
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of fluoro- and seleno-containing D-lactose and D-galactose analogues†
Cecilia
Romanò‡
 ,
Dennis
Bengtsson
,
Angela Simona
Infantino
and
Stefan
Oscarson
*
,
Dennis
Bengtsson
,
Angela Simona
Infantino
and
Stefan
Oscarson
*
Centre for Synthesis and Chemical Biology, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: stefan.oscarson@ucd.ie
First published on 27th February 2023
Abstract
Synthetic deoxy-fluoro-carbohydrate derivatives and seleno-sugars are useful tools in protein–carbohydrate interaction studies using nuclear magnetic resonance spectroscopy because of the presence of the 19F and 77Se reporter nuclei. Seven saccharides containing both these atoms have been synthesized, three monosaccharides, methyl 6-deoxy-6-fluoro-1-seleno-β-D-galactopyranoside (1) and methyl 2-deoxy-2-fluoro-1-seleno-α/β-D-galactopyranoside (2α and 2β), and four disaccharides, methyl 4-O-(β-D-galactopyranosyl)-2-deoxy-2-fluoro-1-seleno-β-D-glucopyranoside (3), methyl 4-Se-(β-D-galactopyranosyl)-2-deoxy-2-fluoro-4-seleno-β-D-glucopyranoside (4), and methyl 4-Se-(2-deoxy-2-fluoro-α/β-D-galactopyranosyl)-4-seleno-β-D-glucopyranoside (5α and 5β), the three latter compounds with an interglycosidic selenium atom. Selenoglycosides 1 and 3 were obtained from the corresponding bromo sugar by treatment with dimethyl selenide and a reducing agent, while compounds 2α/2β, 4, and 5α/5β were synthesized by the coupling of a D-galactosyl selenolate, obtained in situ from the corresponding isoselenouronium salt, with either methyl iodide or a 4-O-trifluoromethanesulfonyl D-galactosyl moiety. While benzyl ether protecting groups were found to be incompatible with the selenide linkage during deprotection, a change to acetyl esters afforded 4 in a 17% overall yield and over 9 steps from peracetylated D-galactosyl bromide. The synthesis of 5 was performed similarly, but the 2-fluoro substituent led to reduced stereoselectivity in the formation of the isoselenouronium salt (α/β ∼ 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.3). However, the β-anomer of the uronium salt could be obtained almost pure (∼98%) by precipitation from the reaction mixture. The following displacement reaction occurred without anomerisation, affording, after deacetylation, pure 5β.
:2.3). However, the β-anomer of the uronium salt could be obtained almost pure (∼98%) by precipitation from the reaction mixture. The following displacement reaction occurred without anomerisation, affording, after deacetylation, pure 5β.
Introduction
Unnatural modifications of carbohydrates, such as synthetic mono-deoxy-derivatives and mono-O-alkylated congeners, are excellent tools to elucidate the binding of glycans with proteins and chemically map their binding epitope.1–4 In the same way, the introduction of fluorine atoms on carbohydrates, yielding deoxy-fluoro derivatives, has also been exploited in chemical mapping strategies. In fact, C–F and C–OH bonds are quite similar in terms of length and polarization, but they differ in their hydrogen bonding abilities. As the fluorine atom prevents hydrogen bond donation, while maintaining weak hydrogen bond acceptance properties, the OH → F substitution is a valid technique to probe hydrogen bond patterns involved in carbohydrate–protein interactions.5,6 Thus, fluorinated carbohydrate derivatives have found application in the elucidation of the activity of enzymes involved in carbohydrate metabolism,7,8 in the inhibition of glycosyltransferases involved in disease,9,10 and in the detection of protein–carbohydrate interactions by 19F NMR.11–21 The 19F nucleus is 100% abundant, naturally absent in biomolecules, and has a very similar sensitivity to the 1H nucleus, with the plus of a broad ppm range (200 ppm) that lowers the chance of observing overlapping signals. This, in turn, leads to an overall simplification of the NMR spectrum, especially beneficial in studies with complex carbohydrate substrates12,16 and/or cocktails of monosaccharide ligands for the detection of the binding preferences and modes of lectins.17 Adding to the list of chemical modifications for the elucidation of carbohydrate–lectin interactions is the introduction of a selenium atom, which has also very favourable physical properties for structural analysis. Selenium derivatives of carbohydrate ligands, most often methyl selenoglycosides, have been used as substrates for glycosidase inhibition (similarly to S-linked carbohydrates22),23 as ligands for plant and mammalian lectins,24–26 in phasing crystal structures of carbohydrate-binding macromolecules, by