
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Precise control of the site selectivity in ruthenium-catalyzed C–H bond amidations using cyclic amides as powerful directing groups†
Yu-Chao
Yuan
 *a,
Qiu-Li
Lu
a,
Xiao-Tong
Zhu
a,
Sergio
Posada-Pérez
b,
Miquel
Solà
b,
Albert
Poater
*b,
Thierry
Roisnel
c and
Rafael
Gramage-Doria
*c
*a,
Qiu-Li
Lu
a,
Xiao-Tong
Zhu
a,
Sergio
Posada-Pérez
b,
Miquel
Solà
b,
Albert
Poater
*b,
Thierry
Roisnel
c and
Rafael
Gramage-Doria
*c
aJiangsu Key Laboratory of New Drug Research and Clinical Pharmacy, School of Pharmacy, Xuzhou Medical University, Xuzhou 221004, China. E-mail: yuchao.yuan@xzhmu.edu.cn
bInstitut de Química Computacional i Catàlisi and Departament de Química, Universitat de Girona, C/M. Aurèlia Capmany, 17003 Girona, Catalonia, Spain. E-mail: albert.poater@udg.edu
cUniv Rennes, CNRS, ISCR – UMR6226, F-35000 Rennes, France. E-mail: rafael.gramage-doria@univ-rennes1.fr
First published on 5th November 2022
Abstract
Selective C–H functionalizations aiming at the formation of new C–N bonds is of paramount importance in the context of step- and atom-economy methodologies in organic synthesis. Although the implementation of noble metal catalysts is prevalent, more benign cobalt pre-catalysts have recently appeared to be promising. However, they sometimes feature selectivity issues that limit their applicability in late-stage functionalization. Herein, we report on a highly reactive ruthenium-based catalytic system displaying excellent levels of mono-, regio- and site-selectivity by exploiting a series of biologically-relevant cyclic amides as weak directing groups. The use of dioxazolone derivatives as amidating reagents overcomes the issues encountered in the use of unstable azide derivatives for such transformations and it enables us to perform these reactions under very mild reaction conditions (air, 40 °C). Moreover, a combination of deuteration experiments and a comparative study with different types of directing groups highlights the relevance of weak amide directing groups for enabling the formation of six-membered cycloruthenate intermediates in the key elementary steps of the catalytic cycle. In addition, DFT computational calculations were carried out for the first time for studying ruthenium-catalyzed C–N bond-forming processes via C–H activation assisted by weak directing groups, thereby elucidating the origin of the regio- and site-selectivity.
Introduction
The selective formation of C–heteroatom bonds is a fundamental transformation with direct consequences in the streaming synthesis of highly elaborated molecules with important applications ranging from pharmacology to materials science.1 In this context, C–N bond-forming processes are significantly relevant as they can be targeted in a sustainable manner using transition metal complexes as homogeneous catalysts.2 From the pioneering discoveries of the Ullmann coupling with copper catalysts3 to the most recent Buchwald–Hartwig aminations with palladium ones,4 these and analogous transformations have completely revolutionized the way of conceiving organic synthesis.5 However, most of these protocols require the use of pre-activated starting materials which impose synthetic limitations together with undesired over-stoichiometric formation of hazardous byproducts.3–5 Consequently, new promising approaches based on transition metal-catalyzed C–H bond functionalizations have appeared in the last few decades for amination reactions that can directly be performed on low functionalized starting materials.6 In this scenario, the selectivity is typically controlled by the presence of a directing group (DG) nearby the C–H bond desired to be functionalized.7 Therefore, this leads typically to ortho-selectivity for aromatic C–H bond aminations7 although few cases of remote meta- and para-C–H bond aminations have been reported.8Among many known C–N bond-forming reactions, transition metal-catalyzed ortho-C–H amidation ones are particularly interesting given the ubiquitous nature of amide bonds.9,10 Catalysts based on Ir, Rh, Pd, Ru or Co are of choice due to their high reactivity11 and amidating agents derived from dioxazolone are preferred instead of azides because of their benchmark stability.12 Taking this into account, it appears that all reports from the literature dealing with transition metal-catalyzed C–H bond amidations focused exclusively on substrates containing only one aromatic C–H bond available for reaction (Scheme 1).7,11,12 However, for implementation in late-stage functionalization for instance, the methodologies may consider also the compatibility with other aromatic C–H bonds prone to react. Unfortunately, the site-selectivity of aromatic C–H bond amidation reactions has been rarely studied to date with low levels of selectivity so far reported. In a first study in 2017, Chang and co-workers demonstrated for a single example (N-phenylbenzamide) that a C–H bond amidation could be site-selective with a cobalt catalyst and unproductive with a ruthenium catalyst, albeit with a modest yield (Scheme 2A).11d Later, Whiteoak and co-workers showed, as it could be expected, that a statistical mixture of amidated products is obtained using a cobalt catalyst with the assistance of an acyclic amide directing group in yields not exceeding 30% (Scheme 2B).13
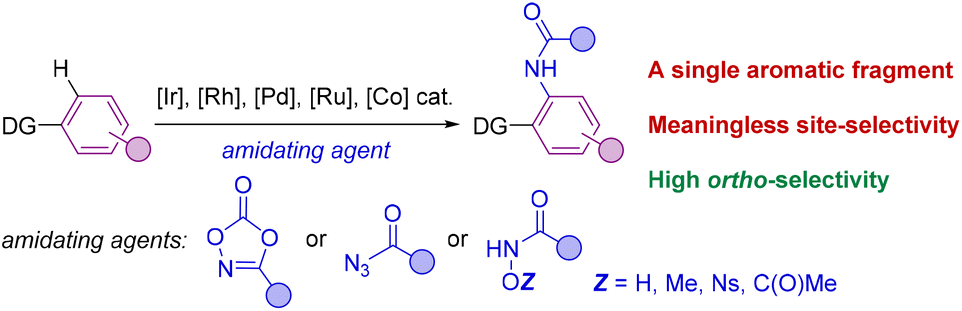 | ||
| Scheme 1 General case of a transition metal-catalyzed C–H bond amidation on aromatic scaffolds. DG = directing groups. | ||
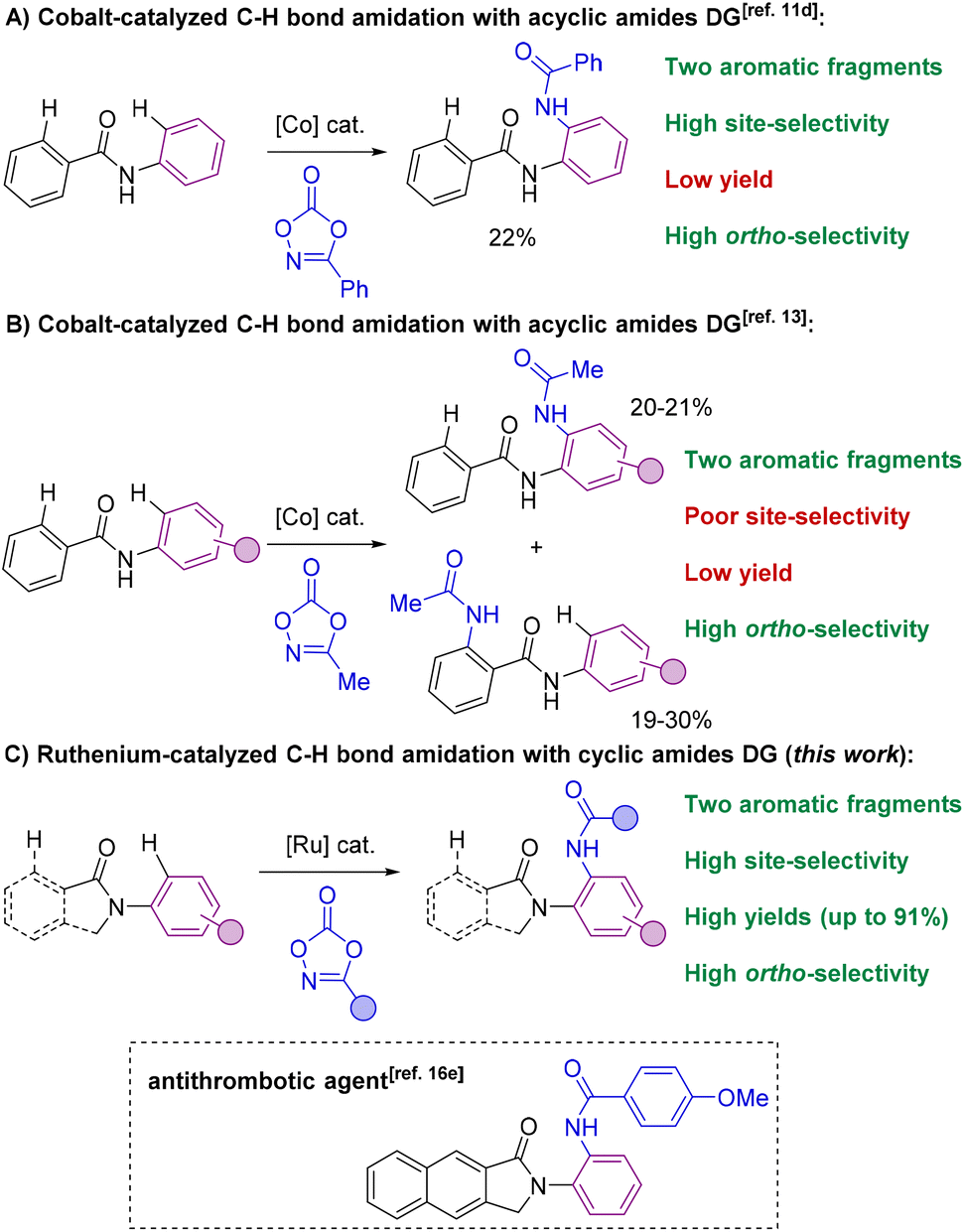 | ||
| Scheme 2 Transition metal-catalyzed C–H bond amidations on aromatic scaffolds using amides as directing groups: the state-of-the-art (A and B) versus present work (C). | ||
These examples undoubtedly show the difficulty in achieving site-selective C–H bond amidations in the presence of multiple aromatic fragments. Herein, we show that both regio- and site-selectivities are achieved when using cyclic amides as directing groups in the presence of two aromatic sites prone to react by means of a ruthenium catalyst that additionally displays broad functional group tolerance (Scheme 2C). We found that ortho-C–H bond amidations selectively took place in the N-aryl fragment rather than in the C(O)-aryl site. Control experiments, preliminary mechanistic studies and thorough DFT calculations unambiguously support that cyclic amides enable the formation of catalytically productive six-membered ruthenacycles as unique intermediates14 as it was evoked but never demonstrated in C–O and C–C bond forming reactions.15 It is noteworthy that cyclic amides, such as isoindolinones employed in this study, are prevalent motifs encountered in several daily-life chemicals (Scheme 2C, framed);16 therefore the presented methodology paves the way towards the use of ruthenium catalysts in C–H bond late-stage functionalization strategies.17 An in-depth comparative study with other types of common directing groups is presented, indicating the suitability of weak amide directing groups over more coordinating ones, such as pyridines, for ruthenium-catalyzed C–H bond amidations. In addition, cyclic amides were found to outperform acyclic ones as directing groups in this relevant transformation.
