
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective synthesis of vinyl nitriles through a Ramberg–Bäcklund approach†
Octavio A.
Valle-González
,
Ángel I.
Salazar-Bello
and
J.
Armando Luján-Montelongo
 *
*
Departamento de Química, Centro de Investigación y de Estudios Avanzados (Cinvestav), Av. Instituto Politécnico Nacional 2508, San Pedro Zacatenco, 07360 Ciudad de México, Mexico. E-mail: jalujanm@cinvestav.mx
First published on 10th March 2023
Abstract
The applicability of vinyl nitriles in the preparation of pharmaceuticals, polymers, and other valuable materials benefits from robust preparative methodologies. In this work, we present a novel approach for the synthesis of vinyl nitriles based on the Ramberg–Bäcklund olefination reaction (RBR). While there are few examples for accessing functionalized olefins using the RBR, we believe that this methodology embodies useful means for installing privileged vinylnitrile building blocks, such as the acrylonitrile fragment of the US FDA-approved antiviral rilpivirine.
Classic olefination reactions, such as the Wittig,1 Julia–Kocienski,2 Horner–Wadsworth–Emmons,3 and Peterson reactions,4 require two complementary reactants with reaction-specific functional groups: a nucleophile (such as phosphorus, sulfonyl, phosphonate, or silicon ylides) and a carbonyl electrophile. In contrast, the Meyer modification5 of the Ramberg–Bäcklund reaction (RBR)6 only requires a sulfonyl group that is flanked by two activated C–H bonds (such as methyl, methylene, or methine). This is significant as the sulfonyl group (either as such or masked as a less oxidized S-functional group) can be brought through a synthetic campaign up to a late-stage.7 The stereochemistry of the RBR has been widely discussed and the consensus is that it depends on the strength of the base used. Strong bases lead to the formation of E isomers, while weak bases lead to Z olefins.8 Although there is a relative scarcity of RBRs for the preparation of functionalized olefins (vinyl groups bearing a functional group),9 some have recently been utilized for synthesizing valuable species that are difficult to access by other methods.10 To the best of our knowledge, there are no reports on the preparation of vinyl nitriles using the Ramberg–Bäcklund reaction.
The vinyl nitrile group is a privileged moiety in many pharmaceuticals (Fig. 1a) and features varied synthetic applications.11,12 In materials science, polymers based on cyanoacrylates are widely used as adhesives13 and support materials.14,15 Vinyl nitriles have also been used as building blocks for synthesizing aryl piperidine carbinols, valuable intermediates in the synthesis of paroxetine-like compounds.16
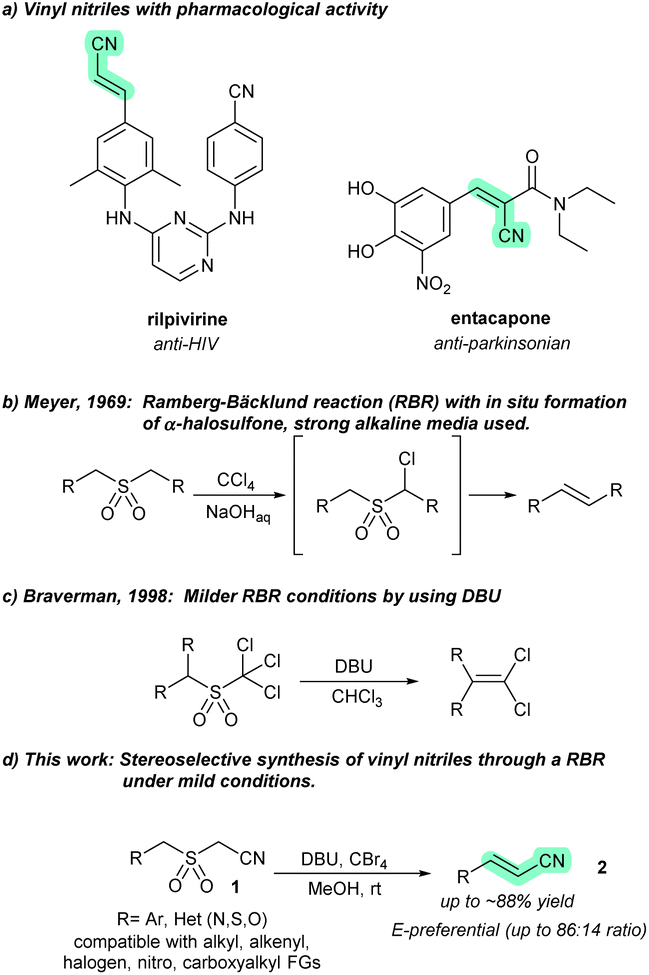 | ||
| Fig. 1 (a) Vinyl nitriles with pharmacological activity. (b) Meyer modification of the Ramberg–Bäcklund reaction (RBR). (c) Use of DBU for milder conditions in the RBR. (d) This work: vinyl nitrile synthesis from α-benzyl-α′-cyano sulfones using a RBR under mild conditions. | ||
