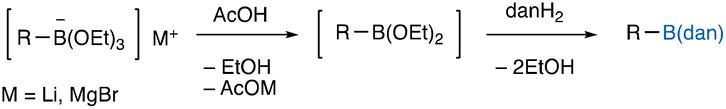DOI:
10.1039/D3OB00613A
(Communication)
Org. Biomol. Chem., 2023,
21, 5347-5350
Efficient one-pot synthesis of dan-substituted organo- and silyl-boron compounds†
Received
21st April 2023
, Accepted 3rd May 2023
First published on 3rd May 2023
Abstract
Direct, transition metal-free B(dan)-installation into organic frameworks has been developed. Heteroaryl–H bonds were transformable into the respective heteroaryl–B(dan) bonds through deprotonation. The resulting heteroaryl–B(dan) compounds, which are otherwise difficult to access, can undergo the direct Suzuki–Miyaura coupling. The method was demonstrated to apply to a silicon nucleophile, giving Lewis acidity-diminished stable silyl–B(dan) and –B(aam) in one pot.
Installation of a naphthalene-1,8-diaminato (dan) substituent on boron centers significantly diminishes the boron-Lewis acidity,1 thus providing transmetalation-resistant properties to the respective organoboron compounds [R–B(dan)] that are conventionally synthesized by dehydration condensation of organoboronic acids [R–B(OH)2] and 1,8-diaminonaphthalene (danH2) (Scheme 1A).2 The characteristic feature has been utilized for the boron-masking strategy in the iterative Suzuki–Miyaura coupling (SMC),2 where the B(dan) moieties remain intact during the cross-coupling event at Lewis acidic B(OH)2 or pinacol boronates [B(pin)]. On the other hand, we have disclosed that the B(dan) moieties can be activated toward transmetalation by treatment with t-BuOK, leading to the direct SMC of aryl–3,4 and cyclopropyl–B(dan).5 In addition to the inactive/active flexible properties in the SMC depending on bases, dan-installation also endows organoboron compounds with air- and water-resistant properties: protodeborylation is substantially suppressed with 2-pyridyl–B(dan),3,6 and PhMe2Si–B(dan)7 becomes stable in air, while their B(OH)2/B(pin) counterparts usually suffer from serious decomposition under these conditions.8,9 In this regard, the development of a direct method of synthesizing dan-substituted organoboron compounds,10 especially heteroaryl ones that are often unstable in their Lewis acidic –B(OH)2 forms,11 would be an important subject, since it could open the way to synthetic transformations with heteroaryl–B(dan). We have previously reported on a reaction of Grignard reagents with H–B(dan) that directly produces various R–B(dan) including 2-pyridyl– and 2-thienyl–B(dan) under transition metal-free conditions (Scheme 1B);6 however, this reaction requires organic bromides (R–Br for preparing R–MgBr) and pre-prepared H–B(dan), which would leave something to be desired.12 Hence, our attention was focused on the use of borates [R–B(OR′)3−], one of the most common intermediates in the synthesis of organoboronic acid derivatives, generated from readily accessible B(OR′)3 and carbon nucleophiles (R–M, M = Li, MgX) for their direct conversion to R–B(dan). Here we disclose an improved approach to prepare R–B(dan) by using only commercially available reagents, wherein deprotonation of R–H moieties is also usable for generating the requisite carbon nucleophiles (Scheme 1C).
 |
| | Scheme 1 Transition metal-free synthesis of Ar–B(dan). | |
We first conducted a reaction (5 mmol scale) of 5-pyrazolyl lithium, prepared readily by deprotonation of 1-methyl-1H-pyrazole (1a), with triethylborate [B(OEt)3]; subsequent treatment of the resulting pyrazoylborate [5-pyrazolyl–B(OEt)3−] with 1,8-diaminonaphthalene and acetic acid, in a similar way to the direct transformation of trialkyl borates [R–B(OR′)3−] into pinacol boronates [R–B(pin)],13 provided 5-pyrazolyl–B(dan) (2a) in 88% yield (Scheme 2).14 Because the one-pot direct conversion of a heteroaryl–H bond into a heteroaryl–B(dan) bond can be carried out under non-aqueous conditions, 5-thiazolyl–B(dan) (2b and 2c) could also be synthesized, avoiding the intermediary formation of their protodeborylation-prone B(OH)2 counterparts.11b An acidic C–H moiety of furan (1d) or thiophene (1e) was also convertible into the respective C–B(dan) (2d: 75% and 2e: 70%), and 4,4-dimethyl-2-phenyl-2-oxazoline (1f) and 1,3-dimethoxybenzene (1g) could participate in the reaction via directed ortho metalation. Owing to the diminished boron Lewis acidity, all the heteroaryl–B(dan) and other products mentioned below exhibit sufficient stability under ambient conditions, allowing for their isolation by column chromatography. The use of 1,1′-dilithioferrocene generated by dual deprotonation of ferrocene (1h) resulted in the formation of 1,1′-B(dan)-substituted ferrocene (2h) in 59% yield, and the procedure was also applicable to phenylacetylene (1i) or 1,3-dithiane (1j), giving phenylethynyl–B(dan) (2i) or 1,3-dithian-2-yl–B(dan) (2j) as air/water-stable compounds.15
 |
| | Scheme 2 Synthesis of R–B(dan) from R–H. | |
Organic bromides (1k–1q) were naturally usable as starting materials (Scheme 3), and thus 2-fluorophenyl–B(dan) (2k), 2-pyridyl–B(dan) (2l) and 2,2′-B(dan)-substituted 1,1′-binaphthyl (2m) were produced through lithium–bromine exchange. In addition, various aryl–B(dan) (2n–2p)16 and alkyl–B(dan) (2q and 2r17) could be synthesized with the respective Grignard reagents.18 A carbon nucleophile used for B(dan)-installation was also available by lithium–tin exchange of 2,5-bis(tributylstannyl)thiophene (1s) to afford a 63% yield of 2s bearing B(dan) and SnBu3 functionalities on the thiophene ring (Scheme 4).