Results and discussion
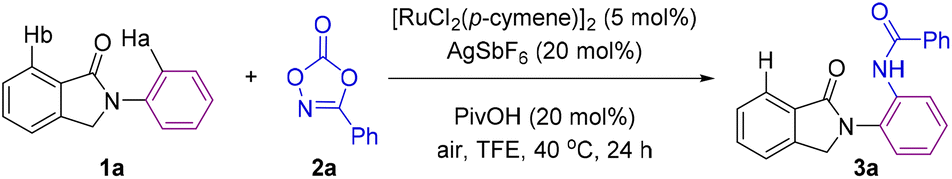
Our initial efforts started by using N-arylisoindolinone (1a) as the model substrate for the optimization of the C–H bond amidation reaction with 3-phenyl-1,4,2-dioxazol-5-one (2a) as the amidating agent in the presence of [RuCl2(p-cymene)]2 as the pre-catalyst. After screening a number of parameters (Table 1 and Table S1 in the ESI†), we found suitable reaction conditions that afforded exclusively the amide derivative 3a, in which the C–H bond amidation occurred in the hydrogen atom Ha (ortho position with respect to the nitrogen atom). The C–H bond functionalization taking place at the other possible hydrogen atom Hb was not observed (Table 1). The reaction conditions consisted of 5 mol% [RuCl2(p-cymene)]2, AgSbF6 as a chloride scavenger, and PivOH as an additive in 2,2,2-trifluoroethanol (TFE) as the solvent at 40 °C under air for 24 hours (Table 1, entry 1). In this way, 3a was obtained in 80% isolated yield. Control experiments indicated the need for both the chloride scavenger AgSbF6 and the ruthenium pre-catalyst (Table 1, entries 2 and 3). Additives influenced in a different manner the reactivity and selectivity of the C–H bond amidation reaction. Among the different protic additives evaluated, PivOH was found to be the most suitable one (Table 1, entries 4–7). Screening other solvents such as 1,2-dichloroethane (DCE), 1,4-dioxane and tetrahydrofuran (THF) was detrimental to the catalysis compared to TFE (Table 1, entries 8–10). Lowering the loading of the amidating agent 2a to 1.2 equivalents afforded the corresponding amidated product 3a in a modest 60% yield (Table 1, entry 11). Gratifyingly, we found that when decreasing the temperature to room temperature, 70% yield of 3a was obtained in a reaction conducted at an ambient temperature of 25 °C (Table 1, entry 12). Swapping the ruthenium pre-catalyst with a cobalt complex largely used in C–H bond amidations13 did not afford any product so far (Table 1, entry 13). The use of tosyl azide (TsN3), which is a well-known nitrene source,7 as an amidating agent led to traces of the corresponding N-tosylated product analogue of 3a (NHTs instead of NHCOPh, Table 1, entry 14). Overall, these findings highlight the suitability of ruthenium catalysts over the more expensive iridium, palladium or rhodium ones,18 as well as dioxazolone 2a as an ideal and safe partner for this type of transformation.|
|
||
|---|---|---|
| Entry | Deviation from the above conditions |
3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (%) b (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.15 mmol), [RuCl2(p-cymene)]2 (5 mol%), AgSbF6 (20 mol%), PivOH (0.02 mmol, 0.2 equiv.), TFE (0.5 mL), 40 °C, 20 h, air. b Determined by 1H NMR spectroscopy against dibromomethane as an internal standard. c Isolated yield shown in the parentheses after purification by column chromatography. d Reaction performed without PivOH. e The product for this reaction is expected to bear a NHTs in place of a NHCOPh fragment in 3a. | ||
| 1 | None | 82 (80)c |
| 2 | No [RuCl2(p-cymene)]2 | 0 |
| 3 | No AgSbF6 | 0 |
| 4 | No PivOH | 73 |
| 5 | AcOH instead of PivOH | 76 |
| 6 | AdCO2H instead of PivOH | 79 |
| 7 | H2O instead of PivOH | 54 |
| 8 | DCE instead of TFE | 65 |
| 9 | 1,4-Dioxane instead of TFE | 0 |
| 10d | THF instead of TFE | 19 |
| 11 | 1.2 equiv. of 2a | 60 |
| 12 | 25 °C instead of 40 °C | 70 |
| 13 | Cp*Co(CO)I2 instead of [RuCl2(p-cymene)]2 | 0 |
| 14e | TsN3 instead of 2a | Traces |
After reaction optimization (Table 1, entry 1), the scope of the catalysis was evaluated with different synthetically useful functional groups at different positions in both compound 1 and the dioxazolone partner 2 (Table 2). para-Substitution patterns in the N-phenyl ring with electron-donating groups, such as methyl and methoxy, were tolerated for the catalysis, yielding the corresponding ortho-amidated products 3b and 3c in 76% and 83% yields, respectively. In addition, X-ray diffraction studies performed on the single crystals of 3c (Table 2) established without ambiguity the site- and regio-selectivity observed in this C–H bond amidation reaction.19 Similarly, introduction of electron-withdrawing groups at the para-position such as fluoro and ester groups was also tolerated by the catalysis and afforded the corresponding ortho-amidated products 3d and 3e in 45% and 66% isolated yields, respectively. Notably, although ester groups have been identified as suitable directing groups in several ruthenium-catalyzed C–H bond activations,20 in the case of 3e, it is the cyclic amide group (and not the ester group) which dictates the exclusive site-selectivity observed in the C–H bond amidation. Other halide groups such as chloro and bromo were compatible leading to products 3f and 3g in 62% and 73% yields, which is relevant for further derivatization sequences by cross-coupling chemistry.21 Methyl- and methoxy-substituted isoindolinones at the meta position of the N-phenyl ring afforded the ortho-amidated products 3h and 3i in 81% and 90% yield, respectively, in a selective manner, without any functionalization occurring at the other ortho-C–H bond positions. Analogously, the catalysis was found to be compatible with a significant number of functional groups attached to the dioxazolone core. For instance, aryl-substituted dioxazolone derivatives bearing either electron-donating or electron-withdrawing groups at the para position such as methyl, tert-butyl, trifluoromethyl and chloro reacted smoothly with 1a, affording the corresponding ortho-C–H bond amidated products 3j–3m in an excellent range of 83–92% isolated yields. Electron deficient substituents on the aryl-substituted dioxazolone such as 3-Cl were also tolerated under the developed catalysis leading to the amidated product 3n in a remarkable 91% yield, which makes the halide site available for post-functionalization.21 Multisubstituted groups on the dioxazolone partner were also employed in this catalysis as shown in the successful synthesis of compound 3o in 66% yield that contains two chloride substituents at both meta positions. Additionally, the reaction was also compatible with a very bulky polycyclic aromatic hydrocarbon fragment such as 1-naphthalene substituted dioxazolone leading to 3p in 38% yield. Interestingly, heteroaromatic-containing amidating agents can also be employed. In the case of a furan-containing dioxazolone, the corresponding product (3q) was obtained in 32% yield. However, the more coordinating thiophene one inhibited the catalysis. Gratifyingly, further exploration of the scope of this C–H bond amidation reaction revealed that the aliphatic-substituted dioxazolones were also reactive. For instance, methyl-, tert-butyl- and n-heptyl-substituted dioxazolones led to the ortho-C–H bond amidated products 3r, 3s and 3t in 80%, 76% and 80% yields, respectively.