In recent times, significant attention has been focused on alcohol ammoxidation,17 the dehydration of oximes18 and amides,19 and the Heck-type coupling between acrylonitriles and aryl halides,20 for the synthesis of vinyl nitriles. However, many of these methods, albeit catalytic, rely on transition metal (TM)-based catalysts. While some of these efforts are an improvement over more traditional approaches, as a result of their catalytic nature and their avoidance of highly toxic species (e.g., cyanide) or multistep sequences, there is a pressing need for reliable methods to access vinyl nitriles with high stereoselectivity. We believe that the RBR has not been extensively explored for synthesizing electron-withdrawing group (EWG)-substituted olefinic products, such as vinyl nitriles. Most RBRs are performed in aqueous, highly alkaline media (Fig. 1b), which limits their applicability in late-stage olefinations due to poor chemoselectivity. This can lead to undesired anionic polymerization on EWG-substituted olefinic products.21 A milder approach involves the use of guanidine-type bases in RBRs, enabling higher tolerance for the preparation of functionalized olefins, as exemplified by the syntheses of 1,1-dichloroalkenes by an RBR under anhydrous conditions, achieving yields of up to 70–90% (Fig. 1c).22
As part of our interest in synthetic methodologies based on sulfur functional groups for advanced synthetic applications,23 including the generation of olefinic blocks for materials,24 we aimed to develop a robust method for the synthesis of vinyl nitriles using the RBR (Fig. 1d). We used 2-(benzylsulfonyl)acetonitrile (1a) as an α-aryl-α′-cyano sulfone probe to investigate RBR-based olefinations. This compound was easily prepared from benzyl mercaptan using an alkylation–oxidation sequence (see the ESI†) and proved to be a convenient choice for our research. The treatment of 1a with tetrabromomethane (CBr4) and 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) delivered, with complete conversion, olefinic products 2a-E and 2a-Z (ratio 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15, respectively) in 69% isolated yield after 2.5 h (entry 1, Table 1).
:15, respectively) in 69% isolated yield after 2.5 h (entry 1, Table 1).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Base | Equiv. | X+ source | Equiv. | Yield % | dr (E):(Z) |
| 1 | DBU | 2 | CBr4 | 1.1 | 69 | 85:15 |
| 2 | DBU | 3 | CBr 4 | 1.1 | 85 |
86![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :14 :14
|
| 3 | iPr2NH | 3 | CBr4 | 1.1 | 28 | 80:20 |
| 4 | iPr2NEt | 3 | CBr4 | 1.1 | 24 | 83:17 |
| 5 | DABCO | 3 | CBr4 | 1.1 | — | — |
| 6 | TMP | 3 | CBr4 | 1.1 | — | — |
| 7 | Et3N | 3 | CBr4 | 1.1 | — | — |
| 8 | Piperidine | 3 | CBr4 | 1.1 | — | — |
| 9 | DMAP | 3 | CBr4 | 1.1 | — | — |
| 10 | DBU | 3 | CCl4 | 1.1 | — | — |
| 11 | DBU | 3 | NBS | 1.1 | 12 | 75:25 |
| 12 | DBU | 3 | DBTCE | 1.1 | 65 | 85:15 |
| 13 | DBU | 3 | p-TsBr | 1.1 | 41 | 83:17 |

