohibited N-nitrosamines in cosmetic products is presented. N-nitrosamines are banned compounds in cosmetic products due to their harmful effects. Therefore, these compounds are not intentionally added to these products but, however, small amounts of them may be present due to unintentional causes, and thus sensitive methods for their analytical control are required. The proposed method is based on vortex-assisted dispersive liquid–liquid microextraction (VA-DLLME) to extract and preconcentrate the analytes, followed by gas chromatography-mass spectrometry (GC-MS) for their determination. The variables involved in the VA-DLLME process were optimized by using a Box–Behnken design and, due to the different polarity of the N-nitrosamines studied, several approaches for sample treatment were compared to achieve the best results. The method was successfully validated, showing a good linearity at least up to 20 ng mL−1, enrichment factors from 2 to 100 depending on the target analyte, limits of detection and quantification at the low μg kg−1 level, and good repeatability values (<13%). Finally, the proposed analytical method was applied to the determination of N-nitrosamines in commercial cosmetic samples of different nature, avoiding the matrix effect by means of standard addition calibration. Significant amounts of some of the N-nitrosamines, even exceeding the established regulatory limit, were found in the samples. The resulting method is fast, simple, and affordable to carry out the quality control of cosmetic products to ensure consumer safety for most laboratories.
Introduction
N-Nitrosamines are N-nitroso derivatives of secondary amines with mutagenic, carcinogenic, and teratogenic effects,1 which can be found in cosmetic products without having been intentionally added during the manufacturing process, thus constituting a risk to consumer health. N-nitrosamines are easily formed when secondary or tertiary amines react with nitrosating agents, such as nitrites or nitrogen oxides.2,3 This implies that ingredients containing or releasing nitrite ions should not be used, but if they are employed, nitrosation reaction inhibition systems should be used (such as the use of α-tocopherol, ascorbic acid and other substances).3For this reason, with the aim of reducing the formation of these compounds in cosmetics, and therefore the health risk of consumers, European legislation has prohibited not only N-nitrosamines in cosmetic products,4 but also secondary alkyl- and alkanolamines. Moreover, the use of fatty acid dialkylamides and dialkanolamides, monoalkylamines, monoalkanolamines, trialkylamines, trialkanolamines, and their salts present restrictions in these products.3,5 Furthermore, in 2012, the European Scientific Committee on Consumer Safety (SCCS) established a maximum content limit of 50 μg kg−1 for traces of N-nitrosamines, both in raw materials used as ingredients in cosmetics and in finished cosmetic products.3 In this regard, according to this scientific opinion, cosmetic industries are required to perform quality control analysis for raw materials and for those products whose constituents may unintentionally cause the formation of nitrosamines, and to avoid impurities and incompatibilities between ingredients to prevent nitrosation reactions.
Different analytical methods for N-nitrosamines determination in cosmetic products can be found in the scientific literature. Most of them are based on the determination of only N-nitrosodiethanolamine (NDELA),6–16 a hydrophilic nitrosamine for whose determination in cosmetic products two official analytical methods have been approved (i.e., ISO 1013017 and ISO 1581918).
Beyond the NDELA, there are few methods in the literature that simultaneously determine several N-nitrosamines in cosmetic products. This is mainly due to the different polar character that exists between them, which hinder to extract them simultaneously in the treatment of the sample. In this context, some studies based on chromatographic techniques have been published by using gas chromatography (GC) coupled to thermal energy analyzer (TEA),19,20 single mass spectrometry (MS)21–23 or in tandem (MS/MS);24 or liquid chromatography (LC) with TEA,25 ultraviolet (UV),26 or MS27 and MS/MS28 detection.
Regarding the pretreatment of the cosmetic samples, both solid- and liquid-phase (micro) extraction techniques have been used for the enrichment of N-nitrosamines. Among the solid-based techniques, solid-phase extraction (SPE),24 dispersive solid-phase extraction (DSPE),29 headspace solid-phase microextraction (HS-SPME),22 stir bar sorptive-dispersive microextraction (SBSDME)28 and micro-matrix solid-phase dispersion (μMSPD)23 have been employed. On the other hand, among liquid-based techniques, liquid–liquid extraction (LLE)21 and vortex-assisted reversed-phase dispersive liquid–liquid microextraction (VA-RP-DLLME)27 are the only applications, to the best of our knowledge. However, the limits of detection of some of these methods are higher than the regulatory limit (i.e., 50 μg kg−1), they consume large amounts of organic solvents and/or they are time-consuming procedures that hinder the sample throughput. Additionally, some of them use unaffordable instruments (e.g., TEA and MS/MS detectors) for the most quality control laboratories of cosmetic manufacturers.
These reasons motivated us to develop an analytical method for the simultaneous determination of nine prohibited N-nitrosamines (see Table 1) at trace level in cosmetic products. The method is based on vortex-assisted dispersive liquid–liquid microextraction (VA-DLLME), followed by gas chromatography-mass spectrometry (GC-MS) analysis. Unlike the conventional DLLME, in which a polar organic solvent is used as disperser solvent, in the VA-DLLME it is the vortex agitation that helps the formation of the cloudy solution, and thus reducing the consumption of additional organic solvents beyond the extraction solvent. Moreover, due to the different polarity of the target nitrosamines (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ko/w from −0.59 to 3.13), different approaches for the sample treatment, such as LLE, SPE, filtration and leaching, were compared to achieve the best results during the procedure.