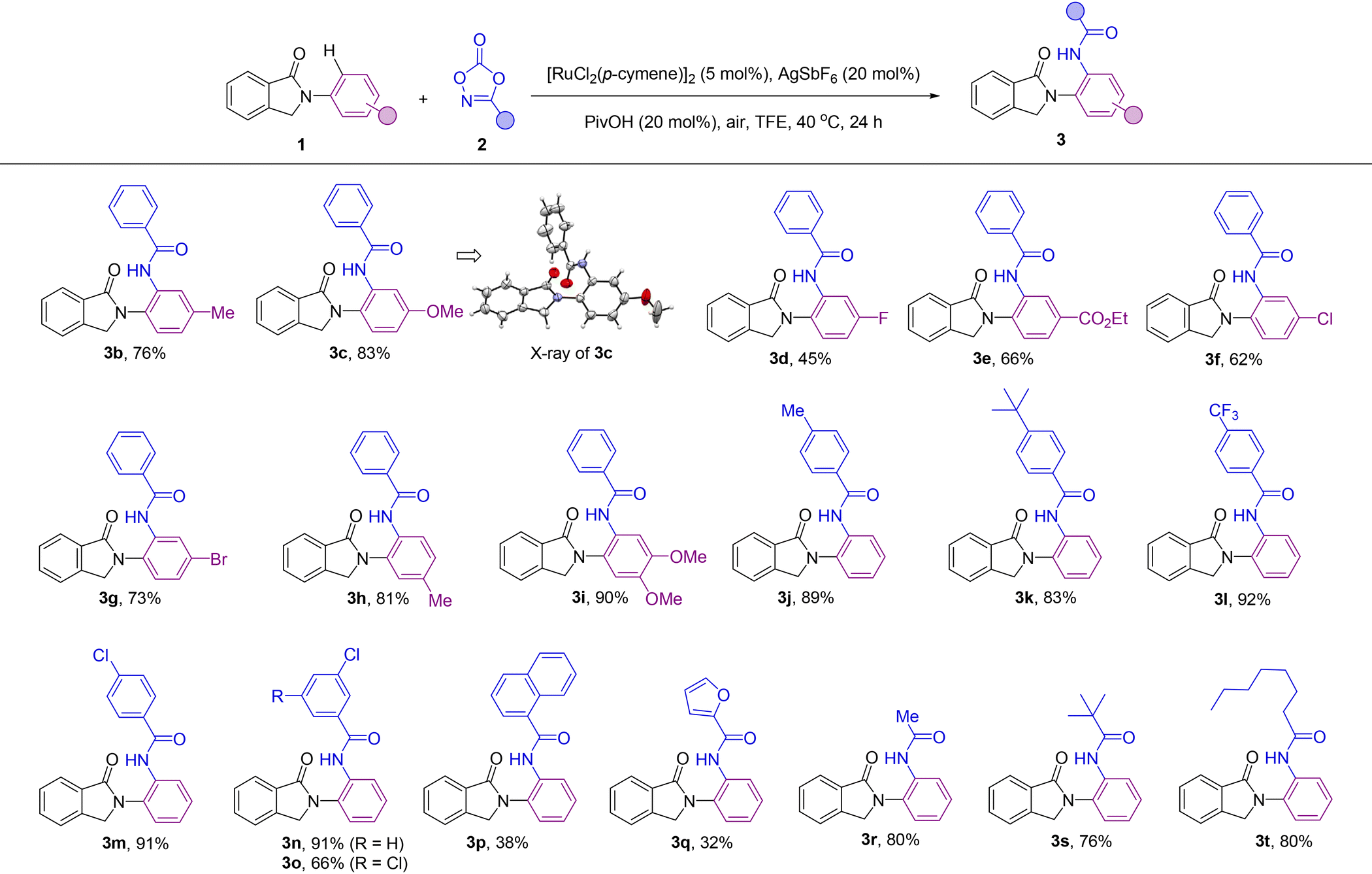
Overall, a panel of more than twenty different functional groups at different positions around both reagents 1 and 2 were tolerated with no products resulting from other site- and/or regio-selectivity. The absence of bis-ortho-amidated products might be rationalized by a plausible intramolecular hydrogen bonding between the NH group from the amide and the carbonyl group from the directing group that forbids the second ortho-C–H bond amidation.22 We additionally noted that the catalysis was sensitive to the steric hindrance found in the coupling partners as evidenced by the lack of reactivity observed for the ortho-tolyl derivative from 1 and the ortho-chloro-containing aryl-substituted dioxazolone derived from 2, respectively.23 Further limitations were encountered when using hydroxyl- or cyano-substituted coupling partners.24
Importantly, the reaction was scalable and the same excellent results in terms of yield and selectivity were obtained when conducting the ruthenium catalyzed C–H bond amidation on a gram scale starting with 5 mmol of 1a leading to ca. 1.30 g of product 3a, thus showing the robustness of the methodology (Scheme 3).
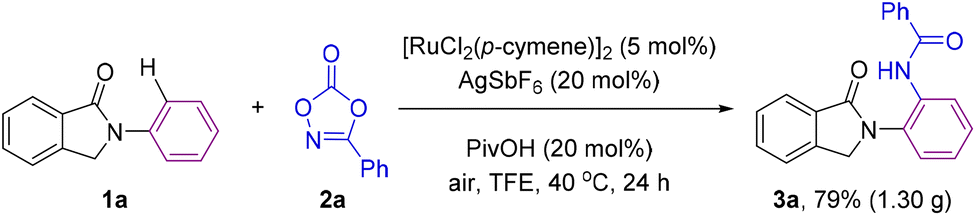 | ||
| Scheme 3 Scale-up reaction for the ruthenium-catalyzed ortho-C–H bond amidation. | ||
For comparison purposes, the catalysis was applied to other substrates featuring cyclic amides as potential directing groups in a view to address the versatility of this transformation (Scheme 4). Using pyrrolidone, the simplest cyclic amide, as the directing group, afforded the corresponding ortho-C–H bond amidated product 4 in 60% yield (Scheme 4). Note that the synthesis of 4 by applying this methodology was more sustainable and efficient compared to previous examples in the literature,25 that required long and tedious reaction sequences using hazardous reagents that have so far limited their exploitation. Interestingly, a remarkable difference in reactivity was encountered exploiting acyclic amide directing groups. Indeed, whilst acetanilide still afforded the corresponding ortho-amidated product 5 in 51% yield, no reactivity was observed for the N-methylated version or for benzanilide as the substrate (Scheme 4). Analogously, the complete absence of reactivity was encountered with N-phenylphthalimide, which features a cyclic imide as a potential directing group, as well as with the more strongly coordinating 2-pyridyl directing group (Scheme 4), which was rather counterintuitive considering previous contributions.26
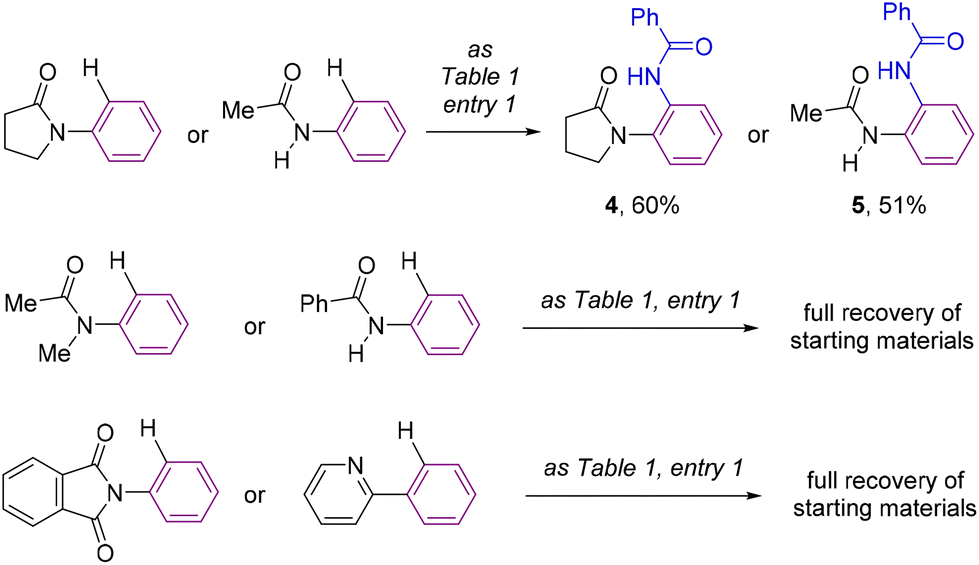 | ||
| Scheme 4 Evaluation of the catalysis with different types of relevant directing groups. | ||
Consequently, the reported ruthenium-catalyzed transformation appears to need a compromise between the geometry, the steric and electronic parameters, and the coordinating ability of the directing groups. Furthermore, these findings establish that the directing ability of cyclic amides outperforms that of acyclic amides following a clear trend as shown in Scheme 5.
 | ||
| Scheme 5 Order of efficiency for amides as directing groups in ruthenium-catalyzed ortho-C–H bond amidations. | ||
After having demonstrated the enhanced efficiency of cyclic amides as superior directing groups in ruthenium-catalyzed aromatic C–H bond amidations, we performed several experiments to better understand the mechanism operating in the catalysis (Scheme 6). Firstly, we verified the necessity of the carbonyl group by attempting a catalytic reaction using a carbonyl-free substrate such as isoindoline (Scheme 6, top). In this case, no C–H bond functionalization was detected, indirectly demonstrating the importance of the carbonyl group in assisting the catalysis likely via coordination to ruthenium.14,15 In addition, deuteration experiments were performed under the catalytic conditions but in the absence of the amidating partner 2 with a mixture of solvents TFE:D2O (Scheme 6, bottom). Under these conditions, 17% deuteration incorporation was observed in the ortho-C–H bonds of the phenyl ring attached to the nitrogen atom with no deuteration observed elsewhere in the molecule (Scheme 6, bottom).
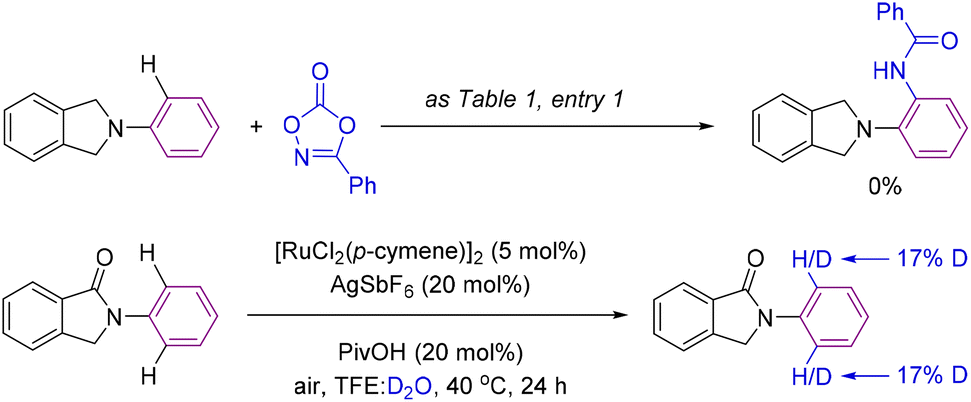 | ||
| Scheme 6 Control experiment in the absence of a carbonyl directing group (top) and a deuteration experiment (bottom). | ||
The overall above-described findings strongly suggest that cyclic amides prefer to accommodate six-membered ruthenacycle species in the catalytic cycle over commonly found five-membered ones for C–N bond forming reactions via C–H bond activation (Scheme 7). This may account for the observed regio- and site-selectivity in the catalysis.