A screening of nitrogenated bases was performed on the same substrate (1a), with diisopropylamine (iPr2NH, DIPA) and diisopropylethylamine (iPr2NEt, DIPEA) proving productive (entries 3 and 4, Table 1) but not as high yielding as DBU (entry 2, Table 1). We observed no correlation between the screened nitrogenated bases and the olefin stereochemistry in any of the cases studied, as the 2e-Z product was the prevailing stereochemical outcome. No clear relationship between base strength and reaction yields was established, although bases with higher pKa values generally showed better results. As anticipated, neither episulfones nor α-halogenated sulfones were detected.25
Besides CBr4, additional halogen sources were also screened including tetrachloromethane (CCl4) (entry 10, Table 1),5N-bromosuccinimide (NBS) (entry 11),26 dibromotetrachloroethane (DBTCE) (entry 12),27 and p-toluenesulfonyl bromide (p-TsBr) (entry 13). While DBTCE, NBS and p-TsBr delivered olefinic products (entries 11–13), CBr4 yielded the best results. Interestingly, no conversion was observed when CCl4 was used (entry 10).
Under the optimized conditions, we next explored the substrate scope to test functional group tolerance and performance (Table 2). All substrates were easily prepared through the S-alkylation of the corresponding thiols or thioacetates with chloroacetonitrile, followed by oxidation using either m-chloroperoxybenzoic (mCPBA) acid or Oxone® (see the ESI†). Substrates bearing electron-donating (e.g., t-Bu and OMe; 1c and 1d) or electron-withdrawing (e.g., NO2 and CO2Me; 1e and 1g) groups underwent complete olefination, with the latter substrates delivering lower yields, presumably because of an increased propensity to react through undesired anionic polymerization pathways as a result of the increased electrophilicity of the olefinic products (2e and 2g; entries 5 and 7, Table 2). In most cases, a strong stereochemical preference for E over Z products was observed,28 except for 2-((2-methylbenzyl)sulfonyl)acetonitrile (1f, entry 8), which gave poorer stereoselectivity (2h-E:2h-Z = 73:27) and inferior yield (22%). In our case, this result can be rationalized through the plausible conformations of diastereoisomeric adducts of 1 with DBU that precede the corresponding W-type transition states (Scheme 1). The presence of an o-substituent such as Me in 1h leads to an increase of destabilizing repulsive interactions on kinetically preferred conformers A andB (especially on B),29 thus diminishing the E:Z ratio. An indole-based substrate (1m, entry 13) showed a similar outcome, conceivably caused by the N-mesyl group albeit more intense. An α,β-unsaturated sulfone (1i, entry 9) led to the corresponding olefin 2i, retaining the original E-stereochemistry, but exhibiting a modest E preference on the second unsaturation. In the case of sulfone 1j (entry 10), we were able to couple a β-dehydrobromination30 for the preparation of a 2-en-4-ynenitrile derivative, although this process seems to have impacted the stereoselectivity as the Z isomer (2j-Z) was obtained preferentially and the product featured partial Br substitution by OMe (see the ESI†). Interestingly, both tertiary α′-ethyl-substituted sulfone (1n, entry 14) and an α-benzyl-substituted substrate (1o, entry 15) exhibited similar stereoselectivity as in the previous cases. It is important to note that the α-tertiary benzylic substrate 1o was prepared through dianion-type alkylation, a novel approach when applied to switchable regioselective alkylations of α-benzyl-α’-cyano sulfones (see the ESI†). Substrates with heterocyclic rings such as furyl (1k), thiophenyl (1l) and indolyl (1m) (entries 11–13, Table 2) were suitable for this method and the stereopreference remained E selective, except for 1m (vide supra). Unfortunately, α-alkyl substrates (1p and 1q) seemed unreactive under our conditions which can be understood by the relatively low α-acidity (entries 16 and 17).
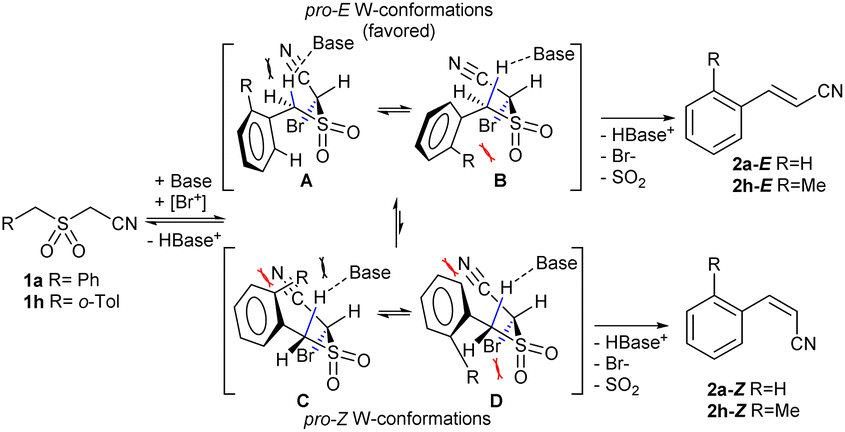 | ||
| Scheme 1 Plausible conformers preceding W-planar transition states leading to stereoisomers 2-E and 2-Z; if o-substituent R = Me (1h), repulsive interactions increase for A–B conformers leading to a decrease of the E:Z ratio. Red marks denote significant steric strain. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Sulfone (1) | Vinyl nitrile (2) | Yield % | E/Z ratio |
| a ca. 7% of (2E,4E)-4-methoxy-5-phenylpenta-2,4-dienenitrile was also detected, see the ESI.† | ||||
| 1 | 1a |

