
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlycosylated quantum dots as fluorometric nanoprobes for trehalase†
Danielle D.
Barnes
a,
Vera
Kuznetsova
b,
Anastasia
Visheratina
c,
Finn
Purcell-Milton
 b,
Mikhail A.
Baranov
d,
Dylan M.
Lynch
a,
Harlei
Martin
a,
Yurii K.
Gun'ko
*b and
Eoin M.
Scanlan
*a
b,
Mikhail A.
Baranov
d,
Dylan M.
Lynch
a,
Harlei
Martin
a,
Yurii K.
Gun'ko
*b and
Eoin M.
Scanlan
*a
aSchool of Chemistry and Trinity Biomedical Sciences Institute, Trinity College, Pearse St, Dublin 2, Ireland. E-mail: eoin.scanlan@tcd.ie
bSchool of Chemistry and CRANN, Trinity College, Pearse St, Dublin 2, Ireland
cDepartment of Chemical Engineering, University of Michigan, Ann Arbor, MI, USA
dITMO University, St. Petersburg, Russia
First published on 17th March 2023
Abstract
Trehalase is an important enzyme in the metabolic cascades of many organisms, catalysing the hydrolysis of the disaccharide trehalose. Herein we describe the first examples of fluorometric nanoprobes for detection of trehalase, based on trehalose-functionalised quantum dots (QDs). QDs cross-linked with trehalose form aggregates, which are released upon enzymatic cleavage of the trehalose glycosidic bond proportionally to the enzyme concentration, offering a unique and efficient approach for specific sensing of this biologically important enzyme.
Trehalase or α,α-trehalose-1-C-glucohydrolase, is the major enzyme involved in the hydrolysis of the biologically ubiquitous disaccharide trehalose,1,2 a process that is essential for numerous organisms, such as bacteria, fungi, plants and invertebrates. Trehalose is a C2-symmetric, non-reducing disaccharide composed of two glucose molecules linked by an unusual α,α-1,1-glycosidic linkage. Trehalase is involved in the production of glucose for cellular respiration, and for the restoration of cellular homeostasis following stress-induced overproduction of trehalose.3,4 In particular, trehalase is an essential enzyme in the survival of several bacteria, such as Mycobacterium tuberculosis, which contain trehalose containing glycolipids in their outer ‘myco-membrane’.5 Also, inhibitors of trehalase such as the amino-sugar derivative trehazolin are of considerable importance for the agri-food sector as potent insecticides, due to the critical role of trehalose as an energy source for insects.6–8
Currently, there are extremely limited options available for assaying trehalase activity and screening potential inhibitors. The unusual non-reducing structure of the α,α-linked disaccharide makes molecular probe design especially challenging, as typical approaches utilizing a reporter moiety at the anomeric site of a substrate carbohydrate are unsuitable.9 To date, the most widely used method for determining trehalase activity in biochemical and clinical experiments is the use of the o-toluidine assay for glucose determination.10,11 This method enables rapid detection and visual indication of the presence of glucose, and thus relies on detection of the enzymatic hydrolysis product. However, a major limitation of this approach is the complete lack of selectivity and consequently the prevalence of false positive results, due to the presence of other reducing sugars.
More recently, a cephalosporinase-dependent green trehalose fluorogenic probe has been reported that allows the labeling and tracking of phagocystosed M. tuberculosis.12 This probe relies on activation by BlaC, a β-lactamase uniquely expressed by M. tuberculosis. A solvatochromic trehalose probe has also been reported that allows rapid detection of M. tuberculosis. The 4-N,N-dimethylamino-1,8-napthalimide conjugated trehalose probe undergoes a large enhancement in fluorescence when it undergoes metabolic conversion and is incorporated into the mycomembrane of the bacteria.13
Although these methods allow detection of pathogenic M. tuberculosis, they do not provide information or direct quantification of trehalase activity in the bacteria. The development of a more accurate and sensitive assay for trehalase detection would enable facile and rapid screening for the presence of trehalase activity, and identification of novel inhibitors.
