
Synthesis and application of mixed-spinel magnesioferrite: structural, vibrational, magnetic, and electrochemical sensing properties†
Aneeta
Manjari Padhan
a,
P.
Mary Rajaitha
a,
Sanjib
Nayak
b,
Sugato
Hajra
a,
Manisha
Sahu
a,
Zvonko
Jagličić
cd,
Primož
Koželj
ef and
Hoe Joon
Kim
 *ag
*ag
aDepartment of Robotics and Mechatronics Engineering, Daegu Gyeongbuk Institute of Science and Technology (DGIST), Daegu-42988, Republic of Korea. E-mail: joonkim@dgist.ac.kr
bDepartment of Physics, Indian Institute of Technology-Madras, Tamilnadu-600036, India
cFaculty of Civil and Geodetic Engineering, University of Ljubljana, Jamova 2, SI-1000 Ljubljana, Slovenia
dInstitute of Mathematics, Physics and Mechanics, Jadranska 19, SI-1000 Ljubljana, Slovenia
eJožef Stefan Institute, Jamova 39, SI-1000 Ljubljana, Slovenia
fFaculty of Mathematics and Physics, University of Ljubljana, Jadranska 19, SI-1000 Ljubljana, Slovenia
gRobotics and Mechatronics Research Center, Daegu Gyeongbuk Institute of Science and Technology (DGIST), Daegu-42988, Republic of Korea
First published on 18th October 2022
Abstract
This article presents an efficient non-enzymatic electrochemical sensor based on catalytic oxidation by the MgFe2O4 magnetic spinel for the sensitive determination of ascorbic acid. MgFe2O4 spinel ferrite is synthesized via the simple and cost-effective solid-state reaction route. X-ray photoelectron spectroscopy and X-ray diffraction studies reveal a mixed spinel structure of the synthesized material with the formula (Mg0.65Fe0.35)A[Fe1.65Mg0.35]BO4 with Fe3+ and Mg2+ occupying both the tetrahedral and octahedral sublattices. The Raman and Fourier-transform infrared spectroscopic analyses confirm the spinel structure formation. The DC magnetic measurements reveal a soft-magnetic behavior of the material due to the cationic distribution in the tetrahedral and octahedral sublattices. A maximum magnetization of 33.3 emu g−1 at 70 kOe is obtained for the hysteresis loop measurement, which is performed at a temperature of 10 K. Furthermore, the magnesioferrite spinel integrated glassy carbon electrode displays an enhanced catalytic activity toward ascorbic acid compared to the bare electrode in the phosphate buffer solution of pH 7.4 owing to the mixed-valence cationic states in the spinel ferrite. The electrochemical performance of the modified electrode under the influence of various parameters such as scan rate, analyte concentration, and interference are studied in detail. The sensor provides a linear increase in the oxidation peak current as a function of increasing concentration with a limit of detection and quantification of 24.09 μM and 80.30 μM, respectively. The synthesized ferrite shows good selectivity toward interfering agents, such as potassium chloride, sulfuric acid, hydrogen peroxide, sodium hydroxide, glucose, and choline chloride.
1. Introduction
Ascorbic acid (AA) or vitamin C is an effective antioxidant and reducing agent which plays a significant role in essential enzymatic reactions and precluding radical-induced disorders. It performs various biochemical activities like gene expression, cell differentiation, normal tissue growth, collagen formation, iron retention, and recuperation of wounds. The standard AA concentration in the human body ranges between 0.6 and 2 mg dL−1, and many diseases are associated with AA concentration changes.1,2 Its deficiency leads to scurvy, rheumatoid arthritis, Parkinson's disease, and even cancer, whereas excessive intake of AA causes impaired kidney functions and stomach irritation. Therefore, AA's quantitative and accurate determination is crucial for healthcare.3 In this context, the cost-effectiveness, fast response, and continuous real-time detection that are associated with the electrochemical instrumentation for AA detection advocate a prime advantage. AA electrochemical detection can be performed in two possible ways, (i) enzymatic and (ii) non-enzymatic.4–6 Among these two, the enzymatic sensors have major disadvantages due to the high cost, limited working conditions, and lack of reproducibility. On the other hand, non-enzymatic detection holds significance due to its stability and high sensitivity.7 Non-enzymatic detection involves the direct oxidation of AA to form Dehydro-L-ascorbic acid as shown in the following reaction:| C6H8O6 = C6H6O6 + 2H+ + 3e− | (1) |
Furthermore, these electrochemical techniques often suffer in real-time applications due to their low selectivity and sensitivity when using bare electrodes.8 Hence, various materials such as functionalized molecules, metals, oxides and composites have been reported to modify the electrodes for enhancing the selectivity and sensitivity toward the detection of AA. Among these, metal oxides act as efficient electrode modifiers due to their superior redox properties, chemical stability, and cost-effectiveness that facilitate the fast, accurate, and increased electron kinetics required for electrochemical sensing.9
In this regard, spinel ferrites are known materials used in electrochemical sensing due to their outstanding accuracy and ability to detect analytes even at trace levels.10 The spinels (denoted as AB2X4) typically crystallize in the hexagonal closed-packed cubic unit cell with space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, where cations are distributed between the tetrahedrally coordinated A site and octahedrally coordinated B-site. Due to their intricate structures with the presence of cations with multiple oxidation states, and tunable magnetic and electronic properties, spinels find potential applications in spintronics, new-generation electronics, energy conversion, catalysis, and sensor technologies.11–16 The spinel ferrites have superior properties such as a higher surface area and porosity in their structure. They are reported to act as electrode-modifying materials because they mediate electron-transfer reactions of electroactive species in solution.17 Due to their good chemical stability, several ferrites have been used for the electrochemical detection of AA. D. Navadeepthy et al.18 developed nano-sized NiFe2O4–PANI as a duo-active electrocatalyst toward the non-enzymatic sensing of AA. Similarly, MnFe2O4@CNT-N was synthesized by Diana M. Fernandes et al.7 through an in situ coprecipitation method, coated over a GCE surface, and used for the simultaneous detection of caffeine, acetaminophen, and ascorbic acid. Phan Thi Kim Thu et al.19 used the hydrothermal method to prepare CoFe2O4 to study their electrochemical activity toward the simultaneous sensing of ascorbic acid, acetaminophen, and caffeine by the voltammetric method. Nevertheless, despite its various physical and chemical properties, the potential of alkaline-earth ceramics in electrochemical sensing of biomolecules and organic pollutants remains unacquainted.
m, where cations are distributed between the tetrahedrally coordinated A site and octahedrally coordinated B-site. Due to their intricate structures with the presence of cations with multiple oxidation states, and tunable magnetic and electronic properties, spinels find potential applications in spintronics, new-generation electronics, energy conversion, catalysis, and sensor technologies.11–16 The spinel ferrites have superior properties such as a higher surface area and porosity in their structure. They are reported to act as electrode-modifying materials because they mediate electron-transfer reactions of electroactive species in solution.17 Due to their good chemical stability, several ferrites have been used for the electrochemical detection of AA. D. Navadeepthy et al.18 developed nano-sized NiFe2O4–PANI as a duo-active electrocatalyst toward the non-enzymatic sensing of AA. Similarly, MnFe2O4@CNT-N was synthesized by Diana M. Fernandes et al.7 through an in situ coprecipitation method, coated over a GCE surface, and used for the simultaneous detection of caffeine, acetaminophen, and ascorbic acid. Phan Thi Kim Thu et al.19 used the hydrothermal method to prepare CoFe2O4 to study their electrochemical activity toward the simultaneous sensing of ascorbic acid, acetaminophen, and caffeine by the voltammetric method. Nevertheless, despite its various physical and chemical properties, the potential of alkaline-earth ceramics in electrochemical sensing of biomolecules and organic pollutants remains unacquainted.
Among several spinel ferrites, MgFe2O4 is of particular interest, which in the bulk state shows ferrimagnetism and crystallizes in a cubic structure. MgFe2O4 experimentally forms an intermediate spinel whose site occupancy can be emphasized as [Mg1−x2+Fex3+]tet[Mgx2+Fe2−x3+]octO4. The value of x in MgFe2O4 is a complex function of processing parameters, which impacts the overall physical and chemical properties of the material. It is reported to have a minimal cytotoxic effect, ease of fabrication, cost-effectiveness, superior chemical stability, high capacity, and biocompatibility, making this ferrite an excellent candidate for electrochemical sensing.20 Therefore, MgFe2O4 and its composites have been studied for various electrochemical sensing applications, such as (i) MgFe2O4 prepared by a solution-based method for electrochemical investigation of dopamine;21 (ii) MgFe2O4 synthesized by a coprecipitation route for electrochemical detection of morphine and diclofenac;22 (ii) MgFe2O4 nanoparticles prepared by a facile nitrate-sucrose decomposition method for trace level detection of mercury(II) from wastewater samples;23 (iii) hydrothermally synthesized MgFe2O4 anchored on reduced graphene oxide to detect the environmentally hazardous 4-cyanophenol;24 (iv) CTAB@MgFe2O4 nanocomposite prepared using a sol-gel route for glucose and H2O2 sensing;25 (v) MoS2@MgFe2O4 composites synthesized via the hydrothermal process for promising colorimetric detection of glucose and H2O2.26 The majority of these reported studies have been performed by time-consuming, expensive methods, follow complicated procedures, and provide less yield. Hence, the conventional ceramic method27 can be advantageous owing to its simple operation, low cost, high yield, and bulk production. Also, though previous works prove that MgFe2O4 can be used as a sensor to detect various electrolytes needed for significant biomedical applications, no considerable reports have been provided for the electrochemical determination of AA using MgFe2O4.
Therefore, this manuscript investigates the electrochemical sensing properties of MgFe2O4 spinel toward AA detection. Several physio-chemical properties like structural, spectroscopic, and magnetic characterizations have also been studied to analyze the as-prepared material. The MgFe2O4 sample was drop-cast to modify the glassy carbon electrode (GCE) in a binder-free protocol and subsequently used for AA detection through the cyclic-voltammetric (CV) approach. The electrochemical response of the material toward AA oxidation was studied for different concentrations, which showed good selectivity toward AA in the presence of various interferent molecules in phosphate buffer.