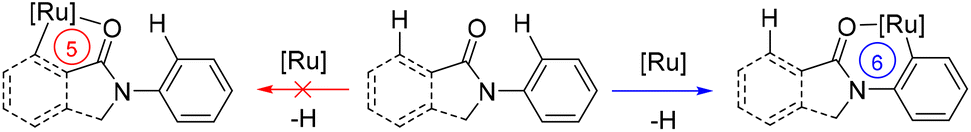 | ||
| Scheme 7 Mechanistic consideration highlighting the stabilization of six-membered ruthenacycles over five-membered ones in the key C–H bond activation event. | ||
Due to the difficulty in obtaining reproducible data for kinetic studies ascribed to solubility issues and the unsuccessful attempts at detecting reaction intermediates, we turned our attention to further unveil the precise reaction mechanism (Fig. 1) by DFT computational calculations at the M06-D3/Def2TZVP∼sdd(smd)//BP86-D3/Def2SVP level of theory.24 In order to keep the whole reaction pathway at the same cationic level, the pre-activation started from the cationic intermediate I0, derived from [RuCl2(p-cymene)]2, after releasing the chlorides by means of the reagent AgSbF6 (a known halide scavenger). As such, SbF6− is considered the weak coordinating anion throughout the whole catalytic cycle. In the next intermediate I0′ the anionic SbF6− ligand is substituted by the PivO− anion that comes from the other reagent, PivOH, with HSbF6 release. This step is thermodynamically unfavoured by 13.1 kcal mol−1, although the formation of complex I0′ is essential to stabilize the intermediate I0′′ by 10.7 kcal mol−1 after the combination of I0′ with substrate 1a. From I0′′ there are two potential aryl C–H activations to explore: from the phenyl on the nitrogen or the aryl ring annulated to the five-membered ring.27 Our simulations show energy barriers of 18.3 and 20.3 kcal mol−1, respectively. Besides the 2 kcal mol−1 difference, the C–H bond activation of the aryl ring leads to a worsening of 9.7 kcal mol−1 for the resulting intermediate I′ as compared to I, as is illustrated in the catalytic cycle in Fig. 1 (red pathway). The resulting ruthenacycle intermediate with the assistance of the cyclic amide as the directing group has literature precedents.7,11–13
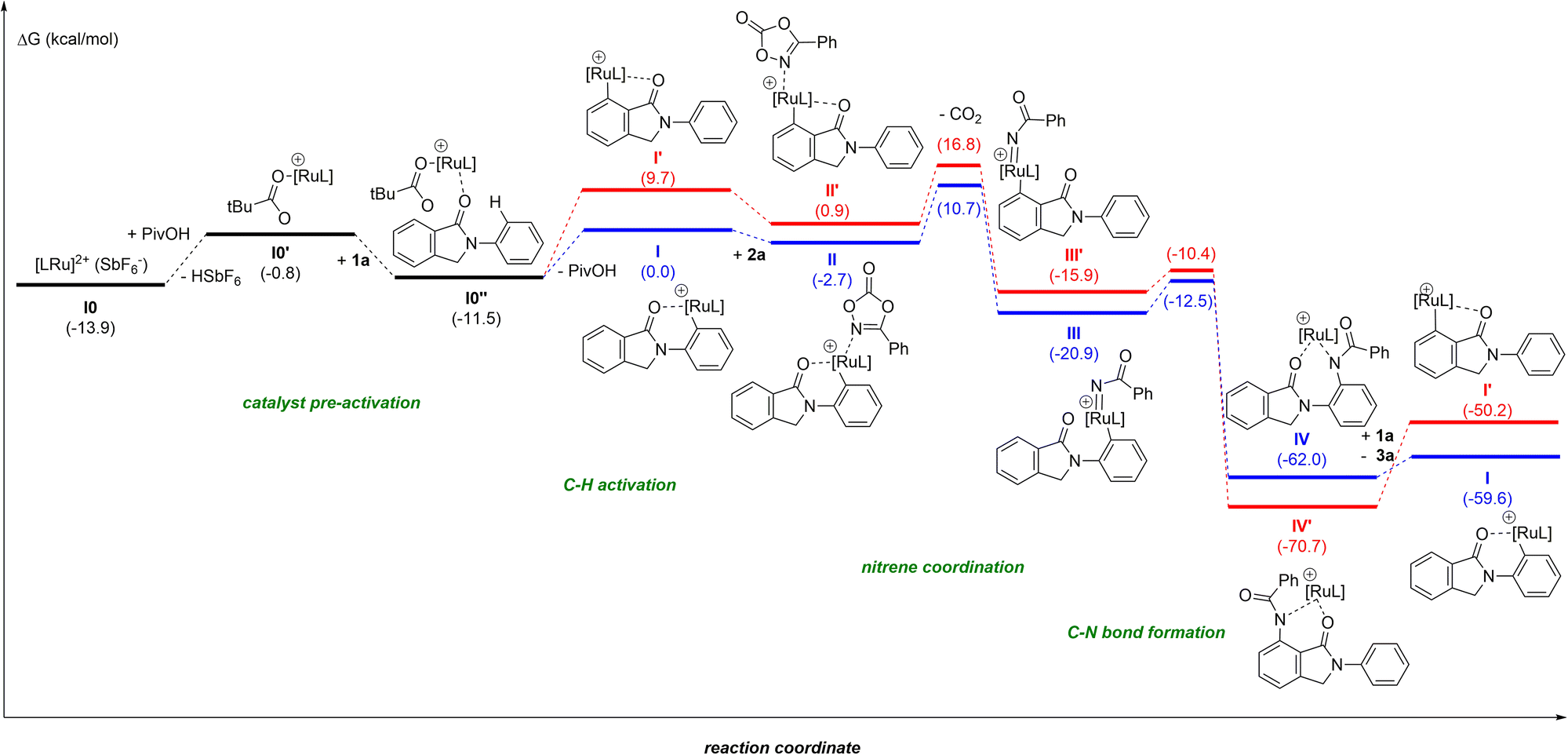 | ||
| Fig. 1 Computed Gibbs energy reaction profile in kcal mol−1 (in brackets) of the ruthenium-catalyzed ortho-C–H bond amidation with cyclic amides as weak directing groups. L = p-cymene. Black pathway = catalyst pre-activation, red color = unfavoured pathway, and blue color = favoured pathway. | ||
In the catalytically productive pathway (Fig. 1, blue pathway), species I coordinates to dioxazolone forming species II followed by extrusion of CO2, overcoming an energy barrier of 13.4 kcal mol−1 and giving rise to the formation of RuIV-imido species III. The migratory insertion of imido species III, with the associated C–N bond formation, leads to the generation of ruthenacycle IV, with a low energy barrier of only 8.4 kcal mol−1. To close the catalytic cycle, the protodemetalation of species IV occurs with a new molecule of substrate 1, which is in excess in the reaction mixture compared to the catalyst. This step could also be performed with HSbF6 or PivOH, furnishing the amidated product, as well; however, instead of the regeneration of the active ruthenium catalyst I, the process would end up in intermediate I0 or I0′, respectively.
The most difficult step in the pre-activation sequence is the C–H bond activation that leads to the ruthenacycle, either I or I′ selectively (Fig. 1). On the other hand, the rate determining step (rds) of the catalytic pathway corresponds to the transition state II-to-III. Considering the catalytic cycle for an elusive C–H bond activation at the aryl ring annulated to the five-membered ring of the substrate 1 (red pathway, Fig. 1), the corresponding transition state II-to-III is 6.1 kcal mol−1 higher in energy compared to the C–H bond activation at the phenyl ring linked to the nitrogen atom in 1 (blue pathway, Fig. 1). Thus, it confirms the selectivity towards the reaction profile with I as the catalytically active species. The reason is not steric hindrance, but it is related to the fact that I forms a rather stable 6-membered ruthenacycle, whereas I′ has a 5-membered ruthenacycle (Fig. 2). In addition, in the transition state the pivalate ligand forms a less strained interaction to remove the proton, and actually the distances in the transition state leading to I are closer to an agostic interaction, with a more activated aryl C–H bond (by nearly 0.1 Å).