Ko/w from −0.59 to 3.13), different approaches for the sample treatment, such as LLE, SPE, filtration and leaching, were compared to achieve the best results during the procedure.
| Analytea | CAS number | Chemical structure | Molecular weight (g mol−1) | LogKo/w |
|---|---|---|---|---|
| a NDMA, N-nitrosodimethylamine; NMEA, N-nitrosoethylmethylamine; NDEA, N-nitrosodiethylamine; NDPA, N-nitrosodipropylamine; NDBA, N-nitrosodibutylamine; NPIP, N-nitrosopiperidine; NPYR, N-nitrosopyrrolidine; NMOR, N-nitrosomorpholine; NDPhA, N-nitrosodiphenylamine. | ||||
| NDMA | 62-75-9 |  |
74.08 | −0.50 |
| NMEA | 10595-95-6 |
 |
88.11 | 0.01 |
| NDEA | 55-18-5 |  |
102.14 | 0.52 |
| NDPA | 621-64-7 |  |
130.19 | 1.54 |
| NDBA | 924-16-3 |  |
158.24 | 2.56 |
| NPIP | 100-75-4 |  |
114.15 | 0.44 |
| NPYR | 930-55-2 |  |
100.12 | −0.09 |
| NMOR | 59-89-2 |  |
116.12 | −0.59 |
| NDPhA | 86-30-6 |  |
198.22 | 3.13 |
Experimental
Apparatus
An 8860-gas chromatography system coupled to a simple quadrupole 5977B mass spectrometer, both from Agilent Technologies (Palo Alto, CA, USA), and a PAL LSI 85 autosampler from CTC Analytics (Zwingen, Switzerland) was used.During the VA-DLLME, a ZX3 vortex mixer from VELP Scientifica (Usmate Velate, Italy) and an EBA 21 centrifuge from Hettich® (Tuttlingem, Germany) were also employed.
Reagents and samples
EPA 8270 Appendix IX Nitrosamine Mix (2000 μg mL−1 of each component in methanol) from Sigma–Aldrich (Steinheim, Germany) was used as standard.LC-MS grade methanol from VWR Chemicals (Fontenay-sous-Bois, France) was used in the preparation of the standard stock and intermediate solutions.
Deionized water (resistivity ≥18 MΩ cm), obtained from a Connect water purification system provided by Adrona (Riga, Latvia), was used in the preparation of working solutions, and analytical reagent-grade chloroform purchased from Scharlau Chemie (Barcelona, Spain) was used as acceptor phase during the VA-DLLME stage.
LC-grade hexane 96% purchased from Scharlau Chemie (Barcelona, Spain) was used for the study of the sample pretreatment.
SPE cartridges (i.e., Discovery® DSC-Diol (50 μm particle size) from Supelco (Bellefonte, PA), and Strata-X™(25 μm particle size) and Strata SDB-L (100 μm particle size), both from Phenomenex (Torrance, CA)) were also used for the study of the sample pretreatment.
Three sample of commercial cosmetic products (i.e., an aftersun gel, and two different body creams) were analysed, and for reasons of confidentiality the brands of these commercial samples are not shown.
Proposed method
Regarding sample preparation, working sample solutions were prepared by standard addition calibration approach as follows: 0.05 g of cosmetic sample were weighed into 5 mL volumetric flasks, then they were spiked with different aliquots (i.e., from 0.5 μL to 1 mL) of the 100 ng mL−1 aqueous standard working solution reaching spiked concentrations of the target analytes from 0.01 to 20 ng mL−1, and filled up to the line with water. Then, they were mixed by vortex stirring (ca. 1 min) until a homogeneous dispersion was obtained.
After that, the sample solutions were transferred to 15 mL polypropylene tube with a conical bottom to perform the VA-DLLME procedure.
Fig. 1 shows a schematic diagram of the whole experimental procedure.
 | ||
| Fig. 1 Schematic diagram of the proposed method. | ||
The GC oven temperature was maintained at 60 °C for 2 min, and then ramped to 160 °C at 10 °C min−1, and to 240 °C at a rate of 40 °C min−1, holding this temperature for 4 min. The carrier gas was helium with a constant flow of 1 mL min−1. The injection was performed at 230 °C in splitless mode, and the injected volume was 2 μL. Ion source operated by electronic ionization at 70 eV at 230 °C, and transfer line and quadrupole temperatures were set at 230 °C and 150 °C, respectively. Acquisition was carried out in both full scan and Selected Ion Monitoring (SIM) mode. To improve the sensitivity, selected ions were acquired in six-time windows depending on their retention times, as shown in Table 2.
