DOI:
10.1039/D2RA06272K
(Paper)
RSC Adv., 2023,
13, 888-894
New boro amino amide organocatalysts for asymmetric cross aldol reaction of ketones with carbonyl compounds†
Received
5th October 2022
, Accepted 20th December 2022
First published on 4th January 2023
Abstract
Distinct types of new boron fused primary amino amide organocatalysts were designed and synthesized from commercially available amino acids. Their catalytic activities were investigated in asymmetric crossed aldol reaction of ketones with aromatic aldehydes to afford the corresponding chiral anti-aldol adducts with good chemical yields, moderate diastereoselectivity and good to excellent enantioselectivities (up to 94% yields, up to 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 dr, up to 94% ee).
:10 dr, up to 94% ee).
1. Introduction
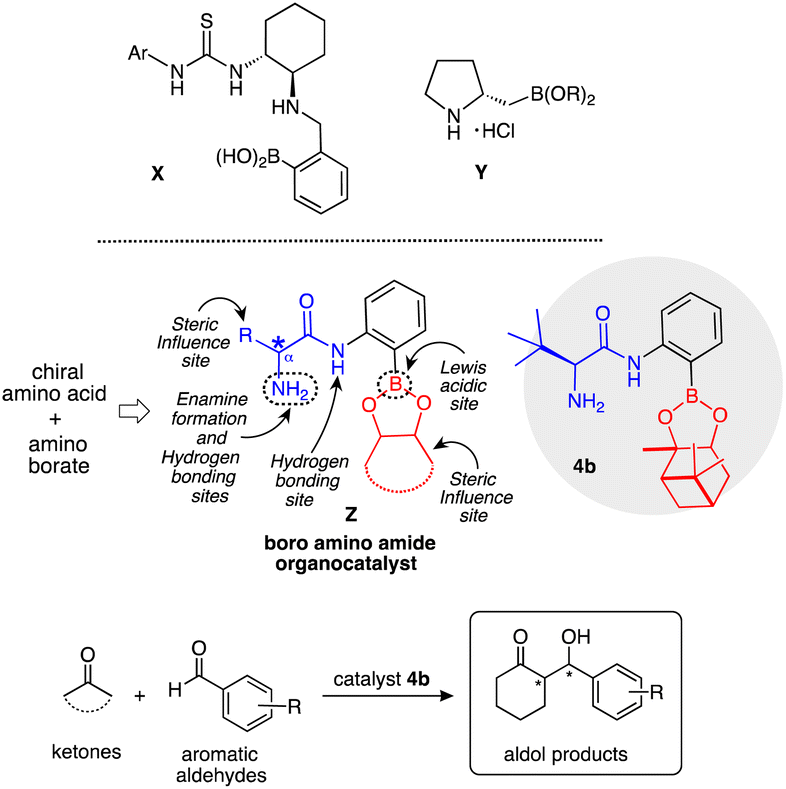
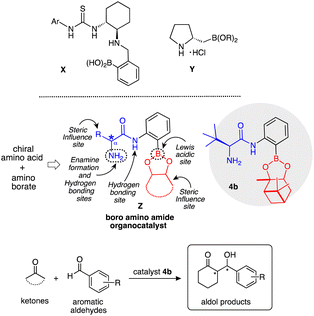
The rapidly growing interest in the area of asymmetric organocatalysis is not only due to the versatile character of small organic molecules which function as efficient and selective catalysts but is also attributed to their important role in the construction of complex and enantiopure molecular skeletons.1 Several contributions to the study of asymmetric aldol reactions were reported earlier2 and with the development of the proline catalysed direct intermolecular asymmetric aldol reaction by List, Lerner and Barbas III,3 the synthesis and application of organocatalysts based on enamine–iminium ion activation and hydrogen bonding activation has received further considerable attention.4 Most organocatalysts currently used are functionally synergic systems which have two or more distinct functionalities within the same molecule. Among the various polyfunctional organocatalysts available, one functional group works as enamine–iminium formation or Brønsted basic sites and the other works as a Lewis acidic site.5 Our group is also exploring new polyfunctional organocatalysts based on primary amino alcohols and amino amides, which are easily derived from commercially available amino acids, in numerous asymmetric reactions.6 However, only a few examples have been reported on the polyfunctional organocatalysts with boron functional groups as Lewis acidic sites, such as thiourea-boronic acid X by Takemoto7 and proline boronate Y by Whiting,8 so far.
