
Dual nickel/photoredox catalyzed carboxylation of C(sp2)-halides with formate†
Ming-Chen
Fu‡
 *ab,
Jia-Xin
Wang‡
b,
Wei
Ge
b,
Fang-Ming
Du
a and
Yao
Fu
*b
*ab,
Jia-Xin
Wang‡
b,
Wei
Ge
b,
Fang-Ming
Du
a and
Yao
Fu
*b
aDepartment of Chemistry, School of Chemistry and Chemical Engineering, Hefei University of Technology, Hefei, China. E-mail: fumingchen@ustc.edu.cn
bHefei National Research Center for Physical Sciences at the Microscale, CAS Key Laboratory of Urban Pollutant Conversion, Anhui Province Key Laboratory of Biomass Clean Energy, University of Science and Technology of China, Hefei, China. E-mail: fuyao@ustc.edu.cn
First published on 8th November 2022
Abstract
Herein, we report an efficient and practical protocol for dehalocarboxylation of C(sp2)-halides with formate by the engagement of a CO2 radical anion in a nickel-mediated bond-forming process. A wide variety of aryl iodides, aryl bromides, and alkenyl bromides bearing a diverse set of functional groups underwent the reaction smoothly through visible-light photoredox nickel dual catalysis. The synthesis of several 13C-labeled drug intermediates and the gram-scale synthesis of commercial drugs highlight the synthetic value of the approach in drug discovery settings.
Introduction
Aromatic carboxylic acids represent a class of important structural motifs that are widespread in numerous drug molecules, natural products, and useful synthetic intermediates (Scheme 1a).1 Palladium-catalyzed carbonylation of aryl halides using carbon monoxide (CO) gas represents an efficient and straightforward approach to synthesize these compounds.2 However, difficulties in handling this highly toxic gas limit its applications in medicinal chemistry research and complex organic syntheses. To circumvent these issues, efforts have been made in the past decades to develop CO-gas-free carbonylation of aryl halides.3 Among the various CO surrogates, formic acid, a renewable green C1-building block that can be obtained through hydrolyzation/oxidation of biomass and hydrogenation of CO2,4 presents an easily operational CO source with a high weight percentage of CO. Many research groups, such as Skrydstrup,5 Shi,6 Wu,7 Zhou,8 and others,9 have developed a series of efficient carbonylation reactions through a thermal pathway using formic acid and its salts. While these efforts have significantly expanded the set of tools available for carbonylation of aryl halides (Scheme 1b), the requirement for precious palladium salts as catalysts, a stoichiometric amount of activator, and heating conditions limits its wide application on a large scale. Therefore, the development of a mild and sustainable protocol to synthesize arene carboxylic acids from aryl halides using formic acid and its salts is still a highly rewarding goal.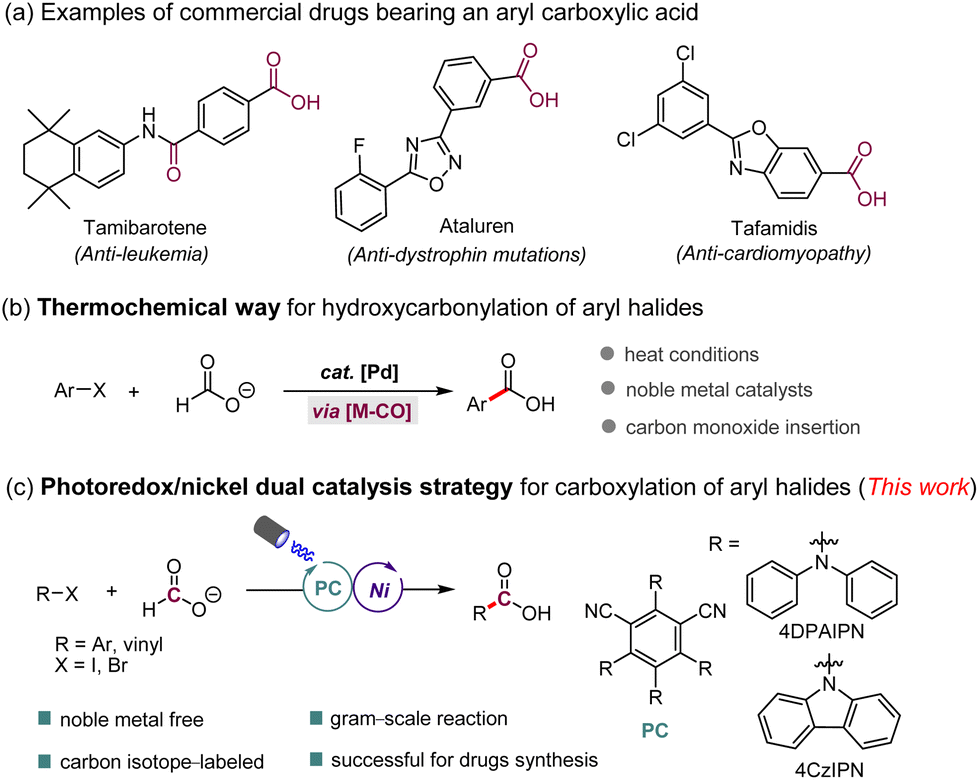 | ||
| Scheme 1 Carboxylation of aryl halides with formate for the synthesis of aryl carboxylic acids. | ||
In recent years, visible-light photoredox catalysis has emerged as a powerful tool for chemists10 because of its potential to allow the access to reactive intermediates that are unavailable to conventional thermal pathways, via single-electron-transfer (SET) and energy-transfer processes under mild reaction conditions. Moreover, the rapid development of photocatalysis has also brought new impetus for studies on the use of formate for the formation of the carbon dioxide radical anion (CO2˙−),11 which has shown broad utility as a reactive intermediate in organic synthesis.12 In particular, the groups of Jui13 and Wickens14 recently reported elegant photochemical processes to synthesize carboxylic acids from the reaction of formate with activated alkenes without the need for a sacrificial electron donor, in contrast to CO2-reduction strategies. Nevertheless, these types of reactions, which involve the participation of the CO2 radical anion, are limited to radical addition to unsaturated substrates or to radical–radical couplings. The synergistic merging of photoredox catalysis and transition-metal catalysis (termed metallaphotoredox catalysis) resulted in many new discoveries that were unfeasible or difficult to achieve with a single catalytic system in recent years.15 To the best of our knowledge, however, the use of a metallaphotoredox strategy to promote formate as a CO2 radical anion for C–C bond formation reactions has not been realized to date. Inspired by these pioneering works11–14 and continuing our interest in photoredox catalysis16 and carboxylation reactions,9a,17 we questioned whether merging visible-light photoredox with nickel catalysis might be used to realize the carboxylation of aryl halides with formate through a CO2 radical anion intermediate (Scheme 1c).