|
85 | 86:14 |
| 2 | 1b |

|
76 | 83:17 |
| 3 | 1c |

|
88 | 82:18 |
| 4 | 1d |
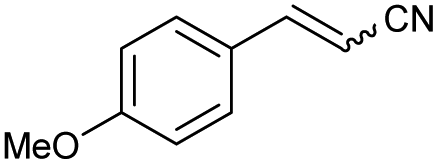
|
61 | 83:17 |
| 5 | 1e |

|
49 | 82:18 |
| 6 | 1f |
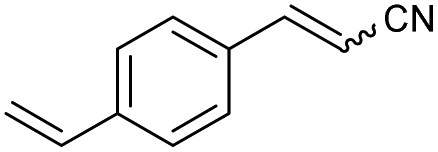
|
76 | 77:23 |
| 7 | 1g |

|
49 | 83:17 |
| 8 | 1h |

|
22 | 73:27 |
| 9 | 1i |

|
60 | 58:42 |
| 10 | 1j |
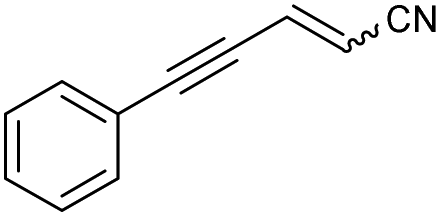
|
48a | 31:69 |
| 11 | 1k |