| Analytea | Acquisition time windows (min) | Retention time (min) | Quantification ion (m/z) | Qualifier ions (m/z) | |
|---|---|---|---|---|---|
| a NDMA, N-nitrosodimethylamine; NMEA, N-nitrosoethylmethylamine; NDEA, N-nitrosodiethylamine; NDPA, N-nitrosodipropylamine; NDBA, N-nitrosodibutylamine; NPIP, N-nitrosopiperidine; NPYR, N-nitrosopyrrolidine; NMOR, N-nitrosomorpholine; NDPhA, N-nitrosodiphenylamine. | |||||
| NDMA | 6.00–6.90 | 6.68 | 74 | 43 | 42 |
| NMEA | 6.90–8.50 | 7.33 | 88 | 56 | 42 |
| NDEA | 7.73 | 102 | 56 | 42 | |
| NDPA | 8.50–11.00 | 9.52 | 130 | 70 | 43 |
| NDBA | 11.00–12.15 | 11.72 | 116 | 84 | 57 |
| NPIP | 12.01 | 114 | 55 | 42 | |
| NPYR | 12.15–14.00 | 12.36 | 100 | 68 | 41 |
| NMOR | 12.81 | 116 | 86 | 56 | |
| NDPhA | 15.00–18.00 | 16.66 | 169 | 168 | 167 |
Under the described conditions, the required time for the chromatographic analysis of all analytes was 18 min.
Results and discussion
Optimization of the VA-DLLME variables
Before the optimization for the extraction step conditions (see Section VA-DLLME), preliminary considerations were made to assess the nature of the extraction solvent. It must be denser than water to remain, after centrifugation, at the bottom of the extraction tube. In addition, from the outset the possibility of avoiding the use of disperser solvent by using instead vortex stirring to provide the cloudy solution formation was evaluated. For this reason, chloroform and dichloromethane were considered as possible extraction solvents, and VA-DLLME procedure was performed by extracting 5 mL of standard solution at 5 ng mL−1 of the analytes. When using dichloromethane, neither the formation of the microemulsion nor the phase separation was obtained, so chloroform was selected as the extractant phase, since a fine microemulsion was formed by vortexing, demonstrating the possibility of assisting the DLLME procedure with vortex and thus avoiding the use of a disperser solvent.Next, the variables involved in the VA-DLLME procedure were optimized through a response surface methodology (RSM), showing the interactions between them. In this work, the studied variables were the volume of the extraction solvent, the ionic strength of the donor phase and the vortex time. The statistical analysis of the results was performed using StatGraphics Centurion XVI software from StatGraphics Technologies, Inc. (The Plains, VA, USA). A Box–Behnken design was performed to assess the three significant extraction variables, performing 15 experimental runs with three levels for each factor (see ESI†). The independent factors (and the ranges) studied were the volume of extraction solvent (60–150 μL), the ionic strength of the donor phase (0–10% NaCl (w/v)), and the vortex time (30–90 s). All experiments were carried out on the same day using 5 mL of a standard solution of the analytes at 5 ng mL−1 in water as donor phase and they are summarized in Table S1.†
To evaluate the suitability of the model, the coefficient of determination (R2) was considered, which a value >0.87 was obtained for all target analytes, indicating that the designed model was efficient for predicting responses. Fig. 2 shows the response surface plots in terms of desirability (estimated as described in ESI†) for the three factors studied.
 | ||
| Fig. 2 Response surface plots of the desirability function representing the relation between the different variables affecting the extraction: (a) vortex time vs. CHCl3 volume, (b) ionic strength vs. CHCl3 volume, and (c) vortex time vs. ionic strength. | ||
Fig. 2a shows that there were no great differences in the vortex time, but the best responses were obtained using 50–70 s, getting the proper dispersion of the extraction solvent in the donor phase. For that reason, 60 s was established for further experiments as mean value. On the other side, as it is shown in Fig. 2a and b, the best response for the volume of extraction solvent was obtained using 60 μL of chloroform. However, a very small droplet (ca. 15 μL) was obtained, which was considered too low to be handled and injected into the GC-MS system, and therefore it was decided to continue selecting the volume of 80 μL for further experiments.
Finally, as can be seen in Fig. 3c, the best response for the ionic strength of the donor phase was showed from 0 to 10% NaCl (w/v). This occurs due to two different effects. On the one hand, the presence of salts decreases the solubility of the extraction solvent (i.e., chloroform), thereby the volume of the extract increases, causing the dilution of the analytes. On the other hand, when a higher salt concentration is used, the solubility of the analytes in the aqueous phase decreases and enhance their transfer to the organic phase (salting-out effect), obtaining a greater extraction of the analytes, with good signals despite the effect of dilution. Based on these results, and to reduce reagent consumption, 0% NaCl (w/v) was selected in further experiments.