virtue of the anomalous dispersion of selenium in response to X-ray irradiation,27–29 and, in more recent years, have been employed in NMR studies with 77Se as the reporting nucleus.30–34 In fact, the 77Se isotope has a ½ spin, 7.6% natural abundance, and it is particularly sensitive to changes in its local environment, with a large chemical shift range around 3000 ppm. Synthetically, several strategies for the introduction of fluorine or selenium atoms in carbohydrates have been optimized over the years. Notably, preparations of deoxy-fluoro carbohydrates can be roughly divided in nucleophilic (e.g. TASF, TBAF, DAST) and electrophilic approaches (e.g. SelectFluor™).35,36 Generally, nucleophilic approaches are preferred for the synthesis of 3-, 4-, and 6-deoxy-fluoro compounds, while 2-deoxy-2-fluoro derivatives are obtained through electrophilic addition to the double bond of glycals. Selenium-containing derivatives are most commonly synthesised as glycosyl selenides via either Koenigs–Knorr type reactions37,38 or via the formation of isoselenouronium salts.39 Se-linked disaccharides, where the selenium atom is introduced at the interglycosidic linkage, have been synthesized either through the formation of suitable selenolates (usually generated in situ)40,41 and selenouronium salts39 or by intermolecular aglycon transfer between a glycosyl trichloroacetimidate and a mixed selenoacetal with TMSOTf activation,42 to cite some of the most popular methodologies.In the search for novel and diverse synthetic tools for probing protein–carbohydrate interactions, we have become interested in the simultaneous incorporation of both fluorine and selenium atoms in D-galactose and D-lactose scaffolds affording bi-functional carbohydrate mimetics to aid the structural analysis of galactose-binding lectins. The disaccharide β-D-Gal-(1→4)-D-Glc (Lac) and the closely related β-D-Gal-(1→4)-D-GlcNAc (LacNAc) are core motifs in mammalian N- and O-glycans, glycosphingolipids (GSLs), and human milk oligosaccharides, where they are found extended by other branching or capping saccharide moieties, and are recognized by a number of mammalian and plant lectins involved in several signalling pathways.43–46 The designed Se/F-containing synthetic saccharides, represented in Fig. 1, are methyl 6-deoxy-6-fluoro-1-seleno-β-D-galactopyranoside (1), methyl 2-deoxy-2-fluoro-1-seleno-α/β-D-galactopyranoside (2α/2β), methyl (β-D-galactopyranosyl)-(1→4)-2-deoxy-2-fluoro-1-seleno-β-D-glucopyranoside (3), methyl (1-seleno-β-D-galactopyranosyl)-(1→4)-2-deoxy-2-fluoro-β-D-glucopyranoside (4), and methyl (2-deoxy-2-fluoro-1-seleno-α/β-D-galactopyranosyl)-(1→4)-β-D-glucopyranoside (5α/5β), the three latter with an interglycosidic selenium atom. This set of compounds maintains the natural anomeric β-configuration found in N- and O-glycans. Additionally, the selenium atom at the β-(1→4) interglycosidic linkage should not substantially affect the recognition process by lectins. In fact, the differences in the van der Waals radius (O 1.52 Å, Se 1.90 Å) and in the angle of the inter-glycosidic bond (C–O–C 112°, C–Se–C 96°) should be minimal.29,32,47 Moreover, the fluorine atom, sitting at the C-2 of the D-glucopyranose ring or the C-2′ of the D-galactopyranose ring should not hinder the binding to galacto-specific lectins, e.g. galectins, but should be close enough to be able to experience variations in its local environment upon protein binding.48 In return, both fluorine and selenium atoms can act as reporters in 19F and 77Se NMR studies, and also help in further structural analysis, e.g. crystallographic investigations.
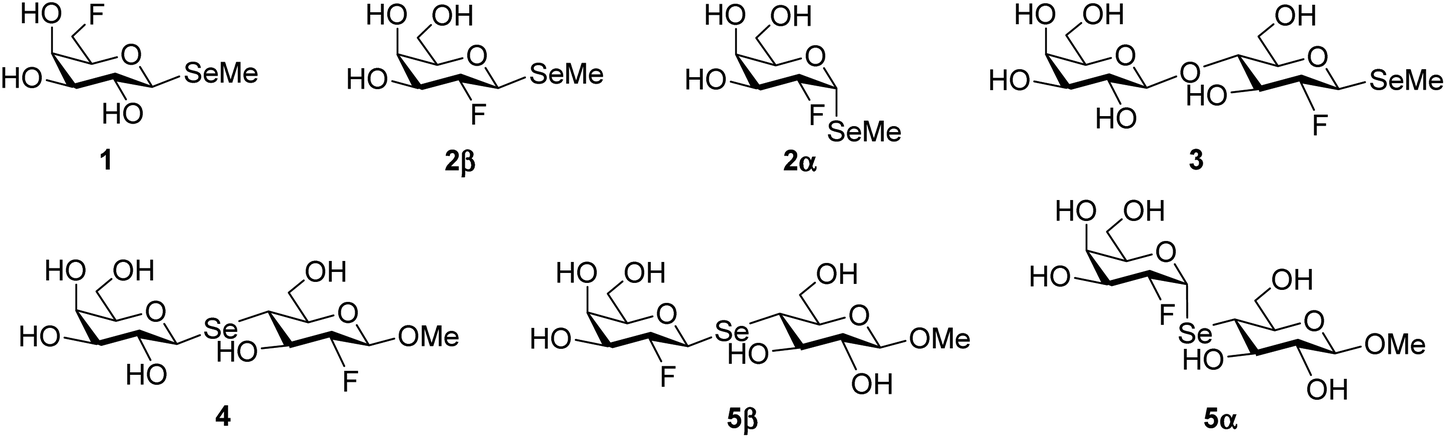 | ||
| Fig. 1 Target F/Se saccharides 1–5. | ||
Results and discussion
The synthesis of methyl selenoglycosides 1 and 3 utilizes the same method as we have used before, i.e., treatment of a glycosyl bromide with methyl diselenide under reducing conditions to form the methyl selenolate in situ.49 After the formation of the known 6-deoxy-6-fluoro-α-bromide 7,50 a 40% yield of the β-methyl selenoglycoside 8 was obtained, which was deprotected under Zemplén conditions to give 1 in 83% yield (Scheme 1). Lactal 951 was treated with Selectfluor™ in nitromethane/water to give a mixture of α/β gluco-/manno-configurations from which, after benzoylation of the anomeric positions, compound 10 could be isolated in a 56% yield (Scheme 2). Treatment with HBr 33% in AcOH afforded the α-bromide (→11, 90%), which was converted to the β-methyl selenide and deprotected under standard conditions to afford target compound 3 in a 24% yield over two steps.