|
55 | 80:20 |
| 12 | 1l |

|
58 | 75:25 |
| 13 | 1m |

|
60 | 54:46 |
| 14 | 1n |

|
70 | 72:28 |
| 15 | 1o |

|
88 | 72:28 |
| 16 | 1p |

|
Trace | — |
| 17 | 1q |

|
— | — |

Rilpivirine (EDURANT™, Fig 1a, Scheme 2a) is an antiviral agent against human immunodeficiency virus 1 (HIV-1), approved by the US FDA in 2011.31 Strategically, rilpivirine synthesis relies on the availability of E-3-(4-amino-3,5-dimethylphenyl)acrylonitrile (3), which is coupled to 4-((4-chloropyrimidin-2-yl)amino)benzonitrile (4) (Scheme 2a). A patent study describes the synthesis of 3 starting with a Pd(OAc)2/P(oTol)3-catalyzed coupling,32 while another, more recent, case uses a Heck reaction catalyzed by Pd/C.33 Both routes require the use of catalysts based on transition metals (e.g., Pd(OAc)2, P(oTol)3, and Pd/C), which are not ideal for industrial setups.34 Our approach to access arylacrylonitriles through the RBR seemed ideal for the development of a transition metal-free formal synthesis of rilpivirine through the preparation of 3 (Scheme 2b). Masked benzylmercaptane 6 was obtained from aminoalcohol 5 by a thio-Mitsunobu reaction, followed by tandem acetyl cleavage/S-alkylation using chloroacetonitrile under basic media, and oxidation with Oxone® to deliver sulfone precursor 7. Finally, the RBR method applied on 7 cleanly delivered acrylonitrile 3 with 68% olefination yield in an 83:17 E/Z ratio. Remarkably, no N-protecting group was needed in the sequence. Efforts on the optimization of the efficiency and stereoselectivity enhancement towards E-3 are currently underway in our laboratory.
 | ||
| Scheme 2 (a) Assembly of rilpivirine by coupling acrylonitrile 3 and chloropyrimidine 4. (b) A transition metal-free synthesis of E-3-(4-amino-3,5-dimethylphenyl)acrylonitrile 3, a strategic intermediary for the synthesis of rilpivirine. | ||
Submission of an isocyanide analog of 1a (((isocyanomethyl)sulfonyl)methyl) benzene (8a)23a delivered olefinic products in comparable yields; however, the stereoselectivity was completely reversed, as the 9a-E:9a-Z ratio was 20:80 (Scheme 3). This stereoselectivity “switch” was also revealed for 4-methoxy isocyanide 9d, supporting a stereodivergency phenomenon triggered by the isocyano group.35
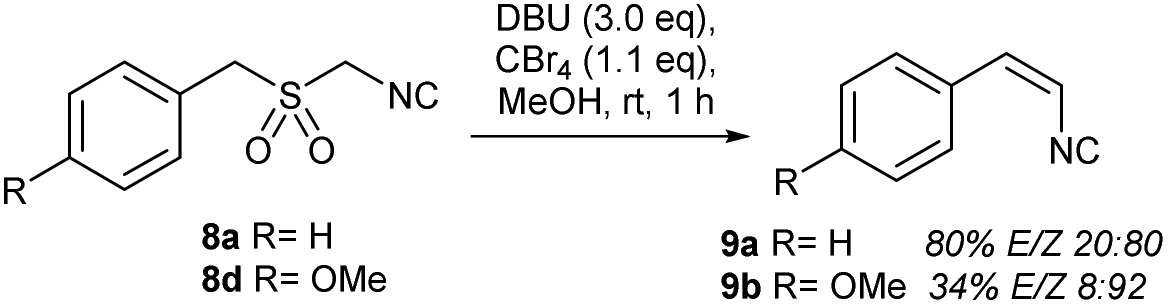 | ||
| Scheme 3 Z-Preferential RBR-based olefination of α-benzyl-α′-isocyano sulfones 8a and 8d. | ||
Conclusions
We have developed a method for the RBR-based olefination of α-benzyl-α′-cyano sulfones (1) with a preference towards E stereoisomeric acrylonitriles (E-2). Our approach is tolerant to substrates bearing both electron-donating and electron-withdrawing groups but limited towards 2-substituted (aryl) substrates that delivered stereoselectivity loss, plausibly caused by steric strain. Heterocycles in general were also suitable for olefination, whereas fully aliphatic sulfones could not be transformed satisfactorily. The method was applied to a transition metal-free synthesis of E-3-(4-amino-3,5-dimethylphenyl) acrylonitrile (3), a strategic block used for the synthesis of the antiviral rilpivirine. The extrapolation of the