2. Materials and methods
Polycrystalline MgFe2O4 was prepared by the conventional solid-state reaction route from the corresponding raw and high-purity binary oxides MgO and Fe2O3 procured from Daejung chemicals, Korea. Stoichiometric mixtures of the oxides were weighed in proportion to the desired composition, mixed and homogenized (thoroughly ground) by an agate mortar and pestle using methanol as a grinding media. The ground powder was calcinated at 1100 °C for 5 hours at a 5 °C minute−1 heating rate in a box furnace to yield the desired product. The schematic depiction of the synthesis methodology is provided in Fig. 1(a). | ||
| Fig. 1 Schematic representation of the synthesis methodology of MgFe2O4 spinel (a); XPS survey spectra (b); XPS high-resolution spectra of Mg-1s (c) Fe-2p (d) and O-1s (e) C-1s; elements (f); room-temperature XRD pattern and the Rietveld refinement analysis of the synthesized MgFe2O4 powder (g); the crystal structure of the mixed spinel MgFe2O4 drawn using VESTA software (h). | ||
The structural information (crystal structure and phase identification/purity) of the synthesized material was characterized using the Empyrean X-ray diffractometer (XRD) instrument (model: Malvern Panalytical, Netherland) with Cu Kα1 (λ = 1.5406 Å) radiation operated at 40 kV and 30 mA. The powder XRD data of the sample was examined in the measurement range of 15° ≤ 2θ ≤ 70° scanned at a step size (Δ2θ) of 0.02° and rate of 0.005° s−1 in the Bragg configuration. The obtained diffraction pattern was refined with the FULLPROF suite program for Rietveld refinement. The surface morphology of the sample was analyzed using a Field-Emission Scanning Electron Microscope (FESEM, SU-8230, Hitachi, Japan). The presence of various elements, elemental mappings, and elemental composition ratio was studied with the help of an energy-dispersive X-ray detector (EDX) unit attached to the FESEM equipment. The compound's chemical composition and electronic structure were probed using X-ray photoelectron spectroscopy (XPS). For the XPS measurement, a Thermo Scientific make Escalab 250Xi instrument equipped with an X-ray source of Al Kα (1486.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) eV) capable of attaining ultrahigh vacuum (∼10−7mbar) was employed. The XPS measurement was performed in the constant analyzer energy (CAE) mode with a pass energy of 20 eV at an energy step of 0.1 eV and a dwell time of 50 ms per data point. The photo-excited electrons were examined over a broad binding energy range of 0–1400 eV. The binding energy calibration for the XPS spectra was done using the binding energy of carbon C-1s orbital (284.5 eV) as the internal reference. The optical absorption in the far-infrared region was measured for the as-prepared sample using a Nicolet Continuum FT-IR spectrometer (ThermoFisher Scientific, USA). The room-temperature Raman spectrum of the ferrite sample was recorded by a Nicolet Almega XR dispersive Raman spectrometer (Thermo Scientific, U.S.) using a 532 nm laser in the frequency range of 100–1000 cm−1. The field- and temperature-dependent dc magnetization measurements were performed employing a Magnetic Property Measurement System (Model: Quantum Design, MPMS3) within 10 K to 300 K. The porous features were measured using an N2 adsorption-desorption measurement tool (Quantachrome Autosorb-iQ2, USA). The samples were kept for degassing at 200 °C for more than 12 hours before the measurement was undertaken.
eV) capable of attaining ultrahigh vacuum (∼10−7mbar) was employed. The XPS measurement was performed in the constant analyzer energy (CAE) mode with a pass energy of 20 eV at an energy step of 0.1 eV and a dwell time of 50 ms per data point. The photo-excited electrons were examined over a broad binding energy range of 0–1400 eV. The binding energy calibration for the XPS spectra was done using the binding energy of carbon C-1s orbital (284.5 eV) as the internal reference. The optical absorption in the far-infrared region was measured for the as-prepared sample using a Nicolet Continuum FT-IR spectrometer (ThermoFisher Scientific, USA). The room-temperature Raman spectrum of the ferrite sample was recorded by a Nicolet Almega XR dispersive Raman spectrometer (Thermo Scientific, U.S.) using a 532 nm laser in the frequency range of 100–1000 cm−1. The field- and temperature-dependent dc magnetization measurements were performed employing a Magnetic Property Measurement System (Model: Quantum Design, MPMS3) within 10 K to 300 K. The porous features were measured using an N2 adsorption-desorption measurement tool (Quantachrome Autosorb-iQ2, USA). The samples were kept for degassing at 200 °C for more than 12 hours before the measurement was undertaken.
A CS150 electrochemical workstation (CorrTest Instruments, China) was used to conduct electrochemical measurements for electrochemical sensing purposes. The sample preparation was started using alumina slurry to polish the GCE. Then the electrode was washed using distilled water and subsequently dried. The sample was drop-cast to modify the GCE. MgFe2O4 solution was prepared by dispersing 2 mg of MgFe2O4 in 1 mL of distilled water. A 10 μL of the prepared solution was then drop-cast on the electrode and dried at room temperature. The CV measurement was performed in a three-electrode configuration where MgFe2O4 modified GCE, platinum, and Ag/AgCl acted as the working, counter, and reference electrodes, respectively.
3. Results and discussion
XPS is a powerful spectroscopic technique for quantitatively analyzing existing chemical elements and their valence states in a material. Fig. 1(b) depicts the photoelectron intensity versus binding energy plot of the synthesized MgFe2O4 sample. From the XPS measurement performed over a wide binding energy range of 0–1400 eV, the presence of elemental Mg, Fe cations, and O anions is identified. The C-1s line in the spectrum comes from the hydrocarbon contamination introduced by the laboratory conditions. Hence, the observed C-1s line in the spectrum was considered the energy reference to determine the core-level spectral positions of the individual elements. The individual peak positions of the Mg-2p, Mg-1s, Fe-2p, O-1s, and C-1s core-level peaks are indexed in the figure. Furthermore, the peaks attained at ∼305 eV and 349 eV can be allocated to the Mg KLL peaks.28 The O KLL peak is observed at 975.8 eV.29 The Auger (KLL) peaks occupying the specified positions in the photoelectron energy scale are produced by the emitted Auger electrons, which come from the relaxation of the excited ions. Furthermore, the high-resolution XPS spectra of individual elements of MgFe2O4 were analyzed to obtain information on the degree of oxidation and electron states. The peaks were fitted using the commercial XPSPEAK 4.1 software by varying the peak positions, the number of peaks, Lorentzian-Gaussian ratios, FWHM, etc.Fig. 1(c)–(f) show the high-resolution scan of the Mg-1s, Fe-2p, O-1s, and C-1s elemental spectra, respectively, depicting their chemical or bonding states. Fig. 1(c) presents the Mg-1s core level, exhibiting a broad peak at 1302.6 eV. The XPS spectrum was further fitted using a Gaussian-Lorentzian peak function, which shows the deconvolution of the peak into two prominent peaks at 1302.5 eV and 1303.9 eV. In general, when cations are distributed in two different sites in ferrites, we expect the existence of two peaks, peak shifting or peak broadening in the XPS spectrum. Therefore, the two observed peaks in the Mg-1s spectrum can be ascribed to the Mg cation distribution in the tetrahedral and octahedral sites. Hence, the high binding energy peak at 1303.9 eV can be related to the octahedral Mg cations, whereas the low energy peak at 1302.5 eV corresponds to the Mg cations present at the tetrahedral sites.30Fig. 1(d) shows the Fe-2p core-level XPS spectrum. Based on the spin-orbit coupling, the Fe-2p orbital splits into the Fe-2p3/2 and Fe-2p1/2 doublet.31 Two characteristic peaks corresponding to the Fe-2p3/2 ∼ 710.6 eV and Fe-2p1/2 ∼ 724.3 eV are observed in the spectrum, along with four satellite peaks at ∼714.8, 719, 732.6, and 741.4 eV, respectively. The binding energy difference or the spin-energy separation between the Fe-2p3/2 and Fe-2p1/2 levels, i.e., ΔE2p3/2–2p1/2 ∼ 13.7 eV corroborates the presence of the trivalent Fe oxidation state in MgFe2O4.32 The fitting analysis using the Gaussian-Lorentzian peak function shows the deconvolution of the major Fe peaks into individual doublets, indicating more than one chemical state. The Fe-2p3/2 peak deconvolutes into two peaks centered at D1 = 710.3 eV and D2 = 712.2 eV. Also, the Fe-2p1/2 peak deconvolutes into two peaks labelled at D3 = 723.8 eV and D4 = 725.5 eV, respectively. The lower binding energy (D1 and D3) peaks of the Fe-2p3/2 and Fe-2p1/2 lines, respectively, are ascribed to the Fe3+ ions situated at the B-sites sublattice, whereas the D2 and D4 peaks can be assigned to the Fe3+ cations present at the A-sublattice of the spinel ferrite, respectively.33,34Fig. 1(e) shows the O-1s spectrum of MgFe2O4, which further splits into three peaks centered at 529.6 eV, 531.4 eV, and 533.4 eV. The peak located at the binding energy of 529.6 eV can be assigned to the lattice oxygen (M–O). It corresponds to the oxygen atoms bound to the Mg and Fe atoms (O–Mg/O–Fe). The peak observed at 531.4 eV represents the existing metal hydroxides or hydroxyl groups (denoted as OH−) present in the material. The third peak at 533.4 eV corresponds to the physically or chemically adsorbed water.35 The C-1s spectra (Fig. 1(f)) could be fitted to three different carbon contributions centered at 284.5 eV, 286.2 eV, and 288.4 eV, which can be associated with the presence of various adventitious carbon species. Furthermore, depending on the percentage of occupancy in both the sublattices, we have determined that the synthesized MgFe2O4 is forming a mixed spinel structure with the chemical formula (Mg0.65Fe0.35)A[Fe1.65Mg0.35]BO4.