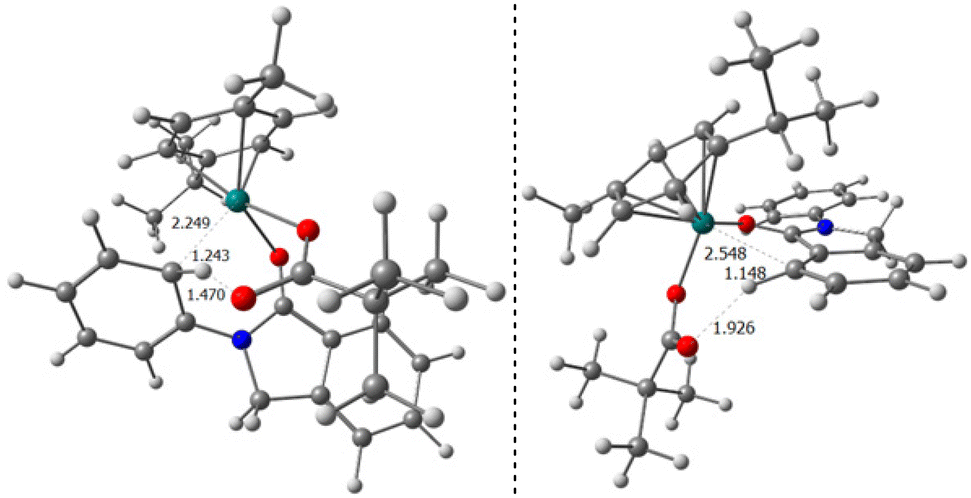 | ||
| Fig. 2 Transition states I0′′ → I (left) and I0′′ → I′ (right) with selected distances given in Å. | ||
For the sake of consistency and to validate mainly the rds, we explored all the possibilities considering the intermediates in the pre-activation and the two substrates. Interestingly, from intermediate I0′ the coordination of the dioxazolone before 1 is omitted since the kinetic cost of the CO2 release increases by 5.3 kcal mol−1 (Fig. 3). Such findings strongly contrast with the initial elementary steps of the reaction mechanism associated with the ruthenium-catalyzed C–H bond amidation with azides, in which the ruthenium-nitrene species are postulated to form before the C–H bond activation step in the substrate.28
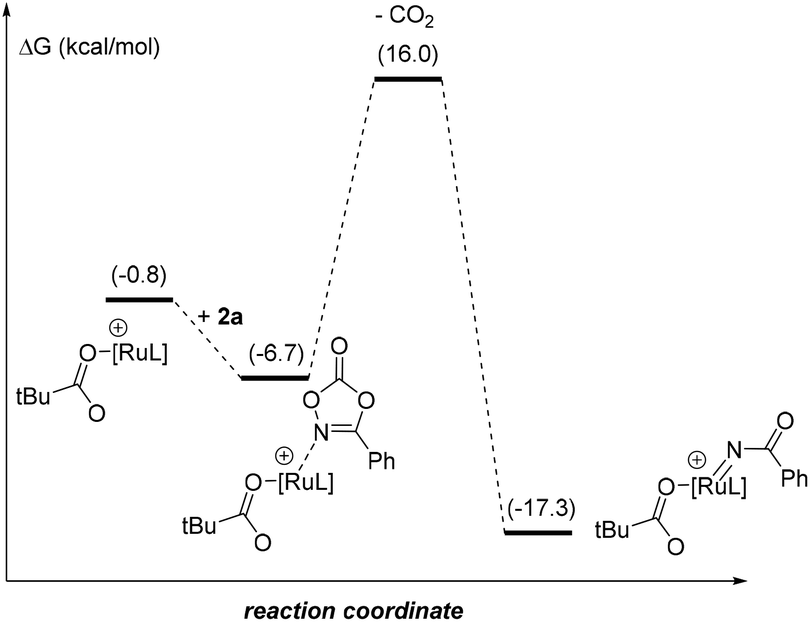 | ||
| Fig. 3 Reaction profile of the extrusion of CO2 with the coordination of dioxazolone in I0′. Relative Gibbs energies relative to I (in kcal mol−1). L = p-cymene. | ||
To further check the importance of the different anions, we performed additional analyses with the initial [RuCl2(p-cymene)]2. The stability of this dimer is not relevant as its cleavage only requires 1.1 kcal mol−1. The substitution of one or both chlorides by SbF6− is affordable, consuming only 3.1 and 2.8 kcal mol−1, respectively. Moreover, for the reaction profile described in Fig. 1, the counter anion SbF6−, that would neutralize the system, was omitted assuming that it does not affect significantly. Preliminary calculations show that the counter anion destabilizes intermediates I and II by 4.1 and 0.5 kcal mol−1, respectively.
Conclusions
In summary, we have developed efficient site-selective C–H bond amidation reactions to form unprecedented C–N bonds by exploiting the directing group ability of cyclic amides via six-membered ruthenacycle formation. A simple ruthenium(II) pre-catalyst and a safe amidating agent have been employed leading to a versatile catalytic system compatible with a large number of synthetically useful functional groups (more than 20 examples). This “close to room temperature” methodology offers a convenient route to access ortho-amidated cyclic amides that might be potentially relevant in medicinal chemistry.16 This study also shows the subtlety associated with C–H bond amidations because in the present case ruthenium outperforms cobalt as a catalyst and dioxazolone outperforms tosylazide as an amidating agent, respectively, which is somehow unexpected with regard to precedents in the literature.6–13,28 Moreover, this contribution establishes that cyclic amides are more powerful directing groups than acyclic ones or other coordinating groups such as imides and pyridines, at least for ruthenium-catalyzed C–H bond amidations. In addition, we provide the first mechanistic considerations of ruthenium-catalyzed C–H bond aminations supported by in-depth DFT calculations. Although the rate-determining step is the extrusion of CO2 in order to form ruthenium species coordinated to both the substrate and nitrene group, the site-selective step is determined by the destabilization associated with the formation of a five-membered ruthenacycle over a six-membered one. In conclusion, further research directed to new carbon–heteroatom bond forming processes via ruthenium-catalyzed C–H bond functionalization strategies should provide appealing methodologies for direct implementation into organic synthesis.Experimental
General procedure for the ruthenium-catalyzed C–H bond amidation
A suspension of substrate 1 (0.3 mmol, 1.0 equiv.), dioxazolone 2 (0.45 mmol, 1.5 equiv.), [RuCl2(p-cymene)]2 (5 mol%), AgSbF6 (20 mol%), and PivOH (0.06 mmol, 0.2 equiv.) in anhydrous TFE (1.5 mL) was stirred at 40 °C for 20 hours under air. At ambient temperature, the solvent was evaporated in vacuo, and the resulting crude reaction mixture was purified by flash column chromatography to afford the corresponding products 3–5 as analytically pure solids.Computational details
All the DFT static calculations were performed with the Gaussian16 set of programs,29 using the BP86 functional of Becke and Perdew,30–32 together with the Grimme D3 correction term to the electronic energy.33 The electronic configuration of the molecular systems was described with the double-ζ basis set with polarization of Ahlrichs for main-group atoms (Def2SVP keyword in Gaussian),34 whereas for ruthenium the small–core quasi–relativistic Stuttgart/Dresden effective core potential with an associated valence basis set (standard SDD keywords in Gaussian16) was employed.35–37 The geometry optimizations were performed without symmetry constraints, with analytical frequency calculations for the characterization of the located stationary points. These frequencies were used to calculate unscaled zero-point energies (ZPEs) as well as thermal corrections and entropy effects at 298.15 K. Energies were obtained by single-point calculations on the optimized geometries with the M06 functional,38 with the Grimme D3 correction term33 and the Def2TZVP basis set.39 The reported Gibbs energies in this work contain electronic energies obtained at the M06-D3/Def2TZVP∼sdd level of theory corrected with zero-point energies, thermal corrections and entropy effects evaluated at 298.15 K, achieved at the BP86-D3/Def2SVP∼sdd level plus a solvation contribution evaluated by means of the SMD continuum solvation model based on the quantum mechanical charge density of the solute interacting with a continuum description of the solvent (2,2,2-trifluoroethanol, TFE).40Conflicts of interest
There are no conflicts to declare.Author contributions
Y–C. Y., Q.-L. L. and X.-T. Z. performed all syntheses, product characterization and catalysis. S. P.-P., M. S. and A. P. performed DFT computational calculations. T. R. performed X-ray diffraction studies. Y.-C. Y. and R. G.-D. conceptualized and directed the study. All authors contributed to manuscript writing.Acknowledgements
Y.-C. Y. acknowledges the Science and Technology of XuZhou (KC19042) for financial support. We also thank the Public Experimental Research Center, Xuzhou Medical University, for NMR determination. R. G-D. acknowledges the ANR-JCJC scheme (ANR-19-CE07-0039), CNRS and the University of Rennes 1 for financial support. S. P. P. thanks the Marie Curie fellowship (H2020-MSCA-IF-2020-101020330). A. P. is a Serra Húnter Fellow and thanks ICREA Academia Prize 2019 and Spanish Mineco for project ref. PGC2018-097722-B-I00. A. P. and M. S. thank the Spanish Ministerio de Ciencia e Innovación for projects PID2021-127423NB-I00 and PID2020-13711GB-I00 and the Generalitat de Catalunya for project 2017SGR39.References
- (a) P. Knochel and G. Molander, Comprehensive Organic Synthesis, Elsevier, Amsterdam, 2nd edn, 2014 Search PubMed; (b) A. K. Yudin, Catalyzed Carbon-Heteroatom Bond Formation, Wiley-VCH, Weinheim, 2010 CrossRef; (c) A. R. Muci and S. L. Buchwald, Practical palladium catalysts for C-N and C-O bond formation, Top. Curr. Chem., 2002, 219, 131–209 CrossRef CAS.
- (a) N. R. Candeias, L. C. Branco, P. M. P. Gois, C. A. M. Afonso and A. F. Trindade, More sustainable approaches for the synthesis of N-based heterocycles, Chem. Rev., 2009, 109, 2703–2802 CrossRef CAS; (b) R. Hili and A. K. Yudin, Making carbon-nitrogen bonds in biological and chemical synthesis, Nat. Chem. Biol., 2006, 2, 284–287 CrossRef CAS.
- F. Ullmann, Ueber eine neue Bildungsweise von Diphenylaminderivaten, Ber. Dtsch. Chem. Ges., 1903, 36, 2382–2384 CrossRef.
- (a) F. Paul, J. Patt and J. F. Hartwig, Palladium-catalyzed formation of carbon-nitrogen bonds. Reaction intermediates and catalyst improvements in the hetero cross-coupling of aryl halides and tin amides, J. Am. Chem. Soc., 1994, 116, 5969–5970 CrossRef CAS; (b) J. F. Hartwig, Evolution of a Fourth Generation Catalyst for the Amination and Thioetherification of Aryl Halides, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS; (c) A. S. Guram and S. L. Buchwald, Palladium-catalyzed aromatic aminations with in situ generated aminostannanes, J. Am. Chem. Soc., 1994, 116, 7901–7902 CrossRef CAS; (d) D. S. Surry and S. L. Buchwald, Biaryl phosphane ligands in palladium-catalyzed amination, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS PubMed.