Quantum dots (QDs) are fluorescent semiconductor nanoparticles, which exhibit unique optical and photophysical properties, such as high photoluminescence (PL), large absorption coefficients and exemplary photo- and chemical stability.14,15 This renders QDs excellent candidates for QD-based bioassays and bio-probes.16–20 Research groups led by Mauro, Medintz and Mattoussi have pioneered the development of QDs for biological sensing.14,21–23 Furthermore, glycosylated QDs have been reported to assist intracellular delivery of non-internalisable glycans,24 and as FRET probes for investigating multivalent protein–ligand interactions.25–29 However, their application as sensors for glycosidase enzymes has not previously been investigated. Herein we report a sensing system for trehalase based on QD complexes modified with synthetic trehalose analogues. A schematic representation of the rationale behind our probe is shown in Fig. 1.
 | ||
| Fig. 1 Schematic illustration of glycosylated QD nanoprobes for the detection of trehalase activity. | ||
Surface functionalization of alloyed CdSe@ZnS/ZnS QDs with a synthetic trehalose derivative possessing two thiol residues results in cross-linking of QDs, due to the binding of thiols with Zn ions on the QDs surface. Enzymatic cleavage of the trehalose results in disaggregation of the QDs and the release of free QDs, the concentration of which can be determined by fluorescence methods.
Overall, this process is somewhat analogous to the methodology developed for the detection of protease activity using QD–peptide conjugates.21 The novel glycosylated nanoprobe reported herein has the potential to function as a powerful sensing tool for detection of trehalase activity in biological samples and for screening novel trehalase inhibitors.30
To prepare our QD-based trehalase probe, we first synthesised a suitably modified trehalose derivative that could cross-link QDs while remaining susceptible to enzymatic hydrolysis. The synthetic route for preparation of the required trehalose glycoconjugate 7 is outlined in Scheme 1.
 | ||
| Scheme 1 . Synthesis of trehalose glycoconjugate 7 for QD functionalisation. | ||
D-trehalose 1 was treated with trimethylsilylchloride (TMSCl), in the presence of triethylamine to furnish 2 in a high yield (85%). Regioselective removal of the O-trimethylsilyl group at both C-6 positions was carried out according to a procedure reported by Kulkarni and co-workers, involving treatment of 2 with catalytic K2CO3, furnishing the symmetrical product 3.31 Subsequent treatment of 3 with pentenoic acid, N,N‘-diisopropylcarbodiimide (DIC) and 4-dimethylaminopyridine (DMAP) in anhydrous conditions furnished diester 4 in a modest yield (52%). The remaining O-TMS protecting groups were removed upon treatment of 4 with TFA/anhydrous CH2Cl2 (10% v/v) to furnish the unprotected diester 5 in quantitative yield. With this material in hand, the terminal alkene groups were modified to S-acetyl residues under radical-mediated, thiol–ene ligation conditions. This involved the treatment of 5 with thioacetic acid in DMF, in the presence of the radical initiator 2,2-dimethoxy-2-phenylacetophenone (DPAP) and the photosensitizer 4-methoxyacetophenone (MAP), under UV irradiation. Following thiol–ene ligation, dithioester 6 was obtained in good yield (79%). Finally, the S-acetyl protecting groups were removed via treatment with N2H4 in EtOH to furnish the desired trehalose derivative 7 in excellent yield (>99%).
Quantitative deprotection of the thioesters was confirmed by the disappearance of the S-acetyl peak at δH 2.33 ppm in the 1H NMR. Moreover, the presence of a peak at δC 173.7 ppm in the 13C NMR, corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, confirmed that the oxoester remained intact during the reaction (see ESI†).
O, confirmed that the oxoester remained intact during the reaction (see ESI†).
