
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective [2 + 2] photodimerization: a viable strategy for the synthesis of enantiopure cyclobutane derivatives†
Fabrizio
Medici
 ,
Alessandra
Puglisi
,
Sergio
Rossi
,
Laura
Raimondi
and
Maurizio
Benaglia
*
,
Alessandra
Puglisi
,
Sergio
Rossi
,
Laura
Raimondi
and
Maurizio
Benaglia
*
Dipartimento di Chimica, Università degli Studi di Milano, Via Golgi, 19, 20133 Milano, Italy. E-mail: maurizio.benaglia@unimi.it
First published on 14th March 2023
Abstract
The [2 + 2] photodimerization of cinnamic acid derivatives to afford enantiopure cyclobutanes has been investigated. The use of a chiral auxiliary represents a convenient and straightforward method to exert enantiocontrol on the reaction. By exploiting Evans oxazolidinones, the stereoselective light-driven cyclisation affords a functionalised cyclobutane ring with up to 99% enantiocontrol after removing the chiral auxiliary. In-flow experiments allowed us to improve further the efficiency of the methodology, leading to high conversion and excellent enantioselectivity.
Strained carbocycles are an intriguing class of molecules, thanks to their unusual structural motifs. They can be found in a variety of complex natural molecules, and many of them are endowed with biological activity. The possibility of opening the rings and releasing the strain energy makes them ideal candidates for further transformations.1 In particular, complex natural products containing the cyclobutane ring can be extensively found in nature and exhibit fascinating architectures and attractive biological properties (Fig. 1).
 | ||
| Fig. 1 Four-membered ring natural products. | ||
The synthesis of such molecules, however, remains quite unexplored compared to their homologues.2 The reason for this gap is mainly due to their high strain energy, about 26.7 kcal mol−1,3 which makes the synthesis of the four-membered ring quite challenging. The development of efficient methods to exert stereocontrol over the formation of the stereocenters of the ring is highly desirable since it would afford enantiomerically pure cyclobutanes.4 [2 + 2] photocatalytic cycloaddition represents one of the most popular strategies to build cyclobutanes,5 and in the last few years, following the seminal studies by Bach6 and Yoon,7 some enantioselective photocatalytic reactions have been reported,8 either using metals or organocatalysts.9 Dual catalysis is another powerful methodology for developing stereoselective photocatalytic reactions, in which a photocatalyst is used to generate reactive intermediates and a second catalytic species is used to direct the reactivity of the photogenerated intermediates.10 This synergy enables new interesting transformations, which would not be otherwise possible.11
Despite the great progress of the last decade, the development of efficient enantioselective catalytic systems is hampered by some issues, such as short lifetime and high reactivity characteristic of the photogenerated reactive intermediates, and, in general, the need for highly precise design of catalyst architectures that can provide effective enantiocontrol in a photochemical reaction. Indeed, for the [2 + 2] photocatalytic cycloaddition of cinnamic ester derivatives, a very limited number of asymmetric catalytic reactions are known. The use of a bisthiourea catalyst in a stereoselective reaction, of a chiral Rh catalyst and of a chiral Lewis acid system, and, more recently, of a chiral phosphoric acid has been reported.12
In 2017 Reiser and co-workers introduced a simple and straightforward methodology13 for building cyclobutane rings by [2 + 2] photodimerization. Cinnamic esters easily underwent cycloaddition after treatment with UV-light (450 nm) in the presence of an Ir(III) catalyst ([Ir{dF(CF3)ppy}2(dtb-bpy)]PF6), affording the desired cyclised products as shown in Scheme 1a. The reaction proceeded smoothly in excellent yields, with high diastereoselectivity in favour of the trans compound.