- (a) F. Monnier and M. Taillefer, Catalytic C-C, C-N, and C-O Ullmann-Type Coupling Reactions, Angew. Chem., Int. Ed., 2009, 48, 6954–6971 CrossRef CAS; (b) G. Evano, N. Blanchard and M. Toumi, Copper-mediated coupling reactions and their applications in natural products and designed biomolecules synthesis, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS PubMed.
- (a) F. Collet, R. H. Dodd and P. Dauban, Catalytic C-H amination: recent progress and future directions, Chem. Commun., 2009, 5061–5074 RSC; (b) L. Van Emelen, M. Henrion, R. Lemmens and D. De Vos, C-N coupling reactions with arenes through C-H activation: the state-of-the-art versus the principles of green chemistry, Catal. Sci. Technol., 2022, 12, 360–389 RSC; (c) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Recent advances in the transition metal-catalyzed twofold oxidative C-H bond activation strategy for C-C and C-N bond formation, Chem. Soc. Rev., 2011, 40, 5068–5083 RSC; (d) V. S. Thirunavukkarasu, S. I. Kozhushkov and L. Ackermann, C-H nitrogenation and oxygenation by ruthenium catalysis, Chem. Commun., 2014, 50, 29–39 RSC; (e) M.-L. Louillat and F. W. Patureau, Oxidative C-H amination reactions, Chem. Soc. Rev., 2014, 43, 901–910 RSC; (f) J. Jiao, K. Murakami and K. Itami, Catalytic Methods for Aromatic C-H Amination: An Ideal Strategy for Nitrogen-Based Functional Molecules, ACS Catal., 2016, 6, 610–633 CrossRef CAS; (g) J. L. Jeffrey and R. Sarpong, Intramolecular C(sp3)-H amination, Chem. Sci., 2013, 4, 4092–4106 RSC; (h) T. A. Ramirez, B. Zhao and Y. Shi, Recent advances in transition metal-catalyzed sp3 C-H amination adjacent to double bonds and carbonyl groups, Chem. Soc. Rev., 2012, 41, 931–942 RSC.
- (a) F. Yu, W. Shen, Y. Sun, Y. Liao, S. Jin, X. Lu, R. He, L. Zhong, G. Zhong and J. Zhang, Ruthenium-catalyzed C-H amination of aroylsilanes, Org. Biomol. Chem., 2021, 19, 6313–6321 RSC; (b) X.-H. Hu, X.-F. Yang and T.-P. Loh, Chelation-Assisted Rhodium-Catalyzed Direct Amidation with Amidobenziodoxolones: C(sp2)-H, C(sp3)-H, and Late-Stage Functionalizations, ACS Catal., 2016, 6, 5930–5934 CrossRef CAS; (c) Y. Park, K. T. Park, J. G. Kim and S. Chang, Mechanistic studies on the Rh(III)-mediated amido transfer process leading to robust C-H amination with a new type of amidating reagent, J. Am. Chem. Soc., 2015, 137, 4534–4542 CrossRef CAS PubMed; (d) J. Y. Kim, S. H. Park, J. Ryu, S. H. Cho, S. H. Kim and S. Chang, Rhodium-catalyzed intermolecular amidation of arenes with sulfonyl azides via chelation-assisted C-H bond activation, J. Am. Chem. Soc., 2012, 134, 9110–9113 CrossRef CAS; (e) J. Ryu, K. Shin, S. H. Park, J. Y. Kim and S. Chang, Rhodium-Catalyzed Direct C-H Amination of Benzamides with Aryl Azides: A Synthetic Route to Diarylamines, Angew. Chem., Int. Ed., 2012, 51, 9904–9908 CrossRef CAS; (f) B. Zhou, J. Du, Y. Yang, H. Feng and Y. Li, Rh(III)-Catalyzed C-H Amidation with N-Hydroxycarbamates: A New Entry to N-Carbamate-Protected Arylamines, Org. Lett., 2014, 16, 592–595 CrossRef CAS PubMed; (g) T. Ryu, J. Min, W. Choi, W. H. Jeon and P. H. Lee, Synthesis of 2-Aryl-2H-benzotrizoles from Azobenzenes and N-Sulfonyl Azides through Sequential Rhodium-Catalyzed Amidation and Oxidation in One Pot, Org. Lett., 2014, 16, 2810–2813 CrossRef CAS; (h) H. Hwang, J. Kim, J. Jeong and S. Chang, Regioselective Introduction of Heteroatoms at the C-8 Position of Quinoline N-Oxides: Remote C-H Activation Using N-Oxide as a Stepping Stone, J. Am. Chem. Soc., 2014, 136, 10770–10776 CrossRef CAS PubMed; (i) C. Zhou, J. Zhao, W. Guo, J. Jiang and J. Wang, N-Methoxyamide: An Alternative Amidation Reagent in the Rhodium(III)-Catalyzed C-H Activation, Org. Lett., 2019, 21, 9315–9319 CrossRef CAS; (j) P. Patel and S. Chang, N-Substituted Hydroxylamines as Synthetically Versatile Amino Sources in the Iridium-Catalyzed Mild C-H Amidation Reaction, Org. Lett., 2014, 16, 3328–3331 CrossRef CAS PubMed; (k) P. Lei and Y. Hailong, Chiral Phosphoric Acid Catalyzed Atroposelective C-H Amination of Arenes, Chin. J. Org. Chem., 2020, 40, 2167–2169 CrossRef.
- (a) B. Berzina, I. Sokolovs and E. Suna, Copper-Catalyzed para-Selective C–H Amination of Electron-Rich Arenes, ACS Catal., 2015, 5, 7008–7014 CrossRef CAS; (b) P. Wang, G.-C. Li, P. Jain, M. E. Farmer, J. He, P.-X. Shen and J.-Q. Yu, Ligand-Promoted meta-C–H Amination and Alkynylation, J. Am. Chem. Soc., 2016, 138, 14092–14099 CrossRef CAS PubMed; (c) C. J. Whiteoak, O. Planas, A. Company and X. Ribas, A First Example of Cobalt-Catalyzed Remote C-H Functionalization of 8-Aminoquinolines Operating through a Single Electron Transfer Mechanism, Adv. Synth. Catal., 2016, 358, 1679–1688 CrossRef CAS; (d) H. Sahoo, M. K. Reddy, I. Ramakrishna and M. Baidya, Copper-Catalyzed 8-Amido Chelation-Induced Remote C-H Amination of Quinolines, Chem. – Eur. J., 2016, 22, 1592–1596 CrossRef CAS; (e) R. Zhao, Y. Yang, X. Wang, P. Ren, Q. Zhang and D. Li, An efficient nickel/silver co-catalyzed remote C–H amination of 8-aminoquinolines with azodicarboxylates at room temperature, RSC Adv., 2018, 8, 37064–37068 RSC; (f) Y. Yin, J. Xie, F.-Q. Huang, L.-W. Qi and B. Zhang, Copper-Catalyzed Remote C−H Amination of Quinolines with N-Fluorobenzenesulfonimide, Adv. Synth. Catal., 2017, 359, 1037–1042 CrossRef CAS; (g) Y. Wang, S. Zhang, X. Feng, X. Yu, M. Yamaguchi and M. Bao, Palladium-Catalyzed Para-C–H Bond Amination of 2-Aryl Chloromethylbenzenes, J. Org. Chem., 2022, 87, 10531–10538 CrossRef CAS PubMed.
- (a) A. Greenberg, C. M. Breneman and J. F. Liebman, The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science, Wiley, New York, 2000 Search PubMed; (b) E. Valeur and M. Bradley, Amide bond formation: beyond the myth of coupling reagents, Chem. Soc. Rev., 2009, 38, 606–631 RSC; (c) R. M. Lanigan and T. D. Sheppard, Recent Developments in Amide Synthesis: Direct Amidation of Carboxylic Acids and Transamidation Reactions, Eur. J. Org. Chem., 2013, 7453–7465 CrossRef CAS; (d) R. M. de Figuereido, J.-S. Suppo and J.-M. Campagne, Nonclassical routes for amide bond formation, Chem. Rev., 2016, 116, 12029–12122 CrossRef; (e) V. R. Pattabiraman and J. W. Bode, Rethinking amide bond synthesis, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- For reviews on amides as directing groups in transition metal-catalyzed C–H bond functionalization, see: (a) R.-Y. Zhu, M. E. Farmer, Y.-Q. Chen and J.-Q. Yu, A simple and versatile amide directing group for C−H functionalizations, Angew. Chem., Int. Ed., 2016, 55, 10578–10599 CrossRef CAS; (b) R. Das, G. S. Kumar and M. Kapur, Amides as Weak Coordinating Groups in Proximal C–H Bond Activation, Eur. J. Org. Chem., 2017, 5439–5459 CrossRef CAS; (c) R. Gramage-Doria, Steering Site-Selectivity in Transition Metal-Catalyzed C−H Bond Functionalization: the Challenge of Benzanilides, Chem. – Eur. J., 2020, 26, 9688–9709 CrossRef CAS PubMed; (d) R. Thakur, Y. Jaiswal and A. Kumar, Primary amides: Sustainable weakly coordinating groups in transition metal-catalyzed C–H bond functionalization reactions, Tetrahedron, 2021, 93, 132313 CrossRef CAS.