The mechanistic details of our proposed dual nickel/photoredox catalyzed carboxylation of aryl halides with formate are outlined in Scheme 2. The formate salt can be oxidized to the radical intermediate by the excited photocatalyst,13,14,18 which then undergoes a hydrogen atom transfer process upon oxidation to deliver formic acid and the CO2 radical anion. It should be noted that the CO2 radical anion has a very negative reduction potential (E1/2 = −2.2 V vs. SCE), which is enough to reduce aryl chlorides smoothly.11c,d At this stage, we hoped that the CO2 radical anion would be rapidly captured by Ln–Ni(0) species A rather than the aryl halide getting reduced by it, to generate the intermediate B (path a). Subsequent oxidative addition of the aryl halide with Ni(I) species B generates the key Ni(III) intermediate C. Alternatively, a catalytic pathway is also plausible involving the oxidative addition of aryl halides with Ni(0) species A to give Ni(II) species E, which is trapped by the CO2 radical anion to provide the same Ni(III) intermediate C (path b). The high-valent Ni(III) species C undergoes a facile reductive elimination to give the desired product and Ni(I) complex D. Reduction of Ni(I) complex D by the highly reducing catalyst species regenerates the Ni(0) species and [PC] to close the two catalytic cycles. Simultaneously, the competing dehalogenation reduction of aryl halides by formate under visible-light irradiation is a challenge for the smooth occurrence of the proposed reaction (path c).
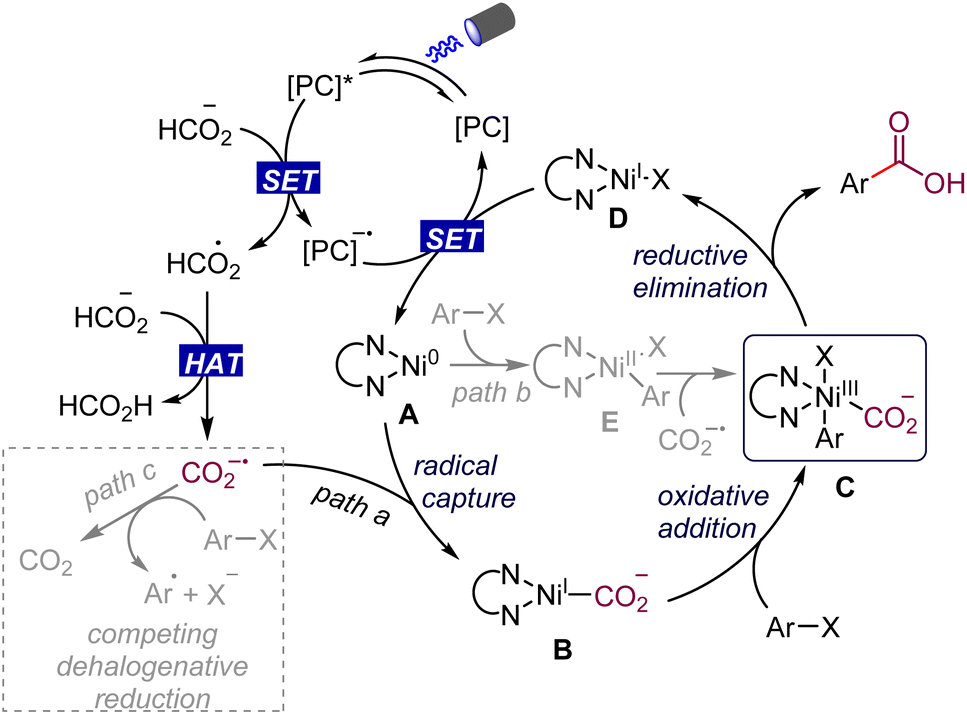 | ||
| Scheme 2 Envisioned catalytic cycle. | ||
Results and discussion
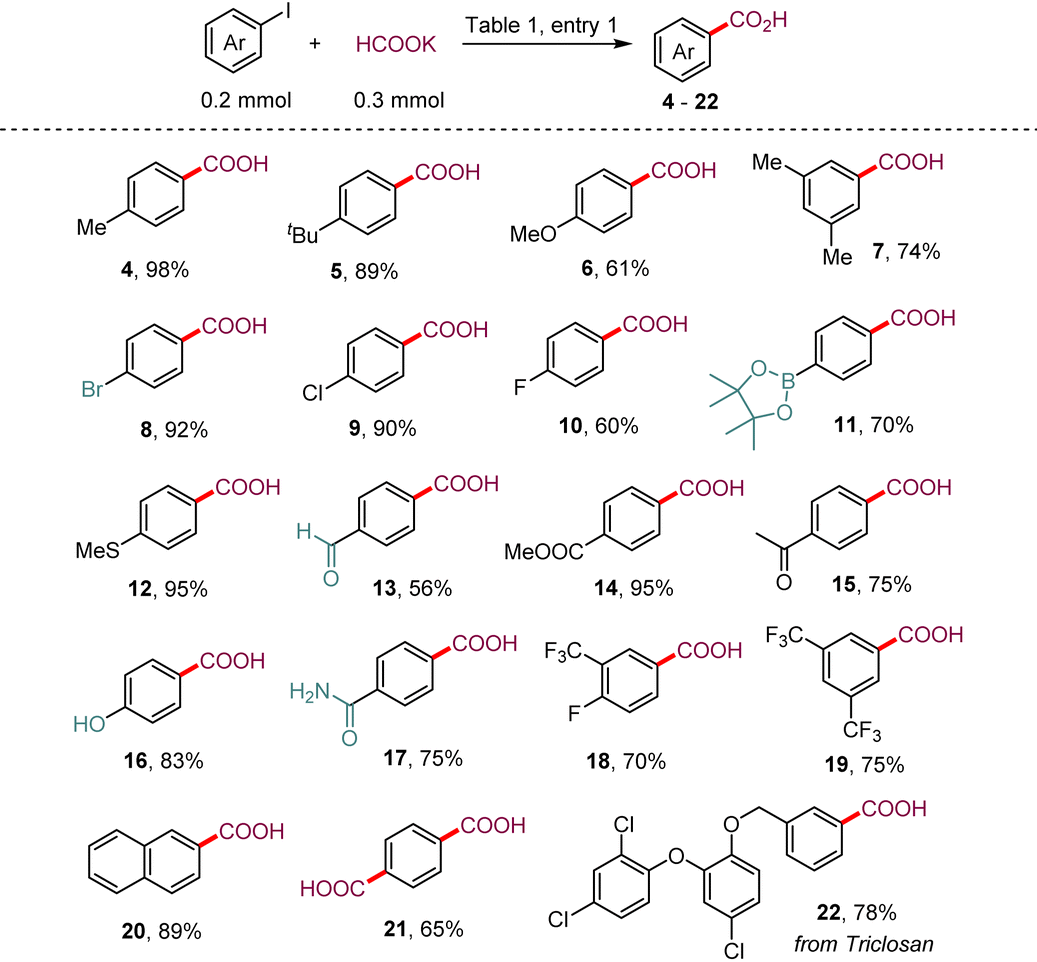
We examined the feasibility of this proposed metallaphotoredox dehalocarboxylation protocol with 4-iodobiphenyl 1 as a model substrate and potassium formate as the carbonyl source. After a comprehensive investigation of all the reaction parameters (see the ESI† for detailed information), we were delighted to find that upon irradiation of a solution of 4-iodobiphenyl 1 and potassium formate with blue light-emitting diodes (LEDs) in the presence of catalytic amounts of 4DPAIPN, Ni(PCy3)2Cl2, and 4,4′-di-tert-butyl-2,2′-dipyridyl (dtbbpy) in N,N-dimethylacetamide (DMA), 98% yield of the desired product 3 was obtained after methyl esterification with iodomethane, without observable formation of the by-product biphenyl (entry 1). The organic photocatalyst 4CzIPN could be used to catalyze the transformation with 92% yield (entries 2 and 3), and iridium-based photocatalysts Ir–I also promoted this transformation with moderate efficiency. Other nickel salts were also investigated (entries 4–7). The yield decreased sharply when other Ni(II) salts were used as the precatalysts, such as NiCl2·glyme, NiBr2·glyme, and Ni(acac)2, but a 77% yield of 3 could be obtained when Ni(PPh3)2Cl2 was used instead of Ni(PCy3)2Cl2. From these results, we