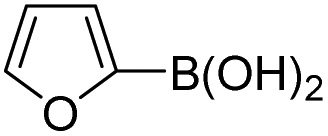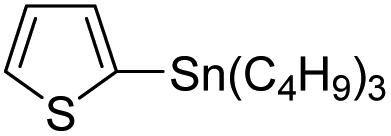DOI:
10.1039/D2QO01732F
(Research Article)
Org. Chem. Front., 2023,
10, 54-61
Ring-expansion from tellurophenes to telluropyrans: inhibition of C–Te bond cleavages in transition metal-catalyzed reactions†
Received
1st November 2022
, Accepted 18th November 2022
First published on 22nd November 2022
Abstract
Tellurium-fused aromatic hydrocarbons have attracted extensive attention due to their unique properties as well as the synthetic challenges. However, the synthesis of organotellurium molecules falls much behind those of other chalcogen-containing compounds since the synthetic methods to build C–S and C–Se bonds always fail to extend the C–Te bonds. Herein, a PtCl2-catalyzed ring-expansion reaction is attempted on tellurophene derivatives. However, tellurium-free vinylogous pentafulvalene analogues instead of telluropyrans are produced unexpectedly. To prevent the Pt–Te interactions and avoid the C–Te bond cleavage, bulky phosphine ligands are incorporated into the PtCl2 catalyst. A ring-expansion cyclization towards neutral telluropyran derivatives from the corresponding tellurophene compounds has been thus developed. The mechanisms of both the ring-opening and the ring-expansion reactions are proposed and the pathways are verified by density functional theory calculations. Furthermore, the tolerance of the obtained telluropyran derivatives in metal-related reagents is further investigated and the conjugation can be facilely extended via various transition metal-catalyzed coupling reactions. Therefore, our work provides not only a synthetic methodology towards telluropyran-fused polycyclic aromatic hydrocarbons, but also an effective pathway to avoid the C–Te bond cleavages in transition metal-catalyzed coupling reactions.
Introduction
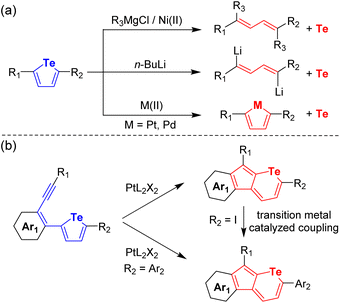
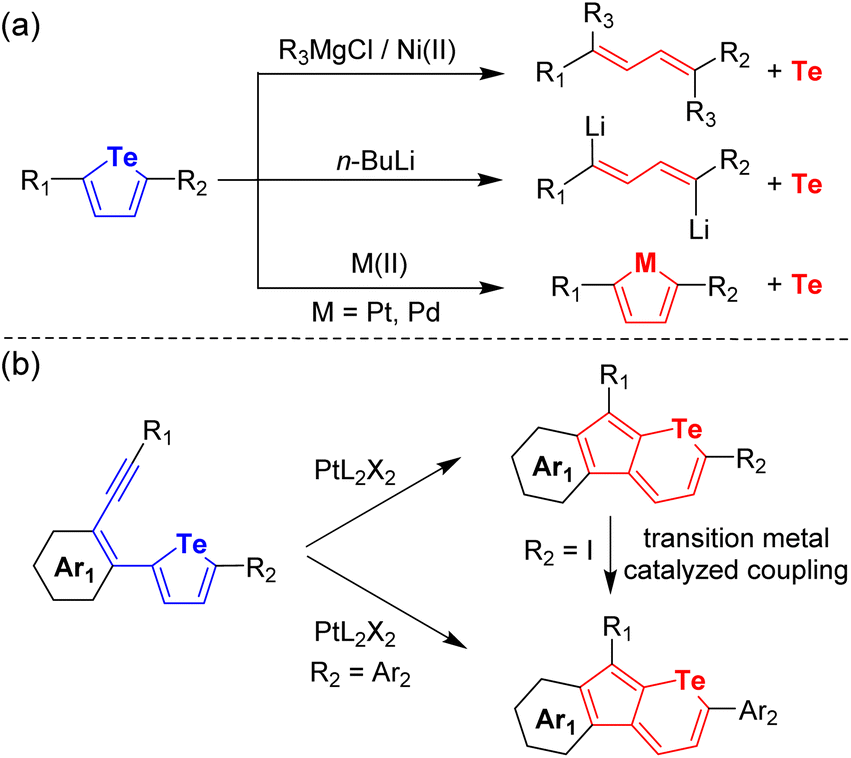
Chalcogen-containing aromatic hydrocarbons have attracted extensive attention due to their increasing demand in both chemistry and material fields.1–3 Among the chalcogen elements, tellurium (Te) is the heaviest non-metallic one with certain metallic properties. Therefore, Te atoms have been incorporated into organic semiconductors due to their electrical conductivity,4,5 strong polarizability,6,7 heavy atom effect,8,9 and intense intermolecular interactions.10 Until now, several methodologies have been established for the synthesis of organotellurium compounds.8,11,20 However, despite these promising advances, the synthetic approaches towards organotellurium molecules fall much behind those for S- and Se-containing compounds since the synthetic methods to build C–S and C–Se bonds always fail to extend the C–Te bond.12 Moreover, the formed C–Te bond can be easily cleaved by Pt(II),13 Pd(II),14 HCl,15–17 LiBu,18–20 and so on (Fig. 1a) due to its lower bond energy (200 kJ mol−1) in comparison with C–S (272 kJ mol−1) and C–Se bonds (234 kJ mol−1).12 Furthermore, it is always unsuccessful to functionalize organotellurium compounds using the conventional methods for S- and Se-containing molecules.21 For example, bromination with N-bromosuccinimide has been proved to be an efficient way for thiophene and selenophene but fails for tellurophene.22 In addition, the strong coordination ability of the Te atom always poisons the transition metal-based catalyst during coupling reactions.23 Consequently, in contrast to the abundant S- and Se-heterocyclic compounds, Te-containing cyclic systems have been much less explored and thus the synthesis thereof remains challenging.
 |
| | Fig. 1 (a) Reported C–Te bond cleavages by metal-related reagents and (b) our ring-expansion reactions from tellurophenes to telluropyrans. | |
Transition metal-catalyzed ring-expansion and skeletal rearrangement have been extensively investigated as efficient pathways to offer direct access towards cyclic molecules which are difficult to construct by conventional synthetic methods.24 Recently, we have developed a facile ring-expansion cyclization from S-fused five-membered thiophene rings to six-membered thiopyran rings catalyzed by PtCl2.25 The incorporated chalcogen atoms endow the heterocyclic systems with not only intense intramolecular charge transfer interactions but also high intermolecular charge transport mobilities.26 Meanwhile, most of the Te-containing six-membered rings are reported in the form of onium salts, such as tellurorhodamine.27 Neutral telluropyran derivatives and the universal synthetic methods have been rarely reported. Therefore, PtCl2-catalyzed ring-expansion cyclization has been attempted on tellurophene derivatives in this work. Unexpectedly, no telluropyran derived product can be synthesized. Instead, Te-free vinylogous pentafulvalene derivatives are discovered by single-crystal X-ray analysis. This can be attributed to the interactions between the Te and Pt atoms and a possible mechanism of the complicated ring-opening, detelluration, and dimerization reaction is proposed. To prevent the harmful interactions, different ligands have been introduced and telluropyran derivatives can be generated (Fig. 1b). Interestingly, the incorporation of electron-deficient P(C6F5)3 is the most effective to prevent the cleavage of the tellurium atoms from the heterocyclic rings and to produce telluropyran derivatives. The scope has been further investigated by tuning the aryl backbones and the alkyl/aryl substituents. Furthermore, the tolerance of the obtained telluropyran derivatives in metal-related reagents is further investigated and the conjugation can be facilely extended via various transition metal-catalyzed coupling reactions. Therefore, the reactivity of telluropyran derivatives is explored and the diversity of Te-containing heterocycles is expanded.