 |
| | Scheme 3 Synthesis of R–B(dan) from R–Br. | |
 |
| | Scheme 4 B(dan)-installation via lithium–tin exchange. | |
It should be noted that the present method could also be applied to capturing a silicon nucleophile: air-resistant PhMe2Si–B(dan) (2t) became straightforwardly accessible in 40% yield by treating PhMe2SiLi with B(OEt)3 (Scheme 5),19,20 while the previous multistep method7 required the use of air/water-sensitive reagents and intermediates [BCl3, (i-Pr2N)2BCl and PhMe2Si–B(Ni-Pr2)2]. A new air-resistant silylborane bearing an anthranilamide substituent on the boron center [PhMe2Si–B(aam) (2u)] was generated by extending the direct synthesis, although the yield was relatively low.21 According to the previous procedure using PhMe2Si–B(Ni-Pr2)2, the yield was improved to 65%.
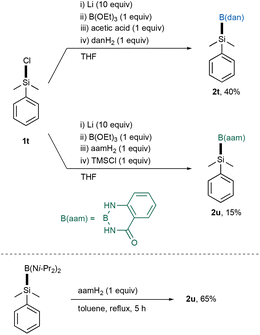 |
| | Scheme 5 Synthesis of PhMe2Si–B(dan) and –B(aam). | |
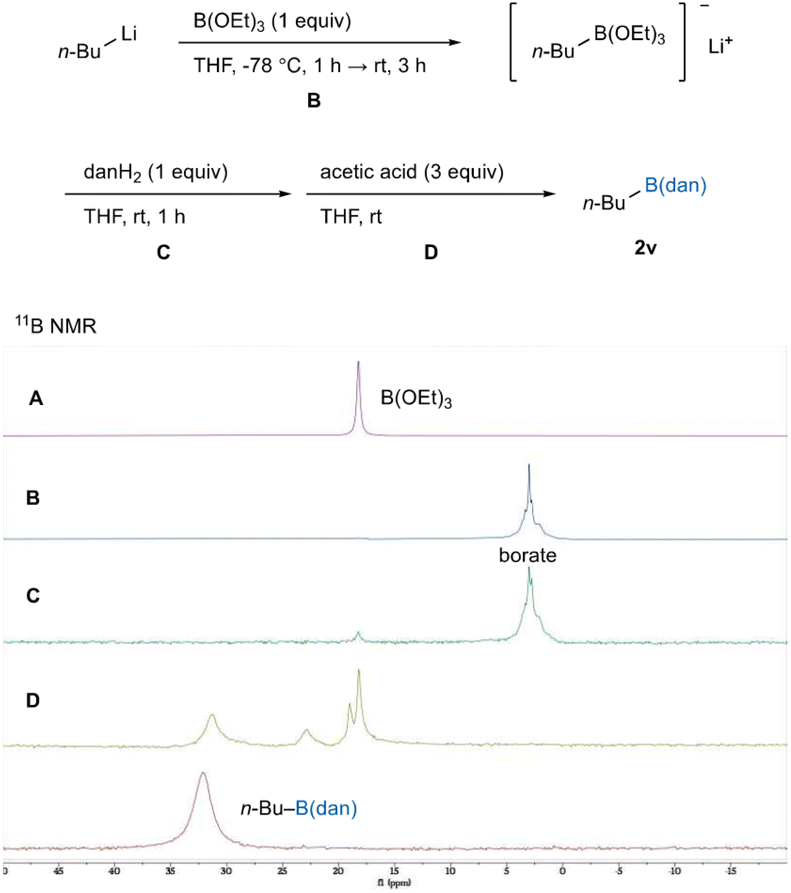
To gain insight into the reaction pathway, we carried out 11B NMR experiments using n-BuLi as a carbon nucleophile (Fig. 1). Treatment of B(OEt)3 (18.3 ppm, Fig. 1A) with n-BuLi resulted in the generation of an upfield-shifted species (3.0 ppm, Fig. 1B), being in a typical chemical shift range of a tetracoordinate borate.22 The resulting borate [n-Bu–B(OEt)3−] remained unchanged upon mixing with 1,8-diaminonaphthalene (Fig. 1C), while further addition of acetic acid led to the formation of n-Bu–B(dan) (2v) with complete disappearance of the borate (Fig. 1D).23 These results imply that dan-installation on the boron center does not occur at the tetracoordinate borate stage; the intermediary formed neutral species [R–B(OEt)2] via protonation of the borates would immediately undergo ligand exchange to afford R–B(dan) (Scheme 6).