Based on the above background, we designed a boron derived amino amide organocatalyst Z, obtained by facile synthetic transformation. Catalyst Z contains an amino group acting as an enamine formation site, amino amide group for hydrogen bonding site, boron atom acting as a Lewis acidic site, and also pinene and the β-position substituent act as steric influence sites in a single molecule (Scheme 1). The cross aldol reaction of ketones with aldehydes was chosen to explore the activity of Z as an organocatalyst. The aldol reaction9 is one of the most versatile and popular method for the formation of C–C bonds10 in modern organic synthesis.
 |
| | Scheme 1 Asymmetric cross aldol reaction of ketones with carbonyl compounds. | |
Herein, we describe new boro amino amide Z as an organocatalyst, which showed an efficient catalytic activity in crossed aldol reaction of ketones with aromatic aldehydes to afford aldol products. Especially, catalyst 4b showed satisfactory catalytic activity in this reaction to afford the corresponding aldol products (up to 94%, up to anti:syn/90:10, up to 94% ee) in eco-friendly sea-H2O-tap H2O solvent condition.
2. Results and discussion
2.1. Catalyst design and synthesis
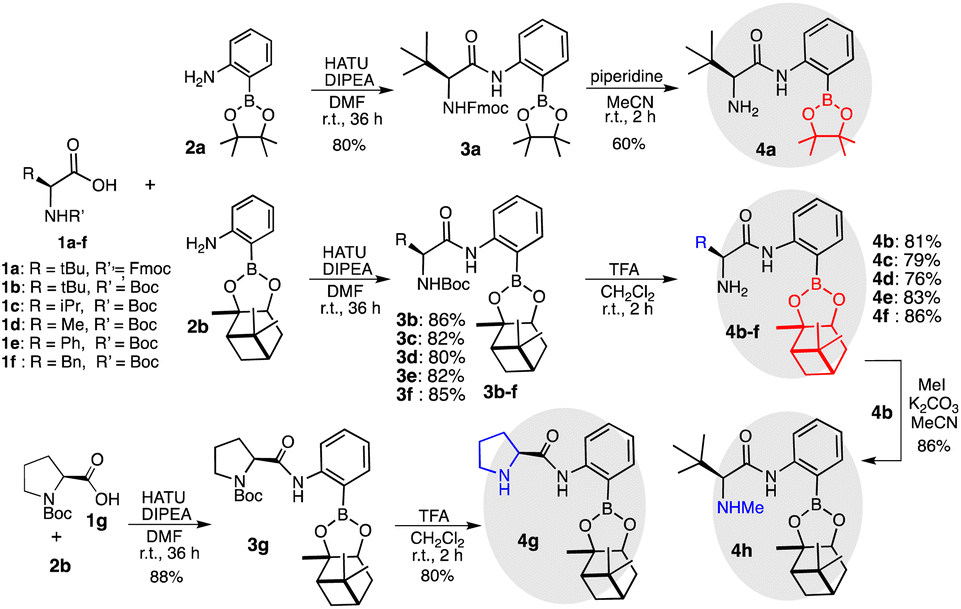
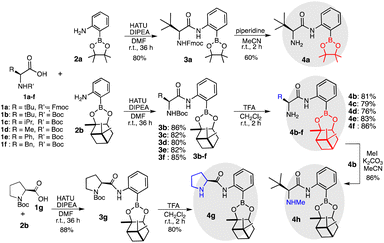
Initially, we synthesized catalysts 4a–f having primary amino group by the coupling of protected amino acids 1a–f with amines 2a and 2b having boron substituents, respectively, followed by the deprotections of protected boro amino amides 3a and 3b–f, respectively, in good yields (Scheme 2). Moreover, secondary amino catalyst 4g having proline ring was synthesized by condensation of 1g with 2b, followed by the deprotection of 3g. Catalyst 4h having secondary amino group was derived from 4b by methylation with MeI and K2CO3.
 |
| | Scheme 2 Preparations of catalysts 4a–h. | |
2.2. Catalytic efficiency
To find out the best catalyst, our preliminary investigations were focused on screening of the synthesized catalysts 4a–h in the crossed aldol reaction of cyclohexanone 5a as an aldol donor with p-nitro benzaldehyde 6a as an aldol acceptor (Table 1). First, the catalytic activity of boro amino amide 4a with primary amino group and pinacolyl boron substituent was examined under THF at room temperature, but the formation of the corresponding aldol product 7a or 8 was not observed (entry 1). Next, this reaction using benzoic acid as a co-catalyst was examined (entries 2–9). The reaction was carried out with 10 mol% of 4a and 20 mol% of benzoic acid in THF at room temperature. Initially, the functionality of catalyst 4a was examined (entry 2). However, this catalyst 4a did not work enough and the corresponding aldol product 7 was obtained in low chemical yield as racemate. Next, catalysts 4b–f with primary amino group, several substituents at α-position and bulky pinenyl boron group on amide amino group were investigated (entries 2–7). As a result, all the catalysts showed catalytic activity. Especially, the use of catalyst 4b with tert-butyl group afforded anti-aldol product 7a as a main product with moderate diastereo and enantioselectivity (anti:syn/60:40, 66% ee), although chemical yield was low (24%) (entry 3). We also screened the activity of secondary amino catalyst 4g having pyrrolidine ring that is valid for forming enamine with ketone 5a (entry 8). However, the sufficient improvement of both chemical yield and stereoselectivities (28%, anti:syn/60:40, 56% ee) in the reaction using 4g was not observed in comparison with the result of catalyst 4b having primary amino group. Based on the above results, the functionality of catalyst 4h which is obtained by a change of primary amino group on 4a to secondary amino group was also examined under similar reaction condition (entry 9). However, contrary to expectation, catalyst 4h did not work in this reaction. In addition, the aldol reaction was also carried out in the similar conditions as reported by Whiting et al., using the HCl salt of our best catalyst 4b, but 4b HCl salt catalyst only showed less catalytic activity and the aldol product 7a was observed as racemate (Scheme 3). From all of the above results, it was observed that catalyst 4b with primary amino group and both tert-butyl group at α-position and bulky pinenyl boron group on amide amino group as a steric influence sites performed better comparatively with other catalysts 4a,c–h. The absolute configuration and anti/syn diastereoselectivity of 7a and 8 were identified based on comparison with literature data.11