Furthermore, the octahedral and tetrahedral site occupancies obtained from the XPS analysis are taken as the initial parameters for the Rietveld refinement of the powder X-ray diffraction patterns to obtain a more detailed structural analysis. Fig. 1(g) shows the room-temperature powder XRD pattern of the polycrystalline (Mg0.65Fe0.35)A[Fe1.65Mg0.35]BO4 spinel-ferrite along with the analyzed Rietveld refinement least-square technique performed using the FULLPROF software package. The background profile was modeled during the refinements using a Chebychev polynomial, and the peaks were fitted with a pseudo-Voight peak-shape function. The lattice parameters, positional coordinates, zero-point correction, scale factor, isotropic thermal parameters, etc., were refined to determine the crystal structure and phase of the sample. The XRD pattern along with the Rietveld refinement displayed in Fig. 1(g), the experimental data (Yobs) are indicated by open circles, the solid red lines present the calculated patterns (Ycal), and the difference between Yobs and Ycal is shown at the bottom of the curve via solid blue lines. Vertical marks represent the Bragg positions just under the XRD profile. From the refinement results, it is observed that the synthesized (Mg0.65Fe0.35)A[Fe1.65Mg0.35]BO4 sample belongs to the family of spinels, crystallizing in a cubic structure (space group Fdm; number 227). The corresponding structural parameters like the lattice parameters, atomic coordinates, and isothermal parameters are summarized in Table 1. The above results confirm the successful preparation of the magnesioferrite sample with high crystallinity. The corresponding unit cell was drawn using commercial VESTA software, as depicted in Fig. 1(h). However, we observed the formation of a small amount of Fe3O4 as the secondary phase in the synthesized material. The structural parameters and the approximated phase percentages are provided in Table 1.
| Atoms | x | y | z | Uiso (Å2) | Occ. |
|---|---|---|---|---|---|
| Phase-I: MgFe2O4; a = b = c = 8.3872 ± 0.0012 Å, α = β = γ = 90°; Fdm; phase percentage = 90.61%; Rp = 4.6, Rwp = 9.3, χ2 = 1.11 |
|||||
| Fe1 | 0.0 | 0.0 | 0.0 | 0.0119(4) | 0.825 |
| Mg1 | 0.0 | 0.0 | 0.0 | 0.0119(4) | 0.175 |
| O | 0.2646(3) | 0.2646(3) | 0.2646(3) | 0.094(3) | 1 |
| Mg2 | 0.375 | 0.375 | 0.375 | 0.00824(6) | 0.65 |
| Fe2 | 0.375 | 0.375 | 0.375 | 0.00824(6) | 0.35 |
| Phase-II: Fe3O4; a = b = c = 8.3959 ± 0.0011 Å, α = β = γ = 90°; Fdm; phase percentage = 9.39% |
|||||
| Fe1 | 0.375 | 0.375 | 0.375 | 0.0229(4) | 1 |
| Fe2 | 0.0 | 0.0 | 0.0 | 0.02412(4) | 1 |
| O | 0.2515(3) | 0.2515(3) | 0.2515(3) | 0.089(3) | 1 |
Furthermore, the crystallite size-strain parameters of the MgFe2O4 sample are also obtained by using the size-strain plot, which considers peaks in the average range and gives less weight to the data from reflections at higher angles where the precision is usually lower.
Accordingly, we have,36
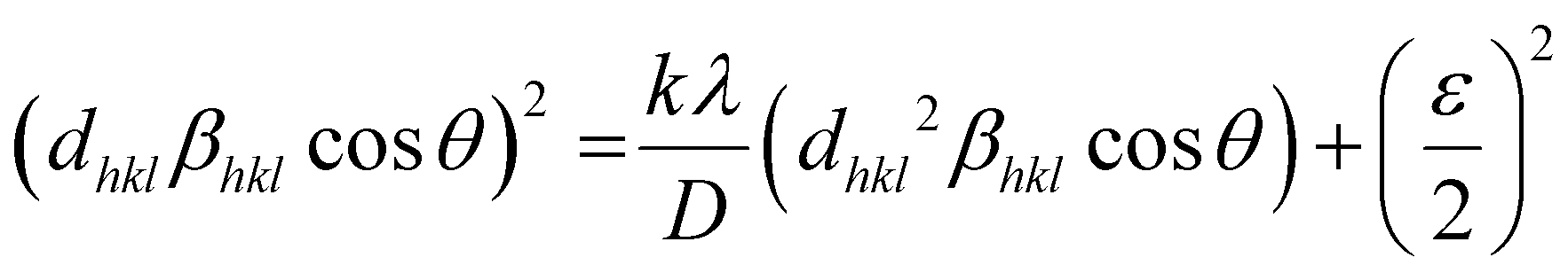 | (2) |
θ)2 is plotted as a function of (dhkl2βhklcosθ), as shown in Fig. S1 (ESI†). The estimated values of D = 61.5 nm and ε = 0.000338 are obtained from the slope of the linearly fitted data and the root of the y-intercept, respectively.
Furthermore, the particle morphology recorded by the FESEM analysis is displayed in Fig. 2(a). The micrograph shows the well-developed and irregular polyhedral grains inhomogeneously distributed throughout the microstructure. The individual elemental mappings illustrate a nearly homogeneous distribution of Mg, Fe, and O, confirming the formation and phase purity of the material, as shown in Fig. 2(b)–(d). Fig. 2(e) shows the EDS compositional analysis illustrating the Mg, Fe, and O spectral lines. The relative concentrations of the requisite elements attained from the measured EDS spectra are provided in the inset of Fig. 2(e) and assent well with the powder XRD results. Fig. S2 (ESI†) shows the nitrogen adsorption–desorption isotherms and pore size distribution curve (inset) of MgFe2O4. The isotherm depicts a type IV adsorption isotherm as per the International Union of Pure and Applied Chemistry classification, which suggests the presence of a mesoporous structure. An average pore diameter of 33.288 nm and an average surface area of 82.7 m2 g−1 were observed.
 | ||
| Fig. 2 (a) FESEM image; (b)–(d) Elemental mappings of the individual elements present in the MgFe2O4 sample; (e) EDS spectral result; (f) Raman spectrum at room-temperature; and (g) FT-IR spectra at room temperature. | ||
Raman spectroscopy is a recognized non-destructive technique for exploring the structural properties of oxides, such as phase transitions and subtle distortions, by probing their vibrational modes. It also provides information about local heterogeneities associated with structural and chemical modifications due to the phonons’ shorter coherence length and time scale. Therefore, the room temperature Raman spectrum of MgFe2O4 is recorded in the wavenumber region of 150–1000 cm−1, as shown in Fig. 2(f). As stated above, the MgFe2O4 crystal is a cubic spinel with Fdm space group symmetry and its unit cell comprises 56 atoms (Z = 8). However, its smallest Bravais cell, i.e., the primitive cell contains only 14 atoms (Z = 2). Hence, the group theory analysis of Fdm crystal symmetry can have optical phonon modes at the Brillouin-zone center Γvib (![[k with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_006b_20d1.gif) = 0) for each atomic displacement in the structural primitive cell denoted by the irreducible representation:37
= 0) for each atomic displacement in the structural primitive cell denoted by the irreducible representation:37
| Γvib = A1g + Eg + F1g + 3F2g + 2A2u + 2Eu + 4F1u + 2F2u | (3) |
Among these modes, the factor group analysis predicts the appearance of only five Raman active modes (A1g + Eg + 3F2g) under ambient conditions. However, we observed seven distinguished Raman modes through a peak fitting performed using the Lorentzian peak function. The Raman modes are centered at ∼215, 331, 380, 482, 548, 658, and 708 cm−1. Similar spectroscopic behavior has also been reported by Wang et al.37 and Nakagomi et al..38 Based on the analysis of Z. Wang and co-workers, the A1g mode in the spectrum is ascribed to the order-disorder effect between the Mg2+ and Fe3+ metal ions.37 Contrastingly, Nakagomi et al. suggested that substituting Fe with Mg ions at the tetrahedral sites results in the Raman A1g mode. Based on their study, the splitting of the A1g mode into a doublet-like peak happens due to the mass difference between the Fe3+ and Mg2+ ions (due to the different ionic radii of Fe3+ and Mg2+ ions). The A1g mode present at a higher wavenumber accounts for the vibrations of oxygen anions around Mg2+ cations, whereas the peak at the lower wavenumber side corresponds to the vibrations of oxygen anions around Fe3+ cations at the tetrahedral sites.38
Furthermore, a prominent A1g Raman mode is observed in cubic ferrites corresponding to the vibration of tetrahedral AO4 in the wavenumber region of the 600–720 cm−1 region, while the low-frequency Raman modes below 600 cm−1 are observed due to the vibration of the octahedral BO6 sublattice.39 A similar spectroscopic behavior is observed for the present (Mg0.65Fe0.35)A[Fe1.65Mg0.35]BO4 spinel.40,41 Therefore, the observed Raman modes for the prepared (Mg0.65Fe0.35)A[Fe1.65Mg0.35]BO4 spinel-ferrite can be described as follows: (i) the modes present at A1g at 708 cm−1 and its shoulder peak A1g,sh at 658 cm−1 relate to the symmetric stretching of oxygen atoms along the Mg2+–O2− and Fe3+–O2− bonds at the tetrahedral site; (ii) the F2g (3) Raman mode present at 548 cm−1 is associated with the asymmetric bending of oxygen; (iii) the F2g (2) band present at 482 cm−1 corresponds to the asymmetric stretching of Fe3+–O2− and Mg2+–O2− bonds at the octahedral site; (iv) the Eg mode at 331 cm−1 is associated with the symmetric bending of oxygen; and (v) the F2g (1) band present at 215 cm−1 is related to the translatory movement of the tetrahedral site.
FTIR is a significant spectroscopic method used to estimate the cubic spinel phase formation and probe the possible functional groups present in the material. Therefore, the FTIR analysis of the as-synthesized MgFe2O4 powder was performed in the wave number range of 400–4000 cm−1, and the spectrum is shown in Fig. 2(g). Two prominent absorption bands corresponding to the vibration of the tetrahedral and octahedral complexes are observed at ∼562 cm−1 and 440 cm−1 which indicate the formation of the spinel ferrite structure.42 The vibrational mode at 562 cm−1 is associated with the intrinsic stretching vibration of cations (Fe–O) of the tetrahedral site, whereas the mode at 440 cm−1 is correlated with the metal-oxygen vibration of the octahedral B-site. The higher wavenumber value of the tetrahedral cluster is indicative of the shorter metal-oxygen bond length of the tetrahedral site as compared to the octahedral site. Furthermore, the vibrational band ∼3429 cm−1 is also observed due to the stretching vibrational absorption of the hydroxyl functional group (O–H) of absorbed water on the surface of the sample, and the mode at ∼1631 cm−1 is attributed to the bending vibrations of the adsorbed water molecules.43 Hence, FTIR studies also confirmed the successful synthesis of MgFe2O4.