 |
| | Fig. 1
11B NMR experiments on the reaction pathway. | |
 |
| | Scheme 6 Proposed reaction pathway. | |
Finally, the synthetic utility of heteroaryl–B(dan) was demonstrated by the direct SMC: treatment of 2a with 4-bromoanisole and Ba(OH)2 under Pd–dppf catalysis3 quantitatively provided the coupling product (3a), which verifies the protodeborylation-resistant yet enough reactive property of the C–B(dan) bond (Scheme 7). Furthermore, chemoselective cross-coupling at the SnBu3 moiety of 2s gave a 95% yield of 3b, whose thienyl–B(dan) bond was then coupled with 4-(trifluoromethyl)bromobenzene efficiently under the Ba(OH)2 conditions.
 |
| | Scheme 7 Direct SMC of heteroaryl–B(dan). | |
Conclusions
We have developed a one-pot direct method for installing the B(dan) moiety without the intermediary formation of sometimes unstable organoboronic acids. The present transition metal-free method is superior to our previous H–B(dan) one6 in that organolithium reagents directly generated by deprotonation are usable as carbon nucleophiles;12 diverse R–B(dan) (R = heteroaryl, aryl, alkynyl, alkyl) endowed with air/water-resistant characters have become accessible in good yields. Moreover, the method was demonstrated to apply to a silicon nucleophile to lead to the direct synthesis of PhMe2Si–B(dan) and –B(aam). Further studies on the synthetic utilization of the resulting R–B(dan) especially for catalytic carbon–carbon bond-forming reactions are in progress.
Author contributions
K.T. and H.Y. conceived the concept and wrote the manuscript. K.T. conducted most of the experiments and data collection. Y.I. synthesized 2u according to the previous method. K.N. performed the X-ray crystal structure analysis of 2u. M.N. provided advice for the research. H.Y. directed the project. All authors have approved the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the Fukuoka Naohiko Memorial Foundation and the Nagase Science and Technology Foundation.
References
-
(a) H. Tanaka, M. Nakamoto and H. Yoshida, RSC Adv., 2023, 13, 2451–2457 RSC;
(b) S. Kamio and H. Yoshida, Adv. Synth. Catal., 2021, 363, 2310–2324 CrossRef CAS;
(c) J. Li and H. Yoshida, Heterocycles, 2021, 102, 1478–1516 CrossRef CAS.
-
(a) H. Noguchi, K. Hojo and M. Suginome, J. Am. Chem. Soc., 2007, 129, 758–759 CrossRef CAS PubMed;
(b) H. Noguchi, T. Shioda, C.-M. Chou and M. Suginome, Org. Lett., 2008, 10, 377–380 CrossRef CAS.
- H. Yoshida, M. Seki, S. Kamio, H. Tanaka, Y. Izumi, J. Li, I. Osaka, M. Abe, H. Andoh, T. Yajima, T. Tani and T. Tsuchimoto, ACS Catal., 2020, 10, 346–351 CrossRef CAS.
- Y. Mutoh, K. Yamamoto and S. Saito, ACS Catal., 2020, 10, 352–357 CrossRef CAS.
- M. Koishi, K. Tomota, M. Nakamoto and H. Yoshida, Adv. Synth. Catal., 2023, 365, 682–686 CrossRef CAS.
- J. Li, M. Seki, S. Kamio and H. Yoshida, Chem. Commun., 2020, 56, 6388–6391 RSC.
- H. Yoshida, Y. Izumi, Y. Hiraoka, K. Nakanishi, M. Nakamoto, S. Hatano and M. Abe, Dalton Trans., 2022, 51, 6543–6546 RSC.