Results and discussion
Cleavage of tellurophene derivatives catalyzed by PtX2
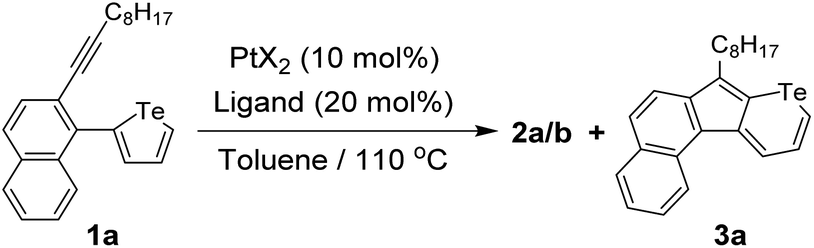
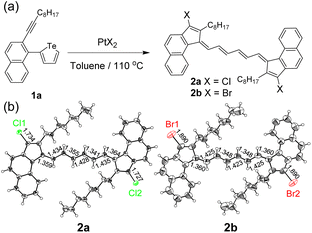
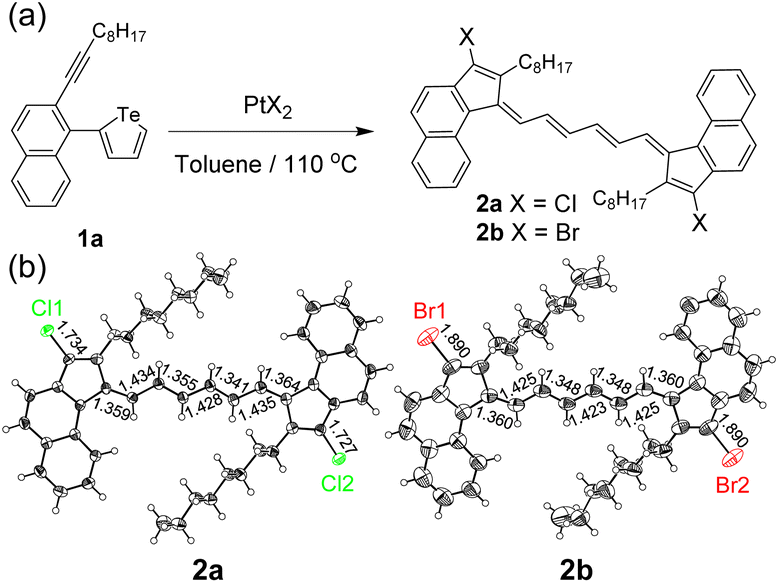
The initial investigation was carried out using easily accessible 2-alkynyl-1-tellurophenyl substituted aromatics. With the assistance of PtCl2 (10 mol%), 2-(1-decynyl)-1-(2-tellurophenyl)naphthalene (1a) could be converted to a dimerized ring-opening product 2a (Fig. 2a). The yield was 14% if calculated from reactant 1a while it was 68% if referenced to PtCl2. The former value increased but the latter almost remained constant with the feed ratio of PtCl2 enhancing. No matter how much PtCl2 was added, no Te-fused heterocyclic compound could be detected. The selection of the naphthalene skeleton was for the facile growth of the single crystals of the products. As shown in Fig. 2b, single crystal X-ray analysis reveals that compound 2a contains an octatetraene-bridged pentafulvalene. The alternating single and double bonds are delocalized and the bond lengths locate between the lengths of the conventional C–C (1.54 Å) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (1.34 Å) bonds.28 Moreover, two chlorine atoms can be found on the pentafulvalene backbone. To figure out the origin of the two chlorine atoms, PtBr2 was further tested and product 2b was obtained. Similarly, two bromine atoms could be found at the same positions (Fig. 2b). Therefore, the two halogen atoms in 2a and 2b come from PtX2 (X = Cl, Br) which serves as a reactant instead of a catalyst.
C (1.34 Å) bonds.28 Moreover, two chlorine atoms can be found on the pentafulvalene backbone. To figure out the origin of the two chlorine atoms, PtBr2 was further tested and product 2b was obtained. Similarly, two bromine atoms could be found at the same positions (Fig. 2b). Therefore, the two halogen atoms in 2a and 2b come from PtX2 (X = Cl, Br) which serves as a reactant instead of a catalyst.
 |
| | Fig. 2 (a) Ring-opening, detelluration, and dimerization reactions of the tellurophene derivatives in the presence of PtX2 and (b) ORTEP diagrams of products 2a and 2b (CCDC 2132835 and 2132837†). | |
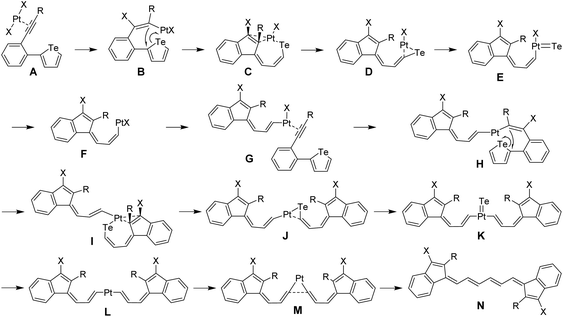
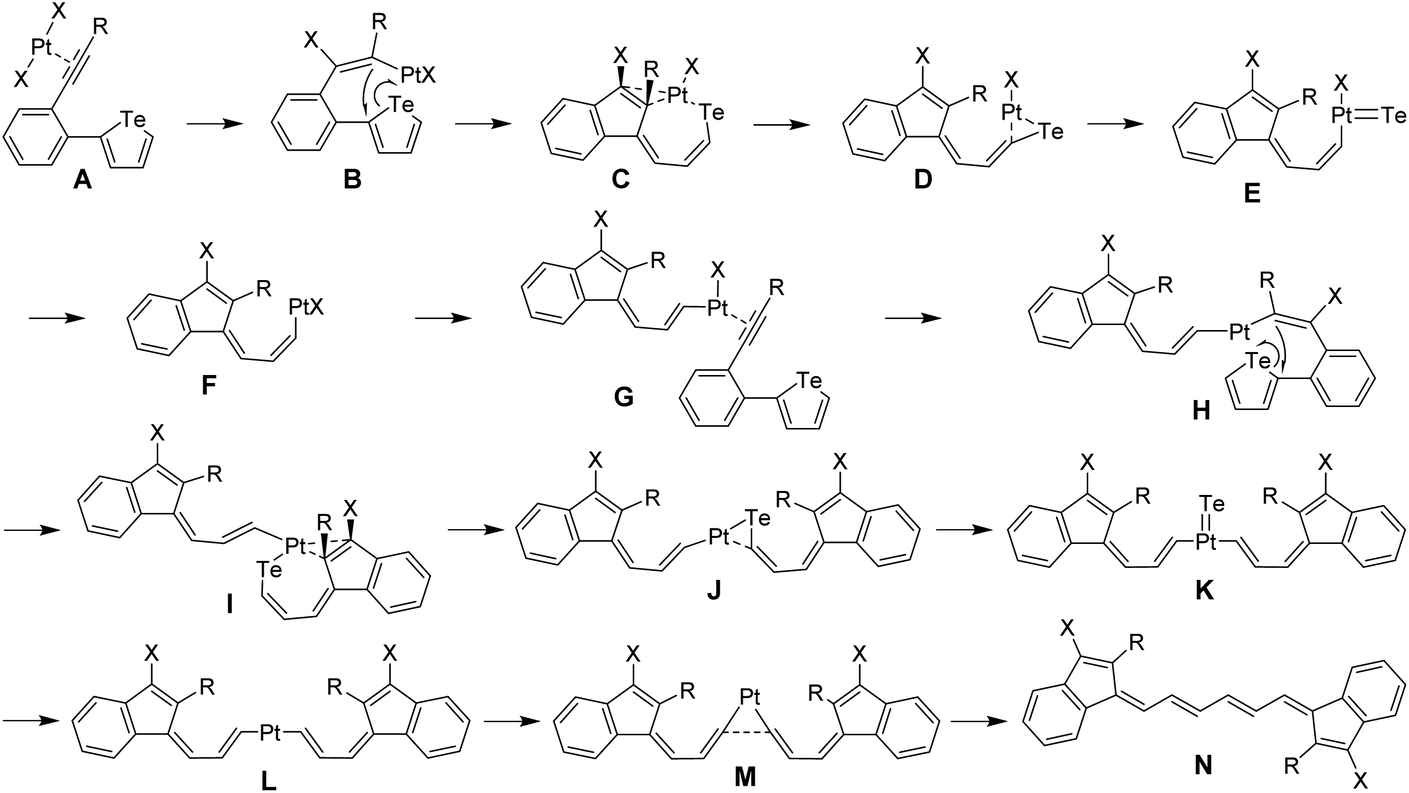
A probable mechanism of this complicated ring-opening, detelluration, and dimerization reaction is proposed and illustrated in Fig. 3. Initially, the complexation of PtX2 with the carbon–carbon triple bond affords intermediate A, followed by trans-addition to give intermediate B. Then an ipso-cyclization occurs and the Pt atom interacts with Te to produce intermediate C. Subsequent Pt-dissociation and Te-extrusion29 afford the key intermediate F which can further react with the alkyne in another molecule to yield intermediate G. After similar procedures, dimerized intermediate L is produced. Upon further dissociation of Pt, the final product M is obtained. Simultaneously, PtX2 is consumed and thus serves as a reactant. To verify the proposed mechanism, the reaction path and the relative free energies for all stationary points were calculated at the ωB97M-V30/def2-TZVPP,31 SMD(toluene)32//ωB97X-D33/6-31G(d) (LANL2DZ for Pt and Te), SMD(toluene) level. The detailed calculation methods and results are given in the ESI.