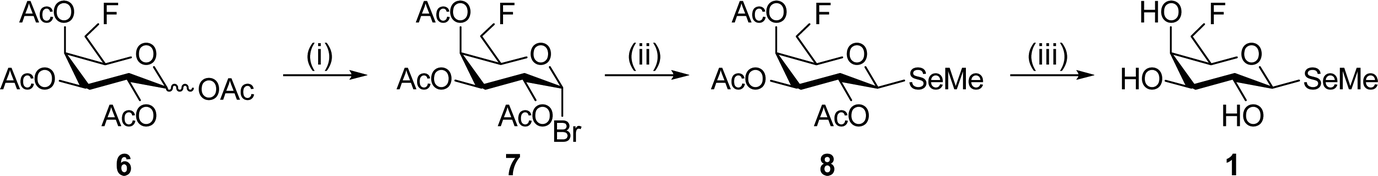 | ||
| Scheme 1 Reagents and conditions: (i) HBr (33% in AcOH), CH2Cl2, RT, 2 h, 94%; (ii) (CH3)2Se2, NaBH4, EtOH/CH3CN, RT, 22 h, 40%; (iii) NaOMe/MeOH, RT, 17 h, 83%. | ||
 | ||
| Scheme 2 Reagents and conditions: (i) SelectFluor™, MeNO2/H2O, RT, 19 h (reflux 1 h); (ii) BzCl, Py, RT, 16 h, 56% (over 2 steps); (iii) HBr (33% in AcOH), RT, 22 h, 90%; (iv) (a) (CH3)2Se2, NaBH4, EtOH/CH3CN, RT, 17 h, (b) NaOMe/MeOH, RT, 20 h, 24% over two steps. | ||
Differently from the synthesis of 1 and 3, the preparation of lactose pseudo-disaccharides 4 and 5α/5β included the coupling between D-galactopyranosyl isoselenouronium salts, as selenyl transfer reagents, and suitably protected methyl 4-O-trifluoromethanesulfonate D-galactopyranosides. For the synthesis of compound 4, acetobromogalactose 12 was treated with selenourea in acetone under reflux, as described by Kumar et al. for D-glucopyranosyl bromide,39 to form the corresponding D-galactosylselenyl transfer reagent 13 in 73% yield (Scheme 3).