To prime the glycosylated nanoprobe for trehalase sensing, alloyed CdSe@ZnS/ZnS QDs were synthesised using the previously reported method.32 The PL wavelength of the QDs was 525 nm and the average diameter 12.9 ± 1.7 nm. More detailed physical and optical characterization is provided in ESI, Fig. S14A and S15A.† Using ligand exchange methodology, the QDs were cross-linked upon treatment with the symmetrical trehalose derivative 7.33 The thiol residues of 7 enabled effective binding to Zn ions onto the ZnS shell and leads to binding of QDs to each other and to the formation of the aggregates with a diameter of about 100 nm and a hydrodynamic radius of about 600 nm (Fig. 2 and Fig. 3B).
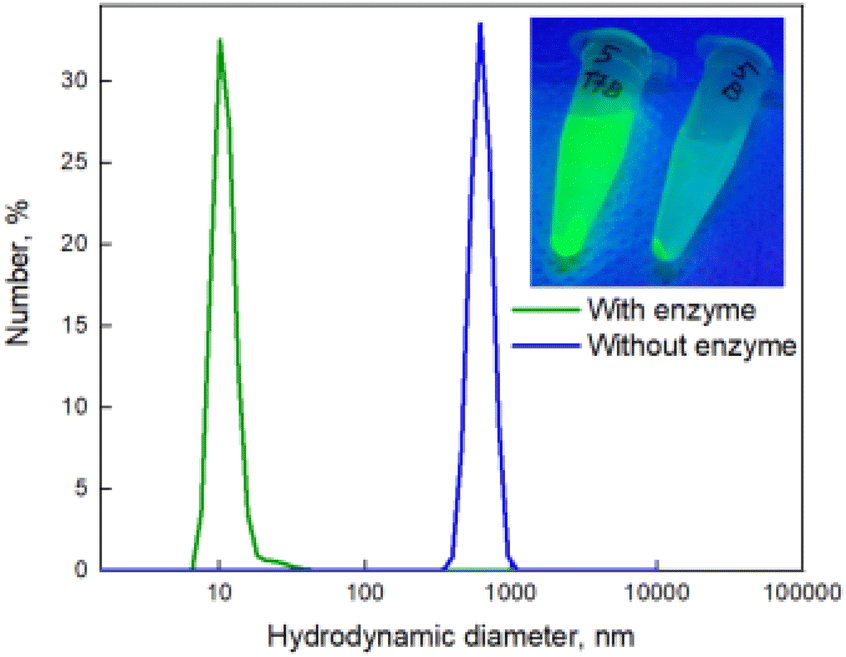 | ||
Fig. 2 DLS data and image under UV lamp (insert) of the trehalose functionalised QDs after incubation with trehalase and centrifugation during 5 min at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 R min−1 (green line, corresponding to the left eppendorf in insert), and negative control (blue line, corresponding to the right eppendorf in insert). 000 R min−1 (green line, corresponding to the left eppendorf in insert), and negative control (blue line, corresponding to the right eppendorf in insert). | ||
 | ||
| Fig. 3 STEM images of (A) QDs after incubation with trehalase and centrifugation during 5 min at 15000 R min−1 and (B) the trehalose functionalized QDs in aggregates (negative control). | ||
In our initial experiments, the resultant QD-trehalose aggregates were dissolved in aqueous buffered solution (140 mM citric acid buffer, pH 4.5) with the QD concentration of 10−7 M. Mildly acidic conditions were employed to ensure optimal activity of the trehalase enzyme. The QD suspension was then incubated with trehalase for 1 h at 37 °C with continuous stirring. The trehalase concentration varied from 2.5 × 10−3 to 2.5 × 10−2 unit per mL. A separate sample of the functionalised QD-trehalose aggregates in the absence of enzyme was employed as a negative control. For the negative control tests, the QDs were incubated with bovine serum albumin (BSA), a close homolog of the most abundant physiological protein, and β-galactosidase (another glycoside hydrolase enzyme, inactive for trehalose) to confirm the selectivity of the nanoprobe.