olefination method to analogous selected α-benzyl-α′-isocyano sulfones (8a and 8d) featured a stereochemical reversal as Z isomers were preferred. We are currently optimizing the route to acrylonitrile 3, and a comprehensive study regarding the synthetic development of the RBR-based olefination of α-benzyl-α′-isocyano sulfones will be released in the near future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Conacyt (Mexico) through a Ciencia Básica grant (CB-2014-241455). O. A. V.-G. wishes to thank Conacyt for a Ph.D. fellowship (614059). A. I. S.-B. also thanks Conacyt for an undergraduate fellowship (27328) within grant CB-2014-241455. Teresa Cortéz, Géiser Cuéllar and Iris Ramos are thanked for their assistance in selected spectra acquisition. Authors thank Dr. Allen Chao for helpful suggestions regarding the manuscript.References
- P. J. Murphy and J. Brennan, Chem. Soc. Rev., 1988, 17, 1–30 RSC.
- P. X. T. Rinu, S. Radhika and G. Anilkumar, ChemistrySelect, 2022, 7, e202200760 CrossRef CAS.
- K. Kobayashi, K. Tanaka and H. Kogen, Tetrahedron Lett., 2018, 59, 568–582 CrossRef CAS.
- L. F. van Staden, D. Gravestock and D. J. Ager, Chem. Soc. Rev., 2002, 31, 195–200 RSC.
- C. Y. Meyers, A. M. Malte and W. S. Matthews, J. Am. Chem. Soc., 1969, 91, 7510–7512 CrossRef CAS.
- L. Ramberg and B. Bäcklund, Ark. Kemi, Mineral. Geol., 1940, 13A, 1–50 Search PubMed.
- (a) J. A. Kozak and G. R. Dake, Angew. Chem., Int. Ed., 2008, 47, 4221–4223 CrossRef CAS PubMed; (b) K. C. Nicolaou, D. Sarlah, T. R. Wu and W. Zhan, Angew. Chem., Int. Ed., 2009, 48, 6870–6874 CrossRef CAS PubMed; (c) L. J. Baird, M. S. M. Timmer, P. H. Teesdale-Spittle and J. E. Harvey, J. Org. Chem., 2009, 74, 2271–2277 CrossRef CAS PubMed.
- R. J. K. Taylor and G. Casy, Org. React., 2003, 62, 357–475 CAS.
- Selected literature: (a) P. A. Grieco and D. Boxler, Synth. Commun., 1975, 5, 315–318 CrossRef CAS; (b) M. G. Ranasinghe and P. Fuchs, J. Am. Chem. Soc., 1989, 111, 779–782 CrossRef CAS; (c) P. Evans and R. J. K. Taylor, Tetrahedron Lett., 1997, 38, 3055–3058 CrossRef CAS; (d) J. Burghart, A. Sorg and R. Brückner, Chem. – Eur. J., 2011, 17, 6469–6483 CrossRef CAS PubMed.
- Y. Mauekawa, M. Nambo, D. Yokogawa and C. M. Crudden, J. Am. Chem. Soc., 2020, 142, 15667–15672 CrossRef PubMed.
- F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902–7917 CrossRef CAS PubMed.
- F. F. Fleming and Q. Wang, Chem. Rev., 2003, 103, 2035–2077 CrossRef CAS PubMed.
- G. M. Bot, K. G. Bot, J. O. Ogunranti, J. A. Onah, A. Z. Sule, I. Hassan and E. D. Dung, J. Surg. Tech. Case Rep., 2010, 2, 44–48 CrossRef CAS PubMed , and references included within.
- S. Olivera, H. B. Muralidhara, K. Venkatesh, K. Gopalakrishna and C. S. Vivek, J. Mater. Sci., 2016, 51, 3657–3674 CrossRef CAS.
- D. Hambali, N. H. Zainol, L. Othman, K. B. Md Isa and Z. Osman, Ionics, 2019, 25, 1187–1198 CrossRef CAS.
- B. Ronsen and S. P. Upadhyaya, Process for Preparing Arylpiperidine Carbinol Intermediates and Derivatives, WO2002076940A2, 2002.