concluded that monodentate phosphine ligands play an important role in controlling the reactivity of the nickel catalysts. The monodentate phosphine ligand could possibly operate in synergy with the bidentate nitrogen ligand to stabilize the Ni(0) species A. Moreover, it should be noted that hydrodehalogenation of aryl halides by formate occurred smoothly under irradiation with blue LEDs, and we found that when DMA was replaced with dimethyl sulfoxide (DMSO) as a solvent, 52% yield of the hydrodeiodination product was detected. Other solvents, such as dioxane, THF, and acetone, failed to give the desired product 3. In addition, we explored other types of bidentate nitrogen ligands (see the ESI†) and found that 4,4′-dimethoxy-2,2′-bipyridine and 4,7-diphenyl-1,10-phenanthroline were also effective for this transformation. Finally, control experiments confirmed that the photocatalyst, nickel salt, and light were all indispensable for the desired reaction.With the optimized conditions in hand, we then investigated the generality of this metallaphotoredox protocol with respect to aryl iodides. As depicted in Table 2, a variety of aryl iodides bearing electron-donating (–Me, –t-Bu, –OMe, and –SMe) or electron-withdrawing (–F, –Cl, –COOMe, and –CF3) substituents reacted smoothly under the optimal conditions, furnishing the corresponding aryl carboxylic acids with good to excellent yields (60–98%). Moreover, aryl bromide (8), aryl aldehyde (13), aryl ketone (15), unprotected phenolic hydroxyl (16), unsubstituted amide (17), and even aryl pinacol boronate (11), which is susceptible to transmetallation under nickel catalysis, were well tolerated under the reaction conditions. This highlights the potential uses of this method in combination with further coupling transformations. 2-Iodonaphthalene also underwent the reaction smoothly and gave the desired product 20 in 89% yield. Notably, 1,4-diiodobenzene successfully underwent deiodocarboxylation smoothly in this system to deliver terephthalic acid (21) in 65% yield. The derivative of triclosan (22), a broad-spectrum antibacterial agent, was also an amenable substrate in this protocol.
|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yield of 3 (%) |
 a Reaction conditions: 1 (0.2 mmol, 1.0 equiv.), HCOOK (0.3 mmol, 1.5 equiv.), 4DPAIPN (1.0 mol%), Ni(PCy3)2Cl2 (5.0 mol%), dtbbpy (6.0 mol%), and DMA (2.0 mL), and irradiation with blue LEDs (440 nm) at room temperature for 20 h under an argon atmosphere. The yield was determined by gas chromatography analysis. Isolated yield in parentheses.
a Reaction conditions: 1 (0.2 mmol, 1.0 equiv.), HCOOK (0.3 mmol, 1.5 equiv.), 4DPAIPN (1.0 mol%), Ni(PCy3)2Cl2 (5.0 mol%), dtbbpy (6.0 mol%), and DMA (2.0 mL), and irradiation with blue LEDs (440 nm) at room temperature for 20 h under an argon atmosphere. The yield was determined by gas chromatography analysis. Isolated yield in parentheses.