Following completion of the incubation period (1 h), the samples were resuspended in aqueous alkaline solution (KOH, pH 11) containing cysteine (0.2 mg mL−1), which binds to the surface of the QDs with high affinity.33 The charged carboxylate groups of cysteine facilitates electrostatic repulsion between the QDs, and thereby facilitates the colloidal stabilisation of free QDs in solution.
Gratifyingly, it was found that the trehalase enzyme cleaved the glycosidic bond of the trehalose units linking the QDs, releasing glucose. The resultant hydrolysis led to the release of free QDs, in a ratio proportional to the concentration of enzyme. Disaggregated QDs could be separated from the remaining aggregates via centrifugation (5 min, 15000 R min−1).
A sample of QDs exposed to trehalase is shown below (Fig. 2, Insert), at a working concentration of 2.5 × 10−2 units per mL. The supernatant of the sample contains free QDs following enzymatic treatment, which are stable in the solution. The negative control precipitated completely (indicating no colloidal stability) and, as expected, the samples incubated with BSA and β-galactosidase also did not appear fluorescent, as they do not contain free QDs in the supernatant (Fig. 2, insert).
Dynamic light scattering (DLS) analysis revealed that after centrifugation, the supernatant of the trehalase-exposed samples possessed a small hydrodynamic diameter of around 10 nm, which corresponds to a non-aggregated QD system (as indicated in Fig. 2). STEM (scanning transmission electron microscopy) data confirmed that treatment with trehalase resulted in disaggregation of QDs (Fig. 3A). QDs functionalised with glucose were in the same range for hydrodynamic diameter (see ESI Fig. S13†). On the other hand, the precipitate from the negative control sample had a much larger hydrodynamic diameter, demonstrating the presence of an aggregated QD system. Aggregates of trehalose-functionalised QDs also can be seen on the STEM image (Fig. 3B).
The concentration of free QDs in the supernatant can be approximated via measurement of the PL intensity. A characteristic PL spectrum of the samples (see ESI Fig. S15A†) shows a well-defined emission peak at 525 nm, which has been assigned as the QDs PL.
In the course of this work, it was determined that free QD concentration in supernatant is proportional to the concentration of trehalase. A representative PL titration curve is shown in Fig. 4, in which the limit of the detection was determined as 5 × 10−3 units per mL. Admittedly, the PL intensity of the sample with the trehalase concentration of 2.5 × 10−3 units per mL appeared to be close to the measurement limitation.
 | ||
| Fig. 4 PL titration curve, which permits estimation of the concentration of free QDs in solution. PL was measured at 525 nm. | ||
In the context of the commercially available test kits for trehalase (MyBioSource, Biorbyt, Arigo), which rely on calorimetric measurements of glucose generated by cleavage of trehalose, the detection limit of glucose is approximately 0.5 mmol L−1 (0.5 μmol mL−1). This corresponds to 0.075 unit per mL of trehalase present in sample (reaction time 10 min, sample volume 40 μL). Hence, the methodology outlined herein can significantly increase the accuracy of determining the concentration of trehalase in solution, with a small penalty in terms of reaction time and sample volume.
We have adapted our method for the qualitative detection of trehalase by the naked eye test. For this, we used QDs with two different PL wavelengths (green and red) and FRET between them. The different luminescence colours under the UV lamp makes it possible to distinguish an aggregated system from non-aggregated one. The method is described in more detail in ESI Fig. S15B.†
As the direct application of this work is within the scope of the large-scale agri-food sector, these caveats do not diminish these promising results. This present work highlights the immense potential of carbohydrate-functionalised QD systems for accurate sensing applications. Our method can potentially be adapted for measurements of intracellular trehalase. But in that case, for colloidal stabilization of free QDs it would be necessary to use biocompatible ligands, such as PEG, instead of KOH and cysteine.