† As shown in Fig. S5–S7,† all the transition states are confirmed to connect the neighboring intermediates, which proves the proposed mechanism for the cleavage of the Te atom and the formation of the Te-free vinylogous pentafulvalene derivatives.
 |
| | Fig. 3 Proposed mechanism of the ring-opening, detelluration, and dimerization reaction of tellurophene derivatives in the presence of PtX2. | |
Ring-expansion from tellurophenes to telluropyrans
To suppress the interactions between Pt(II) and Te, additive phosphine ligands were introduced simultaneously with the reactant and the catalyst. The temperature was then raised to 110 °C. As shown in Table 1, in the presence of PtCl2 (10 mol%), the incorporation of bulky phosphine ligands (20 mol%), such as PPh3, P(o-MePh)3, P(t-Bu)3·HBF4, and P(C6F5)3, successfully reduces the Pt–Te interactions and decreases the yields of by-product 2a (entries 3–6). This means the introduced ligands prevent the Pt–Te coordinate interactions to a certain degree. However, the vinylogous pentafulvalene 2a could still be detected. As discussed above, the obtained by-product consumed the PtCl2 catalyst, which resulted in a relatively low yield of 3a. Interestingly, telluropyran derivative 3a can be isolated when using P(t-Bu)3·HBF4 and P(C6F5)3 as ligands. However, the yields of ring-expansion product 3a are relatively low (<50%) obviously owing to the consumption of the PtCl2 catalyst via the ring-opening pathway. To further improve the yields of telluropyran derivatives, phosphine-coordinated PtCl2 catalysts were directly used in the absence of additional ligands. A perfluoro-substituted phosphine P(C6F5)3 was utilized as the ligand and Pt[P(C6F5)3]2Cl2 (CCDC 2132838†) was crystallized from a toluene solution containing PtCl2 and P(C6F5)3. The chemical structure of the as-prepared catalyst was verified by single-crystal analysis (Fig. S4†). Interestingly, no ring-opening by-product could be detected and the yield of 3a increased to 73% (entry 8). Under the same conditions, Pt[P(C6F5)3]2Br2 was tested to catalyze the ring-opening reaction and a yield of 59% could be achieved (entry 9). These results suggest that the incorporation of ancillary phosphine ligands on PtX2 can effectively weaken the Pt–Te interactions and thus reduce the reaction possibility through the ring-opening pathway. Therefore, further investigation of ring-expansion reactions from tellurophenes to telluropyrans was carried out using Pt[P(C6F5)3]2Cl2 (10 mol%) in toluene at 110 °C.