|
||
| 1 | None | 98 (92) |
| 2 | 4CzIPN instead of 4DPAIPN | 92 |
| 3 | Ir–I instead of 4DPAIPN | 87 |
| 4 | NiCl2·glyme | 21 |
| 5 | NiBr2·glyme | 24 |
| 6 | Ni(acac)2 | 15 |
| 7 | Ni(PPh3)2Cl2 | 77 |
| 8 | DMSO instead of DMA | 36 |
| 9 | Dioxane instead of DMA | Trace |
| 10 | THF instead of DMA | Trace |
| 11 | Acetone instead of DMA | Trace |
| 12 | No PC, or Ni-cat., or light | 0 |

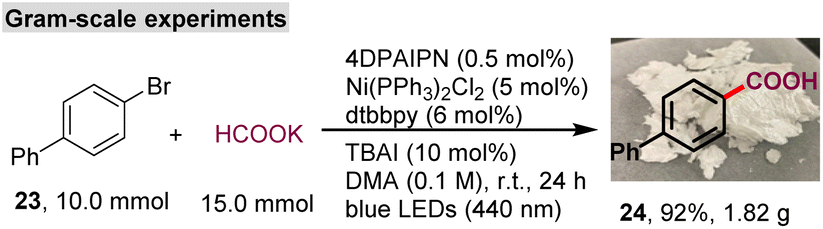
This protocol showed exquisite functional group tolerance (Table 3). A wide range of aryl bromides bearing different functional groups, such as ether (25, 29, 31–33), amine (26), trifluoromethyl (27), cyano (28), ketone (30), amino acid (34), phenolic hydroxyl (35), and sulfone (36), on the arene backbone could be converted into the corresponding aryl carboxylic acid in good to excellent yields (60–95%). Interestingly, the styrene fragment (37) and the electron-deficient C–C π-bond (38), which easily undergo hydrocarboxylation in the presence of formate through a photochemical process,14,18 were compatible in this system, thus demonstrating that this method has excellent chemical selectivity. The ortho substituents of aryl bromide and aryl chloride failed to yield the desired product (see the ESI†). Interestingly, this carboxylation method was compatible with heteroaromatic rings, including quinolone (40), dibenzo[b,d]furan (41), dibenzo[b,d]thiophene (42), pyrimidine (43), and benzo[b]thiophene (44). Furthermore, the protocol was also extended to vinyl bromides. Both styryl bromide (45 and 46) and cyclohexenyl bromide (47 and 48) underwent the debromocarboxylation reaction smoothly. Notably, complex compounds that contained the bromo-substituted 1,3-butadiene fragment were also amenable substrates and gave the desired product 49 in good yields with the butadiene intact. These results highlight the potential synthetic value of this protocol for the synthesis of aryl and vinyl carboxylic acids.
| a Reaction conditions: see the ESI.† Isolated yields. |
|---|
|
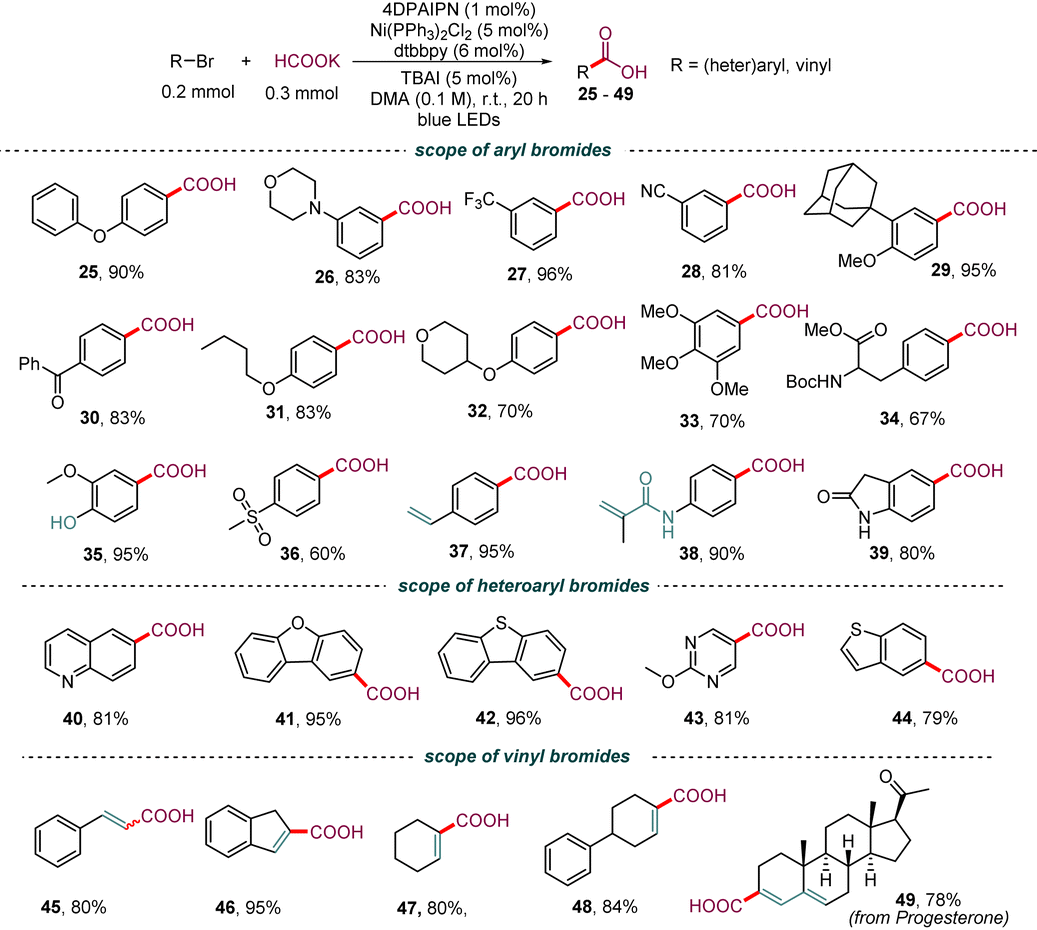
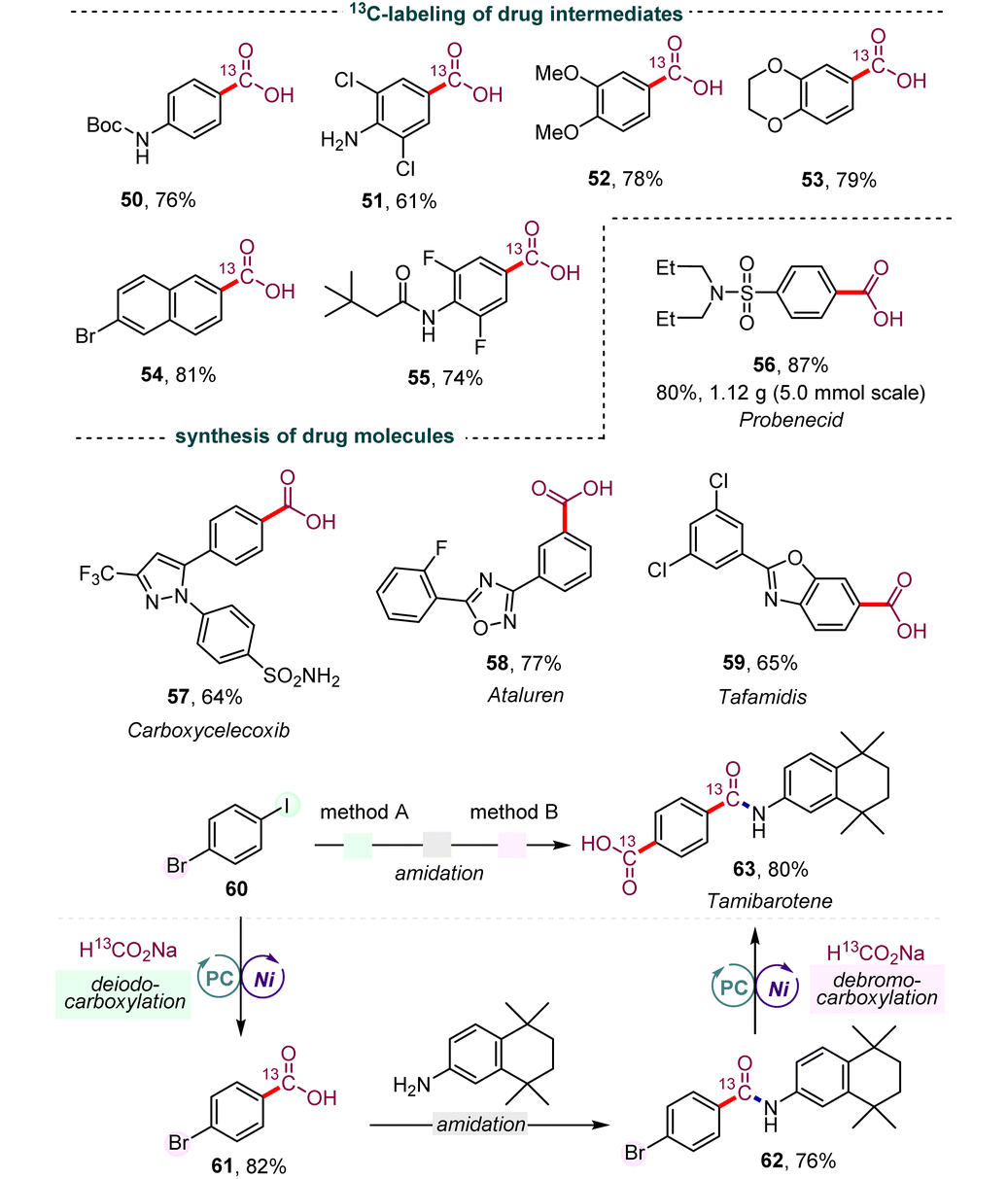
Isotopically labeled active pharmaceutical ingredients are of utmost importance in the field of lead compound drug discovery and the use of such compounds can provide vital information about drug metabolic and pharmacokinetic profiles.20 Thus, we investigated the synthesis of 13C-labeled carboxyl acids to further demonstrate the practicality and versatility of our dehalocarboxylation protocol. As shown in Table 4, several 13C-labeled drug intermediates were obtained in moderate yields (50–55), which offers an operationally simple and practical tool to access isotopically labeled aryl carboxylic acids. In addition, the synthetic value of this strategy was demonstrated through application in several reported medicinally promising scaffolds, such