 | ||
| Scheme 1 [2 + 2] photodimerization of cinnamic esters. | ||
According to the authors, the [2 + 2] photodimerization of cinnamic esters proceeded by energy transfer via diradical formation. The Ir(III) catalyst is photo-excited and transfers energy to the substrate, which dimerizes via the diradical activated species.14 We reasoned that by the introduction of a chiral auxiliary on the cinnamic derivative to be cyclized, a stereoselective synthesis could be realised, which would provide an easy and practical method for the synthesis of enantioenriched cyclobutane derivatives (Scheme 1b).15
We have recently developed a stereoselective visible light-catalyzed cyclization of bis(enones) as a viable approach for the synthesis of enantiomerically enriched cyclopentane rings by inserting a chiral auxiliary on the bisenone to be cyclized.16 This offered a straightforward and convenient option to exert stereocontrol on the light-driven cyclization through a synthetic protocol that was also efficiently implemented under continuous flow conditions,17 in a home-made coil photoreactor and a custom-made, 3D-printed photoreactor.18 The aim of this work was the synthesis of cinnamic derivatives bearing Evans oxazolidinones as chiral auxiliaries, and the study of their [2 + 2] photocatalytic dimerization, both in batch and under flow conditions, to afford, upon chiral auxiliary removal, enantioenriched functionalised cyclobutanes (Scheme 1b).
The insertion of the chiral auxiliary in the scaffold was easily achieved using a well-established procedure (Fig. 2).19
 | ||
| Fig. 2 Synthesis of N-cinnamoyl-oxazolidinones 2a–d. | ||
A small library of cinnamates 2a–d bearing different chiral oxazolidinones was synthesised in excellent yields, by the treatment of cinnamic acid with SOCl2, followed by the reaction with the enantiopure oxazolidinone. Substrates 2a–d were then subjected to [2 + 2] photodimerization, under the reaction conditions previously developed by Reiser,13 using a Penn PhD photoreactor M2, operating with a blue LED at 450 nm, DMF as the solvent and [Ir{dF(CF3)ppy}2(dtb-bpy)]PF6 as the photocatalyst to afford compounds 3a–d (Table 1).

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :
:The reaction afforded the desired products in good yields after 8 hours, which favourably compares to 72 h reaction time of the work by Reiser. The products were obtained as a complex mixture of stereoisomers, which made the reaction crude difficult to analyse. Therefore, in order to remove the chiral oxazolidinone unit, the crude obtained from the photocyclization was rapidly filtered over a short pad of silica, and treated with NaOMe at −5 °C, to afford the corresponding methyl ester in quantitative yields. The product was obtained as a mixture of trans-cyclobutane 4 and cis-cyclobutane 5 (meso form) typically in a 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio (see Table 1).
:10 ratio (see Table 1).
The HPLC analysis of the chiral stationary phase showed that the trans-cyclobutane 4 was obtained with good to excellent ee (Table 1).20 The best result was obtained with (S)-iPr-substituted oxazolidinone (starting compound 3d) that led to the desired cyclobutane with 95% ee. The absolute configuration was determined by the measurement of the optical rotation value of product 4, obtained from entry 4 ([α]20D = −8.26° c = 0.25 in acetone), and comparison with the optical rotation value reported in the literature ([α]20D = −11.1 c = 12.3 in acetone), which allowed us to establish the absolute stereochemistry for the major enantiomer of product 4 to be (1R,2R,3S,4S), as shown in the structure in Table 1. Noteworthily, with (S)-Ph-substituted oxazolidinone 2a product ent-4 was obtained as the major isomer with 91% ee (entry 1, Table 1).
The scope of the reaction was then investigated on different substrates bearing (S)-isopropyl-oxazolidinone as the chiral auxiliary. A collection of cinnamoyl derivatives exhibiting different steric and electronic properties were prepared in high to excellent yields from the corresponding cinnamic acids (Fig. 3). The substrates were subjected to [2 + 2] photodimerization under the previously described conditions. The purified mixture was then treated with NaOMe in MeOH to afford the desired methyl ester derivatives, as described in Fig. 4.
 | ||
| Fig. 3 Synthesis of differently substituted N-cinnamoyl-oxazolidinones 6–17. | ||
 | ||
| Fig. 4 Scope of the reaction. Synthesis of enantioenriched cyclobutanes 18–29. | ||
To maximise the yield, the reactions were performed for 72 h; moreover, to conduct more experiments simultaneously, a home-made, multi-vial reactor, equipped with a blue LED, was fabricated and used (see the ESI† for further details). The multi-vial reactor was first benchmarked with the model substrate, leading to comparable results with those obtained with the Penn PhD photoreactor M2.