Table 1 Optimization of the ring-expansion conditions
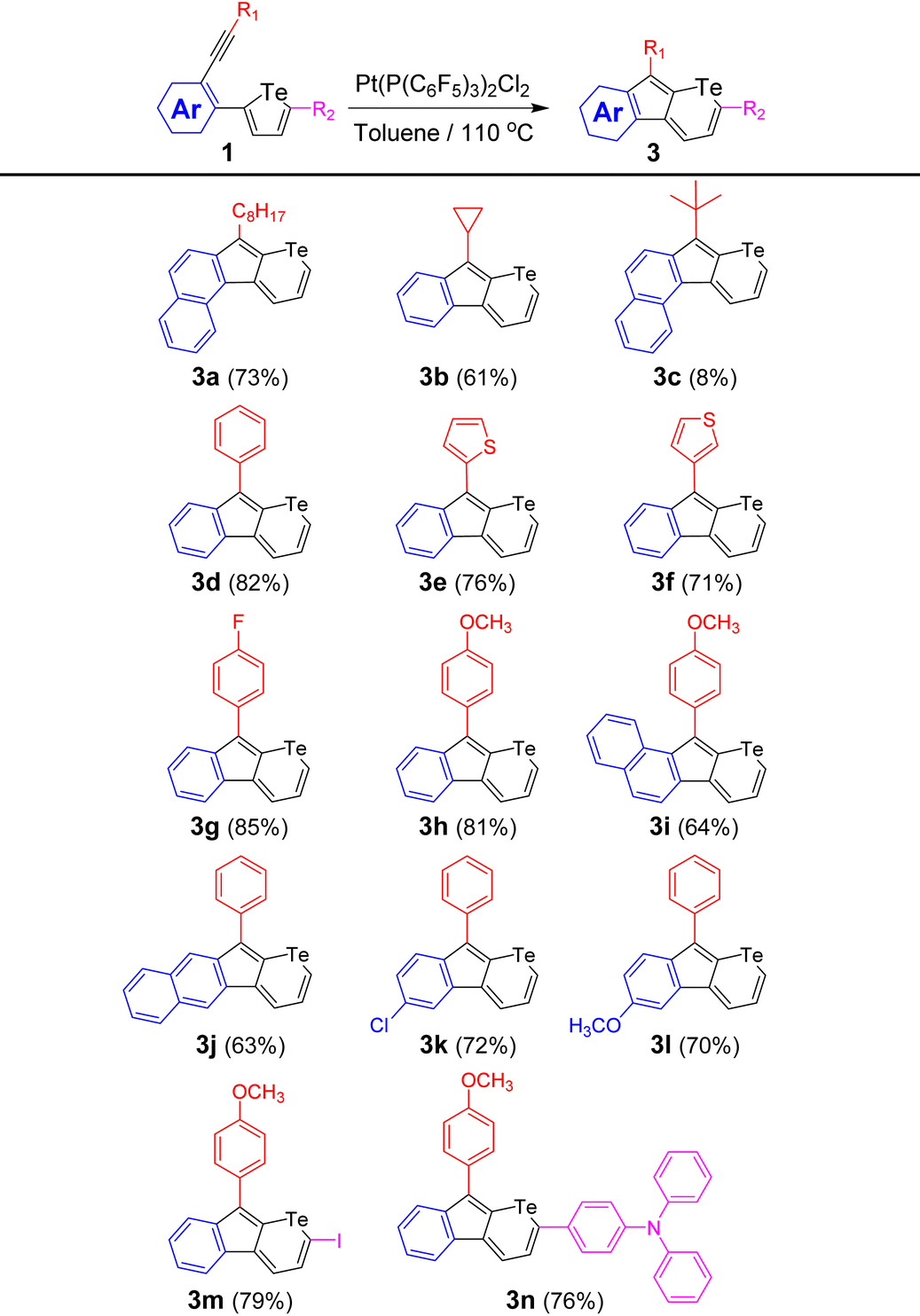
The scope of this Pt(II)-catalyzed ring-expansion cyclization was further investigated by tuning the alkyne (R1), the aryl group in the skeleton (Ar), and the substituents (R2) in compound 1 (Table 2). Since a [1,2]-alkyl/aryl migration is involved in the cyclization mechanism which is discussed below, the scope of the substituted groups attached to the alkyne was first examined. As shown in Table 2, in addition to the linear octyl group, both cyclopropyl and t-butyl groups can migrate in this cyclization. However, the yield decreased from 73% for octyl-based 3a to 61% for cyclopropyl-based 3b, and further to 8% for t-butyl-based 3c. Moreover, when the sp3 hybrid carbon substituted at the alkynyl groups changed to an sp2 hybrid carbon, telluropyrans 3d, 3e, and 3f, with phenyl, 2-thienyl, and 3-thienyl substituents, respectively, could be produced in yields over 70%. Furthermore, when electron-withdrawing fluoride and electron-donating methoxyl groups were attached at the para-position of the phenyl groups, 3g and 3h were obtained in yields of 85% and 81%, respectively, which suggests that the electron density of the substituted aryl group has little effect on this ring-expansion reaction. Secondly, the aryl groups (Ar in 1, Table 2) in the skeleton were explored. When the alkyne and tellurophene were adjusted to different ortho-positions in naphthalene, such as 1-/2-, 2-/1-, and 2-/3-positions, 3a, 3i, and 3j with conjugation extended from different directions were produced in moderate yields. Similarly, when electron-withdrawing chloride and electron-donating methoxyl groups were introduced on the phenyl groups, 3k and 3l were synthesized in yields over 70%. Thirdly, when the 5-position of tellurophene was occupied by iodide, iodo-substituted telluropyran derivative 3m was afforded in 79% yield. Moreover, when the iodide in the precursor was replaced by a triphenylamine, conjugation-extended 3n was provided (76%). The above examples suggest that this approach is a general method to convert tellurophene-containing compounds to the corresponding telluropyran derivatives.
Table 2 Scope of the ring-expansion reaction towards telluropyran derivatives
The chemical structures of the as-prepared telluropyran derivatives were characterized by 1H NMR, 13C NMR, and high-resolution mass spectrometry (HRMS) and were found to be consistent with the proposed structures. In addition, single crystal X-ray diffraction analysis was performed to provide the structural features of the telluropyran ring. The single crystal of 3g (CCDC 2132833†) was obtained as a violet prism-shaped crystal by slow evaporation of its toluene solution at room temperature. As shown in Fig. 4, the lengths of two C–Te bonds in the telluropyran ring of 3g are 2.066 and 2.067 Å, which are close to the value in the five-membered tellurophene ring (2.08 Å).34 This suggests that the lone pair electrons of the tellurium atoms are involved in the π-conjugated backbone. Moreover, the indeno[2,1-b]telluropyran backbone is coplanar with dihedral angles between two neighboring aromatic rings less than 3°. Meanwhile, the benzene ring substituted at the cyclopentadiene moiety is significantly twisted with a dihedral angle of 54° due to the steric repulsion between the hydrogen atoms. Consequently, a relatively large π–π stacking distance of 3.78 Å can be found, indicating their relatively weak intermolecular interactions.