We have performed temperature- and field-dependent magnetic measurements to correlate the structural and magnetic properties. The magnetization measurements were taken with a Quantum Design Magnetic Property Measurement System (MPMS3) magnetometer at applied fields up to H = 7 T and temperatures between T = 10 K and T = 300 K. The temperature-dependent dc magnetic susceptibility, χ(T) =M/H, where M denotes the measured magnetization in an applied magnetic field H, was recorded under the zero-field-cooled (ZFC) and field-cooled (FC) protocols for two applied fields, i.e., H = 100 Oe and 1 kOe. For the ZFC susceptibility, the sample was first cooled down from room temperature to 10 K without any applied magnetic field. After reaching ∼10 K, the magnetization measurement was recorded under external magnetic fields of 100 Oe or 1 kOe during the warming process from 10 K to 300 K. Furthermore, the sample was cooled down from 300 K to 10 K in the presence of the same applied magnetic fields, and the magnetizations were again recorded during this cooling. The corresponding susceptibility data obtained for the ZFC and FC conditions under the applied fields of 100 Oe and 1 kOe are depicted in Fig. 3(a). The ZFC susceptibility in 100 Oe increases with increasing temperature, whereas the FC susceptibility increases upon lowering the temperature. A significant splitting between ZFC and FC susceptibilities measured in 100 Oe is observed at 10 K, which decreases upon increasing temperature. The ZFC and FC curves merge at the highest measured temperature (300 K), suggesting that the transition temperature from the paramagnetic to magnetically ordered phase is above room temperature. On the other hand, there is almost no splitting between ZFC and FC susceptibilities measured in the magnetic field of 1 kOe, suggesting a relatively soft magnetic behavior of the sample. The applied magnetic field of 1 kOe already suppresses any non-ergodicity and remanence of the spin system.
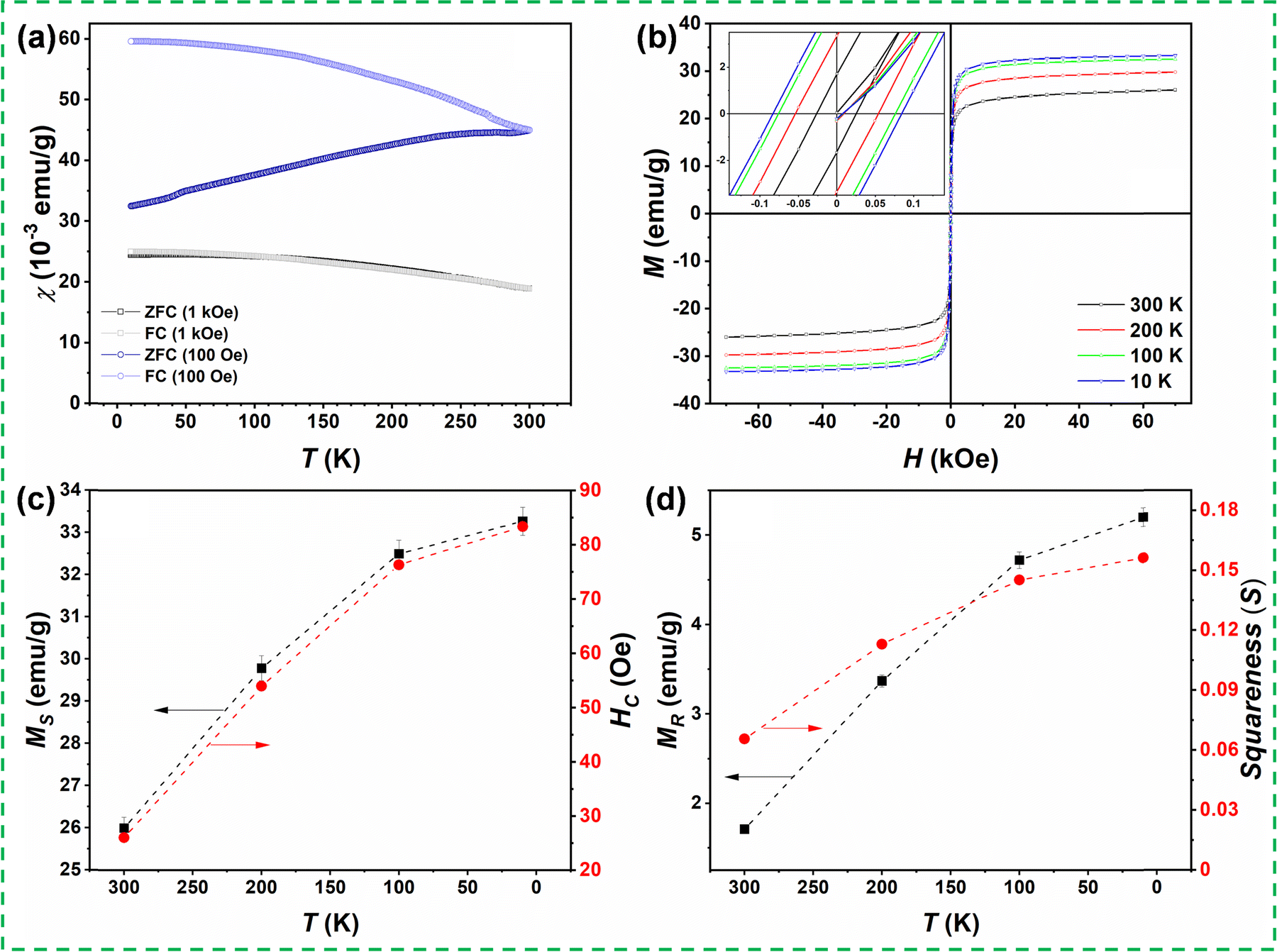 | ||
| Fig. 3 (a) Temperature-dependent susceptibility measured under zero-field-cooled (ZFC) and field-cooled (FC) conditions in 100 Oe and 1 kOe; (b) magnetization (M) versus magnetic field (H) hysteresis loops measured at 10 K, 100K, 200K, and 300K under ZFC conditions. The inset of the M–H loop shows the expanded view close to the origin; (c) saturation magnetization (MS) and coercivity (HC); (d) remanent magnetization (MR) and squareness ratio (S) obtained as a function of temperature. | ||
Fig. 3(b) shows the isothermal magnetization M(H) curves of the as-prepared bulk (Mg0.65Fe0.35)A[Fe1.65Mg0.35]BO4 spinel ferrite measured at different temperatures under the ZFC condition. The various parameters obtained from close analysis of the magnetic hysteresis loops, i.e., saturation magnetization (MS), coercivity (HC); remanent magnetization (MR), and the ratio of remnant magnetization to bulk saturation magnetization (known as Stoner–Wohlfarth ratio or magnetic squareness S) are elucidated in Fig. 3(c) and (d), respectively. The M(H) loops measured at different temperatures (300 K, 200 K, 100 K, and 10 K) show a narrow hysteresis behavior indicating the soft magnetic nature of the magnesioferrite material. The M(H) loops show two distinguished regimes: a nonlinear low field region and a linear high field region. The maximal saturation magnetization of MS = 33.3 emu g−1 was detected at 10 K and 70 kOe. This MS value and the observed M–H hysteresis behavior are in agreement with the expected ferrimagnetic structure and reported values for MgFe2O4 spinel ferrites.44,45 The magnetization in the M(H) loops decreases with increasing temperature. The values of MS and HC (shown in Fig. 3(c)) decrease from 33.3emu g−1 to 26emu g−1 and 83.4 to 26 Oe, respectively, as temperature increases from 10 K to 300 K. Meanwhile, the values of MR and squareness S (Fig. 3(d)) decrease from 5.2 emu g−1 to 1.7 emu g−1 and 0.156 to 0.065, respectively. The S-value is generally used to determine whether a magnetic material comprises single domain or multi-domain particles. In this context, S = 0.5 denotes the existence of single-domain particles, and S < 0.5 corresponds to the materials having multiple domains.46–48 Hence, the obtained value of S indicates the multi-domain nature of the synthesized ferrite. Furthermore, the observed coercivity values indicate the soft-magnetic nature of the (Mg0.65Fe0.35)A[Fe1.65Mg0.35]BO4 compound.49
The electrochemical performance of the MgFe2O4 modified GCE toward AA was studied using cyclic voltammograms in 0.1 M PBS (pH 7.4) solution with 0.1 mM AA at a scan rate of 50 mV s−1. Fig. 4(a) illustrates the preparation of the MgFe2O4 modified GCE for AA detection. Fig. 4(b) and (c) show the electrochemical response of the bare GCE and MgFe2O4 modified GCE. The GCE and the modified electrode exhibited an anodic peak current at 0.64 and 0.52 V, respectively, because of its reaction with AA. The modified GCE showed a smaller oxidation peak potential as compared to the bare GCE. The oxidation peak current of the bare and modified GCE was 1.006 μA and 1.062 μA, respectively. The modified GCE showed no characteristic peak in the absence of AA. The electrochemical response was recorded for different sample loadings (2, 4, and 6 mg mL−1), as shown in Fig. 4(d) and (e). The 2 mg mL−1 sample loading showed the highest electrochemical response and was used for further studies. The thickness of the MgFe2O4 on the GC was varied by increasing the amount of solution (10, 30, 50 μl) used to drop cast on the GC. Fig. S3 (ESI†) shows the CV obtained at various concentrations (10, 30, 50 μl) of MgFe2O4 modified GCE with 0.1 mM of ascorbic acid at 0.1 M PBS buffer (pH 7.4) at a scan rate of 50 mV s−1. It was observed that the peak current reduces with the increase in the amount of solution as well as the increased loading of MgFe2O4 on GC. The aforementioned reduction in the electrochemical activity while increasing the sample loading as well as the sample thickness on GC can be ascribed to the increased agglomeration of the MgFe2O4 particles.