All the substrates afforded the products 18–29 in excellent yields, generally higher than 85%, and trans/cis diastereoselectivities ranging from 70/30 to 99/1 (only heteroaromatic derivatives 28 and 29 were obtained as a 1:1 mixture). Enantiomeric excesses from 80% to 95% were typically observed, but some compounds, like 22, 25, and 29, were obtained in >99% ee. Both electron-donating and electron-withdrawing groups on the aryl moiety were well tolerated in the reaction.
The [2 + 2] photodimerization was then studied under continuous flow conditions. The flow set-up is illustrated in Scheme 2 (see the ESI† for the assembly of the photoreactor and further details): a 76 μl HPFA tubing reactor was wrapped around the blue-LED photoreactor; a DMF solution of substrate 2d and the photocatalyst was fed using a syringe pump for the given residence time.
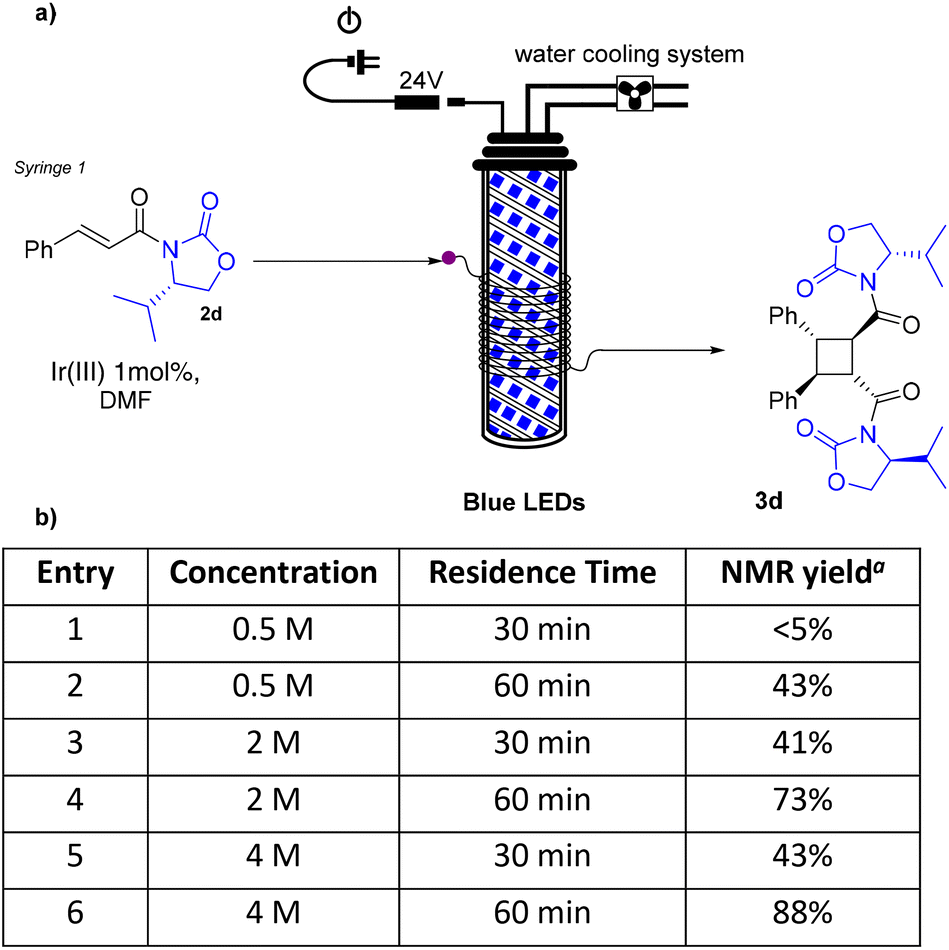 | ||
| Scheme 2 (a) Schematic representation of the flow process; (b) optimisation table. aDetermined by 1H-NMR using 1,3,5-trimethoxybenzene as the internal standard. | ||
In the first attempt, we tried to translate the batch reaction conditions to the flow set-up. The use of the same concentration of the starting material solution (0.5 M) and 30 min residence time led to the isomerization of the double bond only, with no formation of the desired product 3d (entry 1). By increasing the residence time to 60 min, 3d was obtained in 43% yield (entry 2), as assessed by 1H-NMR using 1,3,5-trimethoxybenzene as the internal standard. Better yields were observed by increasing the concentration of the starting material solution. Using a 2 M solution, 3d was produced in 41% yield within 30 min (entry 3) and in 73% yield within 60 min (entry 4). By further increasing the concentration to 4 M we were able to obtain the desired product in 88% yield in 60 min residence time (entry 6). The crude obtained from entry 6 was treated with NaOMe/MeOH solution and the purified methyl ester was analysed by HPLC, allowing us to determine the enantiomeric excess to be 93%.