Table 1 Asymmetric aldol reaction of 5a and 6a with organocatalysts 4a–h
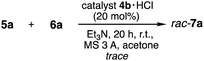 |
| | Scheme 3 Aldol reaction under Whiting et al. condition. | |
2.3. Improving catalytic efficiency
To improve the catalytic efficiency, the reaction conditions such as solvent, co-catalysts, and reaction times were examined by using superior catalyst 4b (Table 2). First, solvent effect was examined with THF, i-PrOH, MeOH and H2O as polar protic solvent, DMF as a polar aprotic solvent, dioxane as ethereal solvent (entries 1–6), neat condition (entry 7) and CH2Cl2 as halogenated solvent (entry 8) in the presence of catalyst 4b (10 mol%) and benzoic acid as a co-catalyst (20 mol%) at room temperature for 96 h. As a result, only tap H2O had a quite influence on the enantioselectivity of the reaction to afford anti-aldol product 7a with moderate chemical yield and diastereoselectivity (50%, anti:syn/64:36, 85% ee) (entry 4). It was observed that based on the previous reports12 of aldol reaction in water, which suggest that water molecule facilitates the ease of the condensation process and enhances the reactivity of the reaction, with no less influence on the transition state, especially in the asymmetric aldol reaction. Next, a series of co-catalysts were examined using superior tap H2O as solvent (entries 9–12). With all the co-catalysts, the results were found to be inferior to benzoic acid in terms of chemical yield and stereoselectivity suggesting benzoic acid as superior co-catalyst. Next, the reaction was carried out in different types of H2O (Table 3). Initially, sea H2O was used in this reaction and the product 7a was obtained with moderate enantioselectivity (60% ee), but chemical yield was increased to 86% with moderate diastereoselectivity (anti:syn/68:32) (entry 1). Using distilled H2O, chemical yield was low (48%), but the enantioselectivity was relatively good (77% ee) with moderate diastereoselectivity (anti:syn/67:33) (entry 2). The use of brine afforded 7a with moderate chemical yield and stereoselectivities (50%, anti:syn/62:38, 67% ee) (entry 3). From those results, it was indicated that sea H2O is effective to increase the chemical yield of the aldol product 7a. This might be due to the high density, higher electrical conductivity, slight basic nature of sea water in combination with mineral contents were assumed to accelerate the reaction yields of the aldol reaction, although the reason is not clear. We then changed the time duration of the reaction using sea water for 72 h and 120 h. As a result, 72 h of reaction time was better for obtaining 7a in good chemical yield, moderate diastereoselectivity and enantioselectivity (80%, anti:syn/68:32, 75% ee) (entry 4). On further increase of reaction time to 120 h resulted in the aldol product in low enantioselectivity (90%, anti:syn/66:34, 11% ee) (entry 5).
Table 2 Optimisation of reaction conditions using catalyst 4b
Table 3 Optimisation of reaction condition using H2O solvent using 4b
Good yield was observed with sea water and relatively good enantioselectivity was observed with tap water. Therefore, we attempted the aldol reaction in a mixture of sea H2O and tap H2O (1:1) while varying the co-catalysts and catalyst mol ratio. Initially, the reaction using mixed sea H2O-tap H2O with 10 mol% of catalyst 4b and 20 mol% of benzoic acid as a co-catalyst was investigated (82%, anti:syn/68:32, 94% ee) (entry 6). To our surprise, the aldol product 7a was obtained in good enantioselectivity (94% ee), good chemical yield and moderate diastereoselectivity (82%, anti:syn/68:32). Further, on changing the catalyst and co-catalyst mol ratios (entries 7–10) with the solvent system as sea H2O-tap H2O, the obtained results were found to be inferior to the result of entry 6. Based on the above results, the catalyst 4b (10 mol%) and benzoic acid (20 mol%) as a co-catalyst in the mixed solvent of sea H2O-tap H2O at 72 h was confirmed as the optimum condition.