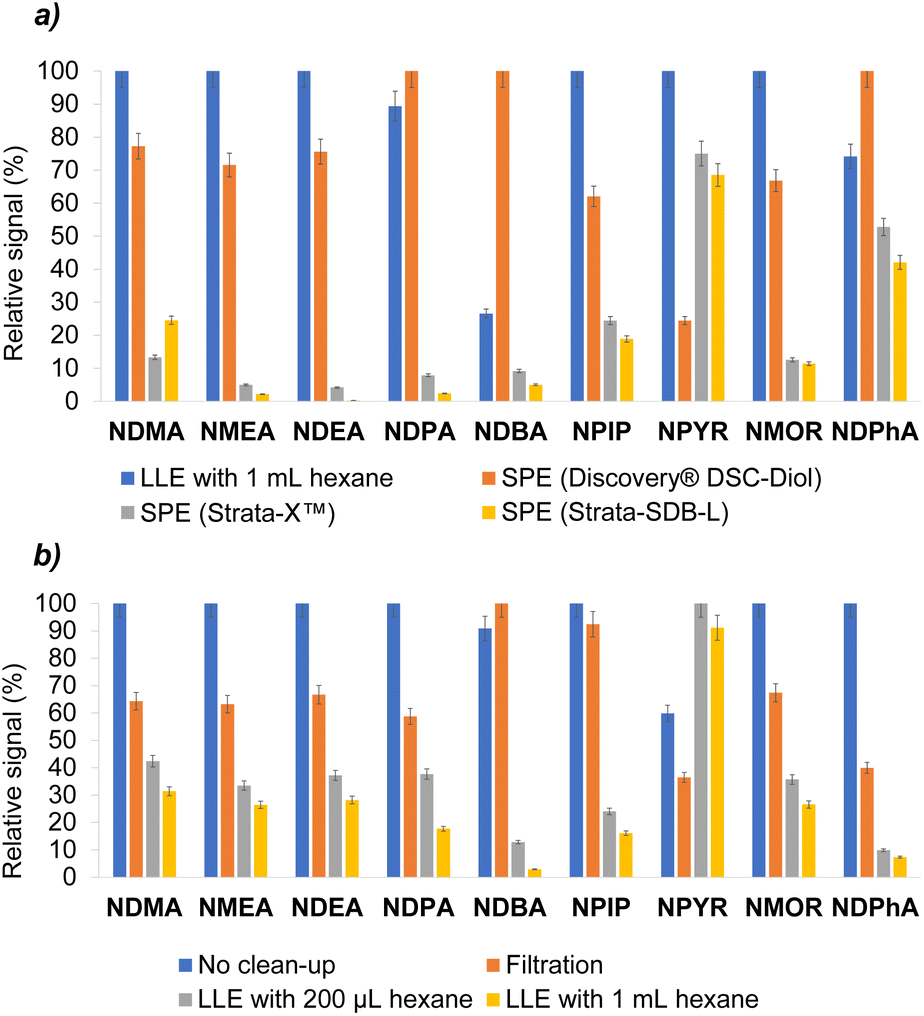 | ||
| Fig. 3 Sample pretreatment studies carried out with a N-nitrosamine free sample solution spiked to 1 ng mL−1 with target analytes: (a) comparison between clean-up through LLE with 1 mL hexane and SPE with different cartridges; (b) comparison between clean-up through LLE with 1 mL and 200 μL of hexane, filtration, and no clean-up step with increasing of the extraction solvent volume (i.e., 120 μL of chloroform). | ||
In summary, the optimized method consisted of 80 μL of chloroform, 60 s of vortex stirring time and no NaCl adjustment. Fig. S1† shows a chromatogram of a standard solution containing the analytes at 10 ng mL−1 subjected to the optimized VA-DLLME procedure, compared to the chromatogram obtained for an unextracted standard solution of same concentration.
Study of the pretreatment of the sample
Once the entire extraction stage was optimized, studies were carried out by performing the VA-DLLME directly on a sample solution obtained from a N-nitrosamines-free cosmetic cream. This type of complex matrices has a high number of lipophilic components and surfactants that could negatively affect the VA-DLLME procedure. Indeed, it was observed that when performing the microextraction, the lipophilic compounds contained in the matrix also passed to the chloroform, precipitating at the bottom of the centrifuge tube, and thus causing the formation of a cloudy drop that was not possible to inject into the GC-MS system. For this reason, different strategies were carried out for the pretreatment of the sample to perform a clean-up step, prior to the VA-DLLME procedure, with the aim of eliminating these impurities. For these studies, a sample solution of a N-nitrosamines-free cosmetic cream was prepared by weighing 0.5 g of sample into a 50 mL volumetric flask and filling to the line with water, after spiking it to 1 ng mL−1 with target analytes.In a first attempt, LLE clean-up was carried out with 1 mL of hexane over 5 mL of the aqueous sample solution. In a second attempt, 5 mL of the aqueous sample solution were percolated throughout different SPE cartridges (i.e., Discovery® DSC-Diol, Strata-X™, and Strata-SDB-L) subjected to vacuum.
As can be seen in Fig. 3a, the best performance was achieved by means of LLE with 1 mL of hexane. Among the SPE cartridges, the best results were obtained with the Discovery® DSC-Diol cartridge, whose performance was the closest to LLE with hexane, obtaining slightly better results only for the more lipophilic nitrosamines (i.e., NDPA, NDBA and NDPhA), which were more likely to be removed with the hexane and therefore lower signal were obtained in these three cases.
Thus, it was considered to continue testing the LLE with hexane, and thus to discard the percolation of the sample solution through the SPE cartridges because the analytes interacted partially with the different sorbents, preventing their immediate elution and causing their loss.