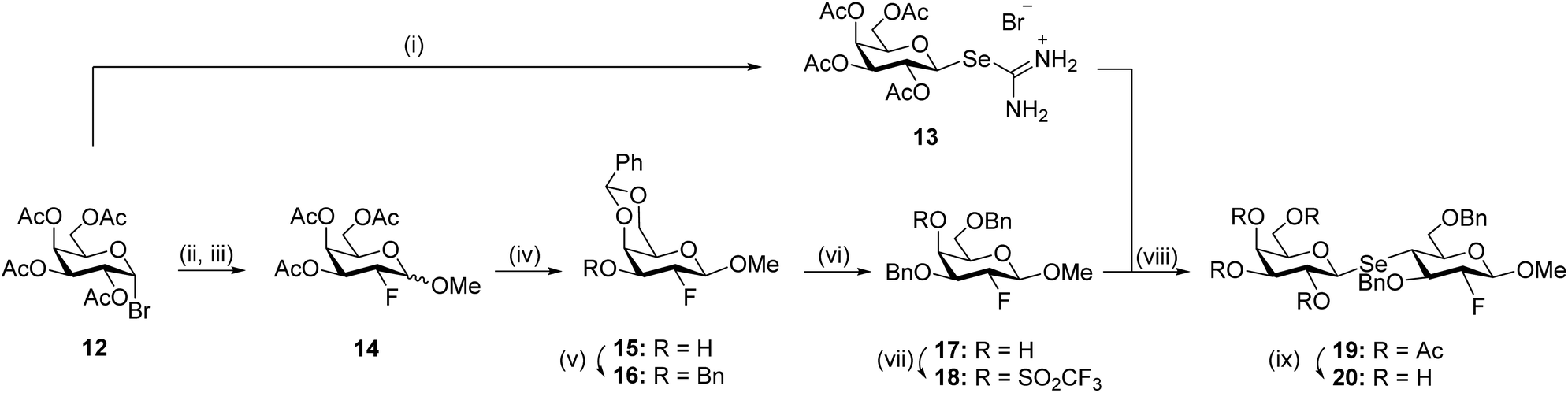 | ||
| Scheme 3 Reagents and conditions: (i) selenourea, acetone, reflux, 1 h, 73%; (ii) Zn, AcOH/H2O, RT, 17 h, 69%; (iii) SelectFluor™, MeNO2/MeOH, RT, 4 h (then 1 h at 90 °C), 84% (α/β = 1:2); (iv) (a) NaOMe, MeOH, RT, 3 h, (b) PhCH(OMe)2, p-TsOH, DMF, 50 °C, 4 h; (v) BnBr, NaH, DMF, 0 °C → RT, 1 h, 56% (over three steps, 74% overall yield; 56% for 16 and 18% for 16α); (vi) NaCNBH3, HCl (1 M in Et2O), RT, 1 h, 91%; (vii) Tf2O, pyridine, 0 °C → RT, 2 h, 87%; (viii) Et3N, CH3CN, RT, 1 h, 90%; (ix) NaOMe, MeOH, RT, 2 h, 90%. | ||
The presence of a characteristic 13C NMR peak at 165.0 ppm for the isoselenouronium carbon and the concurrent shift at 79.7 ppm of the C-1 signal, confirmed the formation of the desired selenouronium moiety. Treatment of galactosyl bromide 12 with activated Zn powder in acetic acid/water gave the corresponding D-galactal in a 69% yield via a reduction–elimination process.52 The newly formed D-galactal was then reacted with SelectFluor™ in nitromethane/methanol to give derivative 1453 as a mixture of anomers (α/β = 1:2) in 84% yield. In particular, the reaction allows for the exclusive equatorial fluorination as a result of the directing properties of the acetate at C-4.53 Although synthesised as a mixture of α/β anomers, the presence of 19F–1H and 19F–13C couplings in both 1H and 13C NMR, the low-field shift of the adjacent protons, and the 19F NMR signals (−207.06, ddd, J = 52.7, 13.3, 2.5 Hz, F-2β and −208.89, ddd, J = 50.0, 10.8, 3.3 Hz, F-2α), confirmed the formation of the galacto 2-deoxy-2-fluoro derivative 14. At this stage the anomers could not be separated and the subsequent reactions were carried out with the α/β mixture. D-Galactopyranoside 14 was readily deacetylated under Zemplén conditions, then reacted with benzaldehyde dimethyl acetal and catalytic amounts of p-toluenesulfonic acid to afford the corresponding 4,6-O-benzylidene acetal protected compound 15, which was finally benzylated under standard conditions to afford a separable mixture of the desired β-anomer 16 and undesired 16α, in 56% and 18% yield, respectively, over three steps. The 4,6-O-benzylidene acetal on 16 was then reductively opened with NaCNBH3 and HCl/Et2O,54 affording the desired 6-O-benzyl product (→17, 91%). Finally, compound 17 was reacted with trifluoromethanesulfonic anhydride in pyridine at low temperature to give derivative 18 in 87% yield. D-Galactosyl isoselenouronium salt 13 and compound 18 were reacted under basic conditions to give the pseudo-lactoside 19 in 90% yield. The formation of the seleno pseudo-disaccharide derivative was confirmed by NMR; the 13C signal at 77.8 ppm for C-1′ and, more interestingly, the upfield shift at 41.8 ppm for C-4, both confirmed the presence of the selenium atom at the interglycosidic linkage. In addition, the H-4 signal (1H NMR 3.27 ppm) showed characteristic satellite peaks corresponding to 2JSe,H = 20.4 Hz. Finally, the large coupling constants observed for the signal at 3.27 ppm in 1H NMR (apparent triplet, J3,4 ∼ J4,5 ∼ 11.1 Hz, H-4) confirmed the inversion of configuration D-Gal → D-Glc. Subsequently, deacetylation under Zemplén conditions (→20, 90%) was followed by benzyl ethers removal attempts as illustrated in Table 1.