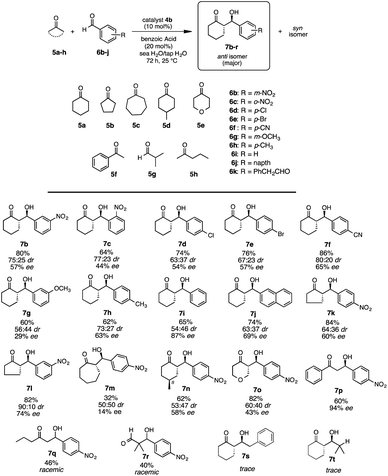
2.4. Substrate scope
After the optimization of reaction conditions, we examined the generality of the superior catalyst 4b in the aldol reactions of various ketones 5a–h with aldehydes 6b–k (Scheme 4). The reactions were carried out in the presence of catalyst 4b (10 mol%) and benzoic acid as a co-catalyst (20 mol%) under the best reaction condition (sea H2O-tap H2O, 72 h, 25 °C).
 |
| | Scheme 4 The aldol reactions of 5a–h with different aromatic aldehydes 6b–k. | |
Initially, substituent influences on the aromatic ring of aldehydes 6b–j were examined. The reactions of 5a with nitro aromatic aldehydes [6b (m-NO2), 6c (o-NO2)] gave the corresponding aldol products 7b,c respectively, with moderate to good chemical yield and diastereoselectivities, but with moderate enantioselectivities (7b: 80%, anti:syn/75:25 dr, anti: 57% ee, syn: 58% ee; 7c: 64%, anti:syn/77:23 dr, anti: 44% ee, syn:25% ee). Whereas the reaction of 5a with halogenated aromatic aldehydes [6d (p-Cl), 6e (p-Br)] gave the corresponding aldol products 7d,e respectively, with good chemical yields, diastereoselectivities and moderate enantioselectivities (7d: 74%, anti:syn/63:37 dr, anti: 54% ee, syn: 10% ee; 7e: 76%, anti:syn/67:23 dr, anti: 57% ee, syn: 16% ee). Furthermore, the reactions of 5a with aldehyde 6f (p-CN), having electron withdrawing group afforded the aldol product 7f in good chemical yield and moderate to good diastereoselectivities and moderate enantioselectivity (7f: 86%, anti:syn/80:20 dr, anti: 65% ee, syn: 60% ee). The influence of electron donating group on aldehydes were also examined by the reactions of 5a with 6g (m-methoxy) and with 6h (p-methyl) to afford the corresponding anti-aldol products 7g and 7h in moderate chemical yields, moderate to good diastereoselectivities and low to moderate enantioselectivities (7g: 60%, anti:syn/56:44 dr, anti: 29% ee, syn: 34% ee; 7h: 62%, anti:syn/73:27, anti: 63% ee, syn: 12% ee), respectively. When benzaldehyde 6i and bulkier napthaldehyde 6j were used, the chemical yields, diastereoselectivities and enantioselectivities were moderate to good (7i: 55%, anti:syn/54:46 dr, anti: 87% ee, syn: 12% ee; 7j: 74%, anti:syn/63:37 dr, anti: 69% ee, syn: 40% ee). Furthermore, the reactions of 5 membered cyclopentanone 5b with 6a,b and 7 membered cycloheptanone 5c with 6a, respectively, was carried out in the presence of catalyst 4b (10 mol%) and benzoic acid as a co-catalyst (20 mol%) under the best reaction conditions. The product 7k derived from the reaction of 5b with 6a was obtained in good chemical yield, moderate diastereoselectivity and enantioselectivity (84%, anti:syn/64:36 dr, anti: 60% ee, syn: 57% ee). The product 7l derived from the reaction of 5b with 6b in good chemical yields and stereoselectivities (82%, anti:syn/90:10 dr, anti: 74% ee, syn: 63% ee). Whereas, the reaction of 7 membered 5c with 6a did not proceed well giving the aldol product 7m in low chemical yield and diastereoselectivity with poor enantioselectivity (32%, anti:syn/50:50 dr, anti: 14% ee, syn: 4% ee). Moreover, the use of substituted cyclohexanone 5d also afforded the product 7n with moderate chemical yield and stereoslectivities (62%, anti:syn/53:47 dr, anti: 58% ee, syn: 49% ee). In addition, the reaction using pyranone 5e as heterocyclic ketone was also examined and the aldol product 7o was afforded in good chemical yield and diastereoslectivity with moderate enantioselectivity (82%, anti:syn/60:40 dr, anti: 43% ee, syn: 42% ee). The reaction of acetophenone 5f with 6a afforded the aldol product 7p in good enantioselectivity but moderate yields (60%, 94% ee). The reaction of acyclic ketones 5g,h with 6a, respectively, gave the aldol products 7q,r as racemate with low chemical yields. The aldol reaction of cyclohexanone with aliphatic aldehydes like 6k and 5g as acceptors, was also tried but the desired product was not observed.