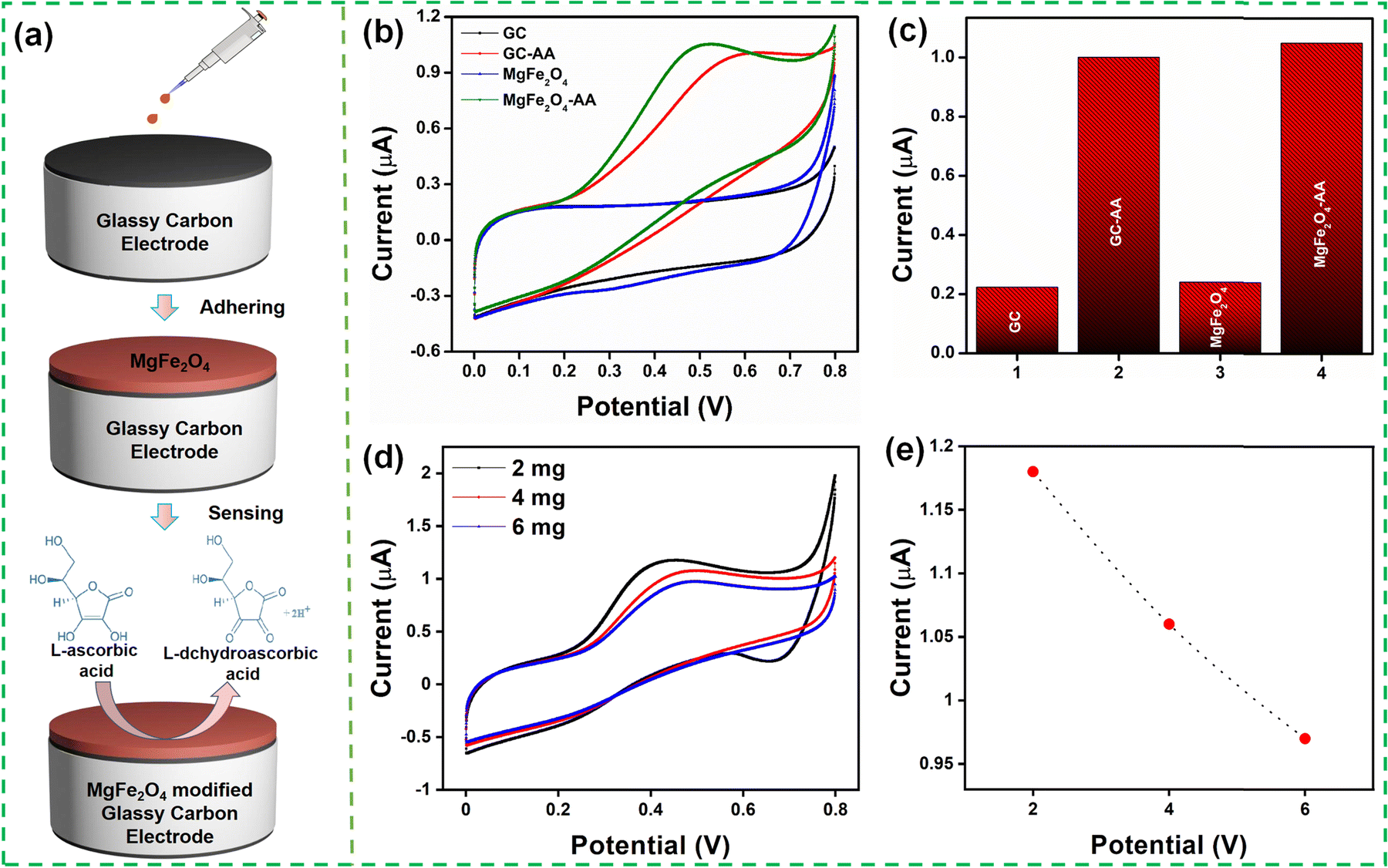 | ||
| Fig. 4 (a) Schematic representation of the preparation of the MgFe2O4 modified GCE for the detection of AA; (b) and (c) cyclic voltammograms of GCE, GCE with 0.1 mM of ascorbic acid, MgFe2O4 and MgFe2O4 with 0.1 mM of ascorbic acid at 0.1 M PBS buffer solution at a scan rate of 50 mV s−1; (d) and (e) cyclic voltammograms obtained for MgFe2O4 (2, 4, 6 mg ml−1) with 0.1 mM of ascorbic acid. | ||
Linear sweep voltammetry (LSV) was recorded for the modified GCE with different concentrations of AA in 0.1 M PBS (pH 7.4) solution at a scan rate of 50 mV s−1. The LSV was recorded for two sets of concentration ranges, 10–100 μM and 100–1000 μM (shown in Fig. 5(a) and (c)). The oxidation peak current increased with increasing concentration, indicating that the catalyst improved the oxidation of the AA in contact with the catalyst. Fig. 5(b) and (d) show the linear range obtained for the concentration ranging from 10 to 100 μM with the correlation coefficient R2 = 0.966 and from 100 to 1000 μM with the correlation coefficient R2 = 0.99. The linear regression for the AA content in the concentration range of 10–100 μM and 100-1000 μM was evaluated as Ipa (μA) = 1.918 + 0.112 (μM) and Ipa (μA) = 0.041 + 0.017 (μM), respectively. The limit of detection (LOD) was calculated using the formula 3SD/S and the limit of quantification (LOQ) was calculated from 10SD/S, where SD denotes the standard deviation, and S is the slope of the calibration curve. The LOD and LOQ were calculated to be 24.09μM and 80.30 μM, respectively. The sensitivity was calculated to be 1578 μA mM−1 cm−2 using the formula, sensitivity = slope of the plot/active surface area of the electrode.
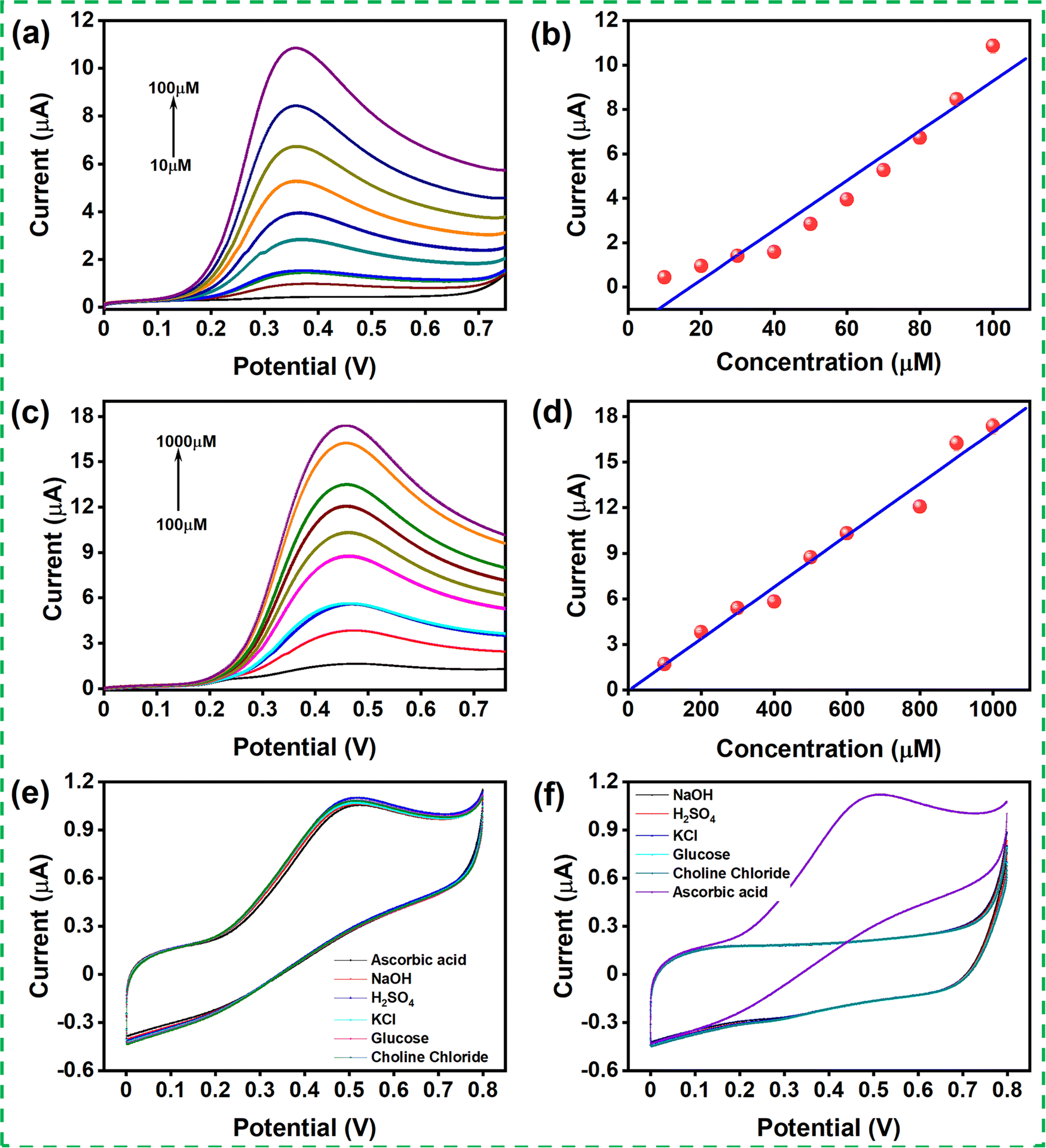 | ||
| Fig. 5 (a) Cyclic voltametric curves of MgFe2O4 modified GCE with various concentrations (10–100 μM) of ascorbic acid in 0.1 M PBS buffer (pH 7.4) at a scan rate of 50 mV s−1; (b) correlation between the peak currents obtained from ‘d’ and the concentration of ascorbic acid; (c) cyclic voltametric curves obtained for MgFe2O4 with various concentrations (100–1000 μM) of ascorbic acid at 0.1 M PBS buffer (pH 7.4) at a scan rate of 50 mV s−1; (d) correlation between the peak currents obtained from ‘f’ and the concentration of ascorbic acid; (e) cyclic voltammograms obtained for MgFe2O4 0.1M PBS (pH 7.4) with 0.1 mM of ascorbic acid initially added with different interferences with a 0.1 mM concentration, respectively; (f) MgFe2O4 0.1M PBS (pH 7.4) with 0.1 mM of ascorbic acid finally added with different interferences with 0.1 mM concentration, respectively. | ||
The selectivity of the MgFe2O4 modified GCE was examined in the presence of various interferent molecules such as potassium chloride, sulfuric acid, hydrogen peroxide, sodium hydroxide, glucose, and choline chloride. Fig. 5(e) and (f) show the LSV response of the modified GCE in the presence of various interferent molecules. The LSV response of the modified GCE upon adding 0.1 mM of AA and the successive addition of other interferent molecules was recorded. It was observed that there was no considerable change in the current response during the addition of the interferent molecules. Here, the interferent molecule was added to the PBS buffer in another set of experiments, and the LSV response was recorded. The modified GCE showed a negligible electrochemical response, although, upon the introduction of 0.1 mM AA, the modified electrode showed its oxidation response. The result indicates that the modified GCE has good selectivity toward AA even in the presence of other interferent molecules.