Then, the possibility of reducing the volume of hexane from 1 mL to 200 μL (minimum amount of solvent to make it easy to collect) was studied. Likewise, the option of not doing an LLE was also evaluated through two other approaches: (1) filtering the sample solution through a 0.45 μm particle size nylon filter to remove impurities from the matrix prior to microextraction, and (2) do not perform any clean-up step and just leach the analytes from the sample into water. The latter approach was also considered because the aforementioned cloudy droplet that formed when VA-DLLME was performed directly on the sample solution with 80 μL of chloroform became a layer between the aqueous donor phase and the extractant phase if the volume of chloroform was increased to 120 μL. In this way, a small clean drop of extractant phase, easy to be collected and injected into the GC-MS system, was obtained at the bottom of the microextraction tube.
Fig. 3b compares these approaches and shows that, in general terms, the best results were achieved by increasing the volume of chloroform without performing a clean-up step, despite not using the previous optimized value.
The results obtained with the selected methodology were very positive compared to all the approaches considered in the sample pretreatment studies described above, not only because no analytes were lost during the procedure and a pretreatment step prior to microextraction was avoided reducing the time of analysis, but also because the amount of waste produced is minimized. Otherwise, the waste would be higher if filters, cartridges, or hexane were used in a clean-up step.
Study of the matrix effect
To evaluate the matrix effect caused by cosmetic matrices during the analytical procedure, an external calibration was performed with aqueous standard solutions from 0.01 to 10 ng mL−1 and subjected to the optimized VA-DLLME process. Likewise, non-spiked samples and spiked with 0.5 and 1 ng mL−1 sample solutions were prepared and subjected to microextraction. The relative recovery (RR) was evaluated as RR% = 100 × (CM+S − CM)/CS added, being CM the concentration of the measurement solution in the original sample, CM+S the concentration of the measurement solution in the fortified sample, and CS added the standard concentration added.The RR values obtained with this study ranged between 11 and 431%, demonstrating that the analytical process was affected by a consistent matrix effect, either positive or negative depending on the analyte and on the sample. Hence, standard addition calibration was employed to correct the observed matrix effects.28
Analytical performance of the proposed method
Different analytical parameters were evaluated to validate the proposed method. These results are summarized in Table 3.| Analytes | LODa (ng L−1) | LOQa (ng L−1) | MLODb (μg kg−1) | MLOQb (μg kg−1) | EFc | Repeatabilityd (%RSD) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Intra-day (N = 5) | Inter-day (N = 5) | ||||||||||
| 0.1 ng mL−1 | 0.5 ng mL−1 | 5 ng mL−1 | 0.1 ng mL−1 | 0.5 ng mL−1 | 5 ng mL−1 | ||||||
| a LOD: limit of detection; LOQ: limit of quantification, calculated as 3 and 10 times, respectively, the signal-to-noise ratio.b MLOD: method limit of detection; MLOQ: method limit of quantification, according to the sample pretreatment.c EF: enrichment factor.d RSD: relative standard deviation.e n.a.: not applicable, since the concentration is below the LOQ. | |||||||||||
| NDMA | 74.3 | 247.5 | 7.4 | 24.8 | 2 | n.a.e | 6.7 | 3.3 | n.a. | 10.9 | 4.3 |
| NMEA | 91.5 | 304.9 | 9.2 | 30.5 | 18 | n.a. | 4.6 | 2.1 | n.a. | 7.2 | 9.1 |
| NDEA | 19.1 | 63.6 | 1.9 | 6.4 | 38 | 3.0 | 3.4 | 3.3 | 7.3 | 4.6 | 4.6 |
| NDPA | 4.4 | 14.7 | 0.4 | 1.5 | 100 | 4.6 | 1.1 | 4.5 | 6.0 | 7.6 | 12.4 |
| NDBA | 0.2 | 0.6 | 0.02 | 0.06 | 73 | 6.0 | 4.4 | 3.7 | 6.6 | 8.4 | 12.5 |
| NPIP | 18.6 | 61.9 | 1.9 | 6.2 | 91 | 5.3 | 4.2 | 4.8 | 9.7 | 12.5 | 7.6 |
| NPYR | 33.3 | 110.9 | 3.3 | 11.1 | 25 | 7.4 | 2.2 | 3.8 | 6.1 | 11.1 | 5.2 |
| NMOR | 83.8 | 279.3 | 8.4 | 27.9 | 10 | n.a. | 2.0 | 2.7 | n.a. | 8.4 | 5.5 |
| NDPhA | 35.0 | 116.8 | 3.5 | 11.7 | 66 | 4.3 | 2.4 | 6.9 | 9.9 | 6.7 | 7.7 |
It should be noticed that, as standard addition calibration is used to correct the matrix effects, and these are different for each analyte in each tested sample, thus affecting the signal in different extension, those parameters depending on the signal, such as linearity, limits of detection (LOD) and quantification (LOQ), and enrichment factor (EF), are matrix-dependent. In this sense, in order to obtain values just to know the magnitude order and to compare with other methods, they were obtained by using aqueous solutions.
The method achieved a good linearity at least up to 20 ng mL−1, with determination coefficients (R2) > 0.990.