Then a preliminary in-batch two-step one-pot procedure was performed, where the NaOMe/MeOH solution was directly added to the crude of the photocyclisation reactor. The reaction worked well, and afforded the expected product in 77% overall yield, 9:1 trans:cis ratio and 91% ee for compound 4 (Scheme 3).
 | ||
| Scheme 3 In batch two-step one-pot reaction. | ||
In an attempt to realize a telescoped process, after the in-flow photodimerization, a CSTR reactor was used to perform the removal of the chiral auxiliary in continuo.21 The representation of the experimental setup is illustrated in Scheme 4: a DMF solution of substrate 2d and the photocatalyst was fed using a syringe pump into the photoreactor; the output of the photoreactor was directed into a 1.5 ml CSTR, where, simultaneously, the NaOMe/MeOH solution and a filling solvent were added. The use of the filling solvent was necessary to limit the amount of the costly Ir(III) photocatalyst used in the reaction. MeOH proved to be the best solvent to fill the CSTR volume; however, this led to a dilution of the reaction mixture and a lower yield. To overcome this inconvenience, a higher amount of NaOMe (7 eq.) was added to the CSTR. Under these conditions dimethyl ester 4 was isolated in 45% yield and 77% ee, while cycloadduct 3d was recovered in 35% yield. Under these harsher conditions, it cannot be excluded that the lower ee (77% ee compared to 91% ee of Scheme 3) might be due to a partial epimerization.22
 | ||
| Scheme 4 A schematic representation of the in-flow two-step reaction, using a combination of the coil reactor and a CSTR. | ||
With the aim of increasing both the yield and enantioselectivity, in another experimental setup, the output of the in-flow photocatalytic reaction was collected in a flask where a 1 M solution of NaOMe in MeOH was continuously fed to maintain a constant 1:2 ratio of 3d:NaOMe (Scheme 5). After 1 h collection time, the reaction mixture was stirred for an additional 1 h. After purification, product 4 was obtained in 72% yield and 87% ee.
 | ||
| Scheme 5 A schematic representation of the successful in continuo two-step flow/batch reaction. | ||
A comparison between the photodimerization conducted in batch and the reaction in the microreactor is reported in Table 2. Productivity and space–time yield were calculated for the two reactors, with a method already established in the literature.23 Thanks to the shorter residence time, the microreactor provided a productivity that was almost 6-times higher than that of the batch reactor. This improvement was further evidenced by considering the space–time-yield (STY), a useful metric to compare reactors of different volumes. With a small microreactor (76 μl) it is possible to obtain product 3d at 3.52 mmol mL−1 h−1, a value that is 73-times higher than the one afforded by the batch reaction.
| Method | Productivitya [mmol h−1] | Rel. factor | STYb [mmol mL−1 h−1] | Rel. factor |
|---|---|---|---|---|
| a Productivity: moles of the product (calculated from the isolated yield) divided by the collection time required to collect the product obtained by the reaction of 0.38 mmol of 2d. b Space–time yield: moles of the product in the reactor, divided by residence time and reactor volume (for details on calculations, see the ESI†). | ||||
| Batch | 3.7 × 10−2 | 1 | 4.8 × 10−2 | 1 |
| Flow | 2.1 × 10−1 | 5.8 | 3.52 | 73 |
Conclusions
In conclusion, we have developed a stereoselective [2 + 2] photodimerization of cinnamic acid derivatives to afford enantioenriched cyclobutanes. The use of the chiral auxiliary provided functionalized cyclobutane rings in up to 99% ee. The synthetic methodology was then implemented under continuous flow conditions, obtaining the products in high yields and enantioselectivity.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SR thanks Università degli Studi di Milano (grant PSR 2020). MB thanks MUR for the project PRIN 2017 “NATURECHEM“. AP thanks MUR for the project PRIN 2017 “SURSUMCAT”. MB thanks Lombardia Region (funding the Regional HUB for the transition toward a circular economy). MB thanks ITN-EID project Marie Sklodowska-Curie Actions Innovative Training Network – TECHNOTRAIN H2020-MSCA-ITN-2018 Grant Agreement 812944, https://www.technotrain-ITN.eu (https://www.technotrain-itn.eu/). The authors thank M. Rossi for building the steel, MultiVial Photoreactor.References
- G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404–9432 CrossRef CAS PubMed.
