
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic artificial nitroalkane oxidases – a way towards organocatalytic umpolung†
Adam
Pokluda
 a,
Ekaterina
Zubova
a,
Josef
Chudoba
b,
Martin
Krupička
a and
Radek
Cibulka
*a
a,
Ekaterina
Zubova
a,
Josef
Chudoba
b,
Martin
Krupička
a and
Radek
Cibulka
*a
aDepartment of Organic Chemistry, University of Chemistry and Technology, Prague, Technická 5, 166 28 Prague, Czech Republic. E-mail: cibulkar@vscht.cz
bCentral Laboratories, University of Chemistry and Technology, Prague, Technická 5, 166 28 Prague, Czech Republic
First published on 7th March 2023
Abstract
Nitroalkane oxidases (NAOs) are flavoenzymes that catalyse the oxidation of nitroalkanes to their corresponding carbonyl compounds while producing nitrite anions. Herein, we present an artificial catalytic system using flavins or ethylene-bridged flavinium salts that works via an NAO-like process. Under conditions optimised in terms of solvent, base, temperature and oxygen pressure, primary nitroalkanes were transformed to aldehydes. In our system, aldehydes immediately reacted with other nitroalkane molecules to form β-nitroalcohols. The reduced flavin catalyst was re-oxidised by oxygen. An alternative mechanism towards β-nitroalcohols via 5-(2-nitrobutyl)-1,5-dihydroflavin was suggested through quantum chemical calculations and by trapping and characterising this dihydroflavin intermediate. Interestingly, 5-(2-nitrobutyl)-1,5-dihydroflavin is an analogue of the flavin adenine dinucleotide adduct previously observed in an NAO X-ray structure. In both mechanistic pathways, flavin-5-iminium species is formed by nitroalkanide addition to flavin. This process represents flavin-based umpolung of an original donor to an acceptor.
Introduction
Nitroalkane oxidases (NAOs) are enzymes with a flavin cofactor – flavin adenine dinucleotide (FAD, 1b) – which enable the oxidative metabolism of nitroalkanes to their corresponding carbonyl compounds.1,2 During this transformation, a nitro group is released in the form of a nitrite anion (Scheme 1A). This makes this biological process somewhat different from the transformation of chemical nitroalkane → carbonyl compound, which is called a Nef reaction.3 In a Nef reaction, HNO (further forming nitrous oxide) is produced via nitro group reduction, which balances the oxidation of carbon possessing a nitro group to carbonyl function (Scheme 1B). In a NAO-mediated reaction, flavin acts as an electron acceptor, and then it is re-oxidised by oxygen, which acts as a stoichiometric oxidant being transformed to hydrogen peroxide.1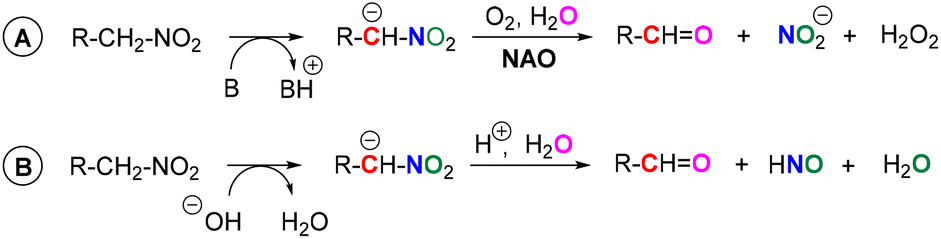 | ||
| Scheme 1 Conversion of nitroalkanes to carbonyl compounds (A) by NAO and (B) via Nef reaction. | ||
A key step in the mechanism of NAOs is flavin-N(5)-adduct formation through nucleophilic attack of a nitroalkanide on a flavin moiety (Scheme 2A).1,4 This is followed by nitrite anion release, which forms electrophilic iminium species. Thus, a change in the polarity (umpolung5) of the nitroalkanide nucleophile is accomplished. The iminium species readily undergo the addition of a nucleophile,6 like deprotonated water, in a reaction leading to a carbonyl compound. The product of another nitroalkanide addition – “5-nitrobutylflavin”‡ – has been observed by X-ray diffraction as a stable form of FAD in NAO (see Scheme 2B).1,7 In another study, cyanide adduct formation was used to prove the presence of an iminium species.8 Finally, a tetrahedral adduct of a nucleophile splits and releases an aldehyde and a reduced flavin.