In conclusion, we have demonstrated that trehalase activity can be detected using trehalose functionalized QDs as photoluminescent nanoprobes. A dithiol-functionalised trehalose derivative was synthesised and fully characterised, enabling, for the first time, binding to the alloyed CdSe@ZnS/ZnS QDs. The resulting change in fluorescence intensity is readily perceived by the naked eye. There are several potential valuable applications for this nanoprobe design, including insecticide screening and the discovery of antibiotics which function by inhibiting trehalase activity. This work will contribute to further developments in the applications of QD-based assays for enzyme detection and the detailed monitoring of enzymatic processes. Ultimately, introducing QD-trehalose nanoprobes into live cells may facilitate real-time monitoring of in vivo glycoside hydrolase activity.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.References
- J. M. Thevelein, Microbiological Reviews, 1984, 48, 42–59 CrossRef CAS PubMed.
- J. A. Jorge, M. T. M. Polizeli, J. M. Thevelein and H. F. Terenzi, FEMS Microbiol. Lett., 1997, 154, 165–171 CrossRef CAS PubMed.
- A. D. Elbein, Y. T. Pan, I. Pastuszak and D. Carroll, Glycobiology, 2003, 13, 17–27 CrossRef.
- J. D. Carroll, I. Pastuszak, V. K. Edavana, Y. T. Pan and A. D. Elbein, FEBS J., 2007, 274, 1701–1714 CrossRef CAS PubMed.
- H. L. Parker, R. M. F. Tomás, C. M. Furze, C. S. Guy and E. Fullam, Org. Biomol. Chem., 2020, 18, 3607–3612 RSC.
- G. Wegener, V. Tschiedel, P. Schloder and O. Ando, J. Exp. Biol., 2003, 206, 1233–1240 CrossRef CAS.
- G. D'Adamio, A. Sgambato, M. Forcella, S. Caccia, C. Parmeggiani, M. Casartelli, P. Parenti, D. Bini, L. Cipolla, P. Fusi and F. Cardona, Org. Biomol. Chem., 2015, 13, 886–892 RSC.
- B. Tang, M. Yang, Q. Shen, Y. Xu, H. Wang and S. Wang, Pestic. Biochem. Physiol., 2017, 137, 81–90 CrossRef CAS PubMed.
- H. M. Burke, T. Gunnlaugsson and E. M. Scanlan, Chem. Commun., 2015, 51, 10576–10588 RSC.
- K. M. Dubowski, Clin. Chem., 2008, 54, 1919 CrossRef CAS.
- C. F. Fasce, R. Rej and A. J. Pignataro, Clin. Chim. Acta, 1973, 43, 105–111 CrossRef CAS PubMed.
- T. Dai, J. Xie, Q. Zhu, M. Kamariza, K. Jiang, C. R. Bertozzi and J. Rao, J. Am. Chem. Soc., 2020, 142, 15259–15264 CrossRef CAS PubMed.
- M. Kamariza, P. Shieh, C. S. Ealand, J. S. Peters, B. Chu, F. P. Rodriguez-Rivera, M. R. Babu Sait, W. V. Treuren, N. Martinson, R. Kalscheuer, B. D. Kana and C. R. Bertozzi, Sci. Transl. Med., 2018, 10, eaam6310 CrossRef PubMed.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS PubMed.
- P. Linkov, V. Krivenkov, I. Nabiev and P. Samokhvalov, Mater. Today: Proc., 2016, 3, 104–108 Search PubMed.
- C. Tang, J. Zhou, Z. Qian, Y. Ma, Y. Huang and H. Feng, J. Mater. Chem. B, 2017, 5, 1971–1979 RSC.
- M. Noh, T. Kim, H. Lee, C.-K. Kim, S.-W. Joo and K. Lee, Colloids Surf., A, 2010, 359, 39–44 CrossRef CAS.
- X. Shi, S. Dong, M. Li, X. Liu, Q. Zhang, W. Zhao, C. Zong, Y. Zhang and H. Gai, Chem. Commun., 2015, 51, 2353–2356 RSC.