- J. Li, K. Gao, M. Bian and H. Ding, Org. Chem. Front., 2019, 7, 136–154 RSC.
- (a) R. D. Bach and O. Dmitrenko, J. Am. Chem. Soc., 2004, 126, 4444–4452 CrossRef CAS PubMed; (b) A. Wilson and D. Goldhamer, J. Chem. Educ., 1963, 40, 504 CrossRef CAS.
- (a) K.-G. Wen, Y.-Y. Peng and X.-P. Zeng, Org. Chem. Front., 2020, 7, 2576–2597 RSC; (b) S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed.
- D. Sarkar, N. Bera and S. Ghosh, Eur. J. Org. Chem., 2020, 1310–1326 CrossRef CAS.
- (a) R. Brimioulle and T. Bach, Science, 2013, 342, 840–843 CrossRef CAS PubMed; (b) C. Brenninger, J. D. Jolliffe and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 14338–14349 CrossRef CAS PubMed.
- (a) T. R. Blum, Z. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Science, 2016, 354, 1391–1395 CrossRef CAS PubMed; (b) Z. D. Miller, B. J. Lee and T. P. Yoon, Angew. Chem., Int. Ed., 2017, 56, 11891–11895 CrossRef CAS PubMed.
- (a) M. J. Genzink, J. B. Kidd, W. B. Swords and T. P. Yoon, Chem. Rev., 2022, 122, 1654–1716 CrossRef CAS PubMed; (b) F.-D. Lu, J. Chen, X. Jiang, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2021, 50, 12808–12827 RSC.
- Selected recent examples: (a) H. Yu, S. Dong, Q. Yao, L. Chen, D. Zhang, X. Liu and X. Feng, Chem. – Eur. J., 2018, 24, 19361–19367 CrossRef CAS PubMed; (b) J. Zheng, W. B. Swords, H. Jung, K. L. Skubi, J. B. Kidd, G. J. Meyer, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 13625–13634 CrossRef CAS PubMed; (c) N. Vallavoju, S. Selva-kumar, S. Jockusch, M. P. Sibi and J. Sivaguru, Angew. Chem., Int. Ed., 2014, 53, 5604–5608 CrossRef CAS PubMed; (d) A. Tröster, R. Alonso, A. Bauer and T. Bach, J. Am. Chem. Soc., 2016, 138, 7808–7811 CrossRef PubMed; (e) S. Stegbauer, C. Jandl and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 14593–14596 CrossRef CAS PubMed; (f) F. Pecho, Y.-Q. Zou, J. Gramüller, T. Mori, S. M. Huber, A. Bauer, R. M. Gschwind and T. Bach, Chem. – Eur. J., 2020, 26, 5190–5194 CrossRef CAS PubMed; (g) T. Rigotti, R. Mas-Ballesté and J. Alemán, ACS Catal., 2020, 10, 5335–5346 CrossRef CAS; (h) E. M. Sherbrook, M. J. Genzink, B. Park, I. A. Guzei, M.-H. Baik and T. P. Yoon, Nat. Commun., 2021, 12, 5735 CrossRef CAS PubMed.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- (a) A. Gualandi, P. G. Cozzi, G. Rodeghiero, T. P. Jansen and R. Perciaccante, Phys. Sci. Rev., 2019, 20180098 Search PubMed; (b) M. M. Mastrandrea and M. A. Pericas, Eur. J. Org. Chem., 2021, 3421–3431 CrossRef.
- (a) R. Telmesani, S. H. Park, T. Lynch-Colameta and A. B. Beeler, Angew. Chem., Int. Ed., 2015, 54, 11521–11525 CrossRef CAS PubMed; (b) X. Huang, T. R. Quinn, K. Harms, R. D. Webster, L. Zhang, O. Wiest and E. Meggers, J. Am. Chem. Soc., 2017, 139, 9120–9123 CrossRef CAS PubMed; (c) M. E. Daub, H. Jung, B. J. Lee, J. Won, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 9543–9547 CrossRef CAS PubMed; (d) see also ref. 9h.