- (a) J. Park and S. Chang, Comparative Catalytic Activity of Group 9 [Cp*MIII] Complexes: Cobalt-Catalyzed C-H Amidation of Arenes with Dioxazolones as Amidating Reagents, Angew. Chem., Int. Ed., 2015, 54, 14103–14107 CrossRef CAS; (b) G. N. Hermann and C. Bolm, Mechanochemical rhodium(III)-catalyzed C–H bond amidation of arenes with dioxazolones under solventless conditions in a ball mill, ACS Catal., 2017, 7, 4592–4596 CrossRef CAS; (c) A. E. Hande and K. R. Prabhu, Ru(II)-Catalyzed C–H Amidation of Indoline at the C7-Position Using Dioxazolone as an Amidating Agent: Synthesis of 7-Amino Indoline Scaffold, J. Org. Chem., 2017, 82, 13405–13413 CrossRef CAS; (d) J. Park, J. Lee and S. Chang, Iterative C−H functionalization leading to multiple amidations of anilides, Angew. Chem., Int. Ed., 2017, 56, 4256–4260 CrossRef CAS PubMed; (e) Y. Hwang, Y. Park and S. Chang, Mechanism-Driven Approach To Develop a Mild and Versatile C−H Amidation through IrIII Catalysis, Chem. – Eur. J., 2017, 23, 11147–11152 CrossRef CAS; (f) H. Xiong, S. Xu, S. Sun and J. Cheng, Cp* Rh(III)-catalyzed annulation of N-methoxybenzamide with 1,4,2-bisoxazol-5-one toward 2-aryl quinazolin-4(3H)-one derivatives, Org. Chem. Front., 2018, 5, 2880–2884 RSC; (g) K. Kawai, Y. Bunno, T. Yoshino and S. Matsunaga, Weinreb Amide Directed Versatile C−H Bond Functionalization under (η5-Pentamethylcyclopentadienyl)cobalt(III) Catalysis, Chem. – Eur. J., 2018, 24, 10231–10237 CrossRef CAS PubMed; (h) Y. Liang, Y.-F. Liang, C. Tang, Y. Yuan and N. Jiao, Cationic Cobalt(III)-Catalyzed Aryl and Alkenyl C-H Amidation: A Mild Protocol for the Modification of Purine Derivatives, Chem. – Eur. J., 2015, 21, 16395–16399 CrossRef CAS PubMed; (i) A. S. Santos, A. M. S. Silva and M. M. B. Marques, Sustainable amidation reactions–recent advances, Eur. J. Org. Chem., 2020, 2501–2516 CrossRef CAS; (j) K.-H. Ng, A. S. C. Chan and W.-Y. Yu, Pd-Catalyzed Intermolecular ortho-C−H Amidation of Anilides by N-Nosyloxycarbamate, J. Am. Chem. Soc., 2010, 132, 12862–12864 CrossRef CAS PubMed.
- (a) J. Ryu, J. Kwak, K. Shin, D. Lee and S. Chang, Ir(III)-Catalyzed Mild C–H Amidation of Arenes and Alkenes: An Efficient Usage of Acyl Azides as the Nitrogen Source, J. Am. Chem. Soc., 2013, 135, 12861–12868 CrossRef CAS; (b) D.-G. Yu, M. Suri and F. Glorius, RhIII/CuII-Cocatalyzed Synthesis of 1H-Indazoles through C–H Amidation and N–N Bond Formation, J. Am. Chem. Soc., 2013, 135, 8802–8805 CrossRef CAS PubMed; (c) Y. Lian, J. R. Hummel, R. G. Bergman and J. A. Ellman, Facile Synthesis of Unsymmetrical Acridines and Phenazines by a Rh(III)-Catalyzed Amination/Cyclization/Aromatization Cascade, J. Am. Chem. Soc., 2013, 135, 12548–12551 CrossRef CAS PubMed; (d) C. Pan, N. Jin, H. Zhang, J. Han and C. Zhu, Iridium-Catalyzed Phosphoramidation of Arene C–H Bonds with Phosphoryl Azide, J. Org. Chem., 2014, 79, 9427–9432 CrossRef CAS; (e) J. Kim and S. Chang, Iridium-Catalyzed Direct C-H Amidation with Weakly Coordinating Carbonyl Directing Groups under Mild Conditions, Angew. Chem., Int. Ed., 2014, 53, 2203–2207 CrossRef CAS PubMed; (f) N. Wang, R. Li, L. Li, S. Xu, H. Song and B. Wang, Rhodium(III)-Catalyzed Intermolecular Amidation with Azides via C(sp3)–H Functionalization, J. Org. Chem., 2014, 79, 5379–5385 CrossRef CAS PubMed; (g) D. Lee, Y. Kim and S. Chang, Iridium-Catalyzed Direct Arene C–H Bond Amidation with Sulfonyl- and Aryl Azides, J. Org. Chem., 2013, 78, 11102–11109 CrossRef CAS PubMed; (h) T. M. Figg, S. Park, J. Park, S. Chang and D. G. Musaev, Comparative Investigations of Cp*-Based Group 9 Metal-Catalyzed Direct C–H Amination of Benzamides, Organometallics, 2014, 33, 4076–4085 CrossRef CAS; (i) K. M. van Vliet and B. de Bruin, Dioxazolones: Stable Substrates for the Catalytic Transfer of Acyl Nitrenes, ACS Catal., 2020, 10, 4751–4769 CrossRef CAS; (j) E. Tosi, R. M. de Figueiredo and J.-M. Campagne, Enantioselective Catalytic CH Amidations: An Highlight, Catalysts, 2021, 11, 471 CrossRef CAS; (k) S. Y. Hong, Y. Hwang, M. Lee and S. Chang, Mechanism-Guided Development of Transition-Metal-Catalyzed C–N Bond-Forming Reactions Using Dioxazolones as the Versatile Amidating Source, Acc. Chem. Res., 2021, 54, 2683–2700 CrossRef CAS; (l) Y. Huang, C. Pi, Z. Tang, Y. Wu and X. Cui, Cp*Co(III)-catalyzed CH amidation of azines with dioxazolones, Chin. Chem. Lett., 2020, 31, 3237–3240 CrossRef CAS; (m) K. Wei, M. Jiang, S. Liang and W. Yu, Iron-Catalyzed Benzene Ring Expansion of α-Azido-N-phenylamides, Synthesis DOI:10.1055/a-1915-7916.
- P. G. Chirila, L. Skibinski, K. Miller, A. Hamilton and C. J. Whiteoak, Towards a Sequential One-Pot Preparation of 1,2,3-Benzotriazin-4(3H)-ones Employing a Key Cp*Co(III)-catalyzed C−H Amidation Step, Adv. Synth. Catal., 2018, 360, 2324–2332 CrossRef CAS.
- For reviews on weak directing groups, see: (a) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Weak coordination as a powerful means for developing broadly useful C–H functionalization reactions, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed; (b) S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Weakly Coordinating Directing Groups for Ruthenium(II)-Catalyzed C–H Activation, Adv. Synth. Catal., 2014, 356, 1461–1479 CrossRef CAS; (c) C. Sambiagio, D. Schoenbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnuerch, A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- (a) Y.-C. Yuan, M. Goujon, C. Bruneau, T. Roisnel and R. Gramage-Doria, C-H Bond Alkylation of Cyclic Amides with Maleimides via a Site-selective-determining Six-membered Ruthenacycle, J. Org. Chem., 2019, 84, 16183–61191 CrossRef CAS PubMed; (b) Y.-C. Yuan, C. Bruneau, T. Roisnel and R. Gramage-Doria, Site-selective Ruthenium-catalyzed C-H Bond Arylations with Boronic Acids: Exploiting Isoindolinones as a Weak Directing Group, J. Org. Chem., 2019, 84, 12893–12903 CrossRef CAS; (c) Y.-C. Yuan, C. Bruneau, T. Roisnel and R. Gramage-Doria, Site-selective Ru-catalyzed C–H bond alkenylation with biologically relevant isoindolinones: a case of catalyst performance controlled by subtle stereoelectronic effects of the weak directing group, Catal. Sci. Technol., 2019, 9, 4711–4717 RSC; (d) Y.-C. Yuan, C. Bruneau, T. Roisnel and R. Gramage-Doria, Ruthenium(II)-catalysed Selective C(sp2)-H Bond Benzoxylation of Biologically Appealing N-arylisoindolinones, Org. Biomol. Chem., 2019, 17, 7517–7525 RSC; (e) Y.-C. Yuan, C. Bruneau, V. Dorcet, T. Roisnel and R. Gramage-Doria, Ru-catalyzed Selective C-H Bond Hydroxylation of Cyclic Imides, J. Org. Chem., 2019, 84, 1898–1907 CrossRef CAS; (f) M. Xiong, Y. Shu, J. Tang, F. Yang and D. Xing, Iridium(I)-Catalyzed Isoindolinone-Directed Branched-Selective Aromatic C–H Alkylation with Simple Alkenes, Molecules, 2022, 27, 1923 CrossRef CAS.
- (a) S. Nomura, K. Endo-Umeda, A. Aoyama, M. Makishima, Y. Hashimoto and M. Ishikawa, Styrylphenylphthalimides as novel transrepression-selective liver X receptor (LXR) modulators, ACS Med. Chem. Lett., 2015, 6, 902–907 CrossRef CAS PubMed; (b) S. Nomura, K. Endo-Umeda, M. Makishima, Y. Hashimoto and M. Ishikawa, Development of Tetrachlorophthalimides as Liver X Receptor β (LXRβ)-Selective Agonists, ChemMedChem, 2016, 11, 2347–2360 CrossRef CAS PubMed; (c) S. Nomura, K. Endo-Umeda, S. Fujii, M. Makishima, Y. Hashimoto and M. Ishikawa, Structural development of tetrachlorophthalimides as liver X receptor β (LXRβ)-selective agonists with improved aqueous solubility, Bioorg. Med. Chem. Lett., 2018, 28, 796–801 CrossRef CAS PubMed; (d) M. R. Lunn, D. E. Root, A. M. Martino, S. P. Flaherty, B. P. Kelley, D. D. Coovert, A. H. Burghes, N. T. Man, G. E. Morris, J. Zhou, E. J. Androphy, C. J. Sumner and B. R. Stockwell, Indoprofen upregulates the survival motor neuron protein through a cyclooxygenase-independent mechanism, Chem. Biol., 2004, 11, 1489–1493 CrossRef CAS PubMed; (e) D. W. Beight, T. J. Craft, J. B. Franciskovich, T. Goodson Jr., S. E. Hall, D. K. Herron, V. J. Klimkowski, J. A. Kyle, J. J. Masters, D. Mendel, G. Milot, J. S. Sawyer, R. T. Shuman, G. F. Smith, A. L. Tebbe, J. M. Tinsley, L. C. Weir, J. H. Wikel, M. R. Wiley and J. K. Yee, Preparation of bis-amides of 1,2-benzenediamines as antithrombotic agents, 1998, WO1998-US13427.