- For protodeborylation of organoboronic acids/esters, see:
(a) P. A. Cox, A. G. Leach, A. D. Campbell and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2016, 138, 9145–9157 CrossRef CAS PubMed;
(b) P. A. Cox, M. Reid, A. G. Leach, A. D. Campbell, E. J. King and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 13156–13165 CrossRef CAS PubMed;
(c) H. L. D. Hayes, R. Wei, M. Assante, K. J. Geogheghan, N. Jin, S. Tomasi, G. Noonan, A. G. Leach and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2021, 143, 14814–14826 CrossRef CAS PubMed.
- For air sensitivity of PhMe2Si–B(pin), see: E. Yamamoto, R. Shishido, T. Seki and H. Ito, Organometallics, 2017, 36, 3019–3022 CrossRef CAS.
- For representative reports on transition metal-catalyzed B(dan)-installing reactions, see:
(a) N. Iwadate and M. Suginome, J. Organomet. Chem., 2009, 694, 1713–1717 CrossRef CAS;
(b) H. Yoshida, Y. Takemoto and K. Takaki, Chem. Commun., 2014, 50, 8299–8830 RSC;
(c) L. Xu and P. Li, Chem. Commun., 2015, 51, 5656–5659 RSC;
(d) H. Yoshida, Y. Takemoto, S. Kamio, I. Osaka and K. Takaki, Org. Chem. Front., 2017, 4, 1215–1219 RSC.
- Heteroarylboronic acids having a C–B(OH)2 bond next to a heteroatom are liable to be protodeborylated. For 5-pyrazolyl, see:
(a) A. V. Ivachtchenko, D. V. Kravchenko, V. I. Zheludeva and D. G. Pershin, J. Heterocycl. Chem., 2004, 41, 931–939 CrossRef CAS. For 5-thiazolyl, see:
(b) P. Stanetty, M. Schnürch and M. D. Mihovilovic, J. Org. Chem., 2006, 71, 3754–3761 CrossRef CAS. For 2-furyl, see:
(c) D. M. Knapp, E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2009, 131, 6961–6963 CrossRef CAS PubMed. For 2-thienyl, see:
(d) K. Billingsley and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 3358–3366 CrossRef CAS PubMed. For 2-pyridyl, see:
(e) X. A. F. Cook, A. de Gombert, J. McKnight, L. R. E. Pantaine and M. C. Willis, Angew. Chem., Int. Ed., 2021, 60, 11068–11091 CrossRef CAS.
- The reaction of organolithium reagents with H–B(dan) was unsuccessful.
-
(a) D. P. Phillion, R. Neubauer and S. S. Andrew, J. Org. Chem., 1986, 51, 1610–1612 CrossRef CAS;
(b) C. Coudret, Synth. Commun., 1996, 26, 3543–3547 CrossRef CAS.
- Depending on the substrates, various reaction conditions (molar ratio, temperature, and time) were employed. See the ESI† for details.
- The starting R–H were recovered when the yields were moderate.
- Ph–B(dan) (2p) could also be synthesized from bromobenzene in 69% yield via lithium–bromine exchange.
- A tert-butyl Grignard reagent was used as the starting material.
- Isopropenyl–B(dan) was prepared according to a similar procedure by using isopropenyl–MgBr, B(OMe)3, 1,8-diaminonaphthalene, and aqueous NH4Cl. See: J. C. Lo, D. Kim, C.-M. Pan, J. T. Edwards, Y. Yabe, J. Gui, T. Qin, S. Gutiérrez, J. Giacoboni, M. W. Smith, P. L. Holland and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 2484–2503 CrossRef CAS PubMed.
- Acetic acid was added first to the silylborate intermediate in this case; the reaction conducted in the usual manner resulted in a slightly decreased yield (38%).
- Using a silyl Grignard reagent (PhMe2SiMgBr) instead of PhMe2SiLi resulted in an almost similar yield (42%). For the generation of silyl Grignard reagents, see: W. Xue, R. Shishido and M. Oestreich, Angew. Chem., Int. Ed., 2018, 57, 12141–12145 CrossRef CAS PubMed.
- The use of acetic acid instead of TMSCl gave 2u in only trace amounts.
- For 11B NMR chemical shifts of n-butyl(trialkoxy)borates, see:
(a) H. C. Brown, M. Srebnik and T. E. Cole, Organometallics, 1986, 5, 2300–2303 CrossRef CAS;
(b) E. Zygadło-Monikowska, Z. Florjańczyk, K. Służewska, J. Ostrowska, N. Langwald and A. Tomaszewska, J. Power Sources, 2010, 195, 6055–6061 CrossRef.
- See the ESI† for the assignment of other 11B NMR signals.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*