The effect of scan rate was examined by recording the electrochemical response of the modified GCE in 0.1 M PBS (pH 7.4) solution in the presence of 0.1mM AA with various scan rates from 10 to 500 mV s−1, as depicted in Fig. 6(a). The oxidation peak current gradually increased with scan rate, which indicates the catalytic activity of the prepared MgFe2O4. Fig. 6(b) shows the variation oxidation peak current as a function of the square root of scan rate with a linear regression of Ipa = 0.115 + 0.152 × (V)1/2 and the correlation coefficient R2 = 0.988. Fig. 6(c) shows the plot of Epvs. logV where Ep(V) = 0.308 + 0.083 × log(V) and the correlation coefficient R2 = 0.993. Both the oxidation peak current versus square root of the scan rate and the plot oxidation peak potential versus the log of scan rate plots show a linear behavior. The Laviron equation was used to calculate the charge transfer coefficient and electron transfer rate constant. The Laviron equation is as follows:50
 | (4) |
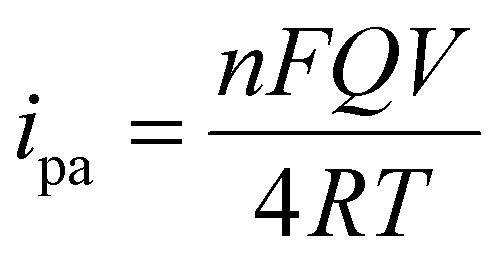 | (5) |
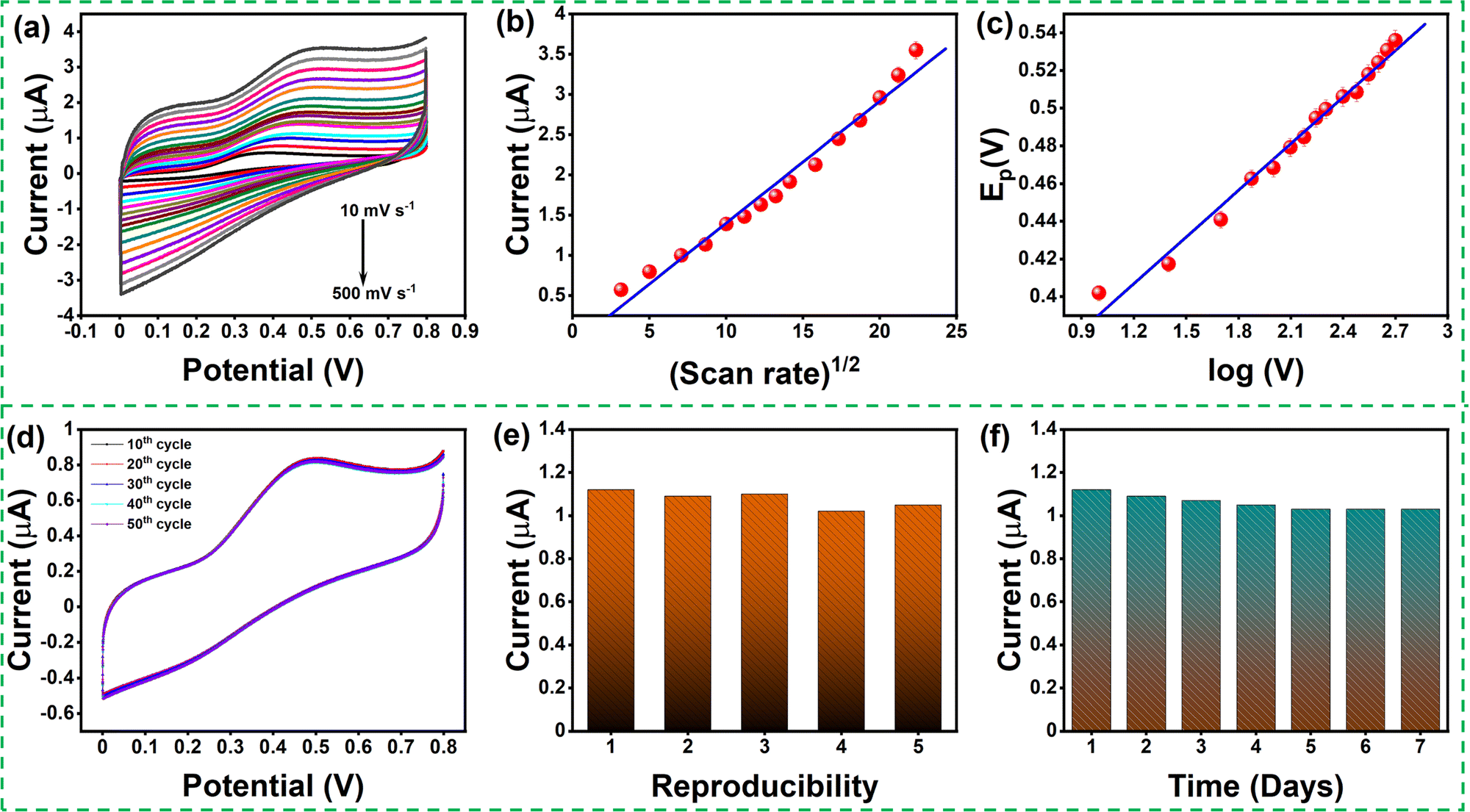 | ||
| Fig. 6 (a) Cyclic voltametric curves of MgFe2O4 in 0.1 M PBS buffer (pH 7.4) with 0.1 mM of ascorbic acid at different scan rates; (b) plot current versus (scan rate)1/2; (c) plot potential versus log (V); (d) MgFe2O4 modified GCE for 50 cycles; (e) MgFe2O4 modified GCE in a different coating; (f) MgFe2O4 on different days. | ||
The stability of the modified GCE was studied by running the CV response for 50 continuous cycles, and no substantial reduction in the oxidation peak current was noticed (Fig. 6(d)). In order to study the electrochemical response of ascorbic acid in the real sample using the MgFe2O4 modified GC, ascorbic acid sensing was studied in orange juice. Fig. S4 (ESI†) shows the LSV response obtained for MgFe2O4-modified GCE in a real sample (orange juice) with the addition of 200, 300, and 400 μM of ascorbic acid. The MgFe2O4 modified GCE showed a satisfactory recovery for the three different concentrations of ascorbic acid shown in Table S1 (ESI†).
Five different modified electrodes were fabricated to study the reproducibility of the modified electrode (Fig. 6(e)). The CV response was recorded for the five modified GCEs upon adding 0.1 mM of AA. The oxidation peak potential of the modified GCE did not show any significant changes, indicating that the modified GCE has good reproducibility. The stability of the modified GCE was studied by continuously analyzing it for seven days (Fig. 6(f)). The attained CV response showed a slight reduction in the oxidation peak current due to the continuous washing after sensing. The MgFe2O4 modified GCE exhibits an outstanding linear sensing range along with respectful measurement sensitivity and LOD compared to previously reported studies, as shown in Table 2.
| Material | Linear range (μM) | Sensitivity (μA mM−1 cm−2) | LOD (μM) | Ref. |
|---|---|---|---|---|
| CuO/rGO | 500–2000 | — | 189.053 | 51 |
| Bi2S3/rGO | 5.0–1200 | 268.8 | 2.9 | 52 |
| Au-MoS2/NiO | 2–50 | 2230 | 0.13 | 53 |
| RuO2/Au | 20–1000 | 342.8 | 11.6 | 54 |
| GO/TmPO4 | 100–1000 | 12.39 | 39 | 55 |
| TiO2-rGO | 25–725 | 1061 | 1.19 | 56 |
| Co3O4/nanoporous carbon | 2–240 | 130 | 0.02 | 57 |
| Mn-SnO2 | 1–900 | 10.92 | 0.058 | 58 |
| ZnO-Au | 100–4000 | 264.16 | 4.699 | 59 |
| NiFe2O4 | 0.7–11 | 37500 |
0.32 | 60 |
| MnFe2O4@CNT-N | 20–1000 | 330 | 1.8 | 7 |
| MnFe2O4/MoS2 | 200–1000 | 700 | 170 | 61 |
| CuGeO3 | 10–5000 | — | 24 | 62 |
| NiFe2O4 | 0.5–100 | — | 0.1 | 63 |
| NiFe2O4-PANI | 0.1–1.0 | — | 0.423 | 18 |
| MgFe 2 O 4 | 10–1000 | 1578 | 24.09 | This Work |
4. Conclusion
This work presents a detailed study of the structural and magnetic behavior of the MgFe2O4 spinel prepared by the cost-effective solid-state reaction method. XPS results and the Rietveld refinement analysis confirm that the prepared system belongs to the mixed spinel family (Mg0.65Fe0.35)A[Fe1.65Mg0.35]BO4 with a cubic crystal structure. The Raman and FT-IR spectroscopic analyses confirmed the successful formation of the magnesioferrite. The field- and temperature-dependent magnetic properties of the material show a robust soft-magnetic behavior. The average pore diameter of 33.288 nm and average surface area of 82.7 m2 g−1 was observed in the N2 adsorption–desorption isotherm plot. The prepared material was further employed for the non-enzymatic electrochemical sensing of AA by modifying the GCE. The (Mg0.65Fe0.35)A[Fe1.65Mg0.35]BO4 modified electrode exhibited a linear increase in the oxidation peak current with concentration. The LOD and LOQ were found to be 24.09 μM and 80.30 μM, respectively. Also, the linear electron transfer rate constant, charge transfer coefficient, and sensitivity were evaluated to be 4.8 s−1, 0.9541, and 1578 μA mM−1cm−2, respectively. The modified electrode showed negligible interference effects toward various molecules, such as potassium chloride, sulfuric acid, hydrogen peroxide, sodium hydroxide, glucose, and choline chloride in phosphate buffer solution, showing outstanding selectivity toward AA. This study reveals that synthesized MgFe2O4 is a promising material for non-enzymatic electrochemical sensing.Author contributions
Aneeta Manjari Padhan: conceptualization, data curation, formal analysis, investigation, writing – original draft; P. Mary Rajaitha: formal analysis, investigation, writing: original draft; Sanjib Nayak: writing: review & editing; Sugato Hajra: writing: review & editing, data curation; Manisha Sahu: visualization; Zvonko Jagličić: data curation; Primož Koželj; data curation; Hoe Joon Kim: funding acquisition, validation, writing: review & editing, supervision.Conflicts of interest
Authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge support from the Basic Science Research Program of the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT of Korea (2021R1C1C1011588), and the National Research Foundation of Korea through the Brain Pool (BP) program (2021H1D3A2A01099471).References
- J. H. H. Rossato, M. E. Oliveira, B. V. Lopes, B. B. Gallo, A. B. La Rosa, E. Piva, D. Barba, F. Rosei, N. L. V. Carreño and M. T. Escote, A Flexible electrochemical biosensor based on NdNiO3 nanotubes for ascorbic acid detection, ACS Appl. Nano Mater., 2022, 5, 3394–3405 CrossRef CAS.