2.5. Reaction mechanism
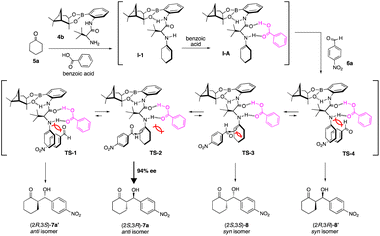
Considering both the good enantioselectivity (94% ee) of the chiral aldol product [2S,3R]-7a that was obtained in the reaction of 5a with 6a and the results of its calculation studies (Fig. 1–3), an enantioselective reaction course is proposed as follows (Scheme 5).
 |
| | Scheme 5 Plausible reaction course of the reaction. | |
For the estimation of enantioselective reaction course, initially the conformational analysis using the scan of total energies for the enamine intermediate I was carried out and the result indicated the conformation of enamine intermediate I-1, with an intramolecular hydrogen bonding between hydrogen atom of amide amino group and oxygen atom adjacent to boron atom, suggesting I-1 is better than I-2, as I-2 does not have similar intramolecular hydrogen bonding (Fig. 1). Furthermore, when benzoic acid as a co-catalyst is coordinated to more stable I-1 by hydrogen bonding, it is suggested that I-A, in which I-1 and benzoic acid are hydrogen bonded at two points to each other, is more stable than I-A′ (Fig. 2). In addition, it is also indicated that in I-1, the olefin of the enamine was anterior, but in I-A, its olefin has a posterior conformation (Fig. 3). Moreover, to examine the regioselectivity of the reaction between I-1 and 6a, the calculation of the energies (Fig. 1 and 3) and coefficients of their frontier orbitals (Fig. 4) were conducted. The energy levels of the orbitals calculation clearly showed the interaction between the LUMO of 6a and the HOMO of I-1, and their orbital phase clearly demonstrated a matching in favor of overlapping to afford the observed configuration of major aldol product 7a (Fig. 4).
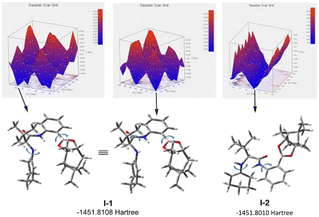 |
| | Fig. 1 Scan of total energies (up) and DFT optimized structures (down, at the B3LYP/6-31G(d) level of theory) of the enamine intermediate I generated by varying the torsion angles (the dihedral scans showed with u-shaped arrows: I-1). | |
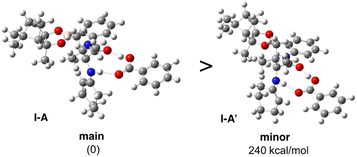 |
| | Fig. 2 The scan of total energies for I-A and I-A′. | |
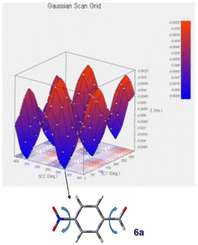 |
| | Fig. 3 Scan of total energies (up) and DFT optimized structures (down, at the B3LYP/6-31G(d) level of theory) of 6a generated by varying the torsion angles (the dihedral scans showed with u-shaped arrows). | |
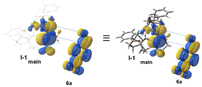 |
| | Fig. 4 The frontier orbital between I-1 and 6a. | |
Based on the above calculation results, the reaction might pass through enamine intermediate I-1 and complex I-A (I-1-benzoic acid) and subsequently, it was indicated that the reaction of I-A with 6a might progress through proposed four different transition states Ts-1–4 as shown in Scheme 5. However, among the proposed Ts-1–4, the reaction might proceed through Ts-2, based on the scan of total energy of I-A (Fig. 2) and the frontier orbital analysis (Fig. 3), having smaller steric interaction both between I-A and aldehyde 6a than those of Ts-1, 3, 4.
In contrary to the expectations, the calculation results suggest that, the boron atom on the catalyst does not coordinate the substrate 6a as a Lewis acid moiety. However, the boron atom has three covalent bonds in the plane, and its configuration may contribute to the formation of preferable transition states in affording aldol product 7a with a high stereoselectivity. Furthermore, it is also suggested that boron atom might coordinate with water solvent, forming an efficient conformation of the transition state, affording high enantioselectivity, although it is not clear why the solvent system is effective.
3. Conclusion
We have developed a new boron fused primary amino amides and their functionality as organocatalysts were examined in the asymmetric cross aldol reactions of various ketones 5a–h with different aromatic aldehydes 6a–k to afford the desired chiral aldol products 7a–r. The developed catalysts showed catalytic activity in this reaction. Particularly, catalyst 4b showed good catalytic activity to afford the various chiral aldol products in satisfactory chemical yields and stereoselectivities (up to 94%, anti:syn/68:32 dr, up to 94% ee) and interestingly, the effective solvent of the reaction was the mixture of sea H2O and tap H2O.