- Selected recent literature: (a) K. Sun, H. Shan, H. Neumann, G. P. Lu and M. Beller, Nat. Commun., 2022, 13, 1848 CrossRef CAS PubMed; (b) T. Senthamarai, V. G. Chandrashekhar, N. Rockstroh, J. Rabeah, S. Bartling, R. V. Jagadeesh and M. Beller, Chem, 2022, 8, 508–531 CrossRef CAS; (c) A. K. Das, S. Nandy and S. Bhar, RSC Adv., 2022, 12, 4605–4614 RSC; (d) K. Kamata, N. Kinoshita, M. Koutani, R. Aono, E. Hayashi and M. Hara, Catal. Sci. Technol., 2022, 12, 6219–6230 RSC; (e) H. Wang, D. Xu, E. Guan, L. Wang, J. Zhang, C. Wang, S. Wang, H. Xu, X. Meng, B. Yang, B. C. Gates and F. S. Xiao, ACS Catal., 2020, 10, 6299–6308 CrossRef CAS.
- Selected recent literature: (a) T. Ghosh, A. Mohammad and S. M. Mobin, ACS Sustainable Chem. Eng., 2019, 7, 13746–13763 CrossRef CAS; (b) H. Gao, J. Y. Chen, Z. Peng, L. Feng, C. H. Tung and W. Wang, J. Org. Chem., 2022, 87, 10848–10857 CrossRef CAS PubMed; (c) D. Zhang, Y. Huang, E. Zhang, R. Yi, C. Chen, L. Yu and Q. Xu, Adv. Synth. Catal., 2018, 360, 784–790 CrossRef CAS; (d) P. N. Borase, P. B. Thale and G. S. Shankarling, ChemistrySelect, 2018, 3, 5660–5666 CrossRef CAS.
- Selected literature: (a) S. Ren, Y. Wang, F. Yang, H. Sun and X. Li, Catal. Commun., 2019, 120, 72–75 CrossRef CAS; (b) G. Chang, X. Li, P. Zhang, W. Yang, K. Li, Y. Wang, H. Sun, O. Fuhr and D. Fenske, Appl. Organomet. Chem., 2020, 34, 1–8 CrossRef; (c) A. B. Wood, J. R. A. Kincaid and B. H. Lipshutz, Green Chem., 2022, 24, 2853–2858 RSC.
- (a) P. Moradi, M. Hajjami and F. Valizadeh-Kakhki, Appl. Organomet. Chem., 2019, 33, 1–13 Search PubMed; (b) M. A. Jani and K. Bahrami, J. Exp. Nanosci., 2020, 15, 182–201 CrossRef CAS.
- Selected literature: (a) Y. I. Estrin, A. E. Tarasov, A. A. Grishchuk, A. V. Chernyak and E. R. Badamshina, RSC Adv., 2016, 6, 106064–106073 RSC; (b) A. Zilkha and B.-A. Feit, J. Appl. Polym. Sci., 1961, 5, 251–260 CrossRef CAS.
- (a) S. Braverman and Y. Zafrani, Tetrahedron, 1998, 54, 1901–1912 CrossRef CAS; (b) C. P. Raj, T. Pichnit and S. Braverman, Tetrahedron Lett., 2000, 41, 1501–1504 CrossRef CAS.
- (a) J. A. Lujan-Montelongo, A. Ojeda Estevez and F. F. Fleming, Eur. J. Org. Chem., 2015, 1602–1605 CrossRef CAS PubMed; (b) C. Silva-Cuevas, C. Perez-Arrieta, L. A. Polindara-García and J. A. Lujan-Montelongo, Tetrahedron Lett., 2017, 58, 2244–2247 CrossRef CAS; (c) C. Silva-Cuevas, E. Paleo, D. F. León-Rayo and J. A. Lujan-Montelongo, RSC Adv., 2018, 8, 24654–24659 RSC.
- (a) A. R. Hernandez-Martínez, J. A. Lujan-Montelongo, C. Silva-Cuevas, J. D. Mota-Morales, M. Cortez-Valadez, A. J. Ruíz-Baltazar, M. Cruz and J. Herrera-Ordonez, React. Funct. Polym., 2018, 122, 75–84 CrossRef; (b) A. Ramos-Jacques, J. A. Lujan-Montelongo, C. Silva-Cuevas, M. Cortez-Valadez, M. Estevez and A. R. Hernandez-Martínez, Eur. Polym. J., 2018, 101, 262–272 CrossRef CAS.
- (a) A. G. Sutherland and R. J. K. Taylor, Tetrahedron Lett., 1989, 30, 3267–3270 CrossRef CAS; (b) E. Vedejs and S. P. Singer, J. Org. Chem., 1978, 43, 4884–4885 CrossRef CAS. For an incomplete RBR that delivered an α-halosulfone: (c) R. J. K. Taylor, Chem. Commun., 1999, 217–227 RSC.
- F. G. Bordwell and E. Doomes, J. Org. Chem., 1974, 39, 2526–2531 CrossRef CAS.