- R. Banavath, A. Abhinav, R. Srivastava and P. Bhargava, Highly sensitive ascorbic acid sensors from EDTA chelation derived nickel hexacyanoferrate/graphene nanocomposites, Electrochim. Acta, 2022, 419, 140335 CrossRef CAS.
- K. Dhara and R. M. Debiprosad, Review on nanomaterials-enabled electrochemical sensors for ascorbic acid detection, Anal. Biochem., 2019, 586, 113415 CrossRef CAS PubMed.
- R. Sha and S. Badhulika, Facile green synthesis of reduced graphene oxide/tin oxide composite for highly selective and ultra-sensitive detection of ascorbic acid, J. Electroanal. Chem., 2018, 816, 30–37 CrossRef CAS.
- E. Erçarıkcı, Z. Aksu, K. Dağcı Kıranşan and E. Topçu, Graphene paper with electrodeposited NiCo2S4 nanoparticles as a novel flexible sensor for simultaneous detection of folic acid and ascorbic acid, Diamond Relat. Mater., 2022, 121, 108713 CrossRef.
- C. Shen, Y. Chen, B. Feng, H. Chi and H. Zhang, Polypyrrole hollow nanotubes loaded with Au and Fe3O4 nanoparticles for simultaneous determination of ascorbic acid, dopamine, and uric acid, Chem. Res. Chin. Univ., 2022, 38, 941–948 CrossRef CAS.
- D. M. Fernandes, N. Silva, C. Pereira, C. Moura, J. M. C. S. Magalhães, B. Bachiller-Baeza, I. Rodríguez-Ramos, A. Guerrero-Ruiz, C. Delerue-Matos and C. Freire, MnFe2O4@CNT-N as novel electrochemical nanosensor for determination of caffeine, acetaminophen and ascorbic acid, Sens. Actuators, B, 2015, 218, 128–136 CrossRef CAS.
- J. Feng, Y. Liu, Y. Shan, Y. Xie, Z. Chu and W. Jin, In-situ growth of Cu@CuFe Prussian blue based core–shell nanowires for non-enzymatic electrochemical determination of ascorbic acid with high sensitivity and reusability, J. Electroanal. Chem., 2021, 900, 115718 CrossRef CAS.
- J. M. George, A. Antony and B. Mathew, Metal oxide nanoparticles in electrochemical sensing and biosensing: A review, Microchim. Acta, 2018, 185, 358 CrossRef.
- A. Manohar, V. Vijayakanth, S. V. P. Vattikuti, P. Manivasagan, E.-S. Jang and K. H. Kim, Electrochemical, oxygen evolution reaction and photoelectrochemical water splitting activity of Ca-doped MgFe2O4 nanoparticles, J. Alloys Compd., 2022, 907, 164566 CrossRef CAS.
- R. Liu, L. Pan, S. Peng, L. Qin, J. Bi, J. Wu, H. Wu and Z. G. Ye, The magnetoelectric effect in a cubic ferrimagnetic spinel LiFe5O8 with high coupling temperature, J. Mater. Chem. C, 2019, 7, 1999–2004 RSC.
- Y. Oh, M. Sahu, S. Hajra, A. M. Padhan, S. Panda and H. J. Kim, Spinel ferrites (CoFe2O4): Synthesis, magnetic properties, and electromagnetic generator for vibration energy harvesting, J. Electron. Mater., 2022, 51, 1933–1939 CrossRef CAS.
- M. Yousaf, N. Mushtaq, B. Zhu, B. Wang, M. N. Akhtar, A. Noor and M. Afzal, Electrochemical properties of Ni0.4Zn0.6Fe2O4 and the heterostructure composites (Ni–Zn ferrite-SDC) for low temperature solid oxide fuel cell (LT-SOFC), Electrochim. Acta., 2020, 331, 135349 CrossRef CAS.
- K. Bindu, K. Sridharan, K. M. Ajith, H. N. Lim and H. S. Nagaraja, Microwave assisted growth of stannous ferrite microcubes as electrodes for potentiometric nonenzymatic H2O2 sensor and supercapacitor applications, Electrochim. Acta, 2016, 217, 139–149 CrossRef CAS.
- M. Shahid, L. Jingling, Z. Ali, I. Shakir, M. F. Warsi, R. Parveen and M. Nadeem, Photocatalytic degradation of methylene blue on magnetically separable MgFe2O4 under visible light irradiation, Mater. Chem. Phys., 2013, 139, 566–571 CrossRef CAS.
- Y. L. Liu, Z. M. Liu, Y. Yang, H. F. Yang, G. L. Shen and R. Q. Yu, Simple synthesis of MgFe2O4 nanoparticles as gas sensing materials, Sens. Actuators, B, 2005, 107, 600–604 CrossRef CAS.
- A. A. Ensafi, A. R. Allafchian and R. Mohammadzadeh, Characterization of MgFe 2 O 4 Nanoparticles as a Novel Electrochemical Sensor: Application for the Voltammetric Determination of Ciprofloxacin, Anal. Sci., 2012, 28, 705–710 CrossRef CAS PubMed.
- D. Navadeepthy, M. Thangapandian, C. Viswanathan and N. Ponpandian, A nanocomposite of NiFe2O4-PANI as a duo active electrocatalyst toward the sensitive colorimetric and electrochemical sensing of ascorbic acid, Nanoscale Adv., 2020, 2, 3481–3493 RSC.
- P. T. K. Thu, N. D. Trinh, N. T. V. Hoan, D. X. Du, T. X. Mau, V. H. Trung, N. H. Phong, T. T. T. Toan and D. Q. Khieu, Synthesis of cobalt ferrite and simultaneous determination of ascorbic acid, acetaminophen and caffeine by voltammetric method using cobalt ferrite modified electrode, J. Mater. Sci.: Mater. Electron., 2019, 30, 17245–17261 CrossRef CAS.
- J. N. Baby, B. Sriram, S.-F. Wang and M. George, Effect of various deep eutectic solvents on the sustainable synthesis of MgFe2O4 nanoparticles for simultaneous electrochemical determination of nitrofurantoin and 4-nitrophenol, ACS Sustainable Chem. Eng., 2020, 8, 1479–1486 CrossRef CAS.
- S. Reddy, B. E. Kumara Swamy, U. Chandra, K. R. Mahathesha, T. V. Sathisha and H. Jayadevappa, Synthesis of MgFe2O4 nanoparticles and MgFe2O4 nanoparticles/CPE for electrochemical investigation of dopamine, Anal. Methods, 2011, 3, 2792–2796 RSC.
- F. Basiri and M. Taei, Application of spinel-structured MgFe2O4 nanoparticles for simultaneous electrochemical determination diclofenac and morphine, Microchim. Acta, 2017, 184, 155–162 CrossRef CAS.
- N. S. Arun Kumar, S. Ashoka and M. Pandurangappa, MgFe2O4 nanoparticles synthesis and characterization: Application to trace level mercury(II) measurement from waste water samples, Mater. Res. Express, 2019, 6, 125049 CrossRef.
- K. Y. Hwa, A. Ganguly and S. K. S. Tata, Influence of temperature variation on spinel-structure MgFe2O4 anchored on reduced graphene oxide for electrochemical detection of 4-cyanophenol, Microchim. Acta, 2020, 187, 633 CrossRef CAS PubMed.
- E. Singh, M. Kaur and S. Sharma, Structural tuning of CTAB@MgFe2O4 nanocomposite as peroxidase mimic for H2O2 and glucose sensing, Mater. Chem. Phys., 2021, 271, 124851 CrossRef CAS.
- Y. Zhang, Z. Zhou, F. Wen, J. Tan, T. Peng, B. Luo, H. Wang and S. Yin, A flower-like MoS2-decorated MgFe2O4 nanocomposite: Mimicking peroxidase and colorimetric detection of H2O2 and glucose, Sens. Actuators, B, 2018, 275, 155–162 CrossRef CAS.
- M. A. Cobos, P. de la Presa, I. Llorente, A. García-Escorial, A. Hernando and J. A. Jiménez, Effect of preparation methods on magnetic properties of stoichiometric zinc ferrite, J. Alloys Compd., 2020, 849, 156353 CrossRef CAS.