4. Experimental
4.1. General information
All reagents and dry solvents were purchased from commercial vendors and used directly without further purification. All reactions were placed in dried sample vials inserted with magnetic beads. 1H and 13C NMR spectroscopic data were recorded using a JEOL JNM-ECA500 instrument with tetramethylsilane as the internal standard. Thin-layer chromatography (TLC) was performed on Merck silica gel 60 F254 plates and the analytes were identified under UV light. Flash column chromatography was performed using silica gel pore size 60 N (40–100 μm). Infrared (IR) spectra were measured with a JASCO FT/IR-4100 spectrophotometer. HPLC data were collected using the TOSOH instrument equipped with (UV-8020, DP-8020, and SD-8022) detectors using Daicel Chiralpak AD-H, AS-H, OD-H columns. Optical rotations were measured with a JASCO-DIP-370 digital polarimeter. MS were taken on a JEOL-JMS-700V spectrometers.
4.2. General procedure for catalytic asymmetric aldol reaction of various ketones 5 with aromatic aldehydes 6
Catalyst 4b (10 mol%) and co-catalyst (20 mol%) were added to a solution of ketones (0.4 mmol) and the respective aldehydes (0.1 mmol) under the sea H2O-tap H2O (1:1) as solvent reaction condition. The reaction is allowed to stir at room temperature for appropriate time until the reaction completion. After the reaction completion, the solvent was removed under reduced pressure and the residue was purified by flash chromatography on silica gel (n-hexane/CH3CO2Et) to give the corresponding aldol products. The compounds are the known compounds and the structures were identified by spectral data which were in good agreement with those reported. The enantiomeric excess (ee) was determined using high pressure liquid chromatography (HPLC) principle by Daicel Chiralpak AD-H, AS-H, OD-H columns.11a–q
Conflicts of interest
There are no conflicts to declare.
References
-
(a) Z. Tang, F. Jiang, L. T. Yu, X. Cui, L. Z. Gong, A. Q. Mi, Y. Z. Jiang and Y. D. Wu, J. Am. Chem. Soc., 2003, 125, 5262 CrossRef CAS PubMed;
(b) Y. Chi and S. H. Gellman, Org. Lett., 2005, 7, 4253 CrossRef CAS PubMed;
(c) A. Lattanzi, Org. Lett., 2005, 7, 2579 CrossRef CAS PubMed;
(d) S. S. chimni and D. Mahajan, Tetrahedron: Asymmetry, 2006, 17, 2108 CrossRef CAS;
(e) R. M. De Figueiredo and M. Christmann, Eur. J. Org. Chem., 2007, 16, 2575 CrossRef;
(f) D. Enders, C. Grondal and R. M. Matthias, Angew. Chem., 2007, 46, 1570 CrossRef CAS PubMed;
(g) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS PubMed;
(h) S. Bertelsen and K. A. Jorgensen, Chem. Soc. Rev., 2009, 38, 2178 RSC;
(i) A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703 CrossRef CAS PubMed;
(j) P. Kasaplar, C. R. Escrich and M. A. Pericas, Org. Lett., 2013, 15, 3498 CrossRef CAS PubMed;
(k) D. L. SIlverio, S. Torker, T. Pilyugina, E. M. Vieria, M. L. Snapper, F. Haeffener and A. H. Hoyeda, Nature, 2013, 494, 7436 CrossRef;
(l) H. X. He and D. M. Du, RSC. Adv., 2013, 3, 16349 RSC;
(m) S. Saravanan, N. H. Khan, R. I. Kureshy, S. H. Abdi and H. C. Bajaj, ACS Catal., 2013, 3, 2873 CrossRef CAS;
(n) C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390 CrossRef CAS;
(o) X. Fang and C. J. Wang, Chem. Commun., 2015, 51, 1185 RSC;
(p) J. Chen, S. Meng, L. Wang, H. Tang and Y. Huang, Chem. Sci., 2015, 6, 4184 RSC;
(q) L. Xu, J. Huang, Y. Liu, Y. Wang, B. Xu, K. Ding, Y. Ding, Q. Xu, L. Yu and Y. Fan, RSC Adv, 2015, 5, 42178 RSC;
(r) T. Sekikawa, T. Kitaguchi, H. Kitaura, T. Minami and Y. Hatanaka, Org. Lett., 2016, 18, 646 CrossRef CAS PubMed;
(s) U. Varal, M. Durmaz and A. Sirit, Org. Chem. Front., 2016, 3, 730 RSC;
(t) J. Kaur, P. Chauhan and S. S. Chimni, Org. Biomol. Chem., 2016, 14, 7832 RSC;
(u) C. S. Evans and L. O. Davis, Molecules, 2018, 23, 33 CrossRef PubMed;
(v) H. Zhang, M. Han, T. Chen, L. Xu and L. Yu, RSC Adv, 2017, 7, 48214 RSC;
(w) Y. Zheng, A. Wu, Y. Ke, H. Cao and L. Yu, Chin. Chem. Lett., 2019, 30, 937 CrossRef CAS;
(x) S. H. Xiang and B. Tan, Advances in asymmetric organocatalysis over the last 10 years, Nat. Commun., 2020, 11, 3786 CrossRef CAS PubMed.