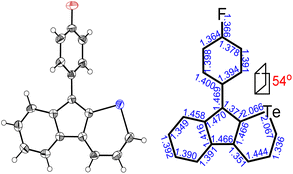 |
| | Fig. 4 ORTEP diagrams with an ellipsoid contour probability level of 50% and selected bond lengths for 3g. | |
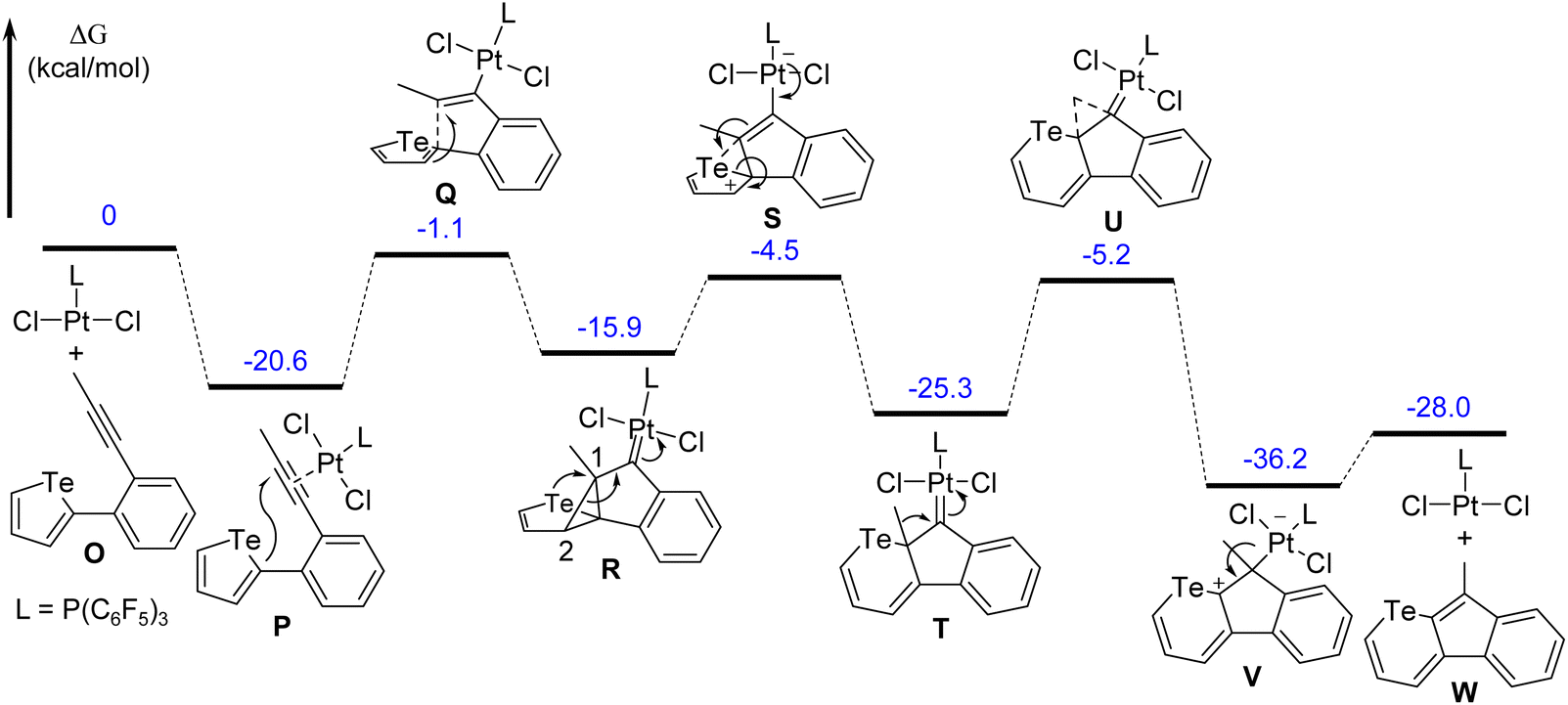
A probable mechanism for the conversion from tellurophene to telluropyran via the ring-expansion reaction was proposed. The reaction pathway and the relative free energies for all stationary points in toluene solutions were also calculated. As shown in Fig. 5, the model reaction starts with the coordination compound N which is converted to intermediate Pvia an ipso-cyclization reaction. Then the C1–C2 bond in the cyclopropanyl unit cleaves due to the huge ring tension. Meanwhile, the C1 atom attacks the close Te atom with simultaneous cleavage of one C–Te bond in the tellurophene unit and intermediate R is produced. Finally, intermediate T is generated via [1,2]-alkyl migration, followed by the dissociation of PtCl2L to give the target telluropyran derivative. The model reaction turns out to be exothermic at 28.0 kcal mol−1.
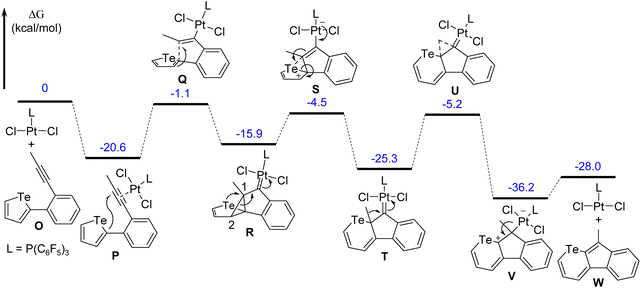 |
| | Fig. 5 Calculated pathway of the ring-expansion reaction and the relative free energies (in kcal mol−1) of the stationary points at the ωB97M-V/def2-TZVPP, SMD(toluene)//ωB97X-D/6-31G(d) (LANL2DZ for Pt and Te), SMD(toluene) level. | |
Tolerance in transition metal-catalyzed coupling reactions and conjugation extension of telluropyran derivatives
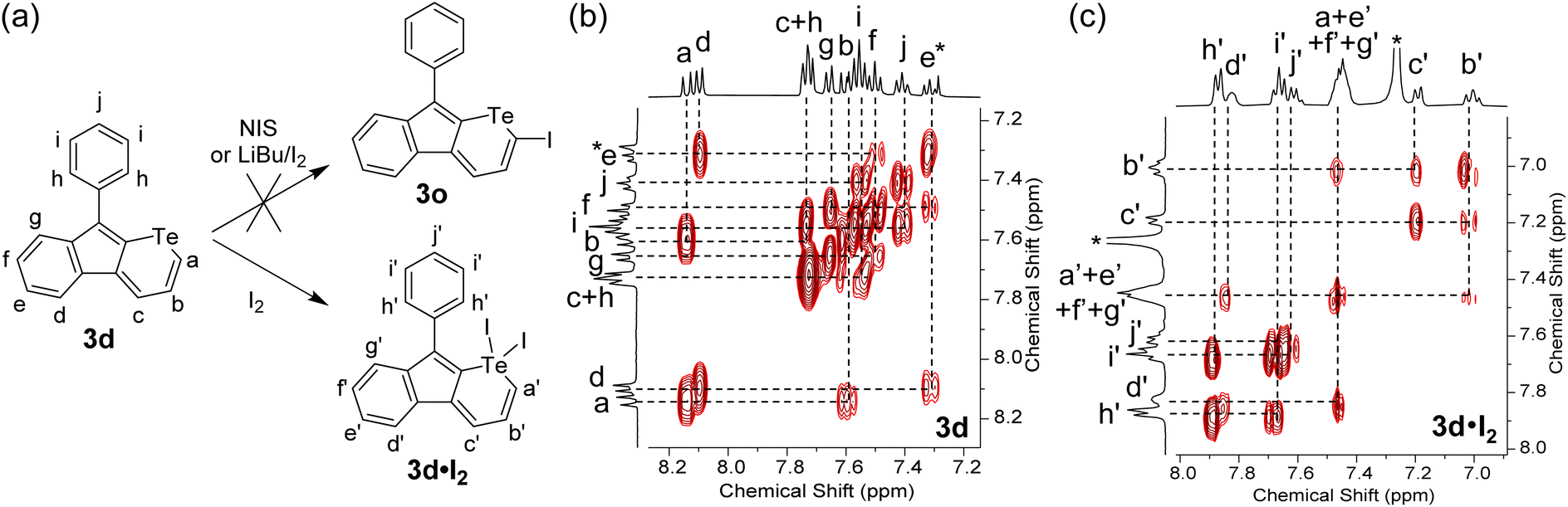
It is well known that tellurophene derivatives can be cleaved by metal-related chemicals, such as Grignard reagents35 and transition metal-based catalysts.13,14 To investigate the tolerance of the telluropyran derivatives in transition metal-catalyzed coupling reactions and extend their conjugation, halogenation of telluropyran derivatives and their further coupling reactions were carried out. In general, a tellurophene can be simply iodinated by N-iodosuccinimide (NIS)36 or LiBu/I2.37 However, when telluropyran derivative 3d was treated with the above-mentioned conditions, the desired product 3o was not produced. As shown in Fig. 6a, when 3d was treated with iodine, whether LiBu was initially added or not, an oxidized product 3d·I2 was produced. Its chemical structure was verified by two-dimensional 1H–1H correlation spectroscopy (2D 1H–1H COSY). As shown in Fig. 6b, the signals of protons a, b, and c on the telluropyran ring in 3d locate in the aromatic region at δ = 8.14 (doublet), 7.59 (triplet), and 7.75 ppm (doublet), respectively. Upon the addition of two iodides, the three proton signals shift to the higher field region at δ = 7.46, 7.01, and 7.20 ppm, respectively. This can be explained by the decreased aromaticity and deshielded effect upon the introduction of iodine on the telluropyran ring.38
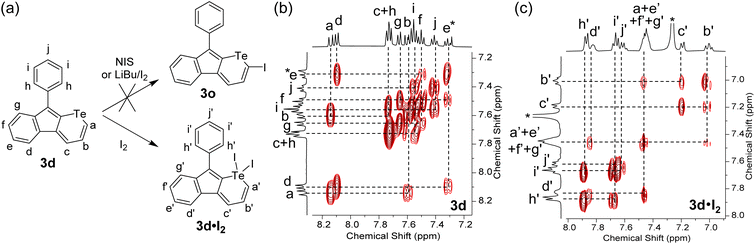 |
| | Fig. 6 (a) Iodation of 3d into 3d·I2 with proton labelling and 2D 1H–1H COSY NMR spectra of (b) 3d and (c) 3d·I2 in CDCl3 with the assignment (* refers to the signal of the solvent). | |