- S. H. Pagire, A. Hossain, L. Traub, S. Kerres and O. Reiser, Chem. Commun., 2017, 53, 12072–12075 RSC.
- (a) X. Guo, Q. Chen, Y. Tong, Y. Li, Y. Liu, D. Zhao and Y. Ma, J. Phys. Chem. A, 2018, 122, 6963–6969 CrossRef CAS PubMed; (b) A. Neubauer, G. Grell, A. Friedrich, S. I. Bokarev, P. Schwarzbach, F. Gärtner, A. E. Surkus, H. Junge, M. Beller, O. Kühn and S. Lochbrunner, J. Phys. Chem. Lett., 2014, 5, 1355–1360 CrossRef CAS PubMed.
- (a) D. A. Evans, G. Helmchem and M. Ruping, Chiral auxiliaries in asymmetric synthesis, in Asymmetric Synthesis-The Essentials, ed. M. Christman, Wiley-VCH, Weinheim, Germany, 2007, pp. 3–9 Search PubMed; (b) M. M. Heravi, V. Zadsirjan and B. Farajpour, RSC Adv., 2016, 6, 30498–30551 RSC; (c) G. Diaz-Muñoz, I. L. Miranda, S. K. Sartori, D. C. de Rezende and M. A. Nogueira Diaz, Chirality, 2019, 31, 776–812 CrossRef PubMed.
- F. Medici, S. Resta, P. Presenti, L. Caruso, A. Puglisi, L. Raimondi, S. Rossi and M. Benaglia, Eur. J. Org. Chem., 2021, 4521–4524 CrossRef CAS.
- Selected reviews on flow chemistry: (a) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed; (b) T. Yu, Z. Ding, W. Nie, J. Jiao, H. Zhang, Q. Zhang, C. Xue, X. Duan, Y. M. A. Yamada and P. Li, Chem. – Eur. J., 2020, 26, 5729–5574 CrossRef CAS PubMed. For recent reviews on flow chemistry technologies applied to APIs synthesis see: (c) B. Gutmann, D. Cantillo and O. K. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed; (d) R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef CAS; (e) M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Org. Process Res. Dev., 2020, 24, 1802–1813 CrossRef CAS.
- Selected reviews on photochemical transformations in flow: (a) D. Cambié, C. Bottecchia, N. J. V. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed; (b) C. Sambiago and T. Noël, Trends Chem., 2020, 2, 92–106 CrossRef; (c) T. H. Rehm, Chem. – Eur. J., 2020, 26, 16952–16974 CrossRef CAS PubMed.
- V. A. Soloshonok, H. Uekia, C. Jianga, C. Caib and V. J. Hruby, Helv. Chim. Acta, 2002, 85, 3616–3623 CrossRef CAS.
- B. S. Green, A. T. Hagler, Y. Rabinsohn and M. Rejto, Isr. J. Chem., 1976, 15, 124–130 CrossRef.
- G. L. Foutch and A. H. Johannes, Reactors in Process Engineering, in Encyclopedia of Physical Science and Technology, ed. R. A. Meyers, Academic Press, New York, 3rd edn, 2003, pp. 23–43 Search PubMed.
- In our previous work (ref. 16), it was verified that the removal of the chiral auxiliary occurred without any racemization and was not detrimental to the stereochemical integrity of the stereocenters. However, in the present work it was not possible to determine d.r. before the cleavage, because of many overlapping signals in the 1H-NMR spectra of the cyclobutane featuring the oxazolidinone ring. Partial epimerization in the reaction with NaOMe cannot be completely excluded, and a fine tuning of the experimental conditions for chiral auxiliary removal for each compound is probably needed. Some preliminary experiments with model compound 2d to afford product 4 showed that a careful optimization allowed to obtain the product in 98% ee, compared to 95% ee obtained in Table 1.
- (a) M. Neumann and K. Zeitler, Org. Lett., 2012, 14, 2658–2661 CrossRef CAS PubMed; (b) F. Herbrik, M. Sanz, A. Puglisi, S. Rossi and M. Benaglia, Chem. – Eur. J., 2022, 28 DOI:10.1002/chem.202200164.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00232b |
| This journal is © The Royal Society of Chemistry 2023 |