as probenecid (56), carboxycelecoxib (57), ataluren (58), and tafamidis (59). Various functional groups, including amides, pyrazoles, unprotected sulfonamides, and oxadiazoles, were well tolerated. Notably, gram-scale synthesis of probenecid (56) was also efficiently realized with 0.5 mol% of the photocatalyst (5.0 mmol scale, 80% yield). Due to the excellent chemical selectivity of this method, we selected the simple 1-bromo-4-iodobenzene (60) as a starting substrate to synthesize tamibarotene (63), a retinoid drug for relapsed or refractory acute promyelocytic leukemia, through the processes of deiodocarboxylation, amidation, and debromocarboxylation. Gratifyingly, the double carbonyl 13C-isotope labeling of tamibarotene proceeded smoothly from a simple commercially available carbonyl source, 13C-labeled sodium formate. This reaction required the use of precious palladium catalysts and MePh2Si13CO2H as the carbonyl source in an earlier study.21
|
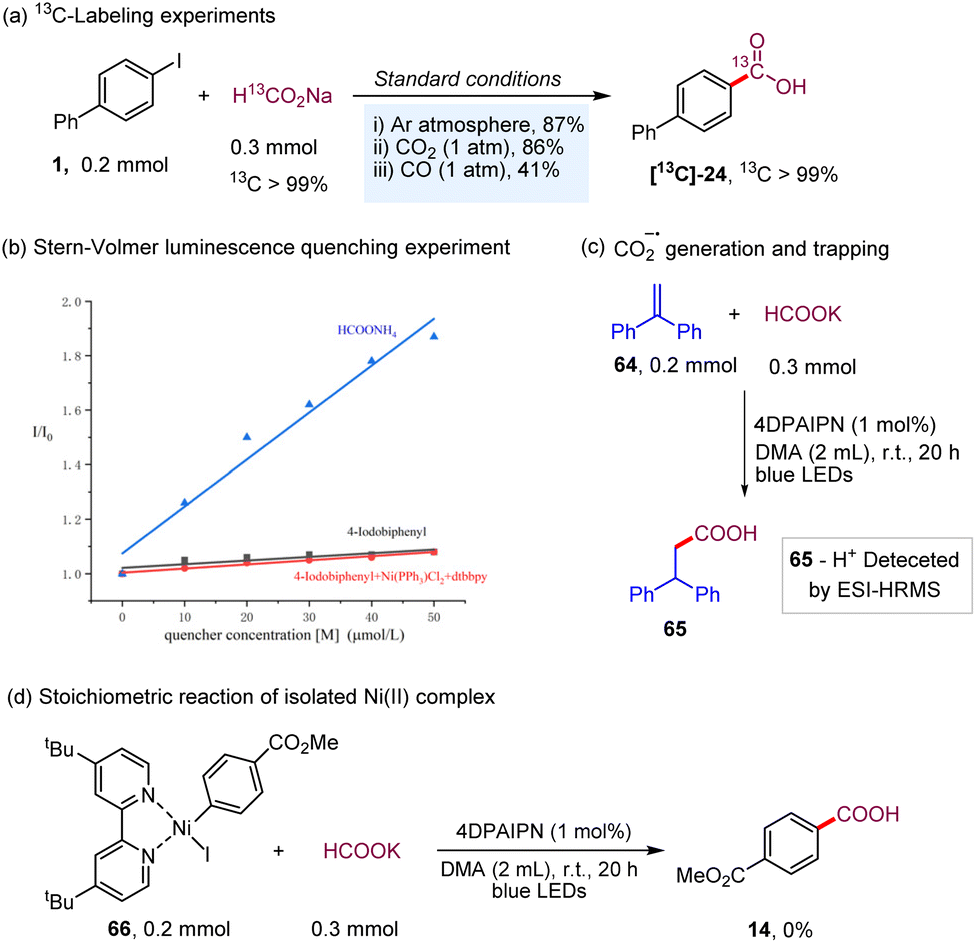
To shed light on the mechanism of the transformation, we conducted a series of preliminary mechanistic experiments. The 13C-labeling experiments were conducted under different gas (Ar, CO2, and CO) atmospheres with 13C-labeled sodium formate as the carbonyl source (Scheme 3a). Deiodocarboxylation afforded 13C-labeled 24 in good-to-high yields with excellent 13C incorporation (>99% 13C incorporation). These results clearly demonstrated that the mechanism of our dehalocarboxylation protocol is distinct from the previously reported systems through visible-light-driven carboxylation of aryl halides with CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 and also excluded the possibility of a carbonylation process with CO in the reaction process.5,16a Consistent with the proposed mechanism shown in Scheme 2, Stern–Volmer luminescence-quenching experiments revealed that the formate salts quenched the excited-state photocatalyst, while 4-iodobiphenyl and a mixture of 4-iodobiphenyl, Ni(PPh3)2Cl2, and dtbbpy did not show a significant quenching effect (Scheme 3b, please see the ESI† for more details). Furthermore, attempts were made to use 1,1-diphenylethylene to trap the CO2 radical anion; the addition product 65 with 1,1-diphenylethylene was also detected. Finally, our stoichiometric study with Ni(II)–ArI 66 failed to give the corresponding product 14. This result indicates that a Ni(II)–aryl species is not involved in the catalytic cycle, making the alternative catalytic Ni0/II/III pathway rather unlikely.23 As a result, based on the above findings of the mechanistic studies and the previous reports,24 a catalytic Ni0/I/III pathway, proceeding via the capture of the CO2 radical anion by Ni(0), followed by oxidative addition and then reductive elimination, is more operative in this catalytic process (Scheme 2).