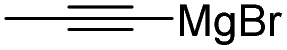
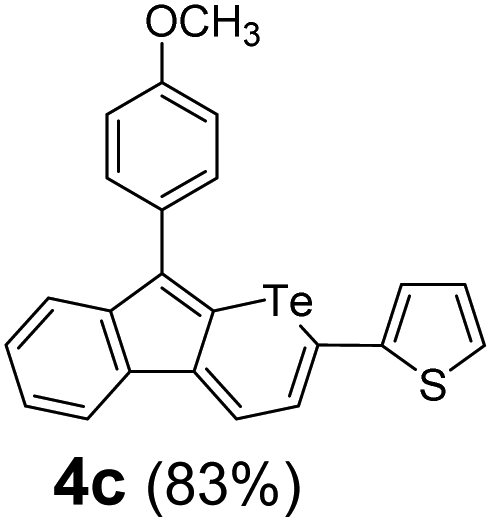
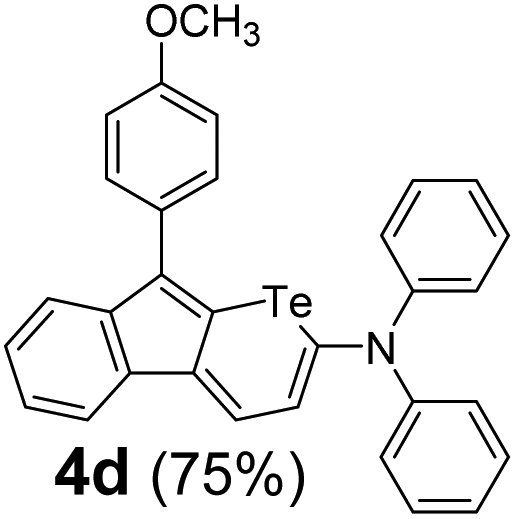
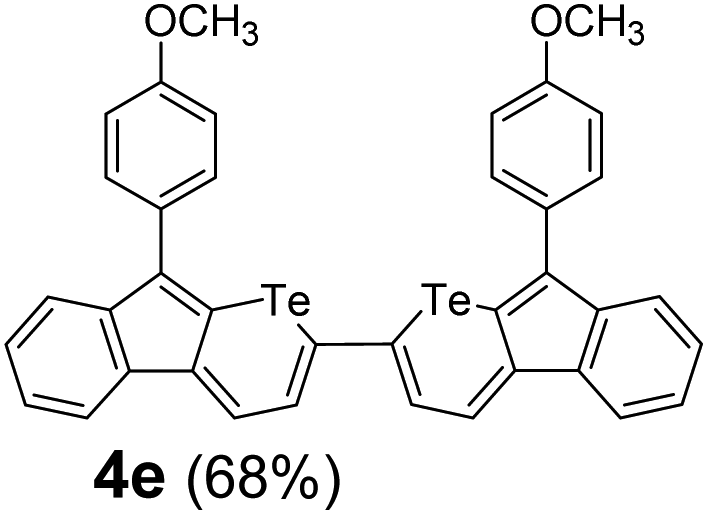
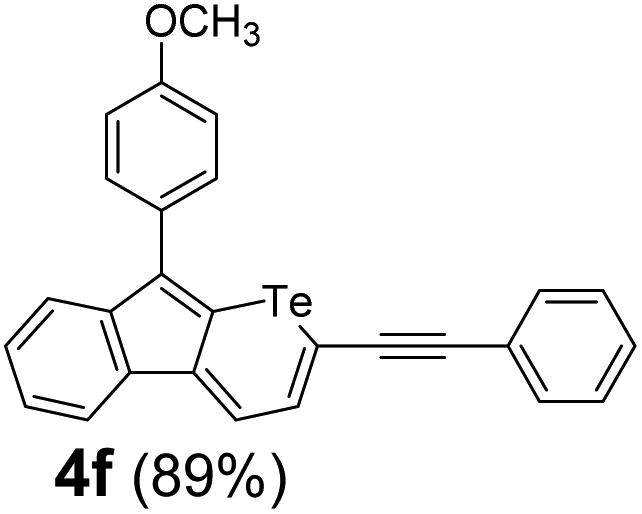
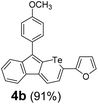
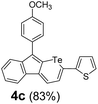
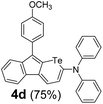
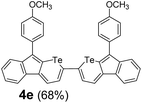
Therefore, to study the tolerance of the telluropyran derivatives in transition metal-catalyzed coupling reactions, compound 3m with iodide was selected and synthesized by ring-expansion of its iodinated tellurophene precursor. Generally, in the presence of a Grignard reagent, tellurophenes always decompose to dienes.39 Interestingly, the as-prepared telluropyran derivatives are chemically stable enough to survive with Grignard reagents. As shown in Table 3, compound 4a could be provided by Ni(II)-catalyzed Kumada40 coupling in 72% yield (entry 1). Moreover, similar to the Ni-containing catalyst, Pd (non- or two-valent) is one of the most important transition metal-based catalysts in coupling reactions. Under the catalyzation of Pd(II) and Pd(0), compounds 4b and 4c were obtained via Suzuki41 and Stille42 coupling reactions in yields of 91% and 83%, respectively (entries 2 and 3). Furthermore, in the presence of a strong base of t-BuONa, a Buchwald43 coupling with diphenylamine afforded compound 4d in 75% yield (entries 4). In addition to Ni- and Pd-based catalysts, telluropyran derivatives can also tolerate Cu-based catalysts. As shown in entries 5 and 6, via Cu-catalyzed Ullmann44 coupling and CuI-cocatalyzed Sonogashira45 coupling reactions, compounds 4e and 4f were produced in high yields. These results suggest that, unlike tellurophenes, the obtained telluropyran derivatives are chemically stable in most of the popular transition metal-catalyzed coupling reactions and their conjugations can be facilely extended via various classic coupling reactions. The better tolerance of the telluropyran derivatives than that of the tellurophene derivatives can be explained by their distinct electronic structures. Unlike the tellurophene with 6 π-electrons, a neutral telluropyran contains 7 π-electrons. Nevertheless, the as-prepared neutral telluropyran derivatives are aromatic since the cyclopenta[b]telluropyran moiety presents the 5π + 7π electron characteristic and tends to form an isoelectronic resonance structure where the telluropyran ring is positively charged and the cyclopentadiene ring is negatively charged. Consequently, the telluropyran derivatives exhibit different reactivity and tolerance from the electron-rich tellurophene analogues. Moreover, the as-prepared telluropyran derivatives consist of the cyclopenta[b]telluropyran moiety which presents the 5π + 7π electron characteristic and is isoelectronic to the non-alternating hydrocarbon azulene. Similarly, separated electron distribution can be achieved between the frontier orbitals, which may result in a relatively low energy band gap. Therefore, the conjugation-expanded compounds are potential candidates for further phosphorescence and photovoltaic applications.