LODs and LOQs, calculated as 3 and 10 times the signal-to-noise ratio of a standard solution subjected to the proposed method, ranged from 0.2 to 91.5 ng L−1 and from 0.6 to 304.9 ng L−1, respectively. Therefore, further considering sample dilution, method LODs (MLODs) ranged from 0.02 to 9.2 μg kg−1, and the method LOQs (MLOQs) ranged from 0.06 to 30.5 μg kg−1, in the cosmetic samples. These values are well below the threshold value of 50 μg kg−1 for traces of N-nitrosamines in cosmetic products established by the European Regulation,3,5 which confirms that the method is suitable for the determination of these compounds in this kind of matrices.
The achieved EFs, defined as EF = Cext/C0, where Cext is the concentration of the analyte in the extract and C0 is the initial concentration of the analyte in the donor phase before the extraction, was calculated using a standard solution at 1 ng mL−1 of the analytes as initial concentration, and ranged from 2 to 100, due to the different polarities of the N-nitrosamines studied that respectively affect the extraction efficiency of each of them.
The repeatability, expressed as relative standard deviation (RSD), was evaluated by applying the proposed VA-DLLME method to five replicates of aqueous standard solution at three different concentrations (i.e., 0.1, 0.5 and 5 ng mL−1) on the same day (intra-day) and for five consecutive days (inter-day). The intra-day repeatability values ranged from 3.0 to 7.4% at 0.1 ng mL−1, from 1.1 to 6.7% at 0.5 ng mL−1, and from 2.1 to 6.9 at 5 ng mL−1. The inter-day repeatability values ranged from 6.0 to 9.9% at 0.1 ng mL−1, from 4.6 to 12.5% at 0.5 ng mL−1, and from 4.3 to 12.5 at 5 ng mL−1. Results show that good repeatability values have been achieved with the proposed method.
Table 4 shows the comparison of the new developed method with those previously published with the same purpose, both solid- and liquid-phase extraction-based methods.
| Target N-nitrosamines | Sample pretreatmenta | Extraction techniqueb | Timec | Organic solvent a | Instrumental techniqued | MLOD (μg g−1) | AGREEprep score | Ref. |
|---|---|---|---|---|---|---|---|---|
| a ACE: acetone; ACN: acetonitrile; EtAc: ethyl acetate; DCM: dichloromethane; HEX: hexane; MeOH; methanol.b DSPE: dispersive solid-phase extraction; HS-SPME: headspace-solid phase microextraction; LLE: liquid–liquid extraction; μ-MSPD: micro matrix solid-phase dispersion; SBSDME: stir bar sorptive dispersive microextraction; SPE: solid-phase extraction; VA-RP-DLLME: vortex-assisted reversed-phase dispersive liquid–liquid microextraction.c n.r.: no reported.d GC: gas chromatography; LC: liquid chromatography; MS: mass spectrometry; MS/MS: tandem mass spectrometry. | ||||||||
| 10 | 1 g + 10 mL MeOH:DCM; sonicated 10 min; centrifuged 15 min |
SPE | n.r. | 6 mL MeOH (conditioning); 3 mL MeOH (30%) (washing); 8 mL MeOH (desorption); evaporation + 1 mL MeOH | GC-MS/MS | 700–3000 | 0.16 | 24 |
| 13 | 0.2 g + 4 mL H2O | LLE | Vortex 1 min (extraction) centrifugation 3 + 2 min | 3 mL ACN × 2 (extraction) evaporation + 1 mL EtAc | GC-MS | 3–15 | 0.50 | 21 |
| 7 | 5 g + 5 mL DCM:MeOH; sonicated 30 min |
HS-SPME | 30 min (extraction) | — | GC-MS | 0.46–36.54 | 0.24 | 22 |
| 11 | 1 g + 7 mL ACN; sonicated 10 min; centrifuged 3 min | DSPE | Vortex 20 min (extraction) centrifugation 3 min | Evaporation + 1 mL MeOH | LC-MS/MS | 7–250 | 0.25 | 29 |
| 7 | g + 5 mL HEX; vortex; centrifuged 5 min | VA-RP-DLLME | Vortex 0.5 min (extraction) centrifugation 5 min | — | LC-MS | 1.8–50 | 0.48 | 27 |
| 8 | 0.5 g + 25 mL H2O; vortex; 1 mL HEX; centrifuged 15 min | SBSDME | 30 min (extraction) 1 min (desorption) | 1 mL ACE (desorption) | LC-MS/MS | 3–13 | 0.37 | 28 |
| 10 | 0.1 g + 0.2 Na2SO4 + 0.4 g Florisil® | μ-MSPD | n.r. | 0.1 g Florisil® 1 + 10 mL EtAC (desorption) | GC-MS | 12–150 | 0.49 | 23 |
| 9 | 0.05 g + 5 mL H2O | VA-DLLME | Vortex 1 min (extraction) centrifugation 5 min | 120 μL CH3Cl (extraction) | GC-MS | 0.06–30.5 | 0.59 | This work |
As can be seen, the MLODs are of the same magnitude order, even that in those methods based on more expensive analytical instruments (e.g., MS/MS detection). Regarding the extraction time, liquid-based extraction techniques, as the proposed method, are much faster since the extraction is achieved practically instantaneously (less than 1 min), which is beneficial to get a high-throughput method. Moreover, no sophisticated equipment or commercial materials (e.g., sorbents, cartridges, or fibers) are needed to perform the extraction, making the method affordable for most laboratories. On the other hand, the main drawback of the proposed method is the use of an organochloride solvent as extraction phase, which is against current trends in Analytical Chemistry, but just a low volume is used.