22 and also excluded the possibility of a carbonylation process with CO in the reaction process.5,16a Consistent with the proposed mechanism shown in Scheme 2, Stern–Volmer luminescence-quenching experiments revealed that the formate salts quenched the excited-state photocatalyst, while 4-iodobiphenyl and a mixture of 4-iodobiphenyl, Ni(PPh3)2Cl2, and dtbbpy did not show a significant quenching effect (Scheme 3b, please see the ESI† for more details). Furthermore, attempts were made to use 1,1-diphenylethylene to trap the CO2 radical anion; the addition product 65 with 1,1-diphenylethylene was also detected. Finally, our stoichiometric study with Ni(II)–ArI 66 failed to give the corresponding product 14. This result indicates that a Ni(II)–aryl species is not involved in the catalytic cycle, making the alternative catalytic Ni0/II/III pathway rather unlikely.23 As a result, based on the above findings of the mechanistic studies and the previous reports,24 a catalytic Ni0/I/III pathway, proceeding via the capture of the CO2 radical anion by Ni(0), followed by oxidative addition and then reductive elimination, is more operative in this catalytic process (Scheme 2).
 | ||
| Scheme 3 Mechanistic studies. | ||
Conclusions
In summary, we have disclosed a novel and practical carboxylation of C(sp2)-halides with formate under photoredox nickel dual catalysis. This method has a broad scope of substrates, high selectivity, and remarkable functional group compatibility. The synthetic value of this protocol was highlighted by the synthesis of several commercial drugs, namely, probenecid, ataluren, tafamidis, and tamibarotene. Moreover, this approach also provides a complementary method that extends the range of the current methodologies available for accessing carbon-13 isotopically labeled carboxylic acids. We expect that engagement of the CO2 radical anion in metal-mediated bond-forming strategies may unlock new platforms that can be used to introduce the carboxyl motif into specific molecular frameworks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support from the National Natural Science Foundation of China (grant numbers 21732006, 51821006, and 22101272). We thank X. Lu (USTC) for the helpful discussions.References
- (a) C. Lamberth and J. Dinges, Different roles of carboxylic functions in pharmaceuticals and agrochemicals, in Bioactive Carboxylic Compound Classes: Pharmaceuticals and Agrochemicals, Wiley-VCH Verlag GmbH &Co. KGaA, 2016, pp. 1–11 Search PubMed; (b) L. J. Goossen, N. Rodriguez and K. Goossen, Carboxylic acids as substrates in homogeneous catalysis, Angew. Chem., Int. Ed., 2008, 47, 3100–3120 CrossRef CAS.
- A. Brennfuhrer, H. Neumann and M. Beller, Palladium-catalyzed carbonylation reactions of aryl halides and related compounds, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef.