Table 3 Tolerance of iodo-substituted telluropyran derivative 3m in transition metal-catalyzed coupling reactions
| Entry |
Reactant |
Catalyst & conditions |
Product (yield) |
| 1 |

|
Ni(dppe)Cl2 |
|
| THF |
| 0 °C to reflux |
| 2 |
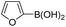
|
Pd(PPh3)2Cl2 |
|
| K2CO3 |
| THF/H2O |
| Reflux |
| 3 |
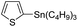
|
Pd(PPh3)4 |
|
| DMF |
| 120 °C |
| 4 |
|
Pd(dba)2 |
|
| P(t-Bu)3·HBF4 |
|
t-BuONa |
| Toluene |
| Reflux |
| 5 |
None |
Cu |
|
| DMF |
| Reflux |
| 6 |
|
Pd(PPh3)2Cl2 |
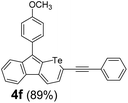
|
| CuI |
| Et3N/DMF |
| r.t. |
Conclusions
In summary, a Pt(II)-catalyzed ring-expansion cyclization towards neutral telluropyran derivatives from the corresponding tellurophene compounds has been developed. Without the assistance of the phosphine ligand, the Te atom coordinates with Pt(II) and cleaves from the tellurophene ring. Consequently, Te-free vinylogous pentafulvalene analogues are produced. However, when bulky phosphine ligands are incorporated into the PtCl2 catalyst, the Pt–Te interactions are suppressed and the cleavage of the C–Te bond is minimized. Consequently, the tellurophene derivatives are converted to the corresponding telluropyran derivatives via a ring-expansion reaction. The substrate scope has been investigated and a possible reaction mechanism for the ring-expansion reaction is proposed. The reactivity of the telluropyran derivatives is further explored and thus their conjugation can be further extended via a series of transition metal-catalyzed coupling reactions. Overall, our work provides not only a synthetic methodology towards telluropyran-fused polycyclic aromatic hydrocarbons, but also an effective pathway to avoid the C–Te bond cleavages in transition metal-catalyzed coupling reactions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank Dr Lingling Li at the Instrumental Analysis Center of SJTU for single crystal analysis. This work was financially supported by the National Key Research and Development Program of China (2018YFA0209401), the National Natural Science Foundation of China (22171053, 21733003), and the Natural Science Foundation of Shanghai (21ZR1409600).
References
- K. T. Mahmudov, M. N. Kopylovich, M. F. C. G. da Silva and A. J. L. Pombeiro, Chalcogen bonding in synthesis, catalysis and design of materials, Dalton Trans., 2017, 46, 10121–10138 RSC.
- J. Lee, D. H. Sin, J. A. Clement, C. Kulshreshtha, H. G. Kim, E. Song, J. Shin, H. Hwang and K. Cho, Medium-Bandgap Conjugated Polymers Containing Fused Dithienobenzochalcogenadiazoles: Chalcogen Atom Effects on Organic Photovoltaics, Macromolecules, 2016, 49, 9358–9370 CrossRef CAS.
- C. L. Wang, H. L. Dong, W. P. Hu, Y. Q. Liu and D. B. Zhu, Semiconducting π-Conjugated Systems in Field-Effect Transistors: A Material Odyssey of Organic Electronics, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- M. J. Wang, T. J. Ko, M. S. Shawkat, S. S. Han, E. Okogbue, H. S. Chung, T. S. Bae, S. Sattar, J. Gil, C. Noh, K. H. Oh, Y. Jung, J. A. Larsson and Y. Jung, Wafer-Scale Growth of 2D PtTe2 with Layer Orientation Tunable High Electrical Conductivity and Superior Hydrophobicity, ACS Appl. Mater. Interfaces, 2020, 12, 10839–10851 CrossRef CAS.
- M. Jeffries-El, B. M. Kobilka and B. J. Hale, Optimizing the Performance of Conjugated Polymers in Organic Photovoltaic Cells by Traversing Group 16, Macromolecules, 2014, 47, 7253–7271 CrossRef CAS.
- E. Pietrasiak and A. Togni, Synthesis and Characterization of Fluorinated Hypervalent Tellurium Derivatives, Organometallics, 2017, 36, 3750–3757 CrossRef CAS.
- X. X. Wu, L. Lv, L. F. Hu, Q. Q. Shi, A. D. Peng and H. Huang, The Synthesis and Optoelectronic Applications for Tellurophene-Based Small Molecules and Polymers, ChemPhysChem, 2019, 20, 2600–2607 CrossRef CAS.
- M. J. Jiang, J. M. Guo, B. X. Liu, Q. T. Tan and B. Xu, Synthesis of Tellurium-Containing π-Extended Aromatics with Room-Temperature Phosphorescence, Org. Lett., 2019, 21, 8328–8333 CrossRef CAS.
- T. Annaka, N. Nakata and A. Ishii, Synthesis, Structures, and Temperature-Dependent Photoluminescence of 1,4-Diphenyl-1-telluro-1,3-butadiene Incorporated in a Dibenzobarrelene Skeleton and Derivatives, Organometallics, 2015, 34, 1272–1278 CrossRef CAS.
- K. Kandasamy, S. Kumar, H. B. Singh and G. Wolmershauser, Influence of both steric effects and Te⋯N center dot center dot center dot N intramolecular nonbonded interactions on the stabilization of organotellurium compounds incorporating [2-[1-(3,5-dimethylphenyl)-2-naphthyl]-4,5-dihydro-4,4-dimethyloxazole], Organometallics, 2003, 22, 5069–5078 CrossRef CAS.
- B. Wu, Melvina, X. Wu, E. K. L. Yeow and N. Yoshikai, Versatile telluracycle synthesis via the sequential electrophilic telluration of C(sp2)-Zn and C(sp2)-H bonds, Chem. Sci., 2017, 8, 4527–4532 RSC.
- T. Chivers and R. S. Laitinen, Tellurium: a maverick among the chalcogens, Chem. Soc. Rev., 2015, 44, 1725–1739 RSC.
- E. Pacholska-Dudziak, G. Vetter, A. Goratowska, A. Bialonska and L. Latos-Grazynski, Chemistry inside a Porphyrin Skeleton: Platinacyclopentadiene from Tellurophene, Chem. – Eur. J., 2020, 26, 16011–16018 CrossRef CAS.
- E. Pacholska-Dudziak, M. Szczepaniak, A. Ksiazek and L. Latos-Grazynski, A porphyrin skeleton containing a palladacyclopentadiene, Angew. Chem., Int. Ed., 2013, 52, 8898–8903 CrossRef CAS.
- E. Pacholska, L. Latos-Grazynski and Z. Ciunik, A direct link between annulene and porphyrin chemistry-21-vacataporphyrin, Chem. – Eur. J., 2002, 8, 5403–5406 CrossRef CAS.
- E. Pacholska-Dudziak, L. Szterenberg and L. Latos-Grazynski, A Flexible Porphyrin-Annulene Hybrid: A Nonporphyrin Conformation for meso-Tetraaryldivacataporphyrin, Chem. – Eur. J., 2011, 17, 3500–3511 CrossRef CAS PubMed.