| Entry | Conditions | Product/outcome |
|---|---|---|
| 1 | Pd/C (10 wt%), H2 (20 bar) | No reaction |
| 2 | 1:1 Pd/C (10 wt%):Pd(OH)2/C (20 wt%), H2 (20 bar) |
Degradation |
| 3 | Na, NH3(l), −78 °C then Ac2O, pyridine |

|
| 4 | FeCl3, CH2Cl2, −30 °C | Degradation |
Standard hydrogenolysis of 20 with Pd/C (10 wt%) under H2 atmosphere (20 bar) gave no conversion after 20 hours (entry 1). Instead, a 1:1 mixture of Pd/C (10 wt%) and Pd(OH)2/C (20 wt%) was tried under the same hydrogen atmosphere affording degradation of the starting material (entry 2). The obtained results are in line with previous reports55 where the selenium atom is described to act as catalyst poison, similarly to sulphur.56 At this point, alternative methodologies were screened to investigate their compatibility with the β-(1→4) Se-linkage. The biphasic oxidative cleavage with NaBrO3/Na2S2O457 was deemed incompatible with the oxidation-sensitive selenium atom, and thus was not tested, Birch reduction conditions58 were envisioned as a good methodology for the removal of the two benzyl ethers. Unexpectedly, when compound 20 was reacted with Na in liquid ammonia at −78 °C, and subsequently acetylated under standard conditions, diselenide 2159 was identified as the main reaction product (entry 3). Supposedly, the reaction conditions promoted the reductive cleavage of the C–Se bond forming a selenolate species prone to oxidation to the corresponding diselenide. Finally, also ferric(III) chloride promoted de-benzylation60 failed to give the desired product (entry 4).
The presented difficulties in removing the benzyl ethers in a clean and efficient way forced a change of strategy, with the design of a differently protected D-galactose building block to couple with transfer reagent 13. As Zemplén conditions are highly compatible with the presence of selenium and fluorine functionalities, it was decided to substitute the benzyl ether protecting groups with acetyl esters. Thus, compound 15 was acetylated instead of benzylated, to give the desired β-anomer (→22, 59%) and α-derivative (→22α, 26%) (Scheme 4). Subsequent cleavage of the 4,6-O-benzylidene acetal (→23, 78%), followed by selective acetylation of the primary C-6 hydroxyl with acetyl chloride and pyridine at low temperature, afforded 24 in 81% yield. Finally, trifluoromethanesulfonyl introduction gave building block 25 in 95% yield. Derivative 25 and isoselenouronium salt 13 were then coupled under basic conditions to give pseudo-disaccharide 26 in 91% yield. Gratifyingly, the substitution of the benzyl ethers for acetyl esters did not lower the reactivity of 25, giving the pseudo-disaccharide 26 in the same yield as compound 19. Again, NMR data confirmed the formation of the desired product: 13C NMR signal at 77.3 ppm for the C-1′ and the upfield shift at 41.2 ppm for C-4 indicated the presence of the selenium atom at the interglycosidic linkage, the coupling pattern of the H-4 signal (1H NMR 2.98 ppm, apparent triplet, J3,4 ∼ J4,5 ∼ 11.2 Hz) confirmed the inversion of configuration D-Gal → D-Glc, and this signal also showed the characteristic satellite peaks corresponding to 2JSe,H = 25.1 Hz. Finally, Zemplén global deacetylation of pseudo-disaccharide 26 gave target 4 in 86% yield.
 | ||
| Scheme 4 Reagents and conditions: (i) Ac2O, pyridine, RT, 18 h, 59% over three steps; (ii) 80% aq. AcOH, 80 °C, 3 h, 78%; (iii) AcCl, pyridine, CH2Cl2/CH3CN, −30 °C → RT, 3 h, 81%; (iv) Tf2O, pyridine, 0 °C → RT, 3 h, 95%; (v) Et3N, CH3CN, RT, 1 h, 91%; (vi) NaOMe, MeOH, RT, 4 h, 86%. | ||
For the synthesis of target compound 5 the same approach was applied, with the 2-deoxy-2-fluoro selenouronium compound 29 formed from the 2-deoxy-2-fluoro galactosyl bromide 28,61 obtained from known compound 2718 (Scheme 5). However, here the 2-fluoro substituent affected the stereoselectivity and rate of the reaction and while selenourea 13 was obtained as the pure β-anomer after 1 hour, compound 29 was obtained as an α/β mixture in a 3:7 ratio in 87% yield after 3 hours. Several attempts to improve the β-selectivity were made but with no success, therefore it was decided to continue with the anomeric mixture of the selenourea salt and try to separate the anomers after the selenide formation. Model experiments were carried out with methyl iodide as the electrophile, instead of a glycosyl triflate, affording the 2-deoxy-2-fluoro methyl selenogalactoside 32 in 66% yield as an α/β mixture with about the same ratio as in 29, which could be separated to give the pure anomers 32α (15%) and 32β (24%). Deprotection under Zemplén conditions gave target compounds 2α and 2β, both in an 87% yield. For the formation of the disaccharide 5, compound 30 was synthesized and converted into the known 4-O-triflate derivative 31.62 The coupling between compound 29 and 31, under basic conditions to form the selenolate from the selenourea salt, was less effective than the formation of 19 and 26 above (Schemes 3 and 4), affording a 61% yield of 33 as an α/β mixture, again with the same α/β ratio as in 29, indicating that there is no anomerisation taking place during the displacement reactions and that the reaction rate of the anomers is about the same. Separation of the anomers turned out to be quite difficult with major loss of material. Finally, employing a long thin silica gel column and slow elution, the anomers could be purified to give 33α (7%) and 33β (15%). Although low yielding, the easy access to the precursors made it possible to obtain good amounts (10–100 mg) of both anomers. However, we then found that if, in the preparation of the selenourea derivative, the product was not precipitated with ethyl ether, but the reaction mixture was allowed to stand and cool, a precipitate was formed, which was much enriched in the (desired) β-anomer, giving a 40–50% yield of almost pure 29β. When this material was used in the formation of the pseudo-saccharide, the yield of 33β was improved to 56%. Deacetylation of 33α and 33β then afforded target compounds 5α and 5β in quantitative yields (→5α, quantitative, →5β, 96%). Compounds 2, 3, 5, 32, and 33 are all containing a novel vicinal fluoro/seleno motif with possible 3JF,Se coupling constants, which are summarized in Table 2. Interestingly, only the α-cis-compounds show any coupling.