-
(a) M. Nakagawa, H. Nakano and K.-I. Watanabe, Chem. Lett., 1985, 391 CrossRef CAS;
(b) Y. Yamada, K.-I. Watanabe and H. Yasuda, Utsunomiya Daigaku Kyoikugakubu Kiyo, Dai-2-bu, 1989, 39, 25 CAS;
(c) Y. M. A. Yamada, N. Yoshikawa, H. Sasai and M. Shibasaki, Angew. Chem., Int. Ed. Engl., 1997, 36, 1871 CrossRef CAS;
(d) Y. M. A. Yamada and M. Shibasaki, Tetrahedron Lett., 1998, 39, 5561 CrossRef CAS.
- B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef CAS.
-
(a) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS PubMed;
(b) W. Notz, F. Tanaka and C. F. Barbas III, Acc. Chem. Res., 2004, 37, 580 CrossRef CAS PubMed;
(c) S. J. Zhang, H. X. Xie, J. Zhu, H. Li, X. S. Zhang, J. Li and W. Wang, Nat. Commun., 2011, 211 CrossRef PubMed;
(d) Y. Q. Zou, F. M. Hormann and T. Bach, Chem. Soc. Rev., 2018, 47, 278 RSC;
(e) X. Fang and C. J. Wang, Chem. Commun., 2015, 51, 1185 RSC;
(f) X. Yu and W. Wang, Chem.–Asian J., 2008, 3, 516 CrossRef CAS;
(g) H. Y. Bae, S. Some, J. Soh, Y. S. Lee and C. E. Song, Chem. Commun., 2011, 47, 9621 RSC.
-
(a) M. Shang, X. Wang, S. M. Koo, J. Youn, J. Z. Chan, W. Yao, B. T. Hastings and M. Wasa, J. Am. Chem. Soc., 2017, 139, 95 CrossRef CAS PubMed;
(b) K. Matsui, S. Takizawa and H. Sasai, Synlett, 2006, 5, 761 Search PubMed;
(c) S. Schenker, A. Zamfir, M. Fraund and S. B. Tsogoeva, Eur. J. Org. Chem., 2011, 12, 2209 CrossRef;
(d) Z. Zhang, H. Y. Bae, J. Guin, C. Rabalakos, M. V. Genmeren, M. Leutzsch, M. Klussmann and B. List, Nat. Commun., 2016, 12478 CrossRef CAS PubMed;
(e) S. Hirashima and H. Yamamoto, J. Synth. Org. Chem., Jpn., 2013, 71, 1116 CrossRef CAS.
-
(a) Y. Kohari, Y. Okuyama, E. Kwon, T. Furuyama, N. Kobayashi, T. Otuki, J. Kumagai, C. Seki, K. Uwai, G. Dai, T. Iwasa and H. Nakano, J. Org. Chem., 2014, 79, 9500 CrossRef CAS PubMed;
(b) T. Otsuki, J. Kumagai, Y. Kohari, Y. Okuyama, E. Kwon, C. Seki, K. Uwai, Y. Mawatari, N. Kobayashi, T. Iwasa, M. Tokiwa, M. Takeshita, A. Maeda, A. Hashimoto, K. Turuga and H. Nakano, Eur. J. Org. Chem., 2015, 33, 7292 CrossRef;
(c) D. Ganesan, M. Chennapuram, Z. Begum, C. Seki, K. Uwai, Y. Okuyama, E. Kwon, M. Tokiwa, M. Takeshita and H. Nakano, Heterocycles, 2019, 98, 1536 CrossRef CAS;
(d) P. Parasuraman, Z. Begum, M. Chennapuram, C. Seki, K. Uwai, Y. Okuyama, E. Kwon, M. Tokiwa, M. Takeshita and H. Nakano, RSC. Adv., 2020, 10, 17486 RSC;
(e) Z. Begum, H. Sannabe, C. Seki, K. Uwai, Y. Okuyama, E. Kwon, M. Tokiwa, M. Takeshita and H. Nakano, RSC. Adv., 2021, 11, 203 RSC;
(f) D. Ganesan, P. Parasuraman, Z. Begum, R. Thiyagarajan, C. Seki, K. Uwai, Y. Okuyama, E. Kwon, M. Tokiwa, M. Takeshita and H. Nakano, Heterocycles, 2022, 105, 369 CrossRef CAS PubMed.
- N. Hayama, R. Kuramoto, T. Foldes, K. Nishibayashi, I. Papai and Y. Takemoto, J. Am. Chem. Soc., 2018, 140, 12216 CrossRef CAS.