- A. K. Visheratina, F. Purcell-Milton, R. Serrano-García, V. A. Kuznetsova, A. O. Orlova, A. V. Fedorov, A. V. Baranov and Y. K. Gun'ko, J. Mater. Chem. C, 2017, 5, 1692–1698 RSC.
- V. A. Kuznetsova, A. K. Visheratina, A. Ryan, I. V. Martynenko, A. Loudon, C. M. Maguire, F. Purcell-Milton, A. O. Orlova, A. V. Baranov, A. V. Fedorov, A. Prina-Mello, Y. Volkov and Y. K. Gun'Ko, Chirality, 2017, 29, 403–408 CrossRef CAS PubMed.
- I. L. Medintz, A. R. Clapp, F. M. Brunel, T. Tiefenbrunn, H. T. Uyeda, E. L. Chang, J. R. Deschamps, P. E. Dawson and H. Mattoussi, Nat. Mater., 2006, 5, 581–589 CrossRef CAS PubMed.
- I. L. Medintz, A. R. Clapp, H. Mattoussi, E. R. Goldman, B. Fisher and J. M. Mauro, Nat. Mater., 2003, 2, 630–638 CrossRef CAS.
- I. L. Medintz, J. H. Konnert, A. R. Clapp, I. Stanish, M. E. Twigg, H. Mattoussi, J. M. Mauro and J. R. Deschamps, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9612–9617 CrossRef CAS PubMed.
- D. Benito-Alifonso, S. Tremel, B. Hou, H. Lockyear, J. Mantell, D. J. Fermin, P. Verkade, M. Berry and M. C. Galan, Angew. Chem., Int. Ed., 2014, 53, 810–814 CrossRef CAS PubMed.
- M. Singh, M. Watkinson, E. M. Scanlan and G. J. Miller, RSC Chem. Biol., 2020, 1, 352–368 RSC.
- Y. Guo, I. Nehlmeier, E. Poole, C. Sakonsinsiri, N. Hondow, A. Brown, Q. Li, S. Li, J. Whitworth, Z. Li, A. Yu, R. Brydson, W. B. Turnbull, S. Pohlmann and D. Zhou, J. Am. Chem. Soc., 2017, 139, 11833–11844 CrossRef CAS.
- Y. Guo, C. Sakonsinsiri, I. Nehlmeier, M. A. Fascione, H. Zhang, W. Wang, S. Poehlmann, W. B. Turnbull and D. Zhou, Angew. Chem., Int. Ed., 2016, 55, 4738–4742 CrossRef CAS PubMed.
- Y. Yang, M. Yu, T.-T. Yan, Z.-H. Zhao, Y.-L. Sha and Z.-J. Li, Bioorg. Med. Chem., 2010, 18, 5234–5240 CrossRef CAS.
- W. Ma, H.-T. Liu, X.-P. He, Y. Zang, J. Li, G.-R. Chen, H. Tian and Y.-T. Long, Anal. Chem., 2014, 86, 5502–5507 CrossRef CAS PubMed.
- S. F. Wuister, C. de Mello Donegá and A. Meijerink, J. Phys. Chem. B, 2004, 108, 17393–17397 CrossRef CAS.
- V. A. Sarpe and S. S. Kulkarni, J. Org. Chem., 2011, 76, 6866–6870 CrossRef CAS PubMed.
- K.-H. Lee, J.-H. Lee, H.-D. Kang, B. Park, Y. Kwon, H. Ko, C. Lee, J. Lee and H. Yang, ACS Nano, 2014, 8, 4893–4901 CrossRef CAS PubMed.
- F. Purcell-Milton, A. K. Visheratina, V. A. Kuznetsova, A. Ryan, A. O. Orlova and Y. K. Gun'ko, ACS Nano, 2017, 9, 9207–9214 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra and physical data. See DOI: https://doi.org/10.1039/d3ob00368j |
| This journal is © The Royal Society of Chemistry 2023 |