- (a) J. Yamaguchi, A. D. Yamaguchi and K. Itami, C-H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS; (b) T. Brueckl, R. D. Baxter, Y. Ishihara and P. S. Baran, Innate and guided C–H functionalization logic, Acc. Chem. Res., 2012, 45, 826–839 CrossRef PubMed; (c) J. F. Hartwig, Evolution of C–H bond functionalization from methane to methodology, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef CAS PubMed; (d) D. J. Abrams, P. A. Provencher and E. J. Sorensen, Recent applications of C–H functionalization in complex natural product synthesis, Chem. Soc. Rev., 2018, 47, 8925–8967 RSC; (e) R. Gramage-Doria and C. Bruneau, Ruthenium-catalysed oxidative coupling of vinyl derivatives and application in tandem hydrogenation, Coord. Chem. Rev., 2021, 428, 21362 CrossRef; (f) Q. Zheng, C.-F. Liu, J. Chen and G.-W. Rao, C–H functionalization of aromatic amides, Adv. Synth. Catal., 2020, 362, 1406–1446 CrossRef CAS.
- Prices accessed on September 1st 2022: iridium (4750 USD/OZ), rhodium
(14200 USD/OZ), palladium (2098 USD/OZ) and ruthenium (585 USD/OZ).
- CCDC 2149748 (3c)† contains the supplementary crystallographic data for this paper.
- (a) R. Manikandan and M. Jeganmohan, Recent advances in the ruthenium(ii)-catalyzed chelation-assisted C–H olefination of substituted aromatics, alkenes and heteroaromatics with alkenes via the deprotonation pathway, Chem. Commun., 2017, 53, 8931–8947 RSC; (b) R. Manikandan and M. Jeganmohan, Recent advances in the ruthenium-catalyzed hydroarylation of alkynes with aromatics: synthesis of trisubstituted alkenes, Org. Biomol. Chem., 2015, 13, 10420–10436 RSC; (c) Y. Yang, Y. Lin and Y. Rao, Ruthenium(II)-catalyzed synthesis of hydroxylated arenes with ester as an effective directing group, Org. Lett., 2012, 14, 2874–2877 CrossRef CAS PubMed; (d) M. Pichette Drapeau and L. J. Goossen, Carboxylic acids as directing groups for C−H bond functionalization, Chem. – Eur. J., 2016, 22, 18654–18677 CrossRef CAS PubMed; (e) G. Shi and Y. Zhang, Carboxylate-Directed C-H Functionalization, Adv. Synth. Catal., 2014, 356, 1419–1442 CrossRef CAS.
- (a) C. R. Le Blond, A. T. Andrews, Y. Sun and J. R. Sowa, Activation of aryl chlorides for Suzuki cross-coupling by ligandless, heterogeneous palladium, Org. Lett., 2001, 3, 1555–1557 CrossRef CAS; (b) P. Orecchia, D. S. Petkova, R. Goetz, F. Rominger, A. S. K. Hashmi and T. Schaub, Pd-Catalysed Suzuki–Miyaura cross-coupling of aryl chlorides at low catalyst loadings in water for the synthesis of industrially important fungicides, Green Chem., 2021, 23, 8169–8180 RSC; (c) R. A. Altman and S. L. Buchwald, Pd-catalyzed Suzuki–Miyaura reactions of aryl halides using bulky biarylmonophosphine ligands, Nat. Protoc., 2007, 2, 3115–3121 CrossRef CAS PubMed.
- H. J. Kim, M. J. Ajitha, Y. Lee, J. Ryu, J. Kim, Y. Lee, Y. Jung and S. Chang, Hydrogen-bond-assisted controlled C–H functionalization via adaptive recognition of a purine directing group, J. Am. Chem. Soc., 2014, 136, 1132–1140 CrossRef CAS PubMed.
- For the absence of reactivity using ortho-substituted patterns in C–H bond functionalizations assisted by cyclic amides as directing groups, see: (a) J. A. Leitch, P. B. Wilson, C. L. McMullin, M. F. Mahon, Y. Bhonoah, I. H. Williams and C. G. Frost, Ruthenium(II)-Catalyzed C–H Functionalization Using the Oxazolidinone Heterocycle as a Weakly Coordinating Directing Group: Experimental and Computational Insights, ACS Catal., 2016, 6, 5520–5529 CrossRef CAS; (b) J. A. Leitch, H. P. Cook, Y. Bhonoah and C. G. Frost, Use of the Hydantoin Directing Group in Ruthenium(II)-Catalyzed C–H Functionalization, J. Org. Chem., 2016, 81, 10081–10087 CrossRef CAS PubMed; (c) W. Ma, H. Dong, D. Wang and L. Ackermann, Late-Stage Diversification of Non-Steroidal Anti-Inflammatory Drugs by Transition Metal-Catalyzed C-H Alkenylations, Thiolations and Selenylations, Adv. Synth. Catal., 2017, 359, 966–973 CrossRef CAS ; and ref. 15.
- See the ESI for details.†.
- (a) R. M. Anderson and D. Harrison, Methanesulphonyl and benzenesulphonyl derivatives of benzimidazolin-2-one, J. Chem. Soc., 1964, 5231–5234 RSC; (b) O. Meth-Cohn, Acylation of benzimidazoles, J. Chem. Soc., 1964, 5245–5247 RSC.
- (a) J. McIntyre, I. Mayoral-Soler, P. Salvador, A. Poater and D. J. Nelson, Insights into mechanism and selectivity in ruthenium(II)-catalysed ortho-arylation reactions directed by Lewis basic groups, Catal. Sci. Technol., 2018, 8, 3174–3182 RSC; (b) D. S. Timofeeva, D. M. Lindsay, W. J. Kerr and D. J. Nelson, A quantitative empirical directing group scale for selectivity in iridium-catalysed hydrogen isotope exchange reactions, Catal. Sci. Technol., 2020, 10, 7249–7255 RSC.
- For examples of activation of aryl C–H bonds in Ru-based catalysts, see: (a) J. A. Fernández-Salas, S. Manzini, L. Piola, A. M. Z. Slawin and S. P. Nolan, Ruthenium catalysed C–H bond borylation, Chem. Commun., 2014, 50, 6782–6784 RSC; (b) A. Poater, N. Bahri-Laleh and L. Cavallo, Rationalizing current strategies to protect N-heterocyclic carbene-based ruthenium catalysts active in olefin metathesis from C–H (de)activation, Chem. Commun., 2011, 47, 6674–6676 RSC; (c) A. Poater and L. Cavallo, Mechanistic insights into the double C–H (de)activation route of a Ru-based olefin metathesis catalyst, J. Mol. Catal. A: Chem., 2010, 324, 75–79 CrossRef CAS; (d) I. Özdemir, S. Demir, B. Çetinkaya, C. Gourlaouen, F. Maseras, C. Bruneau and P. H. Dixneuf, Direct Arylation of Arene C−H Bonds by Cooperative Action of NHCarbene−Ruthenium(II) Catalyst and Carbonate via Proton Abstraction Mechanism, J. Am. Chem. Soc., 2008, 130, 1156–1157 CrossRef PubMed; (e) E. Ferrer Flegeau, C. Bruneau, P. H. Dixneuf and A. Jutand, Autocatalysis for C–H Bond Activation by Ruthenium(II) Complexes in Catalytic Arylation of Functional Arenes, J. Am. Chem. Soc., 2011, 133, 10161–10170 CrossRef PubMed.
- I. Choi, A. M. Messinis and L. Ackermann, C7-Indole Amidations and Alkenylations by Ruthenium(II) Catalysis, Angew. Chem., Int. Ed., 2020, 59, 12534–12540 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. Becke, Density-functional exchange-energy approximation with correct asymptotic behaviour, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- J. P. Perdew, Erratum: Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 7406–7406 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. A. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Schäfer, H. Horn and R. Ahlrichs, Fully optimized contracted Gaussian basis sets for atoms Li to Kr, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- U. Haeusermann, M. Dolg, H. Stoll, H. Preuss, P. Schwerdtfeger and R. M. Pitzer, Accuracy of energy-adjusted quasirelativistic ab initio pseudopotentials: All-electron and pseudopotential benchmark calculations for Hg, HgH and their cations, Mol. Phys., 1993, 78, 1211–1224 CrossRef.
- W. Küchle, M. Dolg, H. Stoll and H. Preuss, Energy-adjusted pseudopotentials for the actinides. Parameter sets and test calculations for thorium and thorium monoxide, J. Chem. Phys., 1994, 100, 7535–7542 CrossRef.
- T. Leininger, A. Nicklass, H. Stoll, M. Dolg and P. Schwerdtfeger, The accuracy of the pseudopotential approximation. II. A comparison of various core sizes for indium pseudopotentials in calculations for spectroscopic constants of InH, InF, and InCl, J. Chem. Phys., 1996, 105, 1052–1059 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- R. A. Kendall, T. H. Dunning Jr. and R. J. Harrison, Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2149748. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qo01434c |
| This journal is © the Partner Organisations 2023 |