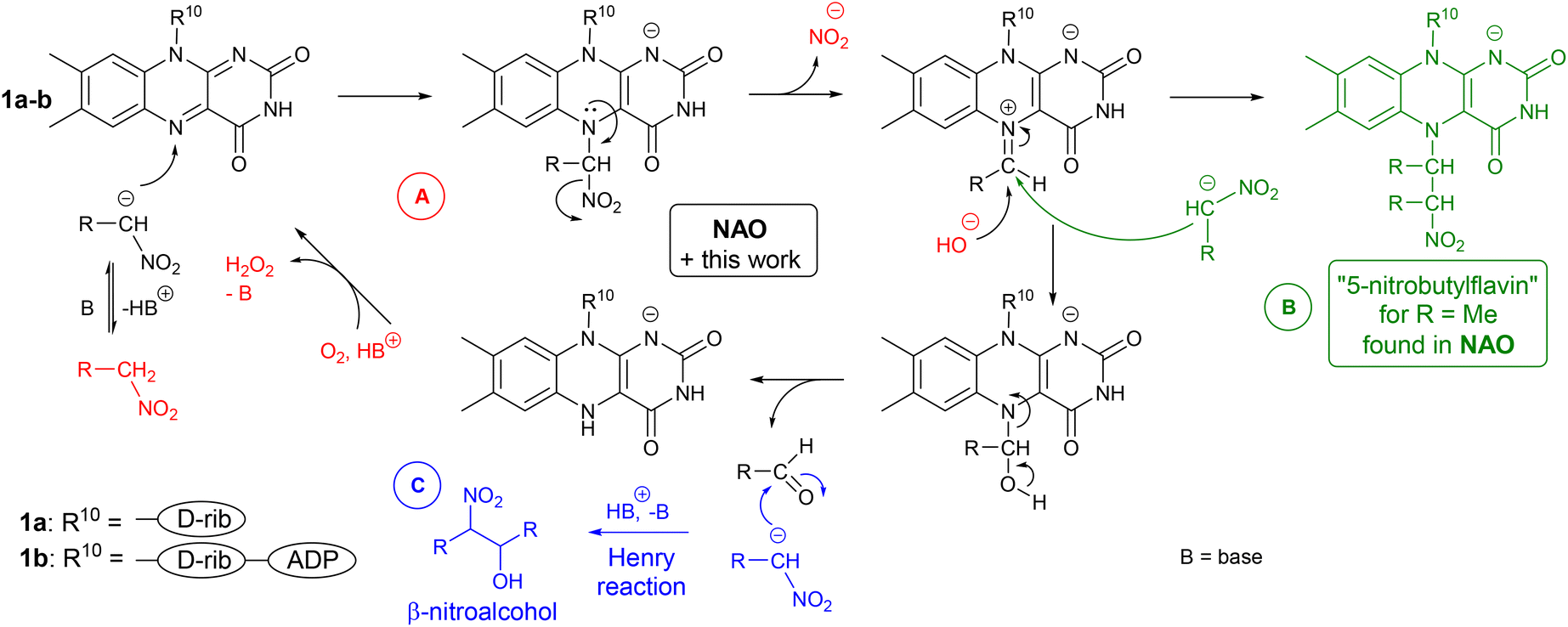 | ||
| Scheme 2 (A) Catalytic cycle in NAO based on a flavin cofactor, (B) formation and structure of 5-nitrobutylflavin, and (C) Henry reaction observed in an artificial system. | ||
The interaction of a nitroalkanide with a flavin resulting in a carbonyl compound and reduced flavin in an artificial system was first reported by Yano.9 Later, Foss and others utilised nitrite, produced from nitromethane in a catalytic process using ethylene-bridged flavinium salt 2, as a nontoxic source of nitrogen dioxide for the stoichiometric agent in the oxidation of alcohols and ethers catalysed by TEMPO.10 Recently, Lupidi, Palmieri and Petrini11 reported a tandem process where a nitroalkane was converted to an aldehyde using riboflavin (1a), which was followed by a Henry reaction12 with another nitroalkane anion (Scheme 2C) to form β-nitroalcohol. There, two equivalents of riboflavin 1a relative to the substrate were used.
Herein, we present a fully catalytic biomimetic process based on flavin derivatives 1 or 2 providing β-nitroalcohols in good preparative yields from the corresponding nitroalkanes via NAO-like umpolung of nitroalkanes (Scheme 2A). Experimental studies and quantum chemical calculations provided evidence for the possible employment of 5-(2-nitroalkyl)-1,5-dihydroflavin (cf.Scheme 2B) as an alternative pathway to that traditionally described for NAO.
Results and discussion
Development of an artificial catalytic NAO-like system
In both natural and artificial systems, flavins are known to be easily re-oxidised from their reduced form by oxygen.13,14 Thus, we suspected that a catalytic amount of a flavin derivative might be sufficient to transform nitroalkanes to β-nitroalcohols. To achieve this, we decided to optimise the reaction conditions using nitroethane (3a) as a model substrate (Table 1). First, we used ethylene-bridged 7,8-dimethoxy-3-methyl flavinium chloride (2c) as a flavin catalyst. Ethylene-bridged salts 2 have been previously shown to likely form adducts with nucleophiles due to a positive charge.15 In our previous study,16 7,8-dimethoxy derivative 2c was shown to be the most stable one among flavinium salts 2; 2c is also stable under slightly basic conditions and readily soluble in water.|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Atmosphere | Temp. (°C) | Reaction time (h) | 3a converted to 4ab (%) |
| a Reactions were performed with 0.6 mmol of nitroethane (3a) in tempered and pressurized Schlenk tubes with 0.55 equiv. of base, 10 mol% of 2c and 1 mL of water as a solvent. b Determined by 1H NMR using dimethyl sulfone as a standard. c In the absence of catalyst. | |||||
| 1 | Bu4N+ OH− | O2 (2.5 atm) | 25 | 16 | 46 |
| 2 | Na2CO3 | O2 (2.5 atm) | 25 | 16 | 54 |
| 3 | Et3N | O2 (2.5 atm) | 25 | 16 | 56 |
| 4 | — | O2 (2.5 atm) | 25 | 16 | 0 |
| 5 | Et3N | Ar | 25 | 16 | 15 |
| 6 | Et3N | O2 (1 atm) | 25 | 16 | 52 |
| 7 | Et3N | O2 (4.5 atm) | 25 | 16 | 68 |
| 8 | Et3N | O2 (4.5 atm) | 5 | 16 | 43 |
| 9 | Et3N | O2 (4.5 atm) | 45 | 2 | 61 |
| 10c | Et3N | O2 (4.5 atm) | 45 | 2 | 5 |
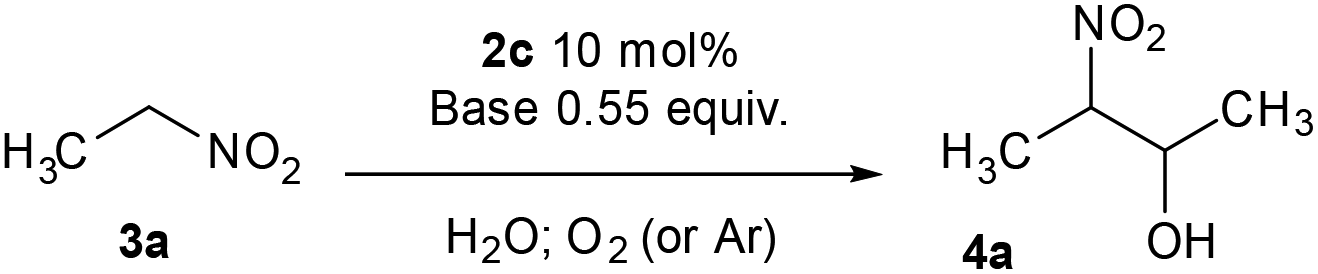
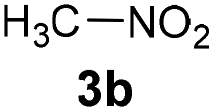
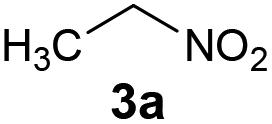
Inspired by a protocol for the reaction with a stoichiometric amount of riboflavin,11 we used 0.55 equiv. of base relative to the substrate and 1 mL of water as a solvent for 0.6 mmol of 3a. On the other hand, we used only 10 mol% of flavin catalyst. The reactions were performed in Schlenk tubes pressurised using oxygen to 2.5 atm and at 25 °C for 16 h. The course of the reaction was monitored using 1H NMR. The first set of experiments (entries 1–5 and ESI S1.7.1†) unambiguously showed that the reaction proceeds in a catalytic manner under oxygen since more than 20% of 3a was converted to β-nitroalcohol 4a, keeping in mind the stoichiometry of the reaction. In the absence of oxygen (under argon), the flavin catalyst did not regenerate, which resulted in a conversion of less than 20% (entry 5). Regarding base, it is critical for the reaction (cf. entry 4). Sodium carbonate and triethylamine were found to be more suitable than tetrabutylammonium hydroxide. However, we observed product loss after 8 hours using sodium carbonate (Fig. 1), which is probably caused by a retro-Henry reaction3d followed by nitroalkane decomposition (see the Mechanistic studies below for details). On the other hand, product loss was not observed when triethylamine was used, so this base was selected for further investigation. The other tested organic and inorganic bases showed worse performance (see ESI S1.7.1†). We also found that a higher oxygen pressure (4.5 atm) was not essential but helpful (cf. entries 4, 6 and 7) and a higher temperature (45 °C) significantly shortened the reaction time to 2 hours (cf. entries 7–9 and Fig. 1). Although the yield at 45 °C after 2 hours was slightly lower than that with a longer time and at 25 °C, we chose the higher temperature and shorter time for further experiments to overcome the potential problem of low solubility of more lipophilic substrates. It is important to note that only a trace amount of product was formed in the control reaction performed without any catalysts (entry 10).
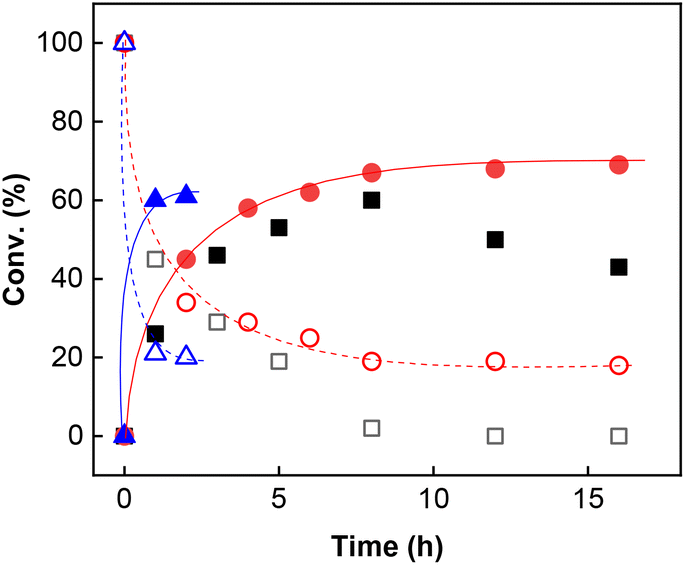 | ||
| Fig. 1 Reaction course of transformation of 0.6 mmol of 3a (empty symbols) to 4a (filled symbols) mediated by 2c (10 mol%) under oxygen (4.5 atm) in 1 mL of water. Squares: sodium carbonate used as a base at RT; circles: Et3N used as a base at RT; triangles: Et3N used as a base at 45 °C. | ||
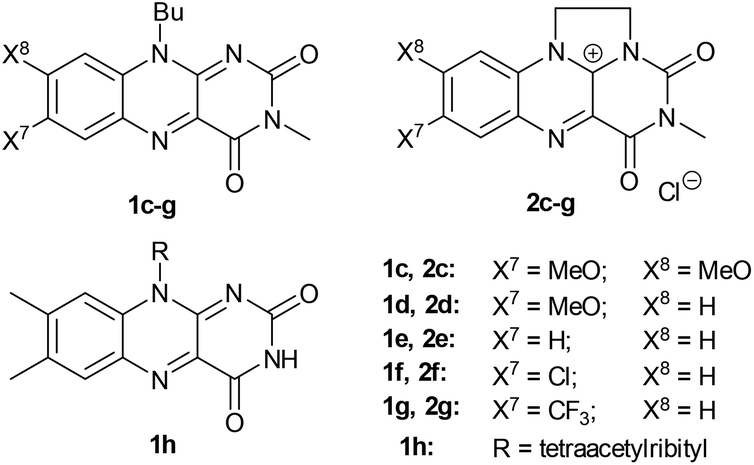
Under the best conditions identified above, we tested other flavinium salts 2 and received similar results, with the exception of the electron-poor derivative 2g possessing a trifluoromethyl group (Table 2). Then, we considered whether other flavin derivatives could be analogously used in the catalytic transformation of 3a to 4a. To our delight, we found that riboflavin derivatives 1d–1g and riboflavin tetraacetate§ (1h) are good catalysts of this reaction. On the other hand, alloxazine and deazaflavin derivatives 5 and 6, and electron-rich dimethoxyflavin 1c were inefficient. As for the neutral catalysts 1, 5 and 6, a water/DMSO (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) mixture was used for their complete solubility. Interestingly, the flavinium catalysts were significantly less efficient in the water/DMSO mixture (see ESI S1.7.3 and S1.7.4†) and in the mixtures of water with other solvents (see ESI S1.7.2† for data on other solvents). Therefore, for further studies, we selected 2c in water as a representative of a catalytic system in a purely aqueous environment (method A). Regarding the neutral flavins, we selected a system with 7-chloro derivative 1f in a mixture of water/DMSO (1:2). Finally, we used 1.1 equivalents of base (Et3N) in the case of 1f, which was found to be beneficial (method B).
:2) mixture was used for their complete solubility. Interestingly, the flavinium catalysts were significantly less efficient in the water/DMSO mixture (see ESI S1.7.3 and S1.7.4†) and in the mixtures of water with other solvents (see ESI S1.7.2† for data on other solvents). Therefore, for further studies, we selected 2c in water as a representative of a catalytic system in a purely aqueous environment (method A). Regarding the neutral flavins, we selected a system with 7-chloro derivative 1f in a mixture of water/DMSO (1:2). Finally, we used 1.1 equivalents of base (Et3N) in the case of 1f, which was found to be beneficial (method B).
|
|
||
|---|---|---|
| 3a converted to 4ab (%) | ||
| Catalyst | In water | In water/DMSO (1:2 v/v) |
| a Reactions were performed with 0.6 mmol of nitroethane in a Schlenk tube tempered at 45 °C and pressurized with O2 at 4.5 atm using 10 mol% of catalyst and 0.55 equiv. of Et3N in 1 mL of solvent. Reaction time was 4 h. b Determined by 1H NMR using dimethyl sulfone as a standard. c 1.1 equiv. of Et3N. | ||
| 1c | — | 15 |
| 1d | — | 45 |
| 1e | — | 50 |
| 1f | 45 | 61 (69c) |
| 1g | — | 51 |
| 1h | — | 60 |
| 2c | 61 | 35 |
| 2d | 51 | — |
| 2e | 66 | 36 |
| 2f | 64 | — |
| 2g | 41 | — |
| 5 | — | 11 |
| 6 | — | 10 |

| Entry | Nitroalkane | Producta | Method | Yield (%) |
|---|---|---|---|---|
|
a 2 mmol of substrates 3a–g were treated in a Schlenk tube pressurized with O2 at 4.5 atm. Products were isolated by column chromatography.
b Product was obtained as a mixture of diastereomers at ratios from 40:60 to 30:70.
c 5 mol% of 1f. Method A: Used 10 mol% of catalyst 2c and 0.55 equiv. of Et3N in water. Reaction performed at 25 °C for 20 h. Method B: Used 10 mol% of catalyst 1f and 1.1 equiv. of Et3N in a water/DMSO (1:2) mixture. Reaction performed at 45 °C for 4 h.
d Ratio of products (4f/4d/4g + 4h) obtained by LC-MS analysis.
|
||||
| 1 |
|
— | A | 0 |
| 2 |
|
|
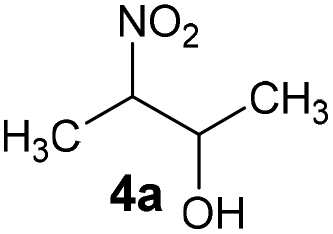
Ab | 62 |
| 3 |
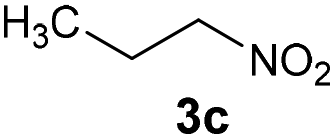
|
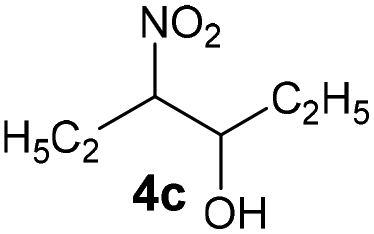
|
Bb | 61 |
| 4 |
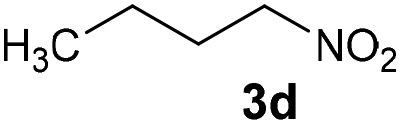
|
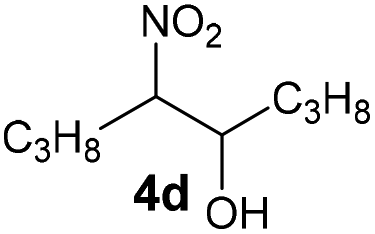
|
Bb | 57 |
| 5 |
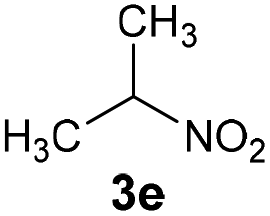
|
— | B | 0 |
| 6 |
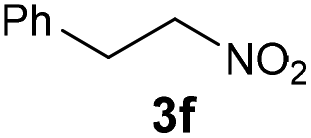
|
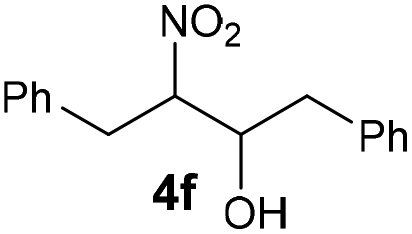
|
Bb | 59 |
| 7 |
|
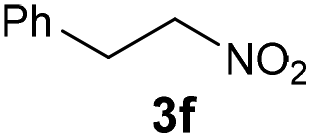
4f | Bb,c | 40 |
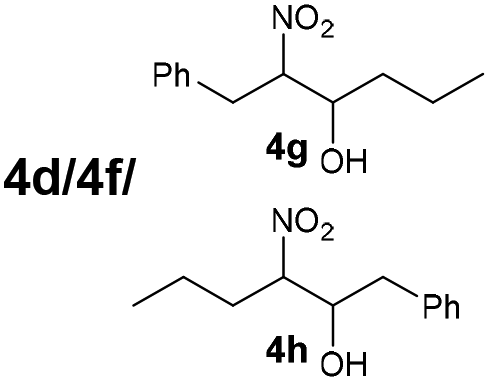
| 8 | 3d + 3f |
|
B | 9/34/57d |
| 9 |
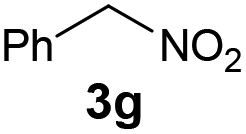
|
|
B | 52 |
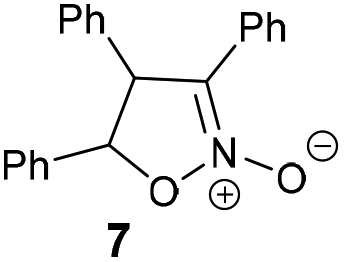
The reaction of different primary nitroalkanes 3d and 3f together under the optimal conditions provided a mixture of four products (Table 3, entry 8 and ESI S2.4†). The use of (nitromethyl)benzene (3g) produced a different product than the expected one, as the primarily formed β-nitroalcohol 4g probably underwent elimination of water and subsequent Michael addition with a third nitroalkanide anion to give a five-membered ring product 7, as described in the literature11 (Table 3, entry 9, Scheme 3).
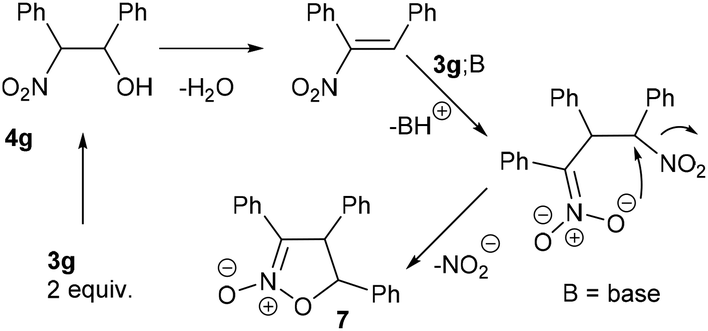 | ||
| Scheme 3 Mechanism of (nitromethyl)benzene 3g “trimerization” to product 7. | ||
Mechanistic studies
To elucidate the overall reaction scheme of nitroalkane → acetaldehyde transformation followed by a Henry reaction, we monitored the main reaction products/intermediates in an experiment using 0.6 mmol of nitroethane in the presence of 10 mol% of 1f (see Fig. 2). We used Griess colorimetry17 to quantify the nitrite anions formed in our reaction, thus confirming the NAO-like process. In an NAO-mediated reaction producing an aldehyde, irreversible formation of NO2− is quantitative with regard to the starting nitro compound amount. However, in the case of the subsequent Henry reaction, there should be only half the amount of NO2− formed in theory (i.e. 0.3 mmol). In the experiment, we observed the formation of ca. 0.4 mmol of nitrite, which is higher than the maximal amount, considering the quantitative conversion of 3a to 4a. This is probably due to the fact that the acetaldehyde produced by the NAO-like process does not completely undergo the Henry reaction, but it is particularly decomposed via self-condensation under basic conditions. The amount of free acetaldehyde was only around 1.5% for the whole time. It is likely that acetaldehyde decomposition is relatively fast. In a similar experiment with 3f, more stable and non-volatile phenylacetaldehyde was found in an amount slightly below 10% (see ESI S2.2†). It should be noted that only traces of carboxylic acid were detected by mass spectroscopy in the reaction mixture (see ESI S2.2†). It means that oxidation of aldehydes is not favourable under our conditions. The aldehyde oxidation did not take place even under irradiation which was proved by an independent experiment (see ESI S2.5†). Thus, aldehydes not consumed by subsequent Henry reactions seem to be the major by-products of the nitroalkane transformations.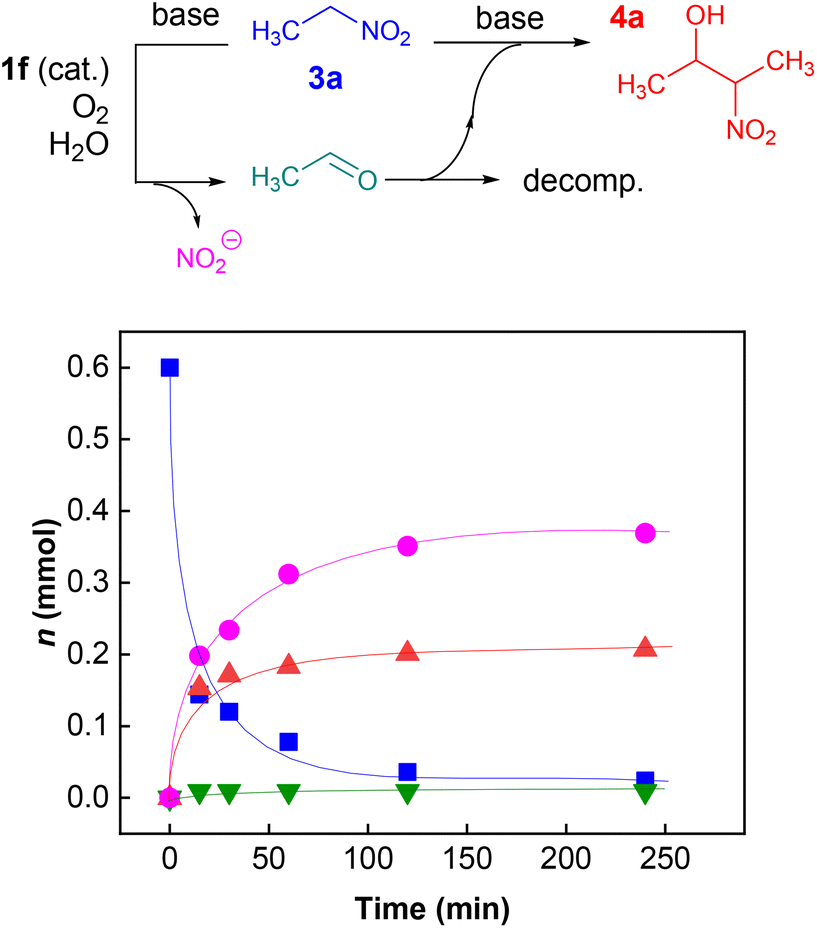 | ||
| Fig. 2 Reaction course of transformation of 0.6 mmol of 3a (squares) to 4a (up-triangles) mediated by 1f (10 mol%) under oxygen (4.5 atm) at 45 °C in 1 mL of water/DMSO (1:2) mixture. Amounts of nitrite (circles) and acetaldehyde (down-triangles) are given. | ||
In addition to the reaction scheme via aldehyde formation and its reaction with nitroalkanes, another reaction pathway to β-nitroalcohol products proceeding through the 5-(2-nitroalkyl)-1,5-dihydroflavin intermediate (F or L, see Fig. 3) could be considered. It can be formed if a nitroalkanide attacks an iminium species (C or J) instead of water/hydroxide. Both the reaction pathways were evaluated using quantum-chemical calculations for flavin 1f and flavinium 2c catalysts, confirming their feasibility from a thermodynamic point of view (see ESI S1.9† for details). For flavinium salts 2, we expected β-nitroalcohols to be released from F through substitution with hydroxide. The dihydroflavin formed is then re-oxidised by oxygen to form a flavinium ion, as is known from several applications of flavinium salts in oxygenation reactions.13 In the case of neutral flavins 1, consideration should be given to an alternative pathway involving the oxidation of L to the corresponding 5-alkylflavinium salt M, which might be dealkylated using a hydroxide/water nucleophile, analogous to procedures described in the literature.18 It is likely that both reaction pathways are involved in the catalytic transformation of nitroalkanes to β-nitroalcohols in a water or water/organic medium, particularly in the case of neutral flavins 1. This interpretation was supported by our observation of adduct 8 formation as a result of the interaction of 1f with sodium 1-nitroethan-1-ide (Scheme 4A). Adduct 8 was fully characterised using NMR and HR-MS techniques, and it represents an artificial analogue of nitrobutyl-FAD found in NAO.7 We also attempted to characterise the primary adducts of nitroalkanide anions with a flavin or flavinium catalyst, but were not successful when using 1-nitroethan-1-ide, probably because of the high reactivity of the corresponding iminium salt formed by its addition to flavin and the release of nitrite. However, we were able to observe and fully characterise the 2-nitropropan-2-ide adducts 9 and 10 as being in enamine form (Schemes 4B–D). This may be why the secondary nitroalkane does not undergo any umpolung with flavin and, consequently, does not provide a product after a Henry reaction.
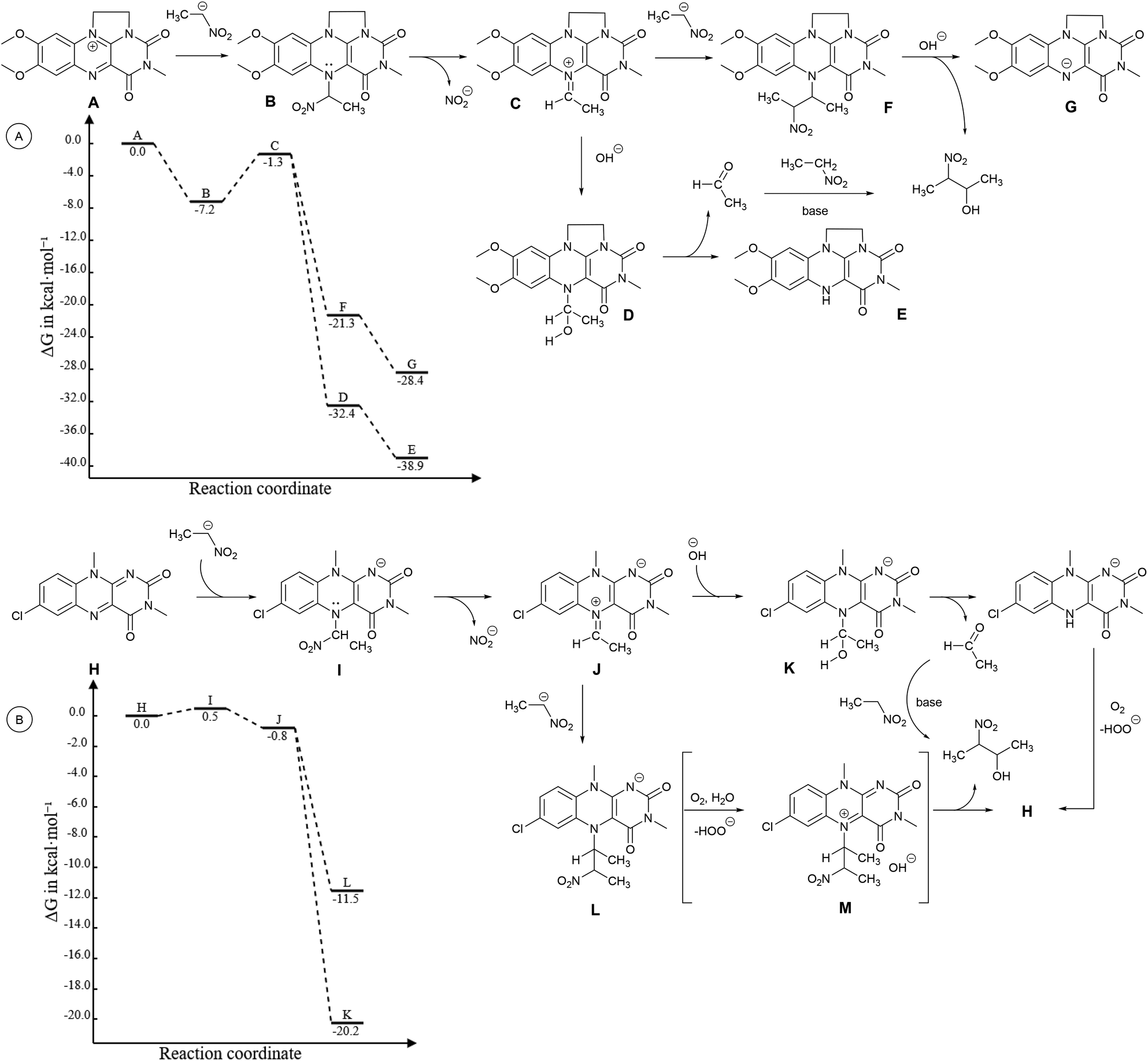 | ||
| Fig. 3 First part of the catalytic transformation of 3a to 4a mediated (A) by 2c and (B) by the 10-methyl analogue of 1f as a series of elementary steps, considered for computational studies. Diagrams for reactions are given. The geometries and Gibbs free energy contributions have been calculated at the B3LYP19/def2-TZVP20 level, solvation free energy contributions using the CPCM21 formalism and parameters for DMSO. Energies have been calculated using the DLPNO-CCSD(T)22 method with def2-QZVP20 basis. Counterions are omitted. | ||
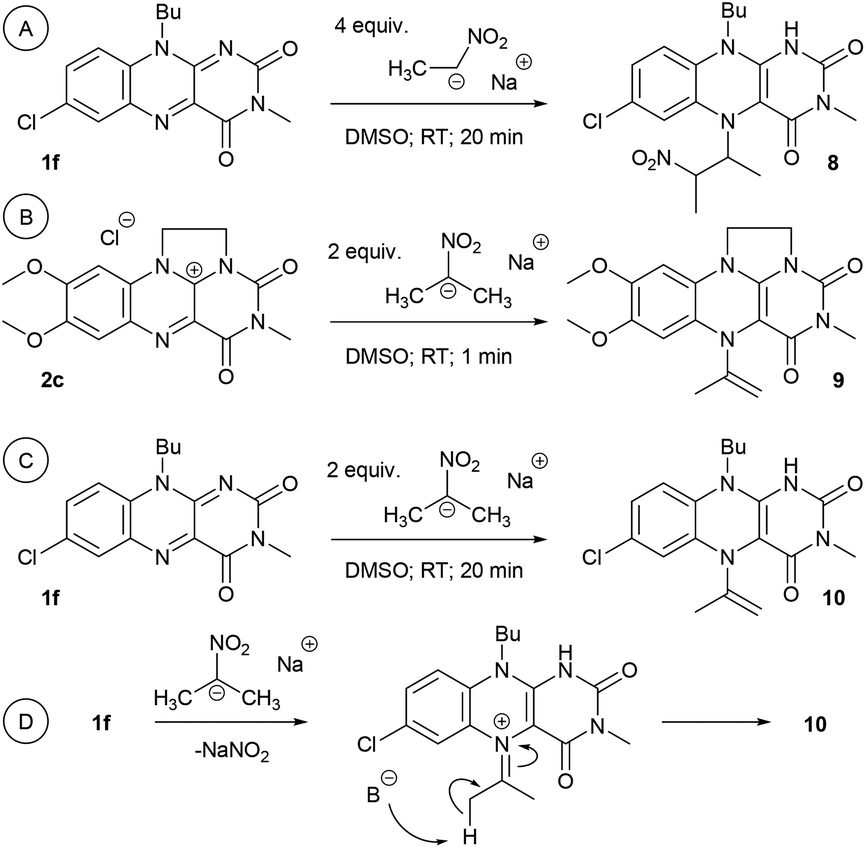 | ||
| Scheme 4 (A) Formation of adduct 8 from 1f and sodium 1-nitroethan-1-ide; (B) adduct 9 from 2c and sodium 2-nitropropan-2-ide; (C) adduct 10 from 1f and sodium 2-nitropropan-2-ide and (D) the corresponding intermediate. See ESI S2.6† for characterization. | ||
Conclusions
Our study clearly showed that flavin derivatives can enable nitroalkane transformation to aldehydes using a catalytic procedure analogous to that occurring with NAO1,2via N(5)-adduct formation with a flavin derivative, producing nitrite and hydrogen peroxide. Under our optimized reaction conditions, the resulting aldehyde readily undergoes the Henry reaction with a second nitroalkane molecule to form a β-nitroalcohol. Using quantum chemical calculations and the isolation of intermediates, we documented an alternative pathway to β-nitroalcohols via nitroalkanide attack on a flavin-iminium intermediate. Importantly, 5-(2-nitrobutyl)-1,5-dihydroflavin, a product of this attack, is an analogue of stable FAD form revealed in NAO by X-ray crystallography.7 We generated and fully characterised it in an artificial system. The results of this study may provide inspiration for other mechanistic studies of NAO-based transformations.Our system for the transformation of nitroalkanes to β-nitroalcohols is catalytic, distinguishing it from previous systems that used more than stoichiometric amounts of riboflavin. Our procedures were based not only on electrophilic flavinium salts 2 but also, most importantly, on neutral flavins 1, which are derivatives of flavin cofactors. From the point of view of flavin-based catalysis, the presented system is the first artificial catalytic system using the flavin-N(5) position to enable C–C bond formation. It is also an artificial example of noncanonical covalent reactivity of flavins.23
We have shown that NAO-like umpolung of nitroalkanes (typically reacting as donors) to any electrophilic species like aldehydes or iminium salts can be used in a catalytic way in an artificial system through various flavin derivatives. The general usage of such a system to change the polarity of reagents can be assumed and is now under investigation in our laboratories.
Author contributions
R. C., A. P. and E. Z. wrote the paper with input from all the authors. A. P. and E. Z. performed the entire synthesis of all used compounds and catalytic experiments. Analysis by mass spectrometry was performed by J. C. E. Z. and M. K. conducted the computational studies. Experimental data were analysed by A. P. The project was conceived by R. C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the Czech Science Foundation (Grant No. 21-14179S). A. P. is thankful for the support from a grant from Specific University Research – grant No. A2_FCHT_2022_113. Computational resources were supplied by the project “e-Infrastruktura CZ” (e-INFRA CZ LM2018140) supported by the Ministry of Education, Youth and Sports of the Czech Republic.References
- P. F. Fitzpatrick, Arch. Biochem. Biophys., 2017, 632, 41–46 CrossRef CAS PubMed.
- G. Gadda and K. Francis, Arch. Biochem. Biophys., 2010, 493, 53–61 CrossRef CAS PubMed.
- (a) W. E. Noland, Chem. Rev., 1955, 55, 137–155 CrossRef CAS; (b) J. U. Nef, Justus Liebigs Ann. Chem., 1894, 280, 263–291 CrossRef CAS; (c) R. Ballini and M. Petrini, Adv. Synth. Catal., 2015, 357, 2371–2402 CrossRef CAS; (d) L. Kurti and B. Czako, Strategic Applications of Named Reactions in Organic Synthesis, Elsevier Academic Press, Burlington, MA, 2005, pp. 202–203 Search PubMed.
- K. A. Kurtz and P. F. Fitzpatrick, J. Am. Chem. Soc., 1997, 119, 1155–1156 CrossRef CAS.
- (a) D. Seebach, Angew. Chem., Int. Ed. Engl., 1979, 18, 239–258 CrossRef; (b) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC.
- M. P. Valley, S. E. Tichy and P. F. Fitzpatrick, J. Am. Chem. Soc., 2005, 127, 2062–2066 CrossRef CAS PubMed.
- A. Nagpal, M. P. Valley, P. F. Fitzpatrick and A. M. Orville, Biochemistry, 2006, 45, 1138–1150 CrossRef CAS PubMed.
- D. J. T. Porter, J. G. Voet and H. J. Bright, J. Biol. Chem., 1973, 248, 4400–4416 CrossRef CAS PubMed.
- (a) Y. Yano, M. Nakazato and E. Ohya, J. Chem. Soc., Perkin Trans. 2, 1985, 77–81 RSC; (b) Y. Yano, M. Nakazato and K. Kasagawa, J. Chem. Soc., Chem. Commun., 1984, 498–499 RSC; (c) Y. Yano, M. Ohshima, I. Yatsu, S. Sutoh, R. E. Vasguez, A. Kitani and K. Sasaki, J. Chem. Soc., Perkin Trans. 2, 1985, 753–758 RSC.
- P. Thapa, S. Hazoor, B. Chouhan, T. T. Vuong and F. W. Foss, J. Org. Chem., 2020, 85, 9096–9105 CrossRef CAS PubMed.
- G. Lupidi, A. Palmieri and M. Petrini, Adv. Synth. Catal., 2021, 363, 742–746 CrossRef CAS.
- F. A. Luzzio, Tetrahedron, 2001, 57, 915–945 CrossRef CAS.
- (a) F. G. Gelalcha, Chem. Rev., 2007, 107, 3338–3361 CrossRef CAS PubMed; (b) E. Romero, J. R. Gómez Castellanos, G. Gadda, M. W. Fraaije and A. Mattevi, Chem. Rev., 2018, 118, 1742–1769 CrossRef CAS PubMed; (c) R. Cibulka, Eur. J. Org. Chem., 2015, 915–932 CrossRef CAS; (d) R. Cibulka and M. W. Fraaije, in Flavin–Based Catalysis, 2021, pp. 97–124 CrossRef; (e) A. Rehpenn, A. Walter and G. Storch, Synthesis, 2021, 53, 2583–2593 CrossRef CAS.
- (a) Y. Imada, H. Iida, S.-I. Murahashi and T. Naota, Angew. Chem., Int. Ed., 2005, 44, 1704–1706 CrossRef CAS PubMed; (b) Y. Imada, H. Iida, S. Ono and S.-I. Murahashi, J. Am. Chem. Soc., 2003, 125, 2868–2869 CrossRef CAS PubMed; (c) Y. Arakawa, K. Yamanomoto, H. Kita, K. Minagawa, M. Tanaka, N. Haraguchi, S. Itsuno and Y. Imada, Chem. Sci., 2017, 8, 5468–5475 RSC; (d) Y. Imada, T. Kitagawa, T. Ohno, H. Iida and T. Naota, Org. Lett., 2009, 12, 32–35 CrossRef PubMed; (e) S. Chen and F. W. Foss, Org. Lett., 2012, 14, 5150–5153 CrossRef CAS PubMed; (f) A. T. Murray, M. J. Dowley, F. Pradaux-Caggiano, A. Baldansuren, A. J. Fielding, F. Tuna, C. H. Hendon, A. Walsh, G. C. Lloyd-Jones, M. P. John and D. R. Carbery, Angew. Chem., Int. Ed., 2015, 54, 8997–9000 CrossRef CAS PubMed; (g) A. T. Murray, P. Matton, N. W. G. Fairhurst, M. P. John and D. R. Carbery, Org. Lett., 2012, 14, 3656–3659 CrossRef CAS PubMed; (h) J. Zelenka, R. Cibulka and J. Roithová, Angew. Chem., Int. Ed., 2019, 58, 15412–15420 CrossRef CAS PubMed.
- (a) M. März, M. Babor and R. Cibulka, Eur. J. Org. Chem., 2019, 3264–3268 CrossRef; (b) J. Žurek, E. Svobodová, J. Šturala, H. Dvořáková, J. Svoboda and R. Cibulka, Tetrahedron: Asymmetry, 2017, 28, 1780–1791 CrossRef; (c) W.-S. Li and L. M. Sayre, Tetrahedron, 2001, 57, 4523–4536 CrossRef CAS.
- A. Pokluda, Z. Anwar, V. Boguschová, I. Anusiewicz, P. Skurski, M. Sikorski and R. Cibulka, Adv. Synth. Catal., 2021, 363, 4371–4379 CrossRef CAS.
- K. R. Paul and V. K. Gupta, Environ. Int., 1981, 5(3), 153–157 CrossRef CAS.
- (a) P. Hemmerich, V. Massey and G. Weber, Nature, 1967, 213, 728–730 CrossRef CAS PubMed; (b) W. H. Walker, P. Hemmerich and V. Massey, Helv. Chim. Acta, 1967, 50, 2269–2279 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98(7), 5648–5652 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102(11), 1995–2001 CrossRef CAS.
- C. Riplinger and F. Neese, J. Chem. Phys., 2013, 138(3), 034106 CrossRef PubMed.
- V. Piano, B. A. Palfey and A. Mattevi, Trends Biochem. Sci., 2017, 42, 457–469 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00101f |
| ‡ “5-nitrobutylflavin” is a simplified name for 5-(2-nitrobutyl)-1,5-dihydroflavin used in the literature for an FAD analogue found in NAO. |
| § Riboflavin tetraacetate can be readily obtained by a one-step procedure from commercially available riboflavin (vitamin B2). |
| This journal is © The Royal Society of Chemistry 2023 |