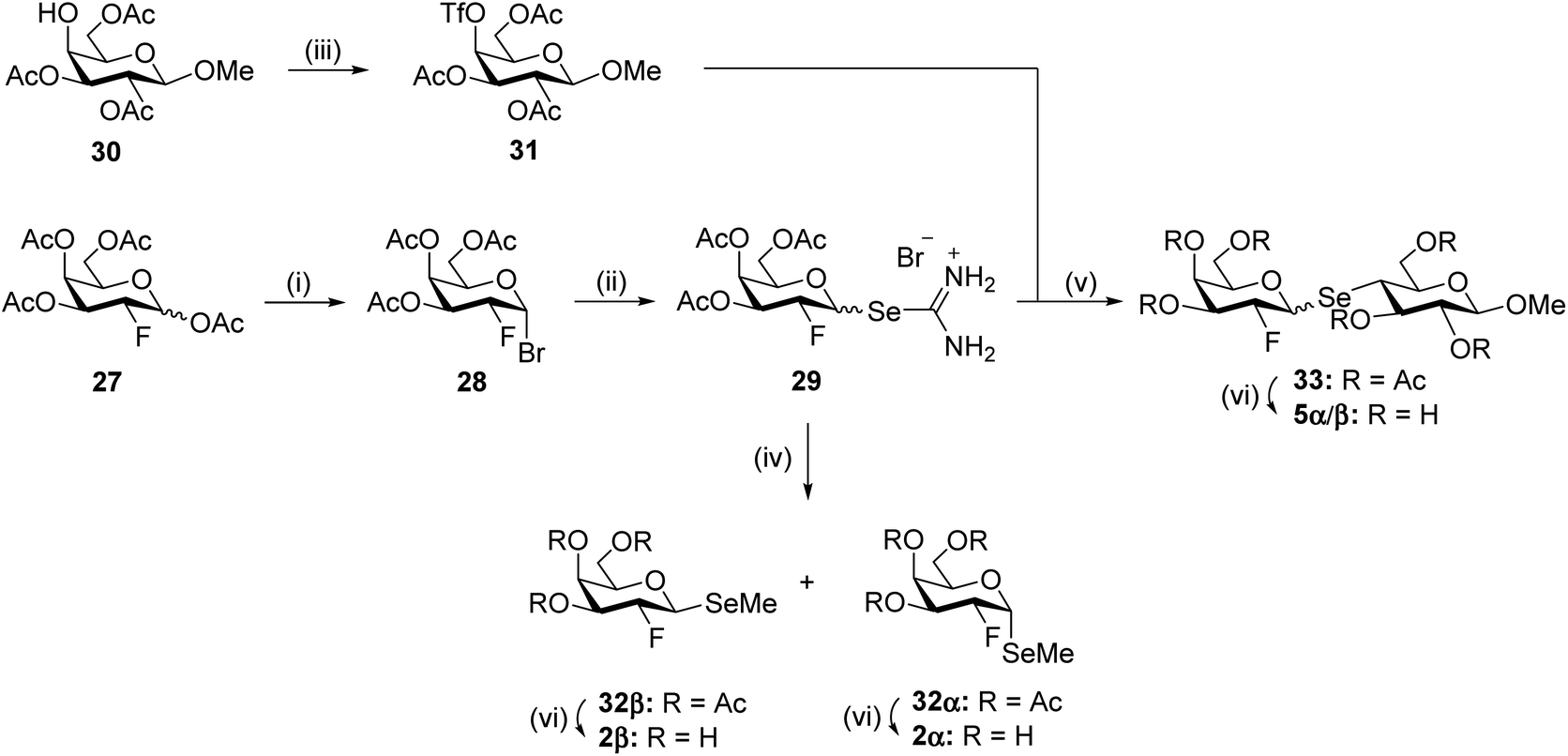 | ||
| Scheme 5 Reagents and conditions: (i) HBr (33% in AcOH), RT; 77% (ii) selenourea, acetone, reflux, 1 h, 87%, α/β = 3:7; (iii) Tf2O, pyridine, 0 °C → RT, 2 h, 85%; (iv) Et3N, MeI, CH3CN, RT, 1 h, 66%, 32α: 15%, 32β: 24%; (v) Et3N, CH3CN, RT, 2 h, 61%, α/β = 0.3:1; (vi) NaOMe, MeOH, RT, 2 h, 2α: 87%, 2β: 87%, 5α: quantitative, 5β: 96%. | ||
| 2α | 2β | 3β | 5a | 5β | 29α | 29β | 32α | 32β | 33α | 33β | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 J Se,F (Hz) | 38 | — | — | 34 | — | 63 | — | 42 | — | 30 | — |
Experimental
General methods
Unless noted, chemical reagents and solvents were used without further purification from commercial sources. Anhydrous solvents as CH2Cl2, Et2O, and THF were obtained from a PureSolv-ENTM solvent purification system (Innovation Technology Inc). Concentration in vacuo was performed using a Buchi rotary evaporator. The 1H/13C/19F NMR spectra (δ in ppm, relative to TMS in CDCl3) were recorded with Varian spectrometers (Varian, Palo Alto, CA, USA) (400/101 MHz or 500/126 MHz) at 25 °C. Assignments were aided by 1H–1H and 1H–13C correlation experiments. HRMS spectra were recorded on a micromass LCT instrument from Waters and LaserToF LT3 Plus MALDI-TOF (DHAP Matrix). LRMS spectra were recorded on a Waters micromass Quattro Micro LC-MS/MS instrument using electrospray ionisation (ESI) in either positive or negative mode. Optical rotations were recorded on a PerkinElmer polarimeter (model 343) at the sodium D-line (589 nm) at 20 °C using a 1 dm cell and are not corrected. Silica gel chromatography was carried out using Davisil LC60A (Grace tech., Columbia, MD, USA) SiO2 (40–63 μm) silica gel. All reactions were monitored by thin-layer