- (a) T. Morimoto and K. Kakiuchi, Evolution of carbonylation catalysis: no need for carbon monoxide, Angew. Chem., Int. Ed., 2004, 43, 5580–5588 CrossRef CAS; (b) L. Wu, Q. Liu, R. Jackstell and M. Beller, Carbonylations of alkenes with CO surrogates, Angew. Chem., Int. Ed., 2014, 53, 6310–6320 CrossRef CAS PubMed.
- (a) M. Grasemann and G. Laurenczy, Formic acid as a hydrogen source – recent developments and future trends, Energy Environ. Sci., 2012, 5, 8171–8181 RSC; (b) J. Albert, R. Wölfel, A. Bösmann and P. Wasserscheid, Selective oxidation of complex, water-insoluble biomass to formic acid using additives as reaction accelerators, Energy Environ. Sci., 2012, 5, 7956–7962 RSC.
- S. Korsager, R. H. Taaning and T. Skrydstrup, Effective palladium-catalyzed hydroxycarbonylation of aryl halides with substoichiometric carbon monoxide, J. Am. Chem. Soc., 2013, 135, 2891–2894 CrossRef CAS.
- (a) W. Ren, W. Chang, J. Dai, Y. Shi, J. Li and Y. Shi, An Effective Pd-Catalyzed Regioselective Hydroformylation of Olefins with Formic Acid, J. Am. Chem. Soc., 2016, 138, 14864–14867 CrossRef CAS; (b) Y. Wang, W. Ren, J. Li, H. Wang and Y. Shi, Facile Palladium-Catalyzed Hydrocarboxylation of Olefins without External CO Gas, Org. Lett., 2014, 16, 5960–5963 CrossRef CAS PubMed.
- (a) F. P. Wu, J. B. Peng, X. Qi and X. F. Wu, Palladium-Catalyzed Carbonylative Transformation of Organic Halides with Formic Acid as the Coupling Partner and CO Source: Synthesis of Carboxylic Acids, J. Org. Chem., 2017, 82, 9710–9714 CrossRef CAS; (b) Z. Chen, W. F. Wang, H. Yang and X. F. Wu, Palladium-Catalyzed Four-Component Carbonylative Cyclization Reaction of Trifluoroacetimidoyl Chlorides, Propargyl Amines, and Diaryliodonium Salts: Access to Trifluoromethyl-Containing Trisubstituted Imidazoles, Org. Lett., 2020, 22, 1980–1984 CrossRef CAS PubMed.
- (a) J. Hou, J. H. Xie and Q. L. Zhou, Palladium-catalyzed hydrocarboxylation of alkynes with formic acid, Angew. Chem., Int. Ed., 2015, 54, 6302–6305 CrossRef CAS; (b) C. Fan, J. Hou, Y. J. Chen, K. L. Ding and Q. L. Zhou, Rhodium-Catalyzed Regioselective Hydroformylation of Alkynes to α,β-Unsaturated Aldehydes Using Formic Acid, Org. Lett., 2021, 23, 2074–2077 CrossRef CAS.
- (a) M. C. Fu, R. Shang, W. M. Cheng and Y. Fu, Efficient Pd-Catalyzed Regio- and Stereoselective Carboxylation of Allylic Alcohols with Formic Acid, Chem. – Eur. J., 2017, 23, 8818–8822 CrossRef CAS PubMed; (b) S. Cacchi, G. Fabrizi and A. Goggiamani, Palladium-Catalyzed Hydroxycarbonylation of Aryl and Vinyl Halides or Triflates by Acetic Anhydride and Formate Anions, Org. Lett., 2003, 5, 4269–4272 CrossRef CAS; (c) R. Sang, P. Kucmierczyk, K. Dong, R. Franke, H. Neumann, R. Jackstell and M. Beller, Palladium-Catalyzed Selective Generation of CO from Formic Acid for Carbonylation of Alkenes, J. Am. Chem. Soc., 2018, 140, 5217–5223 CrossRef CAS PubMed; (d) N. Hussain, A. K. Chhalodia, A. Ahmed and D. Mukherjee, Recent Advances in Metal–Catalyzed Carbonylation Reactions by Using Formic Acid as CO Surrogate, ChemistrySelect, 2020, 5, 11272–11290 CrossRef CAS; (e) J. Cao, Z.-J. Zheng, Z. Xu and L.-W. Xu, Transition-metal-catalyzed transfer carbonylation with HCOOH or HCHO as non-gaseous C1 source, Coord. Chem. Rev., 2017, 336, 43–53 CrossRef CAS.
- M. H. Shaw, J. Twilton and D. W. MacMillan, Photoredox Catalysis in Organic Chemistry, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS.
- (a) D. A. Morgenstern, R. E. Wittrig, P. E. Fanwick and C. P. Kubiak, Photoreduction of carbon dioxide to its radical anion by nickel cluster [Ni3(.mu.3-I)2(dppm)3]: formation of two carbon-carbon bonds via addition of carbon dioxide radical anion to cyclohexene, J. Am. Chem. Soc., 1993, 115, 6470–6471 CrossRef CAS; (b) A. J. Boyington, C. P. Seath, A. M. Zearfoss, Z. Xu and N. T. Jui, Catalytic Strategy for Regioselective Arylethylamine Synthesis, J. Am. Chem. Soc., 2019, 141, 4147–4153 CrossRef CAS PubMed; (c) H. Wang, Y. Gao, C. Zhou and G. Li, Visible-Light-Driven Reductive Carboarylation of Styrenes with CO2 and Aryl Halides, J. Am. Chem. Soc., 2020, 142, 8122–8129 CrossRef CAS PubMed; (d) A. F. Chmiel, O. P. Williams, C. P. Chernowsky, C. S. Yeung and Z. K. Wickens, Non-innocent Radical Ion Intermediates in Photoredox Catalysis: Parallel Reduction Modes Enable Coupling of Diverse Aryl Chlorides, J. Am. Chem. Soc., 2021, 143, 10882–10889 CrossRef CAS.