- L. Kékedy-Nagy, M. Abolhassani, L. F. Greenlee and B. G. Pollet, An electrochemical study of ammonium dihydrogen phosphate on Mg and Mg alloy electrodes, Electrocatalysis, 2021, 12, 251–263 CrossRef.
- H. Farahani, R. Wagiran and G. A. Urban, Barium strontium titanate-based humidity sensors: Microstructure, surface morphology, dopant influence, and transduction mechanism investigations, ACS Appl. Electron. Mater., 2021, 3, 4919–4933 CrossRef CAS.
- R. Tholkappiyan and K. Vishista, Combustion synthesis of Mg–Er ferrite nanoparticles: Cation distribution and structural, optical, and magnetic properties, Mater. Sci. Semicond. Process., 2015, 40, 631–642 CrossRef CAS.
- C. S. Fadley and D. A. Shirley, Multiplet splitting of metal-atom electron binding energies, Phys. Rev. A: At., Mol., Opt. Phys., 1970, 2, 1109–1120 CrossRef.
- L. Li, H. Bi, S. Gai, F. He, P. Gao, Y. Dai, X. Zhang, D. Yang, M. Zhang and P. Yang, Uniformly dispersed ZnFe2O4 nanoparticles on nitrogen-modified graphene for high-performance supercapacitor as electrode, Sci. Rep., 2017, 7, 1–12 CrossRef.
- A. C. Heredia, M. I. Oliva, C. I. Zandalazini, U. A. Agú, G. A. Eimer, S. G. Casuscelli, E. R. Herrero, C. F. Pérez and M. E. Crivello, Synthesis, characterization, and catalytic behavior of Mg–Al–Zn–Fe mixed oxides from precursors layered double hydroxide, Ind. Eng. Chem. Res., 2011, 50, 6695–6703 CrossRef CAS.
- T. Yamashita and P. Hayes, Analysis of XPS spectra of Fe2+ and Fe3+ ions in oxide materials, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- D. Kang, X. Yu, M. Ge and W. Song, One-step fabrication and characterization of hierarchical MgFe2O4 microspheres and their application for lead removal, Microporous Mesoporous Mater., 2015, 207, 170–178 CrossRef CAS.
- D. Nath, F. Singh and R. Das, X-ray diffraction analysis by Williamson-Hall, Halder–Wagner and size-strain plot methods of CdSe nanoparticles- a comparative study, Mater. Chem. Phys., 2020, 239, 122021 CrossRef CAS.
- Z. Wang, P. Lazor, S. K. Saxena and H. S. C. O’Neill, High pressure Raman spectroscopy of ferrite MgFe2O4, Mater. Res. Bull., 2002, 37, 1589–1602 CrossRef CAS.
- F. Nakagomi, S. W. da Silva, V. K. Garg, A. C. Oliveira, P. C. Morais and A. Franco, Influence of the Mg-content on the cation distribution in cubic MgxFe3−xO4 nanoparticles, J. Solid State Chem., 2009, 182, 2423–2429 CrossRef CAS.
- J. Kreisel, G. Lucazeau and H. Vincent, Raman spectra and vibrational analysis of BaFe12O19 hexagonal ferrite, J. Solid State Chem., 1998, 137, 127–137 CrossRef CAS.
- J. Chandradass, A. H. Jadhav, K. H. Kim and H. Kim, Influence of processing methodology on the structural and magnetic behavior of MgFe2O4 nanopowders, J. Alloys Compd., 2012, 517, 164–169 CrossRef CAS.
- P. Prajapat, S. Dhaka and H. S. Mund, Investigation of the influence of annealing temperature on the structural and magnetic properties of MgFe2O4, J. Electron. Mater., 2021, 50, 4671–4677 CrossRef CAS.
- M. Penchal Reddy, R. A. Shakoor, A. M. A. Mohamed, M. Gupta and Q. Huang, Effect of sintering temperature on the structural and magnetic properties of MgFe2O4 ceramics prepared by spark plasma sintering, Ceram. Int., 2016, 42, 4221–4227 CrossRef CAS.
- K. C. Das and S. S. Dhar, Rapid catalytic degradation of malachite green by MgFe2O4 nanoparticles in presence of H2O2, J. Alloys Compd., 2020, 828, 154462 CrossRef CAS.
- S. A. Masti, A. K. Sharma, P. N. Vasambekar and A. S. Vaingankar, Influence of Cd2+ and Cr3+ substitutions on the magnetization and permeability of magnesium ferrites, J. Magn. Magn. Mater., 2006, 305, 436–439 CrossRef CAS.
- V. Šepelák, I. Bergmann, D. Menzel, A. Feldhoff, P. Heitjans, F. J. Litterst and K. D. Becker, Magnetization enhancement in nanosized MgFe2O4 prepared by mechanosynthesis, J. Magn. Magn. Mater., 2007, 316, 764–767 CrossRef.
- K. Pubby, K. Vijay Babu and S. Bindra Narang, Magnetic, elastic, dielectric, microwave absorption and optical characterization of cobalt-substituted nickel spinel ferrites, Mater. Sci. Eng., B, 2020, 255, 114513 CrossRef CAS.
- C. Choodamani, G. P. Nagabhushana, B. Rudraswamy and G. T. Chandrappa, Thermal effect on magnetic properties of Mg–Zn ferrite nanoparticles, Mater. Lett., 2014, 116, 227–230 CrossRef CAS.
- A. Pradeep, P. Priyadharsini and G. Chandrasekaran, Sol–gel route of synthesis of nanoparticles of MgFe2O4 and XRD, FTIR and VSM study, J. Magn. Magn. Mater., 2008, 320, 2774–2779 CrossRef CAS.
- M. Manikandan, P. Manimuthu and C. Venkateswaran, Structural and magnetic properties of MgFe2O4 ceramic, AIP Conf. Proc., 2014, 194–196 CrossRef CAS.
- S. Vinoth, P. Sampathkumar, K. Giribabu and A. Pandikumar, Ultrasonically assisted synthesis of barium stannate incorporated graphitic carbon nitride nanocomposite and its analytical performance in electrochemical sensing of 4-nitrophenol, Ultrason. Sonochem., 2020, 62, 104855 CrossRef CAS PubMed.
- A. Singh, A. Sharma, A. Ahmed and S. Arya, Highly selective and efficient electrochemical sensing of ascorbic acid via CuO/rGO nanocomposites deposited on conductive fabric, Appl. Phys. A: Mater. Sci. Process., 2022, 128, 1–12 CrossRef.
- C. Qu, H. Li, S. Zhou, G. Li, C. Wang, R. Snyders, C. Bittencourt and W. Li, Bi2S3/rGO composite based electrochemical sensor for ascorbic acid detection, Chemosensors, 2021, 9, 1–15 CrossRef.
- K. Atacan, N. Güy and M. Özacar, Preparation of gold decorated MoS2/NiO nanocomposite in the production of a new electrochemical sensor for ascorbic acid detection, Korean J. Chem. Eng., 2022, 39, 2172–2181 CrossRef CAS.
- F. A. Harraz, M. Faisal, A. E. Al-Salami, A. M. El-Toni, A. A. Almadiy, S. A. Al-Sayari and M. S. Al-Assiri, Silver nanoparticles decorated stain-etched mesoporous silicon for sensitive, selective detection of ascorbic acid, Mater. Lett., 2019, 234, 96–100 CrossRef CAS.
- H. Huang, Y. Yue, Z. Chen, Y. Chen, S. Wu, J. Liao, S. Liu and H. Rui Wen, Electrochemical sensor based on a nanocomposite prepared from TmPO4 and graphene oxide for simultaneous voltammetric detection of ascorbic acid, dopamine and uric acid, Microchim. Acta, 2019, 186, 189 CrossRef PubMed.
- F. A. Harraz, M. Faisal, A. A. Ismail, S. A. Al-Sayari, A. E. Al-Salami, A. Al-Hajry and M. S. Al-Assiri, TiO2/reduced graphene oxide nanocomposite as efficient ascorbic acid amperometric sensor, J. Electroanal. Chem., 2019, 832, 225–232 CrossRef CAS.
- Y. Haldorai, S. R. Choe, Y. S. Huh and Y. K. Han, A composite consisting of microporous carbon and cobalt(III) oxide and prepared from zeolitic imidazolate framework-67 for voltammetric determination of ascorbic acid, Microchim. Acta, 2018, 185, 116 CrossRef PubMed.
- N. Lavanya and C. Sekar, Electrochemical sensor for simultaneous determination of epinephrine and norepinephrine based on cetyltrimethylammonium bromide assisted SnO2 nanoparticles, J. Electroanal. Chem., 2017, 801, 503–510 CrossRef CAS.
- C. Hou, H. Liu, D. Zhang, C. Yang and M. Zhang, Synthesis of ZnO nanorods-Au nanoparticles hybrids via in situ plasma sputtering-assisted method for simultaneous electrochemical sensing of ascorbic acid and uric acid, J. Alloys Compd., 2016, 666, 178–184 CrossRef CAS.
- R. Madhuvilakku and S. Piraman, One-dimensional NiFe2O4 nanorods modified with sulfur-rich spherical carbon nanoparticles for simultaneous voltammetric determination of ascorbic acid, dopamine and uric acid, Microchim. Acta, 2019, 186, 434 CrossRef PubMed.
- P. Wu, Y. Huang, X. Zhao, D. Lin, L. Xie, Z. Li, Z. Zhu, H. Zhao and M. Lan, MnFe2O4/MoS2 nanocomposite as Oxidase-like for electrochemical simultaneous detection of ascorbic acid, dopamine and uric acid, Microchem. J., 2022, 181, 107780 CrossRef CAS.
- L. Z. Pei, Y. K. Xie, Z. Y. Cai, Y. Yang, Y. Q. Pei, C. G. Fan and D. G. Fu, Electrochemical behavior of ascorbic acid at copper germanate nanowire modified electrode, J. Electrochem. Soc., 2012, 159, K55–K60 CrossRef CAS.
- S. Jahani, Evaluation of the usefulness of an electrochemical sensor in detecting ascorbic acid using a graphite screen-printed electrode modified with NiFe2O4 nanoparticles, Anal. Bioanal. Electrochem., 2018, 10, 739–750 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qm00628f |
| This journal is © the Partner Organisations 2023 |