chromatography (TLC). TLC was performed on Merck DC-Alufolien plates precoated with silica gel 60 F254. They were visualised with UV-light (254 nm) fluorescence quenching, and/or by charring with an 8% H2SO4 dip and/or ninhydrin dip. Deprotected sugars were lyophilised using a freeze-dryer Alpha 1-2 Ldplus (Christ Ltd), with a pressure of 0.035 mbar and ice condenser temperature −55 °C.Selected procedures
:1, v/v) was added. The mixture was allowed to reach room temperature and left stirring for 22 hours. Afterwards it was neutralized with acetic acid (1 mL), stirred for an additional 10 minutes, and then concentrated in vacuo. The residue was taken up in EtOAc (20 mL), sequentially washed with water (2 × 10 mL), satd. aq. NaHCO3 solution (2 × 10 mL), brine (1 × 10 mL), dried over MgSO4, filtered, and concentrated in vacuo. The crude was purified by flash chromatography (toluene/EtOAc, 10:1 → 7:1, v/v) to give 8 (160 mg, 0.41 mmol, 39%) as a colourless amorphous solid. Rf 0.48, toluene/EtOAc, 3:1; [α20d] = −128 (c 0.5; CHCl3). 1H NMR (500 MHz, CDCl3) δ 5.51 (dd, J = 3.3, 0.8 Hz, 1H, H-4), 5.31 (at, J = 10.0 Hz, 1H, H-2), 5.06 (dd, J = 10.0, 3.4 Hz, 1H, H-3), 4.68 (dd, J = 30.6, 10.1 Hz, 1H, H-1), 4.59–4.29 (m, 2H; H-6a, H-6b), 3.99 (ddd, J = 11.9, 6.4, 1.0 Hz, 1H, H-5), 2.16, 2.14, 2.08, 2.00 (4s, 12H, 3 COCH3, 1 SeCH3); 13C NMR (126 MHz, CDCl3) δ 170.2, 170.2, 169.8 (3 ![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) OCH3), 80.9 (d, J = 172.1 Hz, C-6), 77.5 (C-1), 76.4 (d, J = 23.1 Hz, C-5), 71.7 (C-3), 67.6 (C-2), 67.5 (d, J = 5.8 Hz, C-4), 21.0, 20.8, 20.8 (3 COH3), 2.7 (SeCH3); 19F NMR (376 MHz, CDCl3) δ −230.78 (dd, J = 46.4, 11.9 Hz, F-6); HRMS (ESI+) m/z calcd for C13H19FO7Se: 409.0178 [M + Na]+; found: 409.0197.