- (a) H. Seo, M. H. Katcher and T. F. Jamison, Photoredox activation of carbon dioxide for amino acid synthesis in continuous flow, Nat. Chem., 2017, 9, 453–456 CrossRef CAS PubMed; (b) H. Seo, A. Liu and T. F. Jamison, Direct β-Selective Hydrocarboxylation of Styrenes with CO2 Enabled by Continuous Flow Photoredox Catalysis, J. Am. Chem. Soc., 2017, 139, 13969–13972 CrossRef CAS PubMed; (c) J. H. Ye, M. Miao, H. Huang, S. S. Yan, Z. B. Yin, W. J. Zhou and D. G. Yu, Visible-Light-Driven Iron-Promoted Thiocarboxylation of Styrenes and Acrylates with CO2, Angew. Chem., Int. Ed., 2017, 56, 15416–15420 CrossRef CAS; (d) H. Huang, J.-H. Ye, L. Zhu, C.-K. Ran, M. Miao, W. Wang, H. Chen, W.-J. Zhou, Y. Lan, B. Yu and D.-G. Yu, Visible-Light-Driven Anti-Markovnikov Hydrocarboxylation of Acrylates and Styrenes with CO2, CCS Chem., 2021, 3, 1746–1756 CrossRef CAS.
- C. M. Hendy, G. C. Smith, Z. Xu, T. Lian and N. T. Jui, Radical Chain Reduction via Carbon Dioxide Radical Anion (CO2*−), J. Am. Chem. Soc., 2021, 143, 8987–8992 CrossRef CAS.
- S. N. Alektiar and Z. K. Wickens, Photoinduced Hydrocarboxylation via Thiol-Catalyzed Delivery of Formate Across Activated Alkenes, J. Am. Chem. Soc., 2021, 143, 13022–13028 CrossRef CAS PubMed.
- (a) J. C. Tellis, D. N. Primer and G. A. Molander, Single-electron Transmetalation in Organoboron Cross-coupling by Photoredox/Nickel Dual Catalysis, Science, 2014, 345, 433–436 CrossRef CAS PubMed; (b) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Merging Photoredox with Nickel Catalysis: Coupling of α-Carboxyl sp3-Carbons with Aryl Halides, Science, 2014, 345, 437–440 CrossRef CAS; (c) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed; (d) C. K. Prier, D. A. Rankic and D. W. MacMillan, Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (e) K. L. Skubi, T. R. Blum and T. P. Yoon, Dual Catalysis Strategies in Photochemical Synthesis, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- (a) M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Photocatalytic decarboxylative alkylations mediated by triphenylphosphine and sodium iodide, Science, 2019, 363, 1429–1434 CrossRef CAS PubMed; (b) J. X. Wang, W. Ge, M. C. Fu and Y. Fu, Photoredox-Catalyzed Allylic Defluorinative Alkoxycarbonylation of Trifluoromethyl Alkenes through Intermolecular Alkoxycarbonyl Radical Addition, Org. Lett., 2022, 24, 1471–1475 CrossRef CAS.
- (a) Y. N. Wu, M. C. Fu, R. Shang and Y. Fu, Nickel-catalyzed carboxylation of aryl iodides with lithium formate through catalytic CO recycling, Chem. Commun., 2020, 56, 4067–4069 RSC; (b) M.-C. Fu, R. Shang, W.-M. Cheng and Y. Fu, Nickel-Catalyzed Regio- and Stereoselective Hydrocarboxylation of Alkynes with Formic Acid through Catalytic CO Recycling, ACS Catal., 2016, 6, 2501–2505 CrossRef CAS.
- Y. Huang, J. Hou, L.-W. Zhan, Q. Zhang, W.-Y. Tang and B.-D. Li, Photoredox Activation of Formate Salts: Hydrocarboxylation of Alkenes via Carboxyl Group Transfer, ACS Catal., 2021, 11, 15004–15012 CrossRef CAS.
- I. Colon and D. R. Kelsey, Coupling of Aryl Chlorides by Nickel and Reducing Metals, J. Org. Chem., 1986, 51, 2627–2637 CrossRef CAS.
- (a) C. S. Elmore and R. A. Bragg, Isotope chemistry; a useful tool in the drug discovery arsenal, Bioorg. Med. Chem. Lett., 2015, 25, 167–171 CrossRef CAS; (b) D. U. Nielsen, K. T. Neumann, A. T. Lindhardt and T. Skrydstrup, Recent developments in carbonylation chemistry using [13C]CO, [11C]CO, and [14C]CO, J. Labelled Compd. Radiopharm., 2018, 61, 949–987 CrossRef CAS; (c) P. Hermange, A. T. Lindhardt, R. H. Taaning, K. Bjerglund, D. Lupp and T. Skrydstrup, Ex situ generation of stoichiometric and substoichiometric 12CO and 13CO and its efficient incorporation in palladium catalyzed aminocarbonylations, J. Am. Chem. Soc., 2011, 133, 6061–6071 CrossRef CAS PubMed.
- S. D. Friis, T. L. Andersen and T. Skrydstrup, Palladium-Catalyzed Synthesis of Aromatic Carboxylic Acids with Silacarboxylic Acids, Org. Lett., 2013, 15, 1378–1381 CrossRef CAS.
- K. Shimomaki, K. Murata, R. Martin and N. Iwasawa, Visible-Light-Driven Carboxylation of Aryl Halides by the Combined Use of Palladium and Photoredox Catalysts, J. Am. Chem. Soc., 2017, 139, 9467–9470 CrossRef CAS.
- L. Guo, F. Song, S. Zhu, H. Li and L. Chu, syn-Selective alkylarylation of terminal alkynes via the combination of photoredox and nickel catalysis, Nat. Commun., 2018, 9, 4543 CrossRef PubMed.
- (a) C. Zhu, H. Yue, L. Chu and M. Rueping, Recent advances in photoredox and nickel dualcatalyzed cascade reactions: pushing the boundaries of complexity, Chem. Sci., 2020, 11, 4051–4064 RSC; (b) O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, Nickel-Catalyzed Cross Coupling of Photoredox Generated Radicals: Uncovering a General Manifold for Stereoconvergence in Nickel-Catalyzed Cross-Couplings, J. Am. Chem. Soc., 2015, 137, 4896–4899 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental protocols and spectral data. See DOI: https://doi.org/10.1039/d2qo01361d |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2023 |