Regarding this last matter, the greenness of the sample preparation of each work was evaluated by using the new metric tool termed AGREEprep.30 This metric tool considers various environmental and health impact factors such as the use of reagents, the consumed energy, possible occupational hazards, and generated wastes, among others. This tool scores the sample preparation stage on a scale from 0 to 1, where 0 is the worst score and 1 is the maximum score. As can be seen in Table 4, the score obtained by the proposed method is positively comparable to the other methods since the sample pretreatment is simple (just lixiviation of the analytes in water), in contrast to those methods where higher amounts of sample and solvents such as methanol, dichloromethane and/or hexane are employed.
Application to the analysis of commercial cosmetic products
Three commercially available cosmetic samples, an aftersun gel and two different body creams, were analyzed using the proposed VA-DLLME method. As can be seen in Table 5, the results obtained in the analysis of the three cosmetic samples show that N-nitrosamines have been quantitatively determined in all of them.| Analytes | Found amount (μg kg−1)a | ||
|---|---|---|---|
| Aftersun gel | Body cream 1 | Body cream 2 | |
| a expressed as mean ± standard deviation of three replicates. | |||
| NDMA | < LOD | < LOD | < LOD |
| NMEA | 770 ± 90 | < LOD | 560 ± 20 |
| NDEA | < LOD | < LOD | < LOD |
| NDPA | 1.19 ± 0.01 | < LOD | < LOD |
| NDBA | 16.5 ± 0.1 | < LOD | < LOD |
| NPIP | 50.6 ± 0.3 | < LOD | < LOD |
| NPYR | 114 ± 5 | < LOD | < LOD |
| NMOR | < LOD | 870 ± 60 | < LOD |
| NDPhA | 10.57 ± 0.03 | < LOD | < LOD |
The results obtained in the aftersun gel revealed that six N-nitrosamines were determined, three of which exceed the safety limit of 50 μg kg−1 defined by the European Regulation (i.e., NMEA 770 ± 90 μg kg−1, NPIP 50.6 ± 0.3 μg kg−1, and NPYR 114 ± 5 μg kg−1).
Regarding the creams, only NMOR has been quantified in the first cream, which widely exceeds the safety limit, being found at a concentration of 870 ± 60 μg kg−1, while in the second cream only NMEA was quantified, which was also found to be above the safety limit, at a concentration of 560 ± 20 μg kg−1. Chromatograms of the sample solutions subjected to the proposed VA-DLLME method are shown in Fig. S2.†
It should be noted that, as declared on the labels, these three samples analyzed included two ingredients that, even though they are permitted ingredients as they are a preservative (i.e., bronopol) and a pH regulator (i.e., triethanolamine), their reaction can unintentionally cause the formation of nitrosamines.
Conclusions
A sensitive analytical method for determining trace levels of nine banned N-nitrosamines in cosmetic products has been successfully developed and validated. The proposed method is based on vortex-assisted dispersive liquid–liquid microextraction (VA-DLLME) followed by gas chromatography-mass spectrometry (GC-MS). The variables involved in the microextraction stage have been optimized, and comparative studies of sample pretreatment have been carried out to find the best methodology that allows the analysis of the greatest number of nitrosamines at the same time with the required sensitivity, favoring their extraction from the complex cosmetic matrices without losing analytes during the procedure.The proposed method has good analytical characteristics that, in addition to being a simple and affordable procedure, make it suitable for quality control of cosmetics in order to guarantee the safety of users and compliance with the European Regulation on cosmetic products. It should not be forgotten that one of the requirements of routine analysis methods is that they allow a high sample throughput, i.e., analyze several samples in a short period of time, and this is achieved by DLLME-based methods.
Author contributions
L. Schettino: methodology, validation, investigation, data curation, writing – original draft; J. L. Benedé: methodology, writing – reviewing and editing, supervision; A. Chisvert: conceptualization, methodology, resources, writing – reviewing and editing, supervision, funding acquisition.Conflicts of interest
The authors declare that there are no conflicts to declare, they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This article is based upon work from the National Thematic Network on Sample Treatment (RED-2018-102522-T) of the Spanish Ministry of Science, Innovation and Universities, and the Sample Preparation Study Group and Network supported by the Division of Analytical Chemistry of the European Chemical Society.References
- R. Fernández-Alba and A. Agüera, in Encyclopedia of Analytical Science, ed. P. Worsfold, A. Townshend and C. Poole, Elsevier, Amsterdam, 2nd edn, 2005, vol. 6, pp. 197–202 Search PubMed.
- G. Eisenbrand, G. M. Blankart, H. Sommer and B. Weber, IARC Sci. Publ., 1991, 105, 238–241 CAS.
- Scientific Committee on Consumer Safety, Opinion on Nitrosamines and Secondary Amines in Cosmetic Products, 2012 Search PubMed.