- DBTCE is a non-ozone-depleting source of electrophilic bromine: S. C. Söderman and A. L. Schwan, J. Org. Chem., 2012, 77, 10978–10984 CrossRef PubMed.
- A similar phenomenon was observed in a recent RBR-based olefination developed by Bognar and van Gemmeren. In their synthesis of olefins from thiols and aldehydes through an α-sulfinoyl sulfone intermediate, the stereoselectivity under E-preferential conditions was attributed primarily to the acidic properties of the benzylic α-position, which is assumed to be equilibrated towards the most stable pro-E-conformer by deprotonation (authors used strong alkali in protic media at 120 °C): S. Bognar and M. van Gemmeren, Chem. – Eur. J., 2023, e202203512 CAS.
- We conducted controlled RBR-olefination experiments to rule out post-RBR E/Z equilibration. Initially, 1a was subjected to DBU with extended reaction times of 12 hours. Additionally, an enriched mixture of Z-2a was exposed to DBU in MeOH, which demonstrated no isomerization towards the E isomer even after 24 hours (see the ESI†). Together, these experiments revealed a robust stereochemical E/Z ratio of olefins that are not susceptible to isomerization under the reaction conditions.
- T. Ohgiya, N. Kutsumura and S. Nishiyama, Synlett, 2008, 3091–3105 CrossRef CAS.
- FDA Approves EDURANT™ (rilpivirine) For Use in Treatment-Naïve Adults with HIV-1 https://www.jnj.com/media-center/press-releases/fda-approves-edurant-rilpivirine-for-use-in-treatment-naive-adults-with-hiv-1 (accessed December 1, 2022).
- J. E. G. Guillemont, P. Palandjian, M. R. Dejonge, L. M. H. Koymans, H. M. Vinkers, F. F. D. Daeyaert, J. Heeres, K. J. A. van Aken, P. J. Lewi and P. A. Janssen, HIV Inhibiting Pyrimidine Derivatives, WO2003016306A1, 2003.
- B. Parthasaradhi Reddy, K. Rathnakar Reddy, D. Muralidhara Reddy, A. Venkat Narsimha Reddy and B. Vamsi Krishna, Process for Rilpivirine, WO2012147091A2, 2012.
- T. Zhang, J. Yang, Z. Zhou, Z. Fu, S. Cherukupalli, D. Kang, P. Zhan and X. Liu, BMC Chem., 2021, 15, 22 CrossRef CAS PubMed.
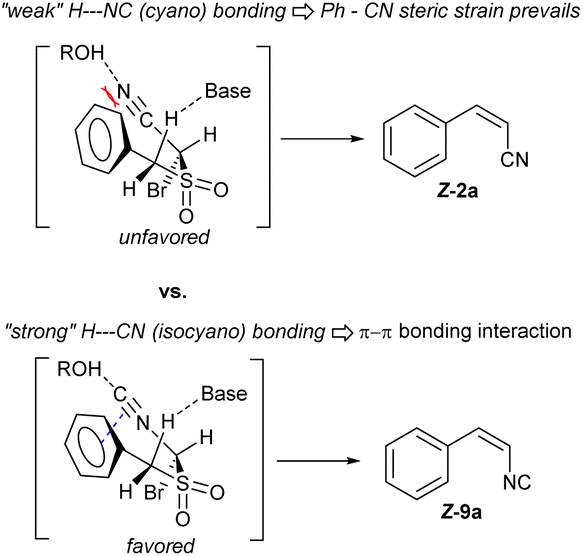
- The Z-preference in the case of α-benzyl-α′-isocyano sulfone olefination is being currently studied in our lab. Early spectroscopic evidence, and additional substrate and condition screenings suggest a non-covalent bonding interaction involving the isocyanide as a significant π-hole acceptor, aided by protic solvents such as MeOH. Selected literature: isocyanide π-hole.
(a) A. S. Mikherdov, M. A. Kinzhalov, A. S. Novikov, V. P. Boyarskiy, I. A. Boyarskaya, M. S. Avdontceva and V. Y. Kukushkin, Inorg. Chem., 2018, 57, 6722–6733 CrossRef CAS PubMed Unexpected Z-selectivity in a RBR by π-stacking:
(b) J. S. Foot, G. M. P. Giblin, A. C. Whitwood and R. J. K. Taylor, Org. Biomol. Chem., 2005, 3, 756–763 RSC Isocyanides as H-bonding bases:
(c) L. L. Ferstandig, J. Am. Chem. Soc., 1962, 84, 1323–1324 CrossRef CAS;
(d) L. L. Ferstandig, J. Am. Chem. Soc., 1962, 84, 3553–3557 CrossRef CAS;
(e) P. v. R. Schleyer and A. Allerhand, J. Am. Chem. Soc., 1962, 84, 1322–1323 CrossRef .
.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00214d |
| This journal is © The Royal Society of Chemistry 2023 |