-
(a) K. Arnold, A. S. Batsanov, B. Davies, C. Grosjean, T. Schutz, A. Whiting and K. Zawatzky, Chem. Commun., 2008, 3879 RSC;
(b) I. Georgiou and A. Whiting, Chem. Commun., 2012, 2422 CAS.
-
(a) B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600 RSC;
(b) Y. Yamashita, T. Yasukawa, W. J. Yoo, T. Kitanasono and S. Kobayashi, Chem. Soc. Rev., 2018, 47, 4388 RSC.
-
(a) A. P. Thankacharu, S. Asha, K. S. Sindhu and G. Anilkumar, RSC Adv., 2015, 5, 62179 RSC;
(b) C. S. Evans and L. O. Davis, Moleclules, 2018, 23, 33 CrossRef PubMed;
(c) P. D. Maria, P. Bracio, L. F. Castelhano and G. Aargeman, ACS Catal., 2011, 1, 70 CrossRef.
-
(a) K. Sakthivel, W. Notz, T. Bui and C. F. Barbas III, J. Am. Chem. Soc., 2001, 123, 5260 CrossRef CAS PubMed;
(b) N. Mase, Y. Nakai, N. Ohara, H. Yoda, K. Takabe, F. Tanaka and C. F. Barbas, J. Am. Chem. Soc., 2006, 128, 734 CrossRef CAS PubMed;
(c) S. Luo, H. Xu, J. Li, L. Zhang and J. P. Cheng, J. Am. Chem. Soc., 2007, 129, 3074 CrossRef CAS PubMed;
(d) S. Guizzetti, M. Benaglia, L. Raimondi and G. Celentano, Org. Lett., 2007, 9, 1247 CrossRef CAS PubMed;
(e) F. Giacalone, R. Noto, P. L. Meo and S. Riela, Adv. Synth. Catal., 2008, 350, 2747 CrossRef CAS;
(f) C. L. Wu, X. K. Fu, X. B. Ma and S. Li, Tetrahedron: Asymmetry, 2010, 21, 2465 CrossRef CAS;
(g) C. L. Wu, X. K. Fu and S. Li, Tetrahedron, 2011, 67, 4283 CrossRef CAS;
(h) V. A. Kumar, C. C. Kumar and A. Siva, Org. Biomol. Chem., 2016, 14, 9021 RSC;
(i) J. Dutta, N. Wakdikar and S. Tiwari, Org. Biomol. Chem., 2017, 15, 6746 RSC;
(j) S. Bhowmick, L. Zhang, G. Ouyang and M. Liu, ACS Omega, 2018, 3, 8329 CrossRef CAS PubMed;
(k) M. G. Emma, A. Tamburrini, A. Martinelli, M. Lombardo, A. Quintavalla and C. Trombini, Catalysts, 2020, 10, 649 CrossRef CAS;
(l) Z. Li, Y. Chen, C. Zheng, Y. Yin, L. Wang and X. Sun, Tetrahedron, 2017, 73, 78 CrossRef CAS;
(m) R. Thiyagarajan, Z. Begum, C. Seki, K. Uwai, Y. Okuyama, E. Kwon, M. Tokiwa, M. Takeshita and H. Nakano, RSC. Adv., 2021, 11, 38925 RSC;
(n) A. karmakar, T. Maji, D. C. S. Wittman and O. Reiser, Chem.–Eur. J., 2011, 17, 11024 CrossRef CAS PubMed;
(o) S. Fotaras, C. G. Kokotos and G. Kokotos, Org. Biomol. Chem., 2012, 10, 5613 RSC;
(p) G. Xie, D. Feng and X. Ma, Mol. Catal., 2017, 434, 8 Search PubMed;
(q) G. D. Yadav and S. Singh, RSC. Adv., 2016, 6, 100459 RSC;
(r) P. V. Milla, J. M. Sansano, C. Najera, B. Fiser and E. G. Bengoa, Eur. J. Org. Chem., 2015, 12, 2614 CrossRef;
(s) B. Lygo, C. Davison, T. Evans, J. A. T. Gilks, J. Leohnard and C. E. Roy, Tetrahedron, 2011, 67, 10164 CrossRef CAS;
(t) R. Kerstin and M. Rainer, Org. Lett., 2012, 8, 2180 Search PubMed.
- Aldol reactions with water as solvent:
(a) A. Lubineau, J. Org. Chem., 1986, 11, 2142 CrossRef;
(b) W. Yao, Y. Cui, P. Wang and Y. Mao, Lett. Org. Chem., 2016, 13, 293 CrossRef CAS;
(c) E. L. Coitino, J. Tomasi and O. N. Ventura, J. Am. Chem. Soc., 1994, 90, 1745 CAS;
(d) D. Gryko and W. J. Saletra, Org. Biomol. Chem., 2007, 5, 2418 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a