- Fifteenth Commission Directive 92/86/EEC of 21 October 1992, Adapting to Technical Progress Annexes II, III, IV, V, VI and VII of Council Directive 76/768/EEC on the Approximation of the Laws of the Member States Relating to Cosmetic Products, 1992 Search PubMed.
- Regulation (EC) No 1223/2009 of the European Parliament and of the Council of 30 November 2009 on Cosmetic Products, and its Successive Amendments, 2009 Search PubMed.
- A. Chisvert, J. L. Benedé, M. Peiró, I. Pedrón and A. Salvador, Talanta, 2017, 166, 81–86 CrossRef CAS PubMed.
- J. L. Ho, H. H. Wisneski and R. L. Yates, J. Assoc. Off. Anal. Chem., 1981, 64, 800–804 CrossRef CAS.
- Y. Fukuda, Y. Morlkawa and I. Matsumoto, Anal. Chem., 1981, 53, 2000–2003 CrossRef CAS.
- R. C. Schothorst and H. H. J. Somers, Anal. Bioanal. Chem., 2005, 381, 681–685 CrossRef CAS.
- C. Flower, S. Carter, A. Earls, R. Fowler, S. Hewlins, S. Lalljie, M. Lefebvre, J. Mavro, D. Small and N. Volpe, Int. J. Cosmet. Sci., 2006, 28, 21–33 CrossRef CAS PubMed.
- D. B. Black, R. C. Lawrence, E. G. Lovering and J. R. Watson, J. AOAC Int., 1981, 64, 1474–1478 CrossRef CAS.
- K. M. Joo, M. S. Shin, J. H. Jung, B. M. Kim, J. W. Lee, H. J. Jeong and K. M. Lim, Talanta, 2015, 137, 109–119 CrossRef CAS PubMed.
- R. Schwarzenbach and J. P. Schmid, J. Chromatogr. A., 1989, 472, 231–242 CrossRef CAS.
- S. K. Vohra and G. W. Harrington, Food Cosmet. Toxicol., 1981, 19, 485–487 CrossRef CAS PubMed.
- S. S. H. Davarani, L. Masoomi, M. H. Banitaba, H. R. L. Z. Zhad, O. Sadeghi and A. Samiei, Talanta, 2013, 105, 347–353 CrossRef CAS PubMed.
- A. Tada, C. Rodrigues-Silva and S. Rath, J. Pharm. Biomed. Anal., 2021, 202, 114132 CrossRef CAS PubMed.
- International Organization for Standardization, ISO 10130, Cosmetics – Analytical methods – Nitrosamines: Detection and determination of N-nitrosodiethanolamine (NDELA) in cosmetics by HPLC, Postcolumn Photolysis and Derivatization, 2009 Search PubMed.
- International Organization for Standardization, ISO 15819, Cosmetics – Analytical methods – Nitrosamines: Detection and determination of N-nitrosodiethanolamine (NDELA) in Cosmetics by HPLC-MSMS, 2014 Search PubMed.
- H. Sommer, H. P. Loeffler and G. Eisenbrand, J. Soc. Cosmet. Chem., 1988, 39, 133–137 CAS.
- H. Sommer and G. Eisenbrand, Z. Lebensm.-Unters. Forsch., 1988, 186, 235–238 CrossRef CAS.
- H. Dong, X. Guo, Y. Xian, H. Luo, B. Wang and Y. Wu, J. Chromatogr. A., 2015, 1422, 82–88 CrossRef CAS.
- N. R. Choi, Y. P. Kim, W. H. Ji, G. S. Hwang and Y. G. Ahn, Talanta, 2016, 148, 69–74 CrossRef CAS PubMed.
- M. Celeiro, L. Rubio, C. Garcia-Jares and M. Lores, Molecules, 2021, 26(9), 2504 CrossRef CAS PubMed.
- Q. Ma, H. W. Xi, C. Wang, H. Bai, G. C. Xi, N. Su, L. Y. Xu and J. B. Wang, Chinese J. Anal. Chem., 2011, 39, 1201–1207 CrossRef CAS.
- S. M. Billedeau, T. M. Heinze, J. G. Wilkes and H. C. Thompson, J. Chromatogr. A., 1994, 688, 55–65 CrossRef CAS.
- L. H. Wang, H. C. Hsia and C. C. Wang, J. Liq. Chromatogr. Relat. Technol., 2006, 29, 1737–1751 CrossRef CAS.
- P. Miralles, A. Chisvert and A. Salvador, J. Sep. Sci., 2018, 41, 3143–3151 CrossRef CAS.
- P. Miralles, I. van Gemert, A. Chisvert and A. Salvador, J. Chromatogr. A., 2019, 1604, 460465 CrossRef CAS PubMed.
- F. Liu, Y. Xian, J. Chen, H. Dong, H. Liu, X. Guo, M. Chen and H. Li, Anal. Methods, 2016, 8, 4245–4253 RSC.
- W. Wojnowski, M. Tobiszewski, F. Pena-Pereira and E. Psillakis, TrAC Trends Anal. Chem., 2022, 149, 116553 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06553c |
| This journal is © The Royal Society of Chemistry 2023 |