- E. Pacholska-Dudziak, J. Skonieczny, M. Pawlicki, L. Szterenberg and L. Latos-Grazynski, Cadmium(II), nickel(II), and zinc(II) complexes of vacataporphyrin: A variable annulene conformation inside a standard porphyrin frame, Inorg. Chem., 2005, 44, 8794–8803 CrossRef CAS PubMed.
- F. Zheng, Y. Komatsuzaki, N. Shida, H. Nishiyama, S. Inagi and I. Tomita, Te-Li Exchange Reaction of Tellurophene-Containing π-Conjugated Polymer as Potential Synthetic Tool for Functional π-Conjugated Polymers, Macromol. Rapid Commun., 2019, 40, 1900171 CrossRef CAS.
- F. Zheng, S. E. Tan, Y. Yanamoto, N. Shida, H. Nishiyama, S. Inagi and I. Tomita, Preparation of a germole-containing π-conjugated polymer by the Te-Li exchange reaction of a tellurophene-containing polymer, Npg Asia Mater., 2020, 12, 41 CrossRef CAS.
- M. J. Dabdoub, V. B. Dabdoub, M. A. Pereira and J. ZukermanSchpector, Iodocyclization of (Z,)-1-(butyltelluro)-1,4-diorganylbut-1-en-3-ynes. Synthesis and reactions of 3-iodotellurophenes, J. Org. Chem., 1996, 61, 9503–9511 CrossRef CAS.
- V. K. Karapala, H. P. Shih and C. C. Han, Cascade and Effective Syntheses of Functionalized Tellurophenes, Org. Lett., 2018, 20, 1550–1554 CrossRef CAS.
- A. A. Jahnke, G. W. Howe and D. S. Seferos, Polytellurophenes with Properties Controlled by Tellurium-Coordination, Angew. Chem., Int. Ed., 2010, 49, 10140–10144 CrossRef CAS PubMed.
- R. Sengupta, K. G. Thorat and M. Ravikanth, Effects of Core Modification on Electronic Properties of para-Benziporphyrins, Inorg. Chem., 2019, 58, 12069–12082 CrossRef CAS.
- V. Michelet, P. Y. Toullec and J. P. Genet, Cycloisomerization of 1,n-enynes: challenging metal-catalyzed rearrangements and mechanistic insights, Angew. Chem., Int. Ed., 2008, 47, 4268–4315 CrossRef CAS.
- Y. Lu, Y. Qiao, H. Xue and G. Zhou, From Colorless to Near-Infrared S-Heteroarene Isomers: Unexpected Cycloaromatization of Cyclopenta[b]thiopyran Catalyzed by PtCl2, Org. Lett., 2018, 20, 6632–6635 CrossRef CAS.
- Y. Qiao, L. Yang, J. Zhu, C. Yan, D. Chang, N. Zhang, G. Zhou, Y. Zhao, X. Lu and Y. Liu, Crystal Engineering of Angular-Shaped Heteroarenes Based on Cyclopenta[b]thiopyran for Controlling the Charge Carrier Mobility, J. Am. Chem. Soc., 2021, 143, 11088–11101 CrossRef CAS.
- M. R. Detty and B. J. Murray, Cyclization of 3-(Arylchalcogeno)Propenoyl Chlorides .2. Chalcogen and Substituent Control in the Regiochemistry of Intramolecular Acylation - Preparation of Benzo[b]Telluropyrones, J. Am. Chem. Soc., 1983, 105, 883–890 CrossRef CAS.
- Y. Li, K. C. Mondal, P. Stollberg, H. Zhu, H. W. Roesky, R. Herbst-Irmer, D. Stalke and H. Fliegl, Unusual formation of a N-heterocyclic germylene via homolytic cleavage of a C-C bond, Chem. Commun., 2014, 50, 3356–3358 RSC.
- U. J. Kilgore, J. A. Karty, M. Pink, X. F. Gao and D. J. Mindiola, Tellus in, Tellus out: The Chemistry of the Vanadium Bis(telluride) Functionality, Angew. Chem., Int. Ed., 2009, 48, 2394–2397 CrossRef CAS.
- N. Mardirossian and M. Head-Gordon, omegaB97M-V: A combinatorially optimized, range-separated hybrid, meta-GGA density functional with VV10 nonlocal correlation, J. Chem. Phys., 2016, 144, 214110 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- J. D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- S. Inoue, T. Jigami, H. Nozoe, T. Otsubo and F. Ogura, 2,2′-Bitellurophene and 2,2′-5′,2′′-Tertellurophene as Novel High Homologs of Tellurophene, Tetrahedron Lett., 1994, 35, 8009–8012 CrossRef CAS.
- E. Wenkert, M. H. Leftin and E. L. Michelotti, A Synthesis of Conjugated Dienes from Aromatic, Five-membered Heterocycles, J. Chem. Soc., Chem. Commun., 1984, 617–618 RSC.
- S. Ye, S. M. Foster, A. A. Pollit, S. Cheng and D. S. Seferos, The role of halogens in the catalyst transfer polycondensation for π-conjugated polymers, Chem. Sci., 2019, 10, 2075–2080 RSC.
- R. B. Panatieri, J. S. Reis, L. P. Borges, C. W. Nogueira and G. Zeni, Synthesis of 2-alkynyl-tellurophene derivatives via palladium-catalyzed cross-coupling, Synlett, 2006, 3161–3163 CAS.
- T. M. McCormick, A. A. Jahnke, A. J. Lough and D. S. Seferos, Tellurophenes with delocalized π-systems and their extended valence adducts, J. Am. Chem. Soc., 2012, 134, 3542–3548 CrossRef CAS.
- E. Wenkert, M. H. Leftin and E. L. Michelotti, A Synthesis of Conjugated Dienes from Aromatic, 5-Membered Heterocycles, J. Chem. Soc., Chem. Commun., 1984, 617–618 RSC.
- K. Tamao, K. Sumitani and M. Kumada, Selective Carbon-Carbon Bond Formation by Cross-Coupling of Grignard-Reagents with Organic Halides-Catalysis by Nickel-Phosphine Complexes, J. Am. Chem. Soc., 1972, 94, 4374 CrossRef CAS.
- N. Miyaura, K. Yamada and A. Suzuki, New Stereospecific Cross-Coupling by the Palladium-Catalyzed Reaction of 1-Alkenylboranes with 1-Alkenyl or 1-Alkynyl Halides, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef.
- D. Milstein and J. K. Stille, General, Selective, and Facile Method for Ketone Synthesis from Acid-Chlorides and Organotin Compounds Catalyzed by Palladium, J. Am. Chem. Soc., 1978, 100, 3636–3638 CrossRef CAS.
- P. Ruiz-Castillo and S. L. Buchwald, Applications of Palladium-Catalyzed C-N Cross-Coupling Reactions, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS.
- J. Hassan, M. Sevignon, C. Gozzi, E. Schulz and M. Lemaire, Aryl-aryl bond formation one century after the discovery of the Ullmann reaction, Chem. Rev., 2002, 102, 1359–1469 CrossRef CAS PubMed.
- K. Sonogashira, Y. Tohda and N. Hagihara, Convenient Synthesis of Acetylenes-Catalytic Substitutions of Acetylenic Hydrogen with Bromoalkenes, Iodoarenes, and Bromopyridines, Tetrahedron Lett., 1975, 4467–4470 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of the telluropyran derivatives and calculated geometries of all the stationary points for the ring-opening and ring-expansion reactions (PDF). CCDC 2132833, 2132835, 2132837 and 2132838. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qo01732f |
|
| This journal is © the Partner Organisations 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: