
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and evaluation of NHC derivatives and 4′-fluorouridine prodrugs†
Li
Xiang
ab,
Tianwen
Hu
bc,
Haitao
Xue
bc,
Wenfang
Pan
bc,
Yuanchao
Xie
*d and
Jingshan
Shen
 *abc
*abc
aSchool of Chinese Materia Medica, Nanjing University of Chinese Medicine, Nanjing 210023, P. R. China. E-mail: shenjingshan@simm.ac.cn
bState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, P. R. China
cUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
dLingang Laboratory, Shanghai 200031, P. R. China. E-mail: xieyuanchao@lglab.ac.cn
First published on 3rd March 2023
Abstract
β-D-N4-Hydroxycytidine (NHC) derivatives with structural modifications at the C4′, O4′ or C6 position and 4′-fluorouridine prodrugs were synthesized and evaluated for their antiviral activities against respiratory syncytial virus (RSV) or influenza virus (IFV) in vitro. The NHC derivatives were found inactive, but 4′-fluorouridine and its prodrugs had potent anti-RSV and anti-IFV activities. 4′-Fluorouridine was proved to be a nucleoside with poor stability, but the tri-ester prodrugs exhibited enhanced stability, especially tri-isobutyrate ester 1a. This prodrug also showed excellent oral pharmacokinetic properties in rats, with potential to be an oral antiviral candidate.
Introduction
The emergence or re-emergence of epidemic viruses poses a serious threat to human health and economic security, exemplified by SARS-CoV-2 which has been circulating for nearly three years since the global outbreak in early 2020.1 Using small-molecule antiviral agents is one of the most effective measures to cope with viral infections. As a well-known class of antiviral agents, nucleoside analogs that function by targeting the conserved active site of viral polymerase have been of research interest in the discovery of antiviral drugs. In recent years, two nucleoside drugs, remdesivir2 and molnupiravir,3 used for treating COVID-19 have been marketed, and their application may be further expanded due to their broad-spectrum antiviral properties.4,5β-D-N4-Hydroxycytidine (NHC, EIDD-1931), a cytidine analog, is the parent nucleoside of molnupiravir (EIDD-2801) (Fig. 1). During the catalysis of cellular kinases, it is converted to NHC triphosphate, which can substitute for cytidine triphosphate or uridine triphosphate by the SARS-CoV-2 RNA dependent RNA polymerase (RdRp) to embed in the growing RNA chain, thus resulting in a lethal mutation.6 NHC also demonstrates antiviral activities against multiple other RNA viruses, including Ebola virus,7 Venezuelan equine encephalitis virus (VEEV),8 respiratory syncytial virus (RSV) and influenza A virus (IAV).9 In BALB/cJ mice infected with RSV or IAV, oral administration of NHC significantly reduced the viral load in the lungs and alleviated disease biomarkers. This nucleoside showed favorable oral pharmacokinetic (PK) properties in mice and ferrets.9
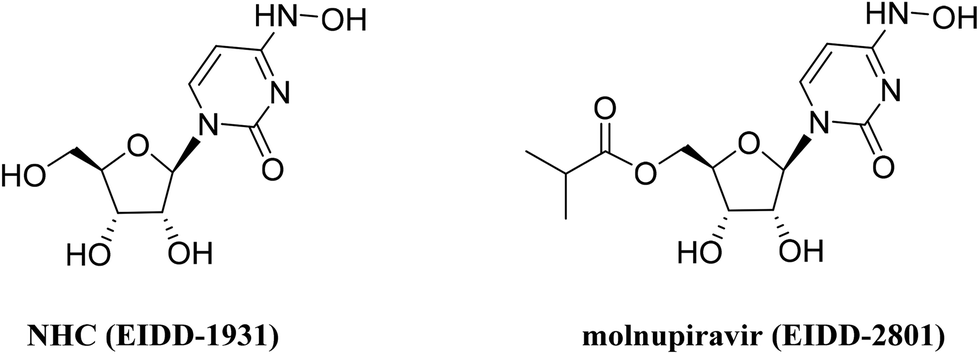 | ||
| Fig. 1 The structures of NHC and molnupiravir. | ||
From a chemical perspective, NHC bearing a hydroxylamine group at the base C4 position is very similar to cytidine and uridine, and able to form hydrogen bonds with adenosine and guanosine, respectively. Because of the high structural similarity, viral polymerase is unable to discriminate NHC triphosphate from natural pyrimidine nucleoside triphosphates, and therefore accepts it as a substrate for viral RNA elongation. For the design of antiviral nucleoside analogs, we envisaged that large structural changes in natural nucleosides may have a negative impact on the antiviral activity. Accordingly, in this study, we designed and synthesized several NHC derivatives with minor structural modifications, including 4′-fluorination, replacement of the 4′-oxygen atom of ribose with a sulfur atom, and replacement of the C6 carbon atom with a nitro atom in the base moiety of NHC. To our disappointment, these nucleoside derivatives did not show inhibitory effects against IFV or RSV. However, 4′-fluorouridine tri-isobutyrate which was used for the synthesis of 4′-fluoro-substituted NHC was found to be a potent replication inhibitor against IFV. We further synthesized several ester prodrugs of 4′-fluorouridine and evaluated their antiviral activities, chemical stability in buffer solution and PK properties, which provided tri-isobutyrate ester 1a as a promising oral antiviral candidate.
Results and discussion
Synthesis of NHC derivatives and 4′-fluorouridine prodrugs
The synthetic approach towards compounds 1a–d is shown in Scheme 1. The methods for introducing a fluorine atom into the C4′ position of ribonucleosides have been well documented.10–12 Starting from uridine 5, the 5′-hydroxyl group was readily converted into iodine through an Appel reaction, and the obtained intermediate 6 was subjected to elimination in the presence of sodium methoxide (CH3ONa)/methanol to yield 7. The synthesis of the key intermediate 10 involved three sequential steps: iodofluorination, isobutyrylation and oxidation. The isobutyrylation of the 5′-hydroxyl group of 10 afforded tri-isobutyrate ester 1a, which subsequently underwent hydroxylamination at the C4 position of the base moiety to provide 1b in 18% yield using 2,4,6-triisopropylbenzenesulfonyl chloride (TPSCl), 4-dimethylaminopyridine (DMAP), triethylamine, and hydroxylamine hydrochloride. Compound 1c was obtained in the same way. We tried to synthesize 1b by direct hydroxylamination of 1c by following a recently reported method,13 but it was not achieved. The reaction gave complicated products which was mainly due to the low stability of the 4′-fluorine skeleton when heated. The removal of the isobutyryl groups of 1b was performed with 0.2 equiv. of potassium carbonate in methanol, but we did not obtain the pure product 1d because it was easily decomposed during the purification process.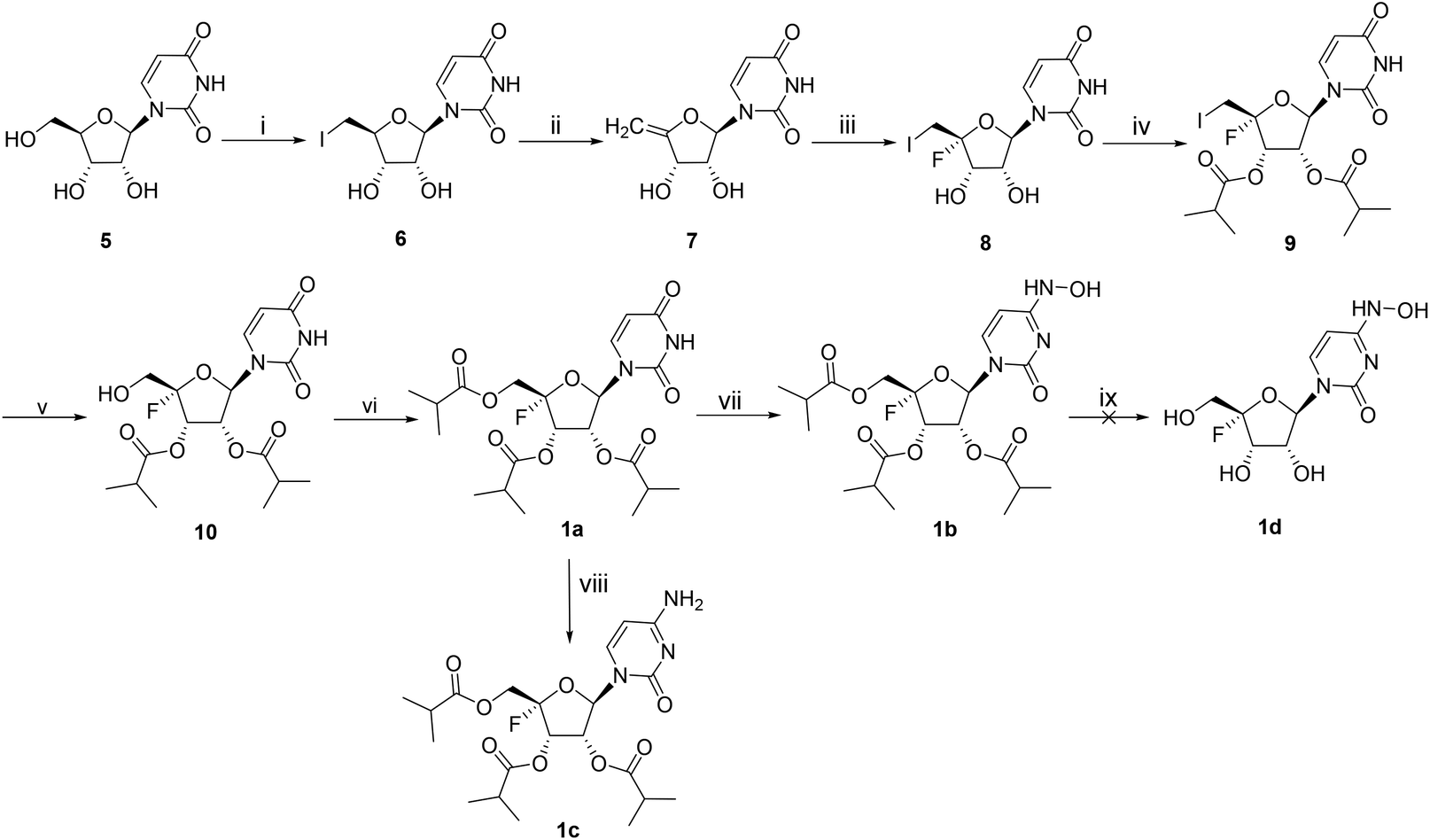 | ||
| Scheme 1 Reagents and conditions: (i) PPh3, I2, pyridine, 0 °C to RT, 67%; (ii) CH3ONa, MeOH, 67 °C, 57%; (iii) NIS, Et3N·3HF, acetonitrile, 0 °C to RT, 77%; (iv) isobutyric anhydride, DMAP, Et3N, CH2Cl2, RT, 47%; (v) TBAOH, TFA, m-CPBA, CH2Cl2, RT, 52%; (vi) isobutyric anhydride, DMAP, Et3N, CH2Cl2, 0 °C to RT, 98%; (vii) TPSCl, DMAP, Et3N, NH2OH·HCl, CH2Cl2, 0 °C to RT, 18%; (viii) TPSCl, DMAP, Et3N, 25% NH3·H2O, CH2Cl2, 0 °C to RT, 14%. | ||
The synthesis of 2a–c is shown in Scheme 2. Compound 12 was synthesized starting from 2,3,5-tri-O-benzyl-D-ribonolactone 11 according to reported methods.14 The reaction of 12 with uracil under Vorbruggen glycosylation reaction conditions yielded an anomeric mixture 13 (α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β = 1:1) in 85% overall yield. After debenzylation with BCl3, the obtained crude was purified by silica gel column chromatography to give β-nucleoside 2a in 76% yield. Compound 2a was then esterified with isobutyric anhydride, followed by hydroxylamination using the aforementioned method to give 2b in 54% yield over two steps. After the removal of the isobutyryl group, compound 2c was obtained.
:β = 1:1) in 85% overall yield. After debenzylation with BCl3, the obtained crude was purified by silica gel column chromatography to give β-nucleoside 2a in 76% yield. Compound 2a was then esterified with isobutyric anhydride, followed by hydroxylamination using the aforementioned method to give 2b in 54% yield over two steps. After the removal of the isobutyryl group, compound 2c was obtained.
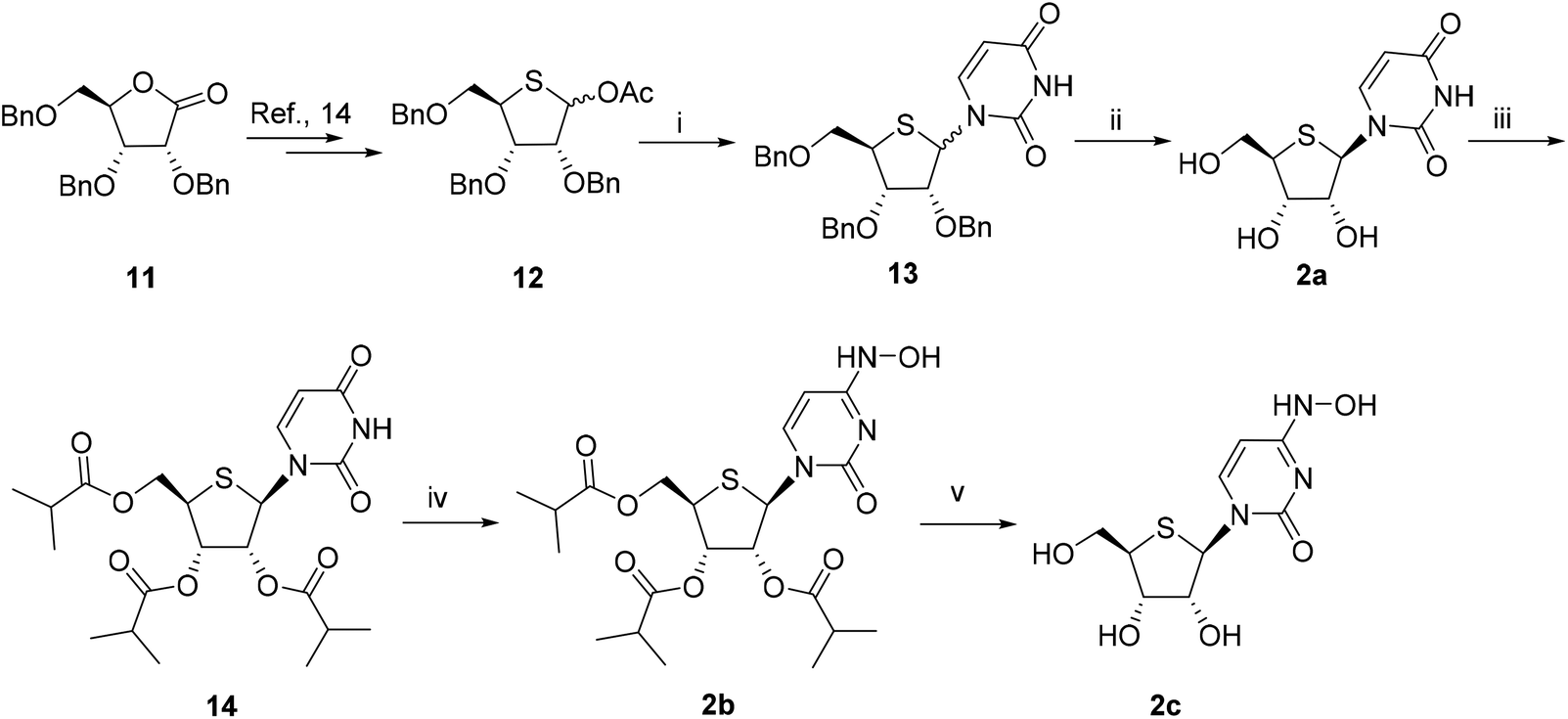 | ||
| Scheme 2 Reagents and conditions: (i) (a) uracil, HMDS, (b) TMSOTf, acetonitrile, −10 °C to RT, 85%; (ii) BCl3, CH2Cl2, −70 °C to −30 °C, 76%; (iii) isobutyric anhydride, DMAP, Et3N, CH2Cl2, RT, 85%; (iv) (a). 2,4,6-Triisopropylbenzenesulfonyl chloride, DIPEA, DMAP, CH2Cl2, 0 °C to RT, (b). hydroxylamine hydrochloride, DIPEA, 64%; (v) NH3/MeOH, RT, 80%. | ||
The 6-azauridine derivatives 3a–d were synthesized as shown in Scheme 3. The reaction of commercial 6-azauracil 16 with peracylated sugar 15 yielded β-nucleoside 17 when using the general glycosylation method. Compound 17 was treated with phosphorus oxychloride/Et3N to generate an active intermediate, which then reacted with hydroxylamine hydrochloride and ammonia to give 3a and 3c, respectively. The removal of the acetyl group of 3a and 17 in ammonia methanol solution afforded nucleosides 3b and 3d that were solidified in methyl tert-butyl ether (MTBE).
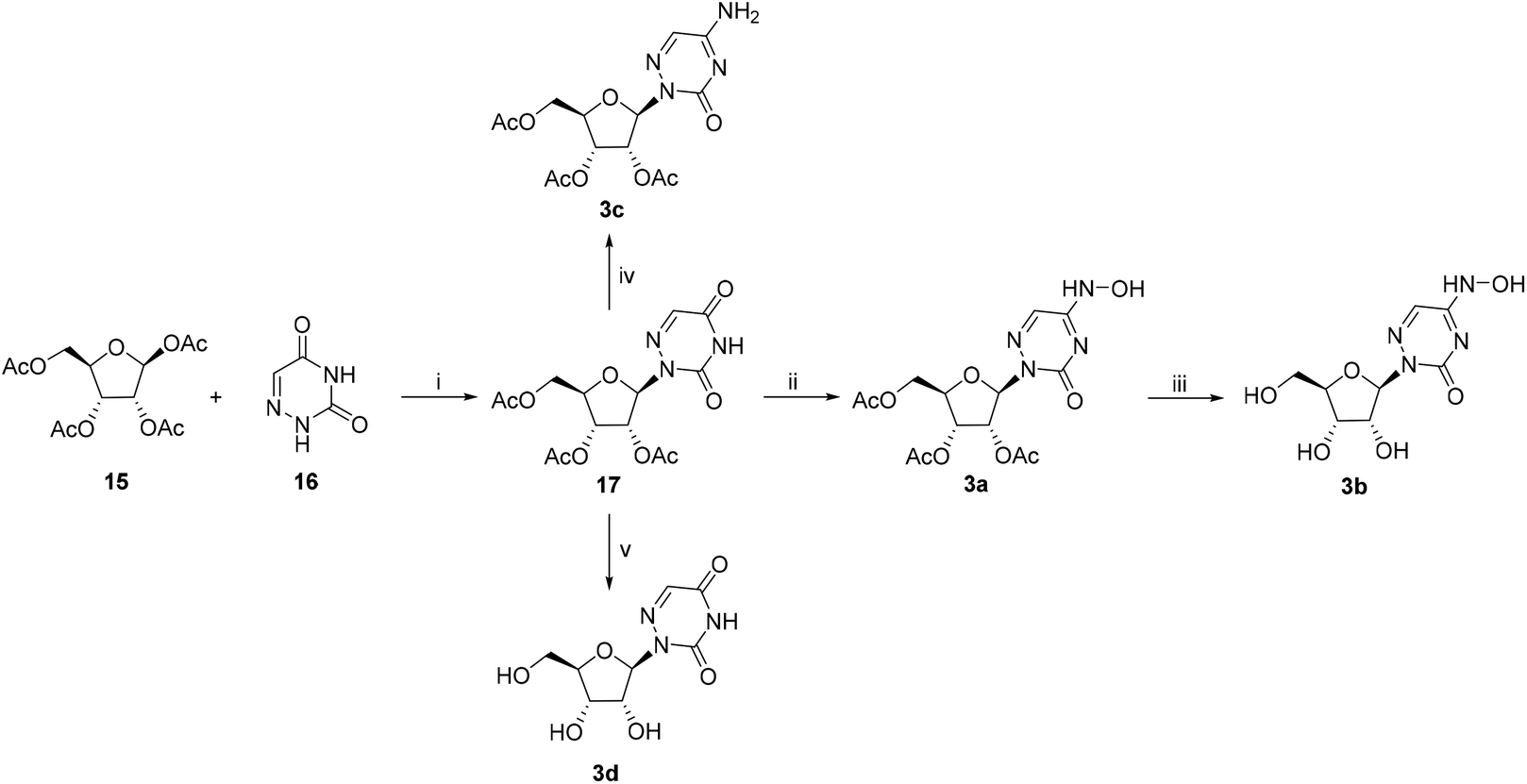 | ||
| Scheme 3 Reagents and conditions: (i) (a) (NH4)2SO4, HMDS, 120 °C, (b) SnCl4, CH2Cl2, RT, 64.5%; (ii) POCl3, Et3N, hydroxylamine hydrochloride, acetonitrile, 0 °C to RT, 17.3%; (iii) NH3/MeOH, RT, 60.2%; (iv) POCl3, Et3N, 25% NH3·H2O, acetonitrile, 0 °C to RT, 45%; (v) NH3/MeOH, RT, 60.2%. | ||
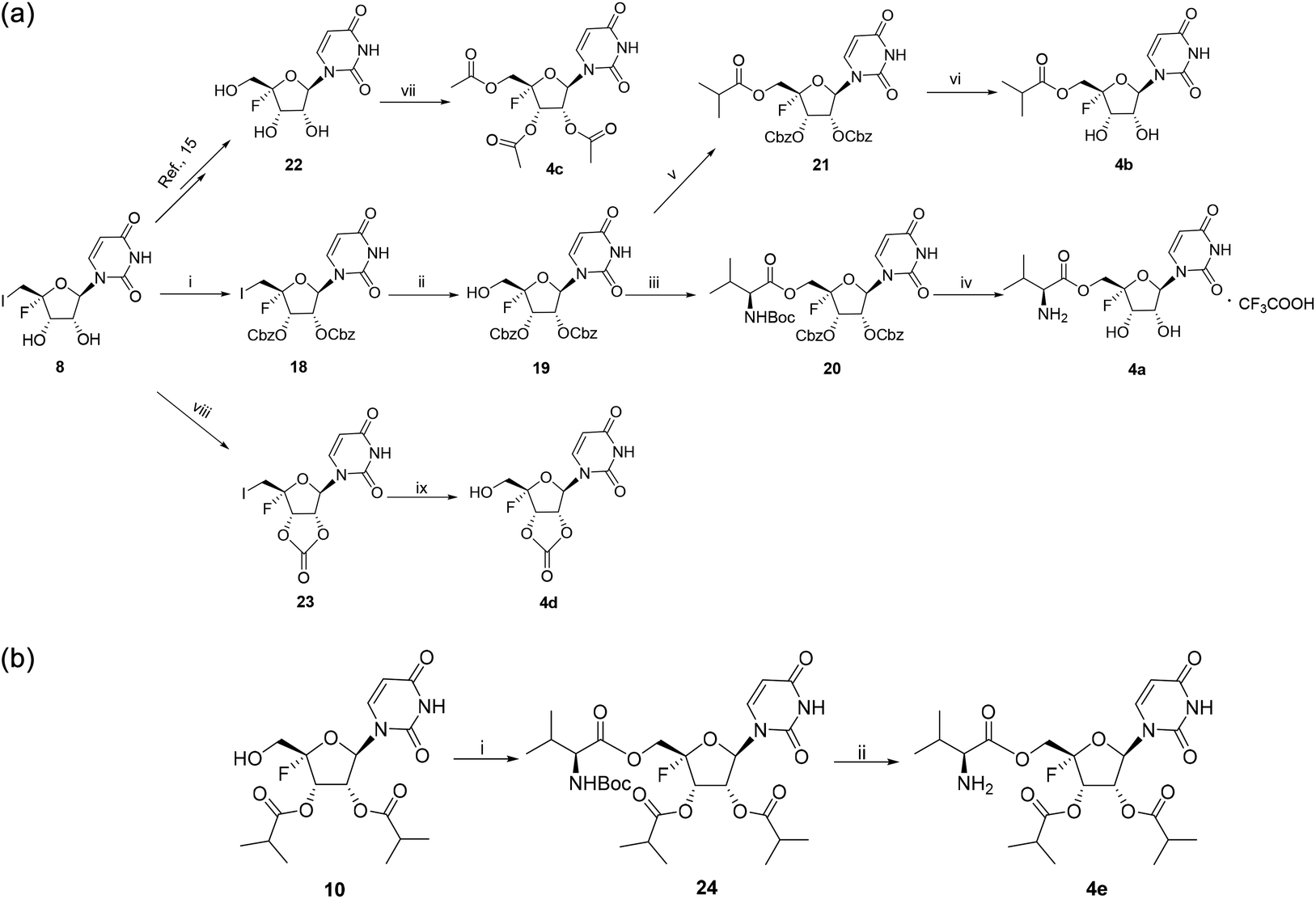
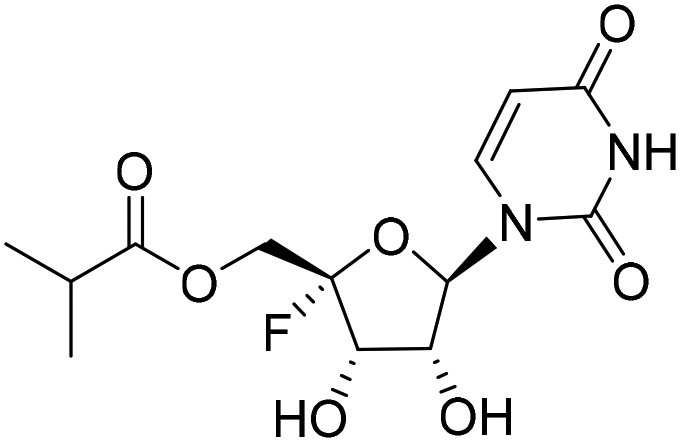
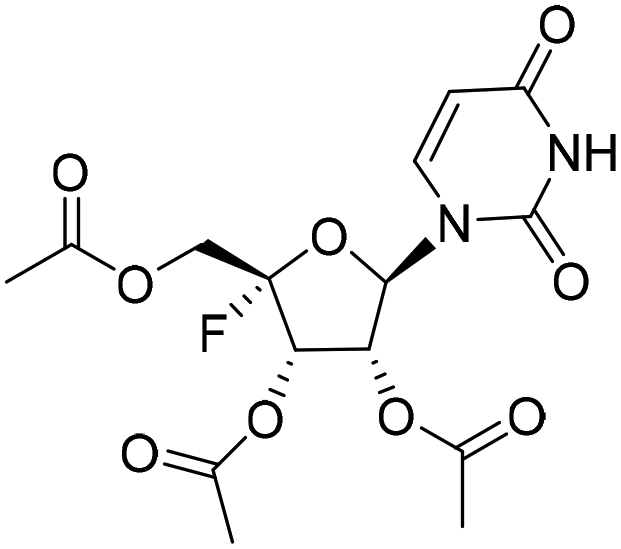
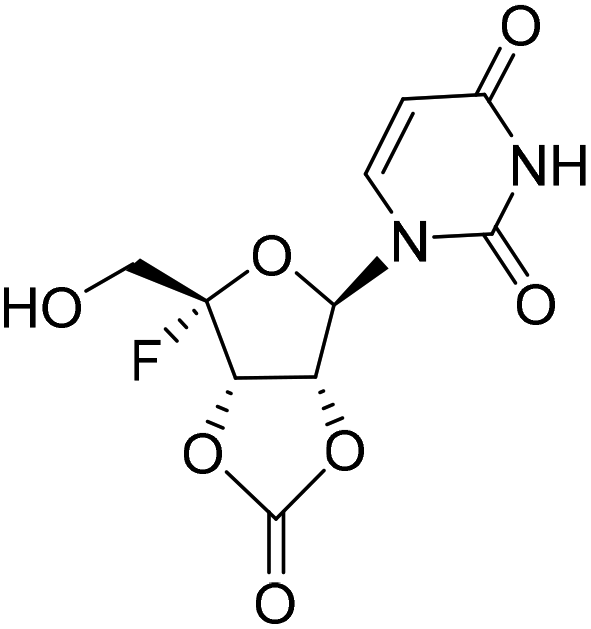
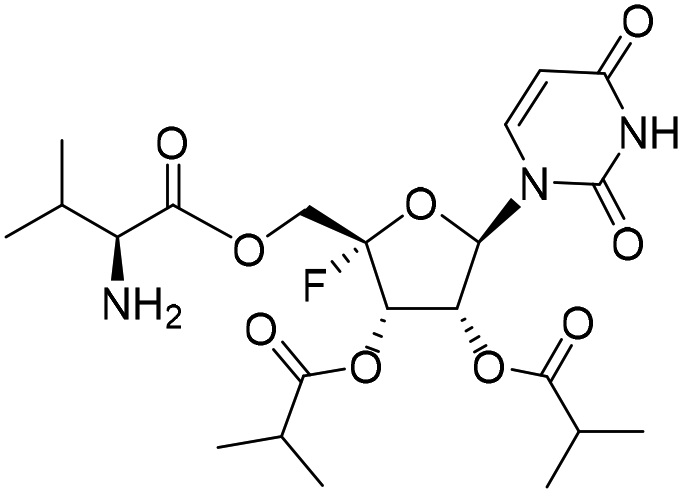
Five ester prodrugs (4a–e) of 4′-fluorouridine were synthesized, and the routes are shown in Scheme 4. The 2′ and 3′ hydroxyl groups of intermediate 8 were protected with benzyloxycarbonyl (Cbz) to give 18, followed by oxidation to yield intermediate 19. The condensation of 19 with N-Boc-L-valine and isobutyric anhydride under general reaction conditions gave 20 and 21, respectively. After the removal of the protecting groups, compounds 4a and 4b were obtained. Notably, more than 50% of 4a was found to be decomposed after being stored for 30 days at room temperature in a sealed tube. The 4′-fluorine substituted uridine 22 was prepared by following the reported procedure15 and acetylation of 22 with acetic anhydride smoothly gave triacetate 4c. The treatment of 8 with 1,1′-carbonyldiimidazole in THF afforded 23 in a good yield, and compound 4d was obtained from 23 by the same reaction used for 19. Compound 4e was easily synthesized from intermediate 10 by condensation with N-Boc-L-valine and Boc-deprotection.
 | ||
| Scheme 4 4a: Reagents and conditions: (i) benzyl chloroformate, 1-methylimidazole, CH2Cl2, 0 °C to RT, 97%; (ii) TBAOH, TFA, CH2Cl2, 0 °C to RT, 32%; (iii) N-Boc-L-valine, HOBT, EDCI, DMAP, CH2Cl2, 0 °C to RT, 76%; (iv) (a) TFA, CH2Cl2, RT; (b) 10% Pd/C, H2, ethanol, RT, 68% over two steps; (v) isobutyric anhydride, DMAP, Et3N, CH2Cl2, RT, 70%; (vi) 10% Pd/C, H2, ethanol, RT, 75%; (vii) acetic anhydride, DMAP, Et3N, EA, 0 °C to RT, 89%; (viii) 1,1′-carbonyldiimidazole, THF, RT, 51%; (ix) TBAOH, TFA, m-CPBA, CH2Cl2, RT, 69%. 4b: (i) N-Boc-L-valine, HOBT, EDCI, DMAP, CH2Cl2, 0 °C to RT, 85%; (ii) TFA, CH2Cl2, RT, 83%. | ||
Antiviral activity assays
The antiviral activities of the synthesized compounds were evaluated by the cytopathic effect (CPE) assay against RSV (ATCC VR-26 strain) and IFV (H1N1 strain) in Hep-2 cells and MDCK cells, respectively. Data were expressed as the inhibition rate, EC50 (concentrations reducing viral replication by 50%) and CC50 (concentrations decreasing cell viability by 50%) values. The results are summarized in Table 1.| Compd. | Structure | RSV (Hep-2 cells) | IFV (MDCK cells) | ||
|---|---|---|---|---|---|
| EC50 (μM) | CC50 (μM) | EC50 (μM) | CC50 (μM) | ||
| a Inhibition rate at 5 μM unless other specified. “—” means undetermined. | |||||
| 1a |
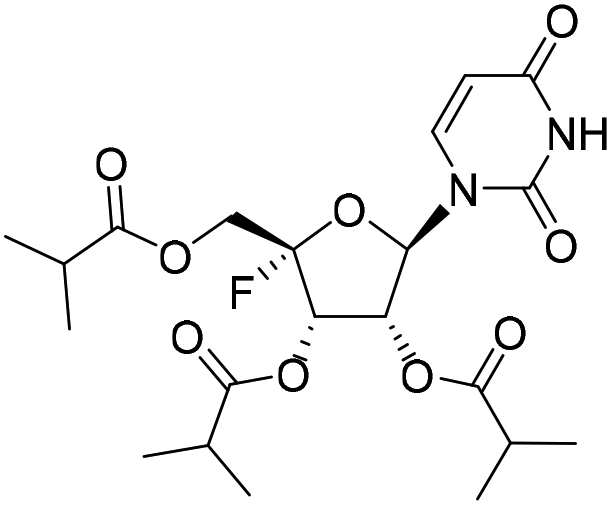
|
1.13 | >10 | 0.34 | >10 |
| 1b |
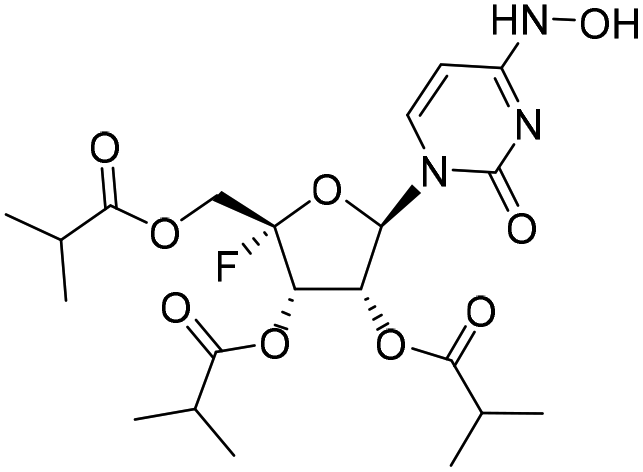
|
−0.1%a |
— | 2.0%a |
— |
| 1c |
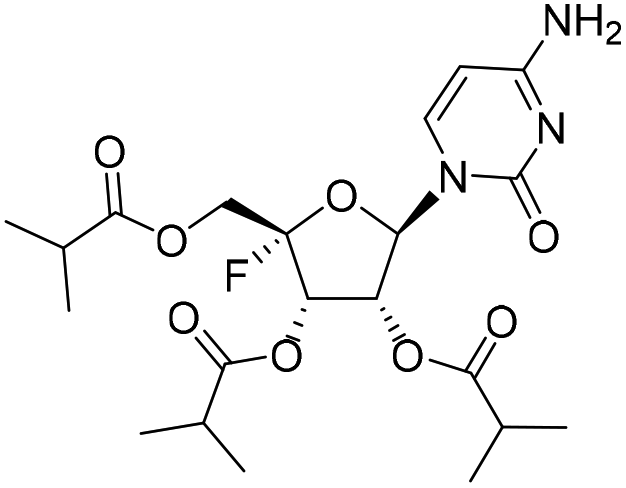
|
— | — | 97.4%a |
— |
| 2a |
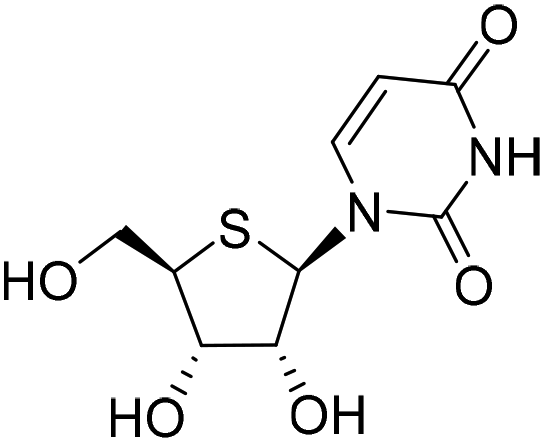
|
−1.6%a |
— | −1.2%a |
— |
| 2b |
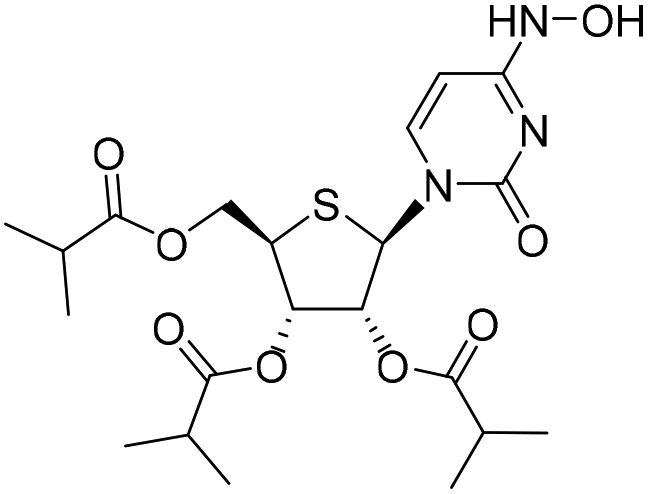
|
— | — | 6.1%a |
— |
| 2c |
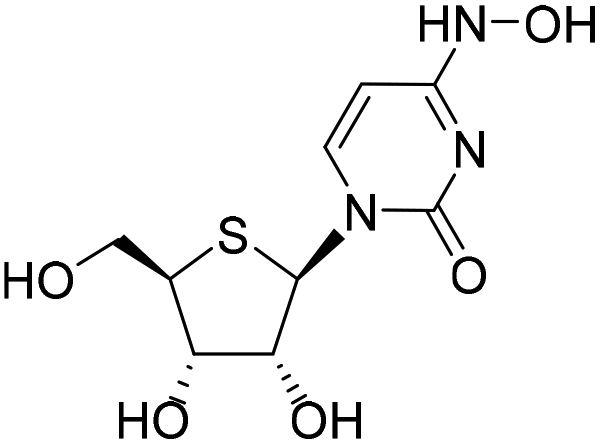
|
−8.9%a |
— | 6.5%a |
— |
| 3a |
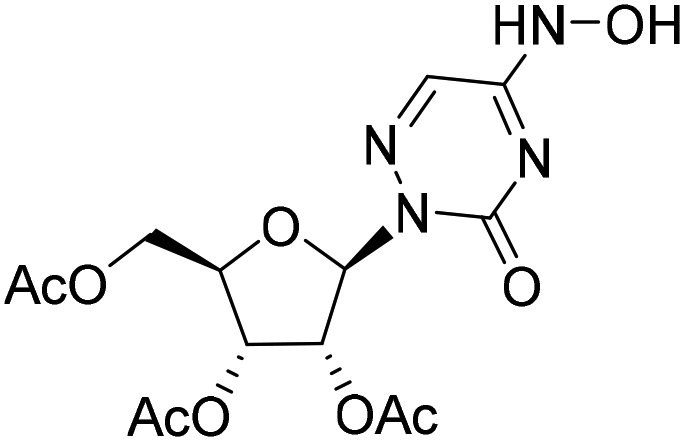
|
— | — | 2.0%a |
— |
| 3b |
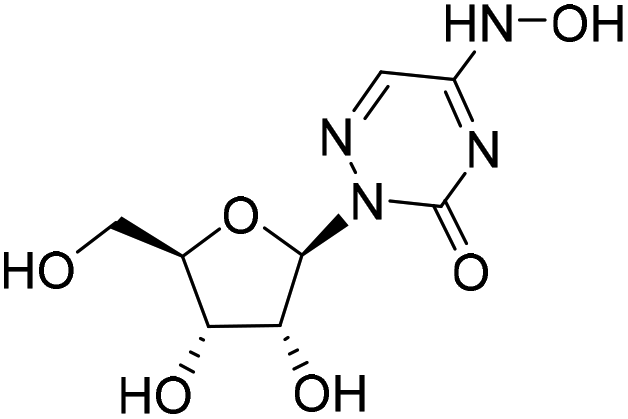
|
2.0%a |
— | 7.5%a |
— |
| 3c |
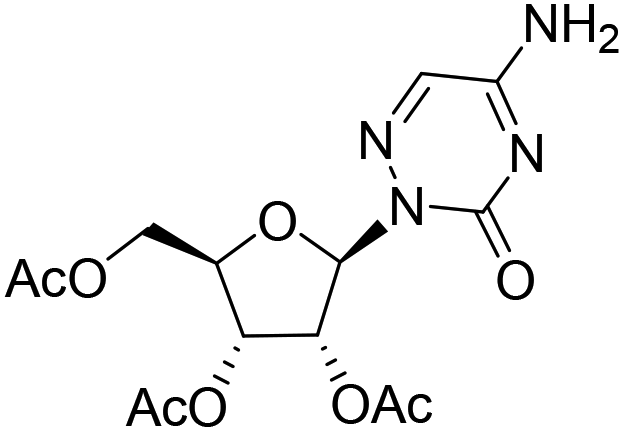
|
— | — | −7.4%a |
— |
| 3d |
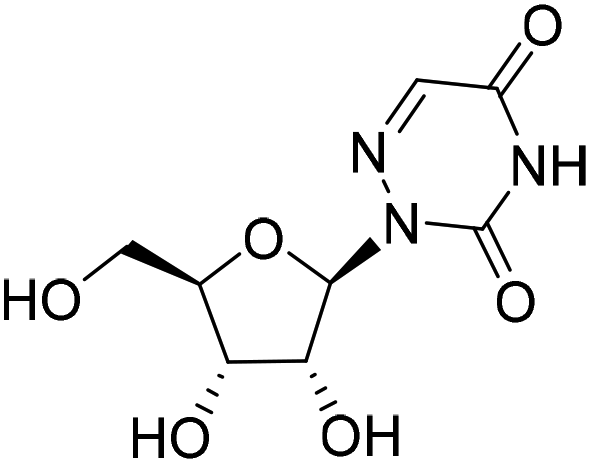
|
— | — | Cytotoxica | — |
| 4a |
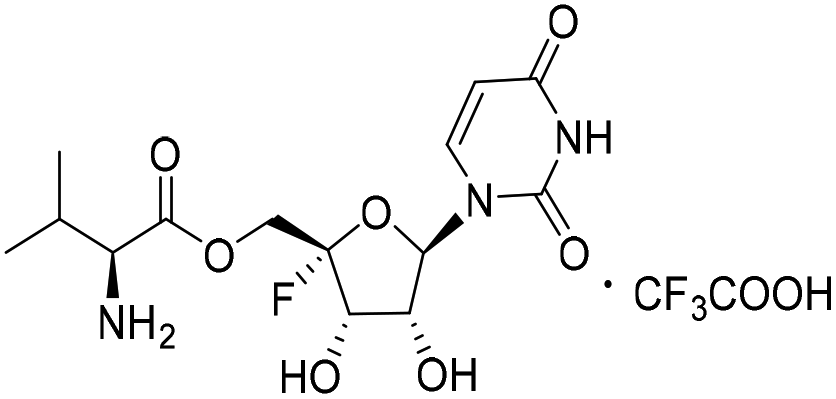
|
4.90 | >10 | <0.12 | >10 |
| 4b |
|
1.46 | >10 | 0.23 | >10 |
| 4c |
|
1.43 | >10 | 0.96 | >10 |
| 4d |
|
3.25 | >10 | <0.12 | >10 |
| 4e |
|
1.86 | >10 | 0.42 | >10 |
| 22 |
|
2.32 | >10 | <0.12 | >10 |
| ALS-8112 |
|
0.48 | >10 | — | — |
In the beginning, we designed and synthesized compounds 1b, 2c and 3b as NHC derivatives all possessing a C4 hydroxylamine group. Compound 1b was a tri-isobutyrate ester of 4′-fluorine-substituted NHC; 2c and 3b were a 4′-thionucleoside and a 6-azanucleoside, respectively. In our study, NHC was found to inhibit the replication of RSV and IFV with an EC50 of 4.6 μM and 0.77 μM, respectively. However, the three compounds didn't show any inhibitory activity against the two viruses, neither did ester prodrugs 2b and 3a. Previous studies have indicated that the steric conformation of nucleoside triphosphates was crucial for binding with the active site of viral polymerase and being incorporated into the growing chain.12 We speculated that the absence of antiviral activities of these NHC derivatives was due to the minor structural modifications which likely altered the steric conformation of nucleosides compared with NHC, thus leading to disfavored binding with the viral polymerase. In addition, the loss of antiviral activities may also be related to reduced phosphorylation efficiency.
Compound 2a is a thionucleoside and its phosphoramidate prodrugs were reported to exhibit anti-hepatitis C activity.16 Recently, a patent disclosed 2a as a potent antiviral agent against SARS-CoV-2 with EC50 values in a low micromolar range.17 In our study, 2a was found inactive against RSV and IFV, indicating that this nucleoside may have a narrow antiviral spectrum. 6-Azauridine and 6-azacytidine had been shown to have anti-cancer and antiviral activities.18,19 However, we observed that the acetate ester prodrug 3c was inactive against IFV and 6-azauridine 3d was highly cytotoxic to MDCK cells, so these nucleosides were not considered for further evaluation.
The 4′-fluorine nucleoside tri-isobutyrate esters 1a and 1c were found to be highly potent against IFV at a concentration of 5.0 μM (with inhibition rates of 99.8% and 97.4%, respectively). Moreover, they did not show cytotoxic effects at this concentration. Because of the presence of the 4′-fluorine atom, the 4′-fluoro-nucleosides were likely susceptible to nucleophilic attack, as evidenced by poor stability in H2O described in previous research.20 In our synthetic study, we found that the stability of 4′-fluorouridine was better than that of the cytidine analog. Therefore, we further synthesized 4′-fluorouridine 22 and its several ester prodrugs with modifications on the hydroxyl groups. Compounds 1a and 4c were two tri-ester prodrugs; 4a and 4b were the 5′-L-valine ester and 5′-isobutyrate ester prodrug, respectively; 4d had a carbonate group masking the 2′ and 3′ hydroxyl groups and 4e contained a 5′-L-valine ester and two isobutyrate ester groups. Nucleoside 22 showed potent antiviral activities against RSV and IFV with an EC50 value of 2.32 μM and less than 0.12 μM, respectively. All these prodrugs exhibited similar activities with anti-RSV EC50 values ranging from 1.13 μM to 4.90 μM and anti-IFV EC50 values ranging from less than 0.12 μM to 0.96 μM. This result suggested that the prodrug moiety could be metabolized in cells to release the parent nucleoside.
Stability studies
4′-Fluorouridine had been shown to be very labile in water and it decomposed slower in phosphate buffer with uracil as the main degradation product.21 The chemical stability of 4′-fluorouridine 22 and four ester derivatives (1a and 4b–d) was determined through the content change by using HPLC in pH 2.0 and 4.0 buffer at 30 °C after 7 days, and the result is shown in Table 2. It was found that compound 22 had poor stability with 45% and 73% decrease in pH 4.0 and pH 2.0 buffer, respectively. Among the four ester derivatives, compound 1a exhibited the best stability profile with only a slight change of the content (0.24% and 0.46% decrease in pH 4.0 and pH 2.0 buffer, respectively). In another experiment, we observed that the level of uracil increased in a nearly linear manner within 7 days for 4′-fluorouridine (22) in pH 2.0 buffer (Fig. 2). Relatively, 4d showed significantly enhanced stability with only a little amount of uracil being generated (1.09% at day 7, ESI†). Based on this result, it could be concluded that modifications of all three hydroxyl groups would greatly improve the stability of the 4′-fluorinated nucleoside.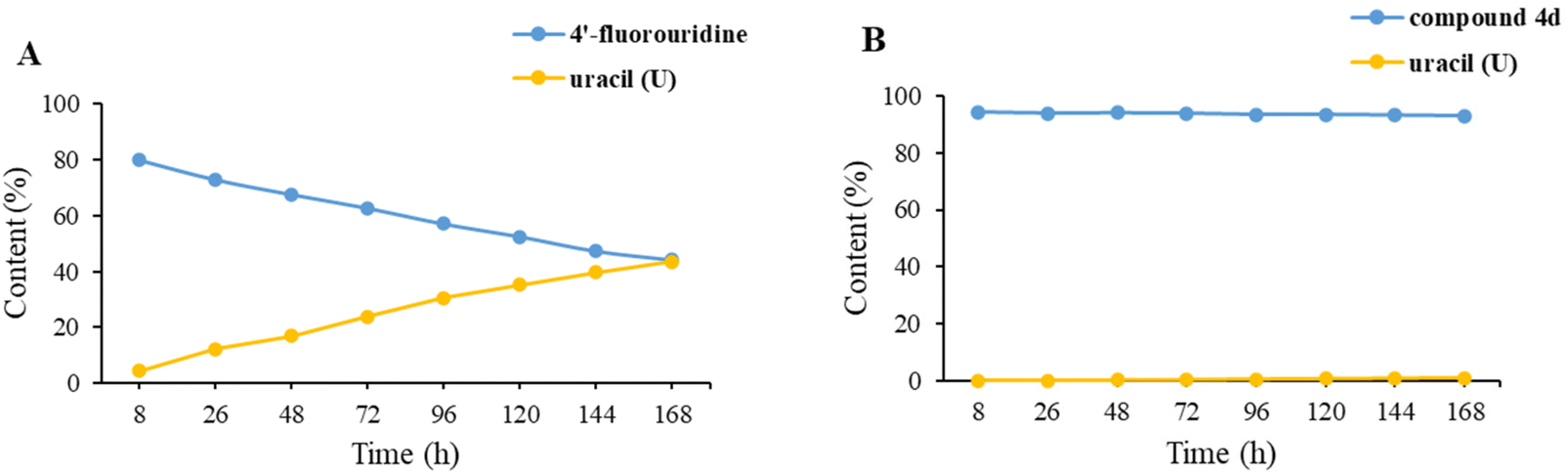 | ||
| Fig. 2 The stability of (A) 4′-fluorouridine and (B) compound 4d and formation of uracil (U) in pH = 2.0 buffer at 30 °C after 7 days. | ||
| Compound | pH 2.0 | pH 4.0 |
|---|---|---|
| Change of content (%) | Change of content (%) | |
| 1a | −0.46 | −0.24 |
| 4b | −7.70 | −6.90 |
| 4c | −2.23 | −0.35 |
| 4d | −5.80 | −7.70 |
| 22 | −73.0 | −45.0 |
Pharmacokinetic studies
Given the low chemical stability of 22, we were curious about the pharmacokinetic (PK) properties of this compound. Therefore, we conducted a PK study of 22 and prodrug 1a in SD rats. Compound 22 was administered at an intravenous dose of 5 mg kg−1 and an oral dose of 10 mg kg−1. The oral bioavailability was 82.9% calculated based on the plasma exposure of the parent nucleoside, suggesting that this nucleoside was efficiently absorbed (Table 3). The half-life of the nucleoside after oral administration was 7.75 h which was a little longer than that after intravenous administration (6.09 h). For the oral administration group at 10 mg kg−1, the maximum concentration (Cmax) reached 2495 ng ml−1 (9.5 μM) which was much higher than the EC50 value against IFV and RSV. Generally, compound 22 showed excellent PK properties. In another experiment, we compared the PK parameters of compound 22 with those of 1a at the same molar dose (10 mg kg−1 for 22 and 18 mg kg−1 for 1a). It was pleasing to find that 1a had better oral PK properties than compound 22 in terms of Cmax, T1/2 and AUC0–t.| Compd. | Route | T max (h) | C max (ng mL−1) | T 1/2 (h) | AUC0–t (ng h mL−1) | AUC0−∞ (ng h mL−1) | MRT0–∞ (%) | F (%) |
|---|---|---|---|---|---|---|---|---|
| a The vehicle for administration is 5% DMSO + 5% ethanol + 40% PEG400 + 50% saline. b The vehicle for administration is 10% DMSO + 10% solutol + 80%(20%HP-β-CD). | ||||||||
| 22 | p.o.a | 2.00 | 2495 | 7.75 | 13757 |
14910 |
7.63 | 82.9 |
| i.v.a | — | 2618 | 6.09 | 8294 | 8691 | 6.89 | ||
| 22 | p.o.b | 0.67 | 2345 | 5.87 | 11555 |
12105 |
5.54 | |
| 1a | p.o.b | 0.83 | 3234 | 8.22 | 15615 |
16936 |
7.26 | |
Conclusions
In summary, we synthesized several NHC derivatives and 4′-fluorouridine prodrugs. The NHC derivatives did not exhibit activities against IFV or RSV in our lab, and innovative work would be warranted for the discovery of novel nucleoside antiviral agents. Recent research has investigated the therapeutic potential of 4′-fluorouridine against RSV and SARS-CoV-2 infection in animal models.15 However, 4′-fluorouridine has poor chemical stability which may significantly limit its application. Our study presented that after esterification, the chemical stability of the derived 4′-fluorouridine prodrugs was improved. Tri-isobutyrate ester 1a with its excellent stability profile and PK properties has the potential to be a new oral antiviral candidate.Experimental section
General methods
All reagents were directly used without further purification. All water-sensitive reactions were carried out under anhydrous conditions using raw materials dried in a vacuum drying chamber overnight; the glassware was kept in an oven at 100 °C for several hours and reagents were dried using molecular sieves. The reactions were monitored by TLC with 0.2 mm silica gel plates (HSGF 254). The purification procedure of some compounds was performed by silica gel column chromatography (200–300 mesh). 1H-NMR spectra and 13C-NMR spectra were determined on Bruker 400 MHz, 500 MHz or 600 MHz and Bruker 126 MHz, 201 MHz or 151 MHz instruments with TMS as an internal standard, respectively. All of the spectra were obtained with MestReNova software. LRMS was measured on a Finnigan LTQ and HRMS was measured on a 1290–6545 UHPLC-QTOF. The raw materials 5, 15 and 16 were purchased from Shanghai Haohong Scientific Co., Ltd, and intermediate 11 was obtained from Topharman Shanghai Co., Ltd.Method A: General procedure for the synthesis of prodrugs with anhydrides (isobutyric anhydride and acetic anhydride)
To a solution of raw material in DCM (EA for the synthesis of 4c), triethylamine, DMAP and anhydrides were sequentially added at 0 °C. After stirring at rt until the raw material was completely consumed, the mixture was diluted with DCM and washed successively with aqueous 1 M hydrochloric acid solution and saturated aqueous NaHCO3 solution. The organic layer was dried (Na2SO4), filtered and concentrated in vacuo. The crude was purified by silica gel column chromatography to afford target compounds.Method B: General procedure for the synthesis of prodrugs with N-Boc-L-valine
To a solution of raw material (1 eq.) in DCM N-Boc-L-valine (1.4 eq.), HOBT (1.5 eq.), EDCI (2.2 eq.) and DMAP (4 eq.) were sequentially added at 0 °C. After stirring at rt until the substrate was completely consumed as monitored by TLC, the mixture was diluted with DCM and washed successively with aqueous 1 M hydrochloric acid solution and saturated aqueous NaHCO3 solution. The organic layer was dried (Na2SO4), filtered and concentrated in vacuo. The crude was purified by silica gel column chromatography (PE:EA = 1:1) to afford the intermediate. After removing the Boc group using trifluoroacetic acid, the target compounds were obtained.
1-((2R,3R,4S,5S)-3,4-Dihydroxy-5-(iodomethyl)tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione (6)
To a solution of triphenylphosphine (9.43 g, 36 mmol) in pyridine (80 mL) at 0 °C, iodine (9.25 g, 36 mmol) was added and the mixture was stirred for 10 min at 20 °C. Uridine 5 (7.32 g, 30 mmol) was added, the mixture was stirred at rt for 6 h, and then quenched with saturated aqueous Na2S2O3 solution. The resulting solution was concentrated and the residue was partitioned between water and tetrahydrofuran. The organic portion was washed with saturated brine, dried with Na2SO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography (DCM:MeOH = 100:1 → 15:1) to give compound 6 as a yellow solid (8.5 g, yield 67%). 1H NMR (500 MHz, DMSO-d6) δ 11.39 (s, 1H), 7.68 (d, J = 8.1 Hz, 1H), 5.80 (d, J = 5.9 Hz, 1H), 5.68 (dd, J = 8.1, 2.1 Hz, 1H), 5.49 (d, J = 5.8 Hz, 1H), 5.37 (d, J = 5.1 Hz, 1H), 4.19 (q, J = 5.7 Hz, 1H), 3.91–3.81 (m, 2H), 3.55 (dd, J = 10.5, 5.5 Hz, 1H), 3.40 (dd, J = 10.5, 6.6 Hz, 1H).
1-((2R,3R,4S)-3,4-Dihydroxy-5-methylenetetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione (7)
A fresh solution of sodium methoxide in MeOH was prepared by adding sodium (830 mg, 36 mmol) to methanol (30 mL) in an ice bath. To a solution of 6 (8.5 g, 24 mmol) in anhydrous MeOH (30 mL), the above prepared solution was added. The reaction mixture was stirred under reflux (67 °C) for 3 h, then adjusted to pH = 8–9 with aqueous 1 M hydrochloric acid solution and concentrated to dryness. Chromatography on a silica gel column (DCM:MeOH = 15:1) afforded 7 as a white solid (3.1 g, yield 57%). 1H NMR (400 MHz, DMSO-d6) δ 11.44 (s, 1H), 7.60 (d, J = 8.1 Hz, 1H), 5.98 (d, J = 5.4 Hz, 1H), 5.65 (d, J = 8.0 Hz, 1H), 5.61 (s, 1H), 5.47 (d, J = 5.4 Hz, 1H), 4.41 (s, 1H), 4.35 (s, 1H), 4.26 (d, J = 5.0 Hz, 1H), 4.19 (s, 1H).
1-((2R,3R,4S,5R)-5-Fluoro-3,4-dihydroxy-5-(iodomethyl)tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione (8)
To a solution of 7 (3.1 g, 13.7 mmol) and triethylamine trihydrofluoride (2.7 g, 16.5 mmol) in anhydrous acetonitrile (60 mL), N-iodosuccinimide (3.7 g, 16.5 mmol) was added in portions at 0 °C. After stirring for 5 min, the mixture was filtered and washed with DCM to give 8 as a white solid (4.1 g, yield 77%). 1H NMR (500 MHz, DMSO-d6) δ 11.50 (s, 1H), 7.67 (d, J = 8.1 Hz, 1H), 5.91 (d, J = 3.0 Hz, 1H), 5.76 (d, J = 5.3 Hz, 1H), 5.68 (dd, J = 8.0, 2.2 Hz, 1H), 5.42 (d, J = 8.9 Hz, 1H), 4.41–4.33 (m, 1H), 4.31–4.26 (m, 1H), 3.62–3.46 (m, 2H).(2R,3S,4R,5R)-5-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-fluoro-2-(iodomethyl)tetrahydrofuran-3,4-diyl bis(2-methylpropanoate) (9)
Compound 9 was prepared by following the same procedure as described for method A. The reaction of 8 (2.0 g, 5.38 mmol) with triethylamine (4.35 g, 43.04 mmol), DMAP (0.33 g, 2.69 mmol) and isobutyric anhydride (1.78 g, 11.29 mmol) gave 9 as a white solid (1.3 g, yield 47%). 1H NMR (500 MHz, DMSO-d6): δ 11.60 (s, 1H), 7.80 (d, J = 8.1 Hz, 1H), 6.02 (d, J = 2.4 Hz, 1H), 5.78 (dd, J = 19.0, 7.3 Hz, 1H), 5.73 (d, J = 8.0 Hz, 1H), 5.59 (dd, J = 7.3, 2.4 Hz, 1H), 3.65–3.49 (m, 2H), 2.65–2.58 (m, 2H), 1.16–1.10 (m, 12H).(2S,3S,4R,5R)-5-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-fluoro-2-(hydroxymethyl)tetrahydrofuran-3,4-diyl bis(2-methylpropanoate) (10)
Tetrabutylammonium hydroxide (3.30 g, 40% aqueous solution, 12.7 mmol) was adjusted to pH ≈ 4 by adding TFA. The resulting solution was added to a solution of 9 (1.3 g, 2.54 mmol) in DCM (10 mL). m-Chloroperbenzonic (2.19 g, 12.7 mmol) was added in portions under vigorous stirring and the resulting reaction mixture was stirred at rt for 7 h. The reaction was quenched with saturated aqueous Na2S2O3 solution and extracted with EA and THF. The combined organic portions were dried (Na2SO4) and concentrated in vacuo. The crude product was purified by silica gel column chromatography (PE:EA = 1:3) to give compound 10 as a white solid (520 mg, yield 52%). 1H NMR (500 MHz, DMSO-d6): δ 11.54 (s, 1H), 7.79 (d, J = 8.1 Hz, 1H), 6.01 (d, J = 2.5 Hz, 1H), 5.74–5.65 (m, 2H), 5.54 (dd, J = 7.1, 2.5 Hz, 1H), 5.50 (t, J = 6.3 Hz, 1H), 2.63–2.53 (m, J = 6.8 Hz, 2H), 1.14–1.06 (m, 12H).
(2S,3S,4R,5R)-5-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-fluoro-2-((isobutyryloxy)methyl)tetrahydrofuran-3,4-diyl bis(2-methylpropanoate) (1a)
Compound 1a was prepared by following the same procedure as described for method A. The reaction of 10 (520 mg, 1.33 mmol) with triethylamine (653 mg, 5.32 mmol), DMAP (67 mg, 0.66 mmol) and isobutyric anhydride (231 mg, 1.46 mmol) gave 1a as a white solid (600 mg, yield 98%). 1H NMR (500 MHz, DMSO-d6): δ 11.59 (s, 1H), 7.78 (d, J = 8.1 Hz, 1H), 6.03 (d, J = 2.2 Hz, 1H), 5.81 (dd, J = 20.0, 7.3 Hz, 1H), 5.70 (dd, J = 8.0, 2.2 Hz, 1H), 5.58 (dd, J = 7.2, 2.1 Hz, 1H), 4.40–4.24 (m, 2H), 2.64–2.52 (m, 3H), 1.13–1.07 (m, 18H). 13C NMR (151 MHz, DMSO-d6): δ 175.27, 175.18, 174.65, 163.18, 150.06, 144.13, 114.58 (d, J = 233.6 Hz), 102.17, 94.02, 70.29, 69.02 (d, J = 19.5 Hz), 61.33 (d, J = 36.9 Hz), 33.08, 33.05, 33.01, 18.62, 18.60, 18.55, 18.55, 18.50, 18.37. HRMS (ESI+) m/z: calcd for C21H29FN2O9 [M + H]+, 473.1930; found, 473.1932.(2S,3S,4R,5R)-2-Fluoro-5-(6-(hydroxyamino)-2-oxo-1l3,3-oxazin-3(2H)-yl)-2-((isobutyryloxy)methyl)tetrahydrofuran-3,4-diyl bis(2-methylpropanoate) (1b)
To a solution of 1a (200.0 mg, 0.43 mmol) in DCM (15 mL), triethylamine (173.7 mg, 1.72 mmol), DMAP (24.4 mg, 0.22 mmol) and TPSCl (260.6 mg, 0.86 mmol) were sequentially added at 0 °C. After stirring at rt for 1 h, TLC was used to monitor that the substrate was completely consumed, then triethylamine (347.4 mg, 3.44 mmol) and hydroxyamine hydrochloride (298.9 mg, 4.30 mmol) were added. After stirring at rt for 4 h, the mixture was diluted with DCM and washed successively with aqueous 1 M hydrochloric acid solution and saturated aqueous NaHCO3 solution. The organic layer was dried (Na2SO4), filtered and concentrated in vacuo. The crude was purified by silica gel column chromatography (PE:EA = 1:1) to afford 1b as a white solid (40 mg, yield 18%). 1H NMR (400 MHz, DMSO-d6): δ 10.15 (s, 1H), 9.90 (s, 1H), 7.00 (d, J = 8.2 Hz, 1H), 5.77 (dd, J = 19.5, 7.3 Hz, 1H), 5.60 (dd, J = 8.1, 1.8 Hz, 1H), 5.52 (dd, J = 7.3, 2.4 Hz, 1H), 4.41–4.21 (m, 2H), 2.66–2.52 (m, 3H), 1.13–1.07 (m, 18H). 13C NMR (126 MHz, DMSO-d6): δ 175.30, 175.16, 174.67, 148.87, 142.98, 133.11, 114.21 (d, J = 233.3 Hz), 98.98, 93.34, 70.12, 69.10 (d, J = 19.7 Hz), 61.58 (d, J = 36.0 Hz), 33.01, 33.01, 32.96, 18.63, 18.57, 18.56, 18.55, 18.51, 18.39. HRMS (ESI−) m/z: calcd for C21H30FN3O9 [M − H]−, 486.1893; found, 486.1900.
(2S,3S,4R,5R)-5-(6-Amino-2-oxo-1λ3,3-oxazin-3(2H)-yl)-2-fluoro-2-((isobutyryloxy)methyl)tetrahydrofuran-3,4-diyl bis(2-methylpropanoate) (1c)
Compound 1c was obtained by following the same procedure as described for 1b. The reaction of 1a (100 mg, 0.215 mmol) with triethylamine (86.9 mg, 0.86 mmol), DMAP (12.2 mg, 0.11 mmol) and 25% ammonium hydroxide (301 mg, 2.15 mmol) gave 1c as a white solid (14 mg, yield 14%). 1H NMR (400 MHz, DMSO-d6): δ 7.68 (d, J = 7.4 Hz, 1H), 7.43 (d, J = 9.0 Hz, 2H), 5.94–5.84 (m, 2H), 5.73 (d, J = 7.4 Hz, 1H), 5.56 (dd, J = 7.2, 1.9 Hz, 1H), 4.39–4.22 (m, 2H), 2.67–2.52 (m, 3H), 1.14–1.06 (m, 18H). 13C NMR (151 MHz, DMSO-d6): δ 175.32, 175.29, 174.70, 166.33, 154.37, 145.28, 114.71 (d, J = 233.0 Hz), 95.75, 94.63, 70.61, 69.69 (d, J = 19.2 Hz), 61.70 (d, J = 36.4 Hz), 33.07, 33.07, 32.99, 18.61, 18.61, 18.58, 18.55, 18.55, 18.40. HRMS (ESI+) m/z: calcd for C21H30FN3O8 [M + H]+, 472.209; found, 472.2092.1-O-Acetyl-2,3,5-tri-O-benzyl-4-thio-α,β-D-ribofuranose (12)
Compound 12 was synthesized according to the literature procedure (α:β = 6:1).14 12α: 1H NMR (500 MHz, CDCl3): δ 7.43–7.26 (m, 15H), 6.26 (d, J = 4.7 Hz, 1H), 4.78–4.45 (m, 6H), 4.17–4.13 (m, 1H), 4.07 (t, J = 4.4 Hz, 1H), 3.80–3.77 (m, 1H), 3.43 (dd, J = 10.0, 5.4 Hz, 1H), 3.40 (dd, J = 10.0, 7.7 Hz, 1H), 2.04 (s, 1H). 12β: 1H NMR (500 MHz, CDCl3): δ 7.40–7.24 (m, 15H), 6.01 (d, J = 1.9 Hz, 1H), 4.81–4.45 (m, 6H), 4.14 (dd, J = 3.3, 2.0 Hz, 1H), 4.01 (dd, J = 8.4, 3.5 Hz, 1H), 3.91–3.86 (m, 1H), 3.82 (dd, J = 9.8, 4.4 Hz, 1H), 3.62 (dd, J = 9.8, 7.0 Hz, 1H), 2.06 (s, 3H). LRMS (ESI+) m/z: calcd for C28H30O5S [M + H]+, 478.18; found, 479.6.
1-((3R,4S,5R)-3,4-Bis(benzyloxy)-5-((benzyloxy)methyl)tetrahydrothiophen-2-yl)pyrimidine-2,4(1H,3H)-dione (13)
Compound 12 (1.4 g, 3.0 mmol, a mixture of α and β) and silylated uracil (6.0 mmol) were dissolved in acetonitrile (20 mL) and then treated with TMSOTf (1.5 g, 6.6 mmol) at −10 °C. After stirring for 30 min, the temperature was raised to rt. When the starting materials were completely consumed, the reaction was quenched with saturated aqueous NaHCO3 solution and then concentrated to remove acetonitrile. The residue was extracted with EA. The organic layer was washed with brine, dried using Na2SO4 and then filtered. After concentration, the residue was purified by flash chromatography (PE:EA = 10:1) to afford 13 (1.4 g, yield 85%, α:β = 1:1). 13α: 1H NMR (500 MHz, CDCl3) δ 8.04 (d, J = 8.2 Hz, 1H), 7.39–7.19 (m, 15H), 6.33 (d, J = 3.4 Hz, 1H), 5.55 (dd, J = 8.2, 2.2 Hz, 1H), 4.75–4.44 (m, 6H), 4.00 (dd, J = 5.9, 3.6 Hz, 1H), 3.96 (t, J = 3.5 Hz, 1H), 3.76–3.73 (m, 1H), 3.69 (dd, J = 10.2, 3.0 Hz, 1H), 3.49 (dd, J = 9.9, 6.2 Hz, 1H). 13β: 1H NMR (500 MHz, CDCl3) δ 8.09 (d, J = 8.2 Hz, 1H), 7.39–7.19 (m, 15H), 6.04 (d, J = 5.9 Hz, 1H), 5.07 (dd, J = 8.2, 2.2 Hz, 1H), 4.75–4.44 (m, 6H), 4.24 (dd, J = 5.4, 3.6 Hz, 1H), 3.91 (t, J = 3.5 Hz, 1H), 3.89 (dd, J = 17.7, 4.0 Hz, 1H), 3.83 (dd, J = 10.2, 2.5 Hz, 1H), 3.56 (dd, J = 9.9, 5.6 Hz, 1H). LRMS (ESI+) m/z: calcd for C30H30N2O5S [M + H]+, 530.19; found, 531.5.
1-((2R,3R,4S,5R)-3,4-Dihydroxy-5-(hydroxymethyl)tetrahydrothiophen-2-yl)pyrimidine-2,4(1H,3H)-dione (2a)
A solution of 13 (700 mg, 1.32 mmol, β anomer) in DCM (5 mL) was treated with 1.0 M BCl3 solution in DCM (5.3 mL, 5.28 mmol) at −78 °C. After the addition, the temperature was slowly increased to −30 °C. When the raw materials were consumed, the reaction solution was quenched with methanol, then the pH of the reaction solution was adjusted to 7–8 with NaHCO3 solution, concentrated and purified by reverse-phase silica gel column chromatography (H2O:MeOH = 100:1 → 50:1) to obtain compound 2a (261 mg, yield 76%). 1H NMR (500 MHz, DMSO-d6): δ 11.32 (s, 1H), 7.99 (d, J = 8.1 Hz, 1H), 5.90 (d, J = 7.4 Hz, 1H), 5.68 (d, J = 8.1 Hz, 1H), 5.49 (d, J = 5.8 Hz, 1H), 5.28 (d, J = 4.3 Hz, 1H), 5.18 (t, J = 5.3 Hz, 1H), 4.16 (dt, J = 8.2, 4.0 Hz, 1H), 4.03 (q, J = 3.1 Hz, 1H), 3.68–3.52 (m, 2H), 3.22–3.18 (m, 1H). 13C NMR (126 MHz, DMSO-d6): δ 162.97, 151.16, 141.52, 102.14, 76.33, 73.00, 63.11, 62.28, 52.98. HRMS (ESI+) m/z: calcd for C9H12N2O5S [M + Na]+, 283.0359; found, 283.0355.
(2R,3R,4S,5R)-2-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-5-((isobutyryloxy)methyl)tetrahydrothiophene-3,4-diyl bis(2-methylpropanoate) (14)
Compound 14 was synthesized according to method A. The reaction of 2a (260 mg, 1.0 mmol) with triethylamine (1.0 g, 10.4 mmol), DMAP (43 mg, 0.35 mmol) and isobutyric anhydride (823 mg, 5.2 mmol) gave 14 as a white solid (400 mg, yield 85%). 1H NMR (500 MHz, DMSO-d6): δ 11.46 (d, J = 1.8 Hz, 1H), 8.02 (d, J = 8.1 Hz, 1H), 6.19 (d, J = 7.8 Hz, 1H), 5.76 (dd, J = 8.1, 2.2 Hz, 1H), 5.68 (dd, J = 7.7, 4.0 Hz, 1H), 5.53–5.45 (m, 1H), 4.46 (dd, J = 11.6, 7.4 Hz, 1H), 4.34 (dd, J = 11.6, 5.9 Hz, 1H), 3.70–3.64 (m, 1H), 2.69–2.56 (m, 2H), 2.50–2.46 (m, 1H), 1.15 (dd, J = 14.0, 7.0 Hz, 12H), 1.03 (dd, J = 7.0, 4.1 Hz, 6H). LRMS (ESI+) m/z: calcd for C21H30N2O8S [M + H]+, 470.17; found, 471.3.(2R,3R,4S,5R)-2-(4-(Hydroxyamino)-2-oxopyrimidin-1(2H)-yl)-5-((isobutyryloxy)methyl)tetrahydrothiophene-3,4-diyl bis(2-methylpropanoate) (2b)
Compound 14 (150.0 mg, 0.32 mmol), DIPEA (207.0 mg, 1.6 mmol) and DMAP (4 mg, cat.) were sequentially added to DCM (10 mL), followed by TPSCl (194.0 mg, 0.64 mmol) at 0 °C for 10 min. After stirring at rt for 2 h, DIPEA (207.0 mg, 1.6 mmol) and hydroxyamine hydrochloride (111.0 mg, 1.6 mmol) were added at 0 °C for 10 min. After stirring at rt for 2 h, the mixture was diluted with DCM and washed successively with aqueous 1 M hydrochloric acid solution and saturated aqueous NaHCO3 solution. The organic layer was dried (Na2SO4), filtered and concentrated in vacuo. The crude was purified by silica gel column chromatography (PE:EA = 1:1) to afford 2b as a foam (100 mg, yield 64%). 1H NMR (600 MHz, DMSO-d6): δ 10.09 (s, 1H), 9.72 (d, J = 2.2 Hz, 1H), 7.18 (d, J = 8.3 Hz, 1H), 6.20 (d, J = 8.3 Hz, 1H), 5.68 (dd, J = 8.2, 2.0 Hz, 1H), 5.58 (dd, J = 8.3, 4.0 Hz, 1H), 5.47 (dd, J = 4.0, 2.2 Hz, 1H), 4.42–4.28 (m, 2H), 3.65–3.60 (m, 1H), 2.69–2.54 (m, 2H), 2.49–2.45 (m, 1H), 1.15 (d, J = 7.0 Hz, 3H), 1.12 (dd, J = 6.9, 3.2 Hz, 9H), 1.02 (dd, J = 7.0, 2.8 Hz, 6H). 13C NMR (126 MHz, DMSO-d6): δ 175.70, 174.89, 174.80, 149.43, 142.89, 129.59, 99.74, 73.68, 72.22, 64.33, 60.19, 46.55, 33.17, 33.17, 33.08, 18.81, 18.66, 18.66, 18.49, 18.41, 18.32. HRMS (ESI+) m/z: calcd for C21H31N3O8S [M + H]+, 486.1905; found, 486.1902.
1-((2R,3R,4S,5R)-3,4-Dihydroxy-5-(hydroxymethyl)tetrahydrothiophen-2-yl)-4-(hydroxyamino)pyrimidin-2(1H)-one (2c)
Compound 2b (50 mg, 0.1 mmol) was added to ammonia solution in methanol (8 mL) and stirred at rt. After the raw material was completely consumed, the reaction solution was concentrated and slurried with methyl tert-butyl ether (MTBE) to obtain 2c as an off-white solid (22 mg, yield 80%). 1H NMR (600 MHz, DMSO-d6): δ 9.96 (s, 1H), 9.47 (s, 1H), 7.15 (d, J = 8.3 Hz, 1H), 5.90 (d, J = 8.0 Hz, 1H), 5.61 (d, J = 8.2 Hz, 1H), 5.33 (d, J = 6.3 Hz, 1H), 5.21 (d, J = 4.1 Hz, 1H), 5.14 (t, J = 5.4 Hz, 1H), 4.12–4.06 (m, 1H), 4.02 (td, J = 3.8, 2.1 Hz, 1H), 3.61–3.50 (m, 2H), 3.17–3.12 (m, 1H). 13C NMR (126 MHz, DMSO-d6): δ 149.89, 143.45, 130.38, 98.86, 75.44, 73.10, 63.56, 61.69, 52.72. HRMS (ESI−) m/z: calcd for C9H13N3O5S [M − H]−, 274.0503; found, 274.0502.(2R,3R,4R,5R)-2-(Acetoxymethyl)-5-(3,5-dioxo-4,5-dihydro-1,2,4-triazin-2(3H)-yl)tetrahydrofuran-3,4-diyl diacetate (17)
6-Azauracil 16 (904 mg, 8 mmol) and (NH4)2SO4 (52.8 mg, 0.4 mmol) were added to HMDS (20 mL) and stirred at 130 °C for 2 h. After the raw material was completely consumed, the reaction solution was concentrated. A solution of 1,2,3,5-tetra-O-acetyl-D-ribofuranose 15 (1.27 g, 4 mmol) in DCM (30 mL) was treated with the concentrate, followed by the addition of SnCl4 (2.5 g, 9.6 mmol) at 0 °C. The reaction was stirred at rt for 3 h and extracted with EA. The organic layers were washed with saturated aqueous NaHCO3 solution and brine, then dried using Na2SO4. After concentration, the residue was purified by flash chromatography (PE:EA = 2:1 → 1:1) to afford 17 as a white foamy solid (950 mg, yield 64.5%). 1H NMR (500 MHz, DMSO-d6): δ 12.36 (s, 1H), 7.67 (d, J = 7.2 Hz, 1H), 6.13 (d, J = 3.2 Hz, 1H), 5.54 (dd, J = 5.5, 3.3 Hz, 1H), 5.35 (t, J = 5.9 Hz, 1H), 4.31 (ddd, J = 27.5, 10.8, 4.1 Hz, 2H), 4.10–4.02 (m, 1H), 2.10 (s, 3H), 2.07 (s, 3H), 2.03 (s, 3H).
(2R,3R,4R,5R)-2-(Acetoxymethyl)-5-(5-(hydroxyamino)-3-oxo-1,2,4-triazin-2(3H)-yl)tetrahydrofuran-3,4-diyl diacetate (3a)
To a solution of 17 (100 mg, 0.27 mmol) and triethylamine (136 mg, 1.35 mmol) in acetonitrile (6 mL), POCl3 (82.9 mg, 0.54 mmol) was added at 0 °C. After stirring at rt for 10 min, a solution of hydroxylamine hydrochloride (187.7 mg, 2.7 mmol) and triethylamine (273 mg, 2.7 mmol) in acetonitrile (1 mL) was added. The mixture was stirred at rt for 1 h, and treated with the same post-processing operation. The residue was purified by thin layer chromatography (PE:EA = 2:1), and then slurred in a mixed solution of petroleum ether and dichloromethane to obtain compound 3a as a white solid (18 mg, yield 17.3%). 1H NMR (500 MHz, DMSO-d6): δ 11.10 (s, 1H), 10.79 (s, 1H), 7.54 (d, J = 1.8 Hz, 1H), 6.03 (d, J = 3.7 Hz, 1H), 5.49 (dd, J = 5.6, 3.7 Hz, 1H), 5.28 (t, J = 5.8 Hz, 1H), 4.32 (dd, J = 12.0, 3.5 Hz, 1H), 4.21 (q, J = 5.3 Hz, 1H), 4.03 (dd, J = 12.0, 5.3 Hz, 1H), 2.07 (s, 3H), 2.06 (s, 3H), 2.02 (s, 3H). 13C NMR (151 MHz, DMSO-d6): δ 170.05, 169.49, 169.46, 146.60, 139.05, 135.60, 86.91, 78.22, 71.75, 70.24, 62.87, 20.54, 20.34, 20.34. HRMS (ESI+) m/z: calcd for C14H18N4O9 [M + H]+, 387.1147; found, 387.1152.
2-((2R,3R,4S,5R)-3,4-Dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-(hydroxyamino)-1,2,4-triazin-3(2H)-one (3b)
Compound 3b was prepared by following the same procedure as described for 2c. 3a (59 mg, 0.15 mmol) was treated with ammonia solution in methanol (2 mL) to yield 3b as a white solid (24 mg, yield 60.2%). 1H NMR (500 MHz, DMSO-d6): δ 10.95 (s, 1H), 10.62 (d, J = 2.0 Hz, 1H), 7.43 (d, J = 1.7 Hz, 1H), 5.80 (d, J = 4.2 Hz, 1H), 5.17 (d, J = 5.5 Hz, 1H), 4.97 (d, J = 5.8 Hz, 1H), 4.67 (t, J = 5.8 Hz, 1H), 4.21–4.13 (m, 1H), 3.92 (q, J = 5.5 Hz, 1H), 3.75–3.69 (m, 1H), 3.52–3.43 (m, 1H), 3.39–3.35 (m, 1H). 13C NMR (126 MHz, DMSO-d6): δ 147.03, 139.28, 134.55, 88.62, 84.06, 71.61, 70.44, 62.25. HRMS (ESI−) m/z: calcd for C8H12N4O6 [M − H]−, 259.0684; found, 259.0684.(2R,3R,4R,5R)-2-(Acetoxymethyl)-5-(5-amino-3-oxo-1,2,4-triazin-2(3H)-yl)tetrahydrofuran-3,4-diyl diacetate (3c)
Compound 3c was obtained by following the same procedure as described for 3a. The reaction of 17 (20 mg, 0.054 mmol) with triethylamine (27.3 mg, 0.27 mmol), POCl3 (16.9 mg, 0.11 mmol) and 25% ammonium hydroxide (78 mg, 0.57 mmol) gave 3c as a white solid (9 mg, yield 45%). 1H NMR (500 MHz, DMSO-d6): δ 8.19 (s, 1H), 8.04 (s, 1H), 7.56 (s, 1H), 6.16 (d, J = 3.5 Hz, 1H), 5.51 (dd, J = 5.6, 3.4 Hz, 1H), 5.34 (t, J = 5.9 Hz, 1H), 4.32 (dd, J = 12.0, 3.5 Hz, 1H), 4.27–4.22 (m, 1H), 4.03 (dd, J = 12.0, 5.1 Hz, 1H), 2.09–2.05 (m, 6H), 2.01 (s, 3H). 13C NMR (201 MHz, DMSO-d6): δ 169.97, 169.48, 169.41, 158.84, 152.68, 128.54, 88.42, 78.53, 72.18, 70.34, 62.93, 20.48, 20.32, 20.30. HRMS (ESI+) m/z: calcd for C14H18N4O8 [M + Na]+, 393.1017; found, 393.1019.2-((2R,3R,4S,5R)-3,4-Dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-1,2,4-triazine-3,5(2H,4H)-dione (3d)
Following the procedure for the synthesis of 2c, 3d (24 mg, yield 60.2%) was obtained as a white solid from 17 (59 mg, 0.15 mmol). 1H NMR (400 MHz, DMSO-d6): δ 12.24 (s, 1H), 7.53 (d, J = 4.1 Hz, 1H), 5.88 (d, J = 3.8 Hz, 1H), 5.26 (s, 1H), 5.04 (d, J = 5.2 Hz, 1H), 4.66 (s, 1H), 4.22 (s, 1H), 3.99 (d, J = 6.3 Hz, 1H), 3.78 (d, J = 5.3 Hz, 1H), 3.54–3.34 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 157.25, 148.97, 136.31, 89.37, 84.53, 72.25, 70.36, 62.02. HRMS (ESI−) m/z: calcd for C8H11N3O6 [M − H]−, 244.0575; found, 244.0573.Dibenzyl ((2R,3S,4R,5R)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-fluoro-2-(iodomethyl)tetrahydrofuran-3,4-diyl) bis(carbonate) (18)
Benzyl chloroformate (68.08 g, 399.11 mmol) and 1-methylimidazole (43.66 g, 532.14 mmol) were added to a solution of 8 (33.0 g, 88.69 mmol) in DCM (330 mL) at 0 °C. The reaction was stirred for 2 h at rt, after which the solution was washed successively with aqueous 1 M hydrochloric acid solution and saturated aqueous NaHCO3 solution. The organic layer was dried (Na2SO4), filtered and concentrated in vacuo. The crude was slurried in propan-2-ol for 2 h to afford 18 as an off-white solid (55 g, yield 97%). 1H NMR (400 MHz, DMSO-d6): δ 11.63 (d, J = 2.2 Hz, 1H), 7.79 (d, J = 8.1 Hz, 1H), 7.44–7.28 (m, 10H), 6.14 (d, J = 2.1 Hz, 1H), 5.81 (dd, J = 19.0, 7.2 Hz, 1H), 5.71 (dd, J = 8.0, 2.2 Hz, 1H), 5.63 (dd, J = 7.1, 2.1 Hz, 1H), 5.25–5.06 (m, 4H), 3.68 (dd, J = 11.9, 8.3 Hz, 1H), 3.53 (dd, J = 22.8, 11.9 Hz, 1H). LRMS (ESI−) m/z: calcd for C25H22FIN2O9 [M − H]−, 640.04; found, 639.06.Dibenzyl ((2S,3S,4R,5R)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-fluoro-2-(hydroxymethyl)tetrahydrofuran-3,4-diyl) bis(carbonate) (19)
Tetrabutylammonium hydroxide (60.77 g, 40% aqueous solution, 93.69 mmol) was treated with trifluoroacetic acid (10.68 g, 93.69 mmol) at 0 °C, and the mixture was stirred for 10 min. A solution of 18 (20 g, 31.23 mmol) in DCM (160 mL) was added and the reaction mixture was stirred overnight at rt. After completion, the reaction was quenched by the addition of saturated aqueous Na2S2O3 solution (160 mL). The organic phase was washed twice with water, dried with Na2SO4 and filtered. Solvent was evaporated and the residue was purified by column chromatography (PE:acetone = 1:1) to give compound 19 as an off-white solid (5.3 g, yield 32%). 1H NMR (400 MHz, DMSO-d6): δ 11.54 (s, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.38–7.34 (m, 10H), 6.12 (d, J = 2.3 Hz, 1H), 5.75 (dd, J = 19.2, 7.0 Hz, 1H), 5.68 (d, J = 8.0 Hz, 1H), 5.60 (dd, J = 7.0, 2.2 Hz, 1H), 5.48 (t, J = 6.3 Hz, 1H), 5.23–5.09 (m, 4H), 3.62 (t, J = 7.9 Hz, 2H).
((2S,3S,4R,5R)-3,4-Bis(((benzyloxy)carbonyl)oxy)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-fluorotetrahydrofuran-2-yl)methyl (tert-butoxycarbonyl)-L-valinate (20)
Compound 20 (314 mg, yield 76%) was synthesized according to method B from 19 (300 mg, 0.57 mmol). 1H NMR (500 MHz, DMSO-d6): δ 11.61 (s, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.40–7.33 (m, 10H), 7.15 (d, J = 8.2 Hz, 1H), 6.19 (d, J = 2.1 Hz, 1H), 5.84 (dd, J = 19.4, 7.2 Hz, 1H), 5.70 (dd, J = 8.0, 2.2 Hz, 1H), 5.61 (dd, J = 7.2, 2.0 Hz, 1H), 5.24–5.10 (m, 4H), 4.39 (d, J = 12.4 Hz, 2H), 3.89 (dd, J = 8.2, 5.9 Hz, 1H), 2.06–1.99 (m, 1H), 1.37 (s, 9H), 0.82 (dd, J = 6.9, 5.1 Hz, 6H). LRMS (ESI−) m/z: calcd for C35H40FN3O13 [M − H]−, 729.25; found, 728.1.((2S,3S,4R,5R)-5-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-fluoro-3,4-dihydroxytetrahydrofuran-2-yl) methyl L-valinate trifluoroacetate (4a)
To a stirred solution of 20 (270.3 mg, 0.37 mmol) in DCM (3 mL), trifluoroacetic acid (0.3 mL, 3.7 mmol) was added. The reaction mixture was stirred at rt for 3 h and then concentrated to obtain the crude product. Then the crude product (233.2 mg, 0.37 mmol, yield calculated for 100%) was dissolved in ethanol (10 mL) and treated with 10% Pd/C (78.7 mg, 0.07 mmol) under hydrogen at rt for 2 h. The reaction solution was filtered and concentrated. The residue was slurried in MTBE and DCM to give 4a as a brown solid (120 mg, yield 68% over two steps). 1H NMR (400 MHz, methanol-d4): δ 7.61 (d, J = 8.0 Hz, 1H), 5.78 (d, J = 2.1 Hz, 1H), 5.70 (d, J = 8.0 Hz, 1H), 4.65–4.53 (m, 3H), 4.47 (dd, J = 7.1, 2.1 Hz, 1H), 4.01 (d, J = 4.4 Hz, 1H), 2.42–2.31 (m, 1H), 1.09 (s, 3H), 1.08 (s, 3H). LRMS (ESI+) m/z: calcd for C14H20FN3O7 [M + H]+, 361.13; found, 362.2.((2S,3S,4R,5R)-3,4-Bis(((benzyloxy)carbonyl)oxy)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-fluorotetrahydrofuran-2-yl)methyl isobutyrate (21)
21 was obtained by following method A. The reaction of 19 (800 mg, 1.51 mmol) with triethylamine (229.2 mg, 2.67 mmol), DMAP (36.9 mg, 0.30 mmol) and isobutyric anhydride (286.5 mg, 1.81 mmol) gave 21 as a white solid (623 mg, yield 70%). 1H NMR (500 MHz, DMSO-d6): δ 11.61 (s, 1H), 7.77 (d, J = 8.1 Hz, 1H), 7.41–7.32 (m, 10H), 6.18 (d, J = 2.0 Hz, 1H), 5.86 (dd, J = 19.9, 7.1 Hz, 1H), 5.69 (dd, J = 8.0, 2.2 Hz, 1H), 5.62 (dd, J = 7.1, 1.9 Hz, 1H), 5.23–5.09 (m, 4H), 4.42–4.29 (m, 2H), 2.49–2.43 (m, 1H), 1.05–1.02 (m, 6H). LRMS (ESI−) m/z: calcd for C29H29FN2O11 [M + Cl]−, 600.18; found, 635.0.((2S,3S,4R,5R)-5-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-fluoro-3,4-dihydroxytetrahydrofuran-2-yl) methyl isobutyrate (4b)
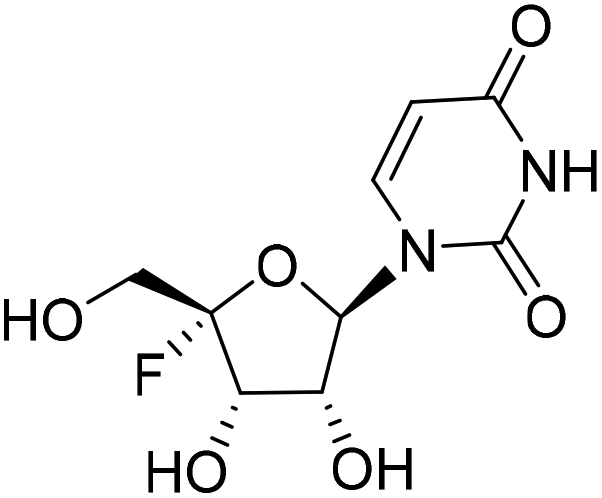
By following the procedure for the synthesis of 4a, 4b (250 mg, yield 75%) was obtained as a white solid from 21 (606.3 mg, 1.01 mmol). 1H NMR (500 MHz, methanol-d4): δ 7.59 (d, J = 8.0 Hz, 1H), 5.88 (d, J = 2.1 Hz, 1H), 5.70 (d, J = 8.0 Hz, 1H), 4.52 (dd, J = 19.2, 6.8 Hz, 1H), 4.42–4.38 (m, 1H), 4.38–4.24 (m, 2H), 2.70–2.58 (m, 1H), 1.19–1.16 (m, 6H). 13C NMR (151 MHz, DMSO-d6): δ 175.46, 163.05, 150.22, 142.38, 115.45 (d, J = 231.1 Hz), 102.37, 93.46, 70.30, 70.00 (d, J = 21.0 Hz), 61.59 (d, J = 39.1 Hz), 33.11, 26.86, 18.71. HRMS (ESI+) m/z: calcd for C13H17FN2O7 [M + Na]+, 355.0912; found, 355.0912.1-((2R,3R,4S,5S)-5-Fluoro-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione (22)
Compound 22 was synthesized starting from 8 by a reported method.151H NMR (400 MHz, DMSO-d6): δ 11.14 (s, 1H), 7.69 (d, J = 8.1 Hz, 1H), 5.99 (d, J = 2.7 Hz, 1H), 5.67 (dd, J = 10.2, 6.5 Hz, 2H), 5.48 (d, J = 6.1 Hz, 1H), 5.18 (d, J = 8.7 Hz, 1H), 4.25 (dt, J = 17.1, 7.2 Hz, 1H), 4.14 (d, J = 4.3 Hz, 1H), 3.56 (t, J = 5.2 Hz, 2H). 13C NMR (126 MHz, DMSO-d6): δ 163.03, 150.34, 140.91, 117.46 (d, J = 230.8 Hz), 102.18, 91.28, 71.45, 68.76 (d, J = 20.1 Hz), 60.02 (d, J = 42.0 Hz). HRMS (ESI+) m/z: calcd for C9H11FN2O6 [M + H]+, 263.0674; found, 263.0676.1-((2R,3R,4S,5S)-5-Fluoro-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione (4c)
Compound 4c was obtained by following method A. To a solution of 22 (100 mg, 0.38 mmol) in EA (2 mL), triethylamine (192.3 mg, 1.90 mmol), DMAP (9.3 mg, 0.08 mmol) and acetic anhydride (136.3 mg, 1.34 mmol) were sequentially added at 0 °C. After stirring at rt for 2 h, the mixture was diluted with EA and washed successively with aqueous 1 M hydrochloric acid solution and saturated aqueous NaHCO3 solution. The organic layer was dried (Na2SO4), filtered and concentrated in vacuo. The crude was slurried to afford 4c as a white solid (132 mg, yield 89%). 1H NMR (400 MHz, DMSO-d6): δ 11.60 (s, 1H), 7.77 (d, J = 8.0 Hz, 1H), 6.06 (s, 1H), 5.79 (dd, J = 19.9, 7.3 Hz, 1H), 5.71 (d, J = 8.0 Hz, 1H), 5.56 (d, J = 7.4 Hz, 1H), 4.43 (t, J = 11.7 Hz, 1H), 4.25 (t, J = 11.5 Hz, 1H), 2.10 (s, 6H), 2.04 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 169.64, 169.49, 168.99, 163.12, 150.00, 143.97, 114.41 (d, J = 233.9 Hz), 102.18, 93.79, 70.27, 68.98 (d, J = 19.5 Hz), 61.37 (d, J = 35.7 Hz), 20.30, 20.26, 20.04. HRMS (ESI+) m/z: calcd for C15H17FN2O9 [M + H]+, 389.0991; found, 389.0990.1-((3ªR,4R,6R,6ªS)-6-Fluoro-6-(iodomethyl)-2-oxotetrahydrofuro[3,4-d][1,3]dioxol-4-yl)pyrimidine-2,4(1H,3H)-dione (23)
A solution of 8 (950 mg, 2.55 mmol) in THF (5 mL) was treated with 1,1′-carbonyldiimidazole (616 mg, 3.80 mmol) at rt for 30 min and then concentrated. The residue was purified by flash chromatography (PE:EA = 1:1 → 1:2) to obtain compound 23 as a white foamy solid (520 mg, yield 51%). 1H NMR (500 MHz, DMSO-d6): δ 11.64 (s, 1H), 7.79 (d, J = 8.0 Hz, 1H), 6.31 (s, 1H), 5.77 (d, J = 7.3 Hz, 1H), 5.72 (d, 1H), 5.56 (dd, J = 11.4, 7.3 Hz, 1H), 3.62 (d, J = 18.2 Hz, 2H).
1-((3aR,4R,6S,6aS)-6-Fluoro-6-(hydroxymethyl)-2-oxotetrahydrofuro[3,4-d][1,3]dioxol-4-yl)pyrimidine-2,4(1H,3H)-dione (4d)
By following the procedure for the synthesis of 10, 4d (220 mg, yield 69%) was obtained as a white solid from 23 (440 mg, 1.1 mmol). 1H NMR (500 MHz, DMSO-d6): δ 11.59 (s, 1H), 7.78 (d, J = 8.0 Hz, 1H), 6.27 (s, 1H), 5.73 (d, J = 7.3 Hz, 1H), 5.70 (d, J = 8.0 Hz, 1H), 5.58 (t, J = 6.5 Hz, 1H), 5.53 (dd, J = 12.1, 7.3 Hz, 1H), 3.76–3.55 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 163.35, 153.65, 150.23, 144.13, 117.55 (d, J = 234.6 Hz), 102.08, 93.52, 81.28, 78.32 (d, J = 20.0 Hz), 61.55 (d, J = 32.0 Hz). HRMS (ESI+) m/z: calcd for C10H9FN2O7 [M + H]+, 289.0467; found, 289.0466.(2S,3S,4R,5R)-2-((((tert-Butoxycarbonyl)-L-valyl)oxy)methyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-fluorotetrahydrofuran-3,4-diyl bis(2-methylpropanoate) (24)
Compound 24 (254.6 mg, yield 85%) was synthesized according to method B from 10 (200.0 mg, 0.50 mmol). 1H NMR (500 MHz, DMSO-d6): δ 11.58 (s, 1H), 7.78 (d, J = 8.1 Hz, 1H), 7.16 (d, J = 8.2 Hz, 1H), 6.05 (d, J = 2.3 Hz, 1H), 5.76 (dd, J = 19.4, 7.3 Hz, 1H), 5.71 (dd, J = 8.1, 2.1 Hz, 1H), 5.56 (dd, J = 7.3, 2.4 Hz, 1H), 4.41–4.29 (m, 2H), 3.91 (dd, J = 8.2, 5.9 Hz, 1H), 2.65–2.56 (m, 2H), 2.08–2.01 (m, 1H), 1.37 (s, 9H), 1.14–1.05 (m, 12H), 0.85 (t, J = 7.4 Hz, 6H). LRMS (ESI−) m/z: calcd for C27H40FN3O11 [M + Cl]−, 601.26; found, 636.2.(2S,3S,4R,5R)-2-(((L-Valyl)oxy)methyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-fluorotetrahydrofuran-3,4-diyl bis(2-methylpropanoate) (4e)
To a stirred solution of 24 (50.0 mg, 0.08 mmol) in DCM (1 mL), trifluoroacetic acid (0.1 mL, 0.83 mmol) was added. The reaction mixture was stirred at rt for 30 min and then concentrated. The residue was purified by thin layer chromatography (DCM:MeOH = 20:1) to obtain 4e as a white foamy solid (34.6 mg, yield 83%). 1H NMR (500 MHz, DMSO-d6): δ 7.78 (d, J = 8.0 Hz, 1H), 6.03 (d, J = 2.2 Hz, 1H), 5.81 (dd, J = 19.7, 7.3 Hz, 1H), 5.70 (d, J = 8.0 Hz, 1H), 5.58 (dd, J = 7.3, 2.2 Hz, 1H), 4.41–4.27 (m, 2H), 3.14 (d, J = 5.2 Hz, 1H), 2.59 (pd, J = 7.0, 2.5 Hz, 2H), 1.92–1.81 (m, 1H), 1.15–1.08 (m, 12H), 0.86 (d, J = 6.9 Hz, 3H), 0.79 (d, J = 6.8 Hz, 3H). 13C NMR (151 MHz, DMSO-d6): δ 175.21, 174.85, 168.41, 163.23, 150.18, 144.02, 114.26 (d, J = 233.9 Hz), 102.31, 93.94, 70.41, 69.26 (d, J = 18.9 Hz), 62.77 (d, J = 36.4 Hz), 57.34, 33.07, 33.02, 29.30, 18.63, 18.59, 18.53, 18.40, 17.85, 17.56. HRMS (ESI+) m/z: calcd for C22H32FN3O9 [M + H]+, 502.2195; found, 502.2200.
Antiviral assays and cytotoxicity
Cytopathic effect (CPE) assay was used for testing the inhibitory activity of the compounds against respiratory syncytial virus (RSV) and influenza virus (IFV) replication.Anti-RSV activity assay
Hep-2 cells were plated on 96-well plates (6 × 103 cells per well) and incubated with 5% CO2 overnight at 37 °C. The following day, serial dilutions of the test compounds were added, with ALS-8112 as the positive control, followed by the addition of RSV/A Long (ATCC VR-26). After incubating for 5 days, the cell viability and cytotoxicity were evaluated using a spectrophotometric plate reader and calculated using GraphPad Prism software. The results are expressed as EC50 (concentrations reducing viral replication by 50%) and CC50 (concentrations decreasing cell viability by 50%) values.Anti-IFV activity assay
Pre-seeded MDCK cells (1.5 × 104 cells per well) were treated with indicated concentrations of the test compounds, and then infected with IFV A (California/07/2009 (H1N1) pdm09 (ATCC, VR-1894)). After 5 days of incubation, CCK-8 was added, and the absorbance of each well was read on a spectrophotometric plate reader. The EC50 and CC50 values were calculated with GraphPad Prism software.Stability assays
Ammonium dihydrogen phosphate (0.115 g) was dissolved in pure water (100 mL) to prepare the buffer solution (30 mL), and phosphoric acid was added to adjust the pH to 4.0. The pH 2.0 buffer solution was prepared in the same way. Compound 1a, 4b–d or 22 (0.5 mg) was added to a 1:1 (v/v) mixture of acetonitrile and buffer solutions (pH = 2.0/4.0) to prepare the incubation mixture (0.5 mg mL−1), which was incubated at rt for 7 days. The samples (5 μL) were analyzed by HPLC. The determination of the purity of compounds 1a, 4b–d and 22 was performed on a YMC Pack Pro C4 column (250 × 4.6 mm, 5 μM). The wavelength of the UV detector was 210 nm. Eluent A: H2O; B: acetonitrile. Gradient: from 0 to 5 min: 95% of A; from 20 to 25 min: 10% of A. The flow rate is 1.0 mL min−1.
Pharmacokinetic studies
The PK studies of compound 22 in SD rats were conducted at HQ Bioscience Co., Ltd. Before the experiment, SD rats (N = 3 for each group, male, 180–280 g) were fasted for 12 h, except the intravenous group, and allowed to drink water. Four hours after administration, the rats were fed. The compound dissolved in 5% DMSO + 5% ethanol + 40% PEG400 + 50% saline was administered orally at 10 mg kg−1 and intravenously at 5 mg kg−1, respectively. 0.2 mL blood sample was collected from the jugular vein and placed in heparin sodium anticoagulation tubes at 0.25, 0.5, 1.0, 2.0, 4.0, 6.0, 8.0, and 24 h after administration, and centrifuged at 2000 g at 4 °C within 10 min. After centrifugation, the plasma samples (30 μL) were accurately aspirated into a plastic tube, which contained 600 μL methanol solution with the internal standard (tolbutamide, 100 ng mL−1), and frozen in a refrigerator at an ultra-low temperature for testing. The concentration of 22 in plasma was determined by LC-MS/MS.The tri-isobutyrate ester 1a and compound 22 dissolved in 10% DMSO + 10% solutol + 80% (20% HP-β-CD) were orally administered at the same molar dose (10 mg kg−1 for 22, and 18 mg kg−1 for 1a). The procedure for PK studies in rats was the same as described above.
The analysis was performed on a system of LC-MS010 (5500) and data processing was performed using Analyst1.6.3 (Sciex). The analytes were separated using an Agela Venusil ASB-C18 column (150 Å, 3 μM, 4.6 × 50 mm) with an injection volume of 3.00 μL. Mobile system: (A) water with 0.5 mM ammonium acetate; B: methanol. Gradient: from 0 to 0.40 min: 35% of B; from 0.40 to 1.20 min: 35%–90% of B; from 1.20 to 2.40 min: 90% of B; from 2.40 to 2.50 min: 90%–35% of B; from 2.50 to 3.50 min: 35% of B. Flow rate: from 0 to 0.20 min: 0.5500 mL min−1, from 0.20 to 3.5 min: 0.700 mL min−1 with the column oven temperature at 40 °C. General MS parameters were set as follows: ion source, ESI; injection voltage: −4500 V; Gas1: 50 psi; Gas2: 50 psi; ion source temperature: 500 °C; dwell time: 80 ms. Auto-MS negative mode was used.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Shanghai Municipal Science and Technology Major Project and Lingang Laboratory Emergent Project (LG202103-04-04).References
- R. Ochani, A. Asad, F. Yasmin, S. Shaikh, H. Khalid, S. Batra, M. R. Sohail, S. F. Mahmood, R. Ochani, M. Arshad Hussham, A. Kumar and S. Surani, Infez. Med., 2021, 29, 20 CAS.
- S. Sunny, J. Samaroo-Campbell, M. Abdallah, A. Luka and J. Quale, Infection, 2023, 51, 247 CrossRef CAS PubMed.
- Y. Y. Syed, Drugs, 2022, 82, 455 CrossRef CAS PubMed.
- E. Tchesnokov, J. Feng, D. Porter and M. Götte, Viruses, 2019, 11, 326 CrossRef CAS PubMed.
- Y. Li, M. Liu, Y. Yan, Z. Wang, Q. Dai, X. Yang, X. Guo, W. Li, X. Chen, R. Cao and W. Zhong, Viruses, 2022, 14, 1142 CrossRef CAS PubMed.
- E. Zarenezhad and M. Marzi, Med. Chem. Res., 2022, 31, 232 CrossRef CAS PubMed.
- O. Reynard, X. N. Nguyen, N. Alazard-Dany, V. Barateau, A. Cimarelli and V. E. Volchkov, Viruses, 2015, 7, 6233 CrossRef CAS PubMed.
- G. R. Painter, R. A. Bowen, G. R. Bluemling, J. DeBergh, V. Edpuganti, P. R. Gruddanti, D. B. Guthrie, M. Hager, D. L. Kuiper, M. A. Lockwood, D. G. Mitchell, M. G. Natchus, Z. M. Sticher and A. A. Kolykhalov, Antiviral Res., 2019, 171, 104597 CrossRef CAS PubMed.
- J. J. Yoon, M. Toots, S. Lee, M. E. Lee, B. Ludeke, J. M. Luczo, K. Ganti, R. M. Cox, Z. M. Sticher, V. Edpuganti, D. G. Mitchell, M. A. Lockwood, A. A. Kolykhalov, A. L. Greninger, M. L. Moore, G. R. Painter, A. C. Lowen, S. M. Tompkins, R. Fearns, M. G. Natchus and R. K. Plemper, Antimicrob. Agents Chemother., 2018, 62, e00766 CrossRef CAS PubMed.
- I. D. Jenkins, J. Verheyden and J. G. Moffatt, J. Am. Chem. Soc., 1976, 98, 3346 CrossRef CAS PubMed.
- S. Martínez-Montero, G. F. Deleavey, A. Kulkarni, N. Martín-Pintado, P. Lindovska, M. Thomson, C. Gonzalez, M. Gotte and M. J. Damha, J. Org. Chem., 2014, 79, 5627 CrossRef PubMed.
- G. Wang, N. Dyatkina, M. Prhavc, C. Williams, V. Serebryany, Y. Hu, Y. Huang, J. Wan, X. Wu, J. Deval, A. Fung, Z. Jin, H. Tan, K. Shaw, H. Kang, Q. Zhang, Y. Tam, A. Stoycheva, A. Jekle, D. B. Smith and L. Beigelman, J. Med. Chem., 2019, 62, 4555 CrossRef CAS PubMed.
- V. Gopalsamuthiram, A. L. Kadam, J. K. Noble, D. R. Snead, C. Williams, T. F. Jamison, C. Senanayake, A. K. Yadaw, S. Roy, G. Sirasani, B. F. Gupton, J. Burns, D. W. Cook, R. W. Stringham, S. Ahmad and R. Krack, Org. Process Res. Dev., 2021, 25, 2679 CrossRef CAS PubMed.
- J. M. Madern, T. Hansen, E. R. van Rijssel, H. A. V. Kistemaker, S. van der Vorm, H. S. Overkleeft, G. A. van der Marel, D. V. Filippov and J. D. C. Codee, J. Org. Chem., 2019, 84, 1218 CrossRef CAS PubMed.
- J. Sourimant, C. M. Lieber, M. Aggarwal, R. M. Cox, J. D. Wolf, J. J. Yoon, M. Toots, C. Ye, Z. Sticher, A. A. Kolykhalov, L. Martinez-Sobrido, G. R. Bluemling, M. G. Natchus, G. R. Painter and R. K. Plemper, Science, 2022, 375, 161 CrossRef CAS PubMed.
- M. Guinan, N. Huang, M. Smith and G. J. Miller, Bioorg. Med. Chem. Lett., 2022, 61, 128605 CrossRef CAS PubMed.
- S. Raymond, A. Frank, Z. Che and Z. Keivan, US pat., 2022217155A2, 2022 Search PubMed.
- I. Alexeeva, N. Dyachenko, L. Nosach, V. Zhovnovataya, S. Rybalko, R. Lozitskaya, A. Fedchuk, V. Lozitsky, T. Gridina, A. Shalamay, L. Palchikovskaja and O. Povnitsa, Nucleosides, Nucleotides Nucleic Acids, 2001, 20, 1147 CrossRef CAS PubMed.
- F. Sorm and J. Skoda, Acta Unio Int. Cancrum, 1964, 20, 37 CAS.
- Q. Li, J. Chen, M. Trajkovski, Y. Zhou, C. Fan, K. Lu, P. Tang, X. Su, J. Plavec and Z. Xi, J. Am. Chem. Soc., 2020, 142, 4739 CrossRef CAS PubMed.
- S. Lee, C. Uttamapinant and G. L. Verdine, Org. Lett., 2007, 9, 5007 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00268c |
| This journal is © The Royal Society of Chemistry 2023 |