:2:1; [α20d] = −2.3 (c 0.3; H2O). 1H NMR (400 MHz, CD3OD) δ 4.54 (dd, 2H, J = 47.3, 5.8 Hz, H-6a, H-6b), 4.54 (d, J = 9.7 Hz, 1H, H-1), 3.89 (dd, J = 3.3, 0.8 Hz, 1H, H-4), 3.84–3.75 (m, 1H; H-5), 3.65 (at, J = 9.7 Hz, 1H, H-2), 3.47 (dd, J = 9.2, 3.4 Hz, 1H, H-3), 2.09 (s, 3H; SeCH3); 13C NMR (101 MHz, CD3OD) δ 83.8 (d, J = 167.6 Hz, C-6), 81.9 (C-1), 79.7 (d, J = 21.2 Hz, C-5), 75.7 (C-3), 71.7 (C-2), 70.3 (d, J = 6.6 Hz, C-4), 1.7 (SeCH3). 19F NMR (376 MHz, CD3OD) δ −231.58 (ddd, J = 47.3, 14.0 Hz); HRMS (ESI+): m/z calcd for C7H13FO5Se: 282.9861 [M + Na]+; found: 282.9869.
:1, v/v) was added. The mixture was allowed to reach room temperature and left stirring for 22 hours, then it was neutralized by adding acetic acid (1 mL), left stirring for additional 10 minutes, and concentrated in vacuo. The residue was taken up in EtOAc (30 mL), sequentially washed with water (2 × 20 mL), satd. aq. NaHCO3 solution (2 × 20 mL), brine (1 × 20 mL), dried over MgSO4, filtered, and concentrated in vacuo. The crude product was dissolved in anhydrous MeOH (3 mL) and a freshly prepared methanolic solution of NaOMe 1 M (100 μL) was added. The mixture was left stirring at room temperature 17 hours, then it was neutralized with Dowex 50WX80 cationic resin, filtered, and concentrated in vacuo. The crude was purified by flash chromatography (EtOAc/MeOH/H2O, 7:2:1, v/v) to give 3 (40 mg, 0.095 mmol, 24%) as a white amorphous solid. Rf 0.58 EtOAc/MeOH/H2O, 7:2:1; [α20d] = +7.25 (c 0.4; H2O). 1H NMR (400 MHz, D2O) δ 4.97 (d, J = 9.1 Hz, 1H, H-1), 4.50–4.25 (m, 2H, H-1′, H-2), 4.01–3.90 (m, 3H), 3.86–3.72 (m, 5H), 3.71–3.62 (m, 2H), 3.60–3.52 (m, 1H, H-2′), 2.17 (s, 3H; SeCH3); 13C NMR (101 MHz, D2O) δ 102.7 (C-1′), 90.5 (d, J = 183.4 Hz, C-2), 79.8, 77.4 (d, J = 8.1 Hz, C-4), 75.7 (d, J = 25.7 Hz, C-1), 75.3, 73.9 (d, J = 18.4 Hz, C-3), 72.4, 70.8 (C-2′), 68.5, 60.9, 59.9, 1.8 (SeCH3); 19F NMR (376 MHz, D2O) δ −188.19 (dd, J = 49.4, 15.4 Hz); HRMS (ESI+) m/z calcd for C13H23FO9Se: 445.0389 [M + Na]+; found: 445.0384.
63 (700 mg, 1.70 mmol) was dissolved in acetone (1.7 mL), then selenourea (209 mg, 1.70 mmol) was added and the mixture was heated to reflux for 1 hour. The formed white precipitate was filtered out, washed with acetone, and dried to give 13 (665 mg, 1.24 mmol, 73%) as a white powder. [α20d] = +11.5 (c 1.0, H2O) 1H NMR (500 MHz, D2O) δ 5.63 (ad, J = 3.2 Hz, 1H, H-4), 5.58–5.54 (m, 2H, H-1, H-2), 5.35 (dd, J = 8.2, 3.2 Hz, 1H, H-3), 4.42 (at, J = 6.0 Hz, 1H, H-5), 4.35–4.26 (m, 2H, H-6a, H-6b), 2.26 (s, 3H, OCOCH3), 2.18 (s, 3H, OCOCH3), 2.13 (s, 3H, OCOCH3), 2.08–2.05 (m, 3H, OCOCH3); 13C NMR (126 MHz, D2O) δ 173.4, 172.9, 172.8, 172.4 (4 OOCH3), 165.0 (SeC
OCH3), 80.9 (d, J = 172.1 Hz, C-6), 77.5 (C-1), 76.4 (d, J = 23.1 Hz, C-5), 71.7 (C-3), 67.6 (C-2), 67.5 (d, J = 5.8 Hz, C-4), 21.0, 20.8, 20.8 (3 COH3), 2.7 (SeCH3); 19F NMR (376 MHz, CDCl3) δ −230.78 (dd, J = 46.4, 11.9 Hz, F-6); HRMS (ESI+) m/z calcd for C13H19FO7Se: 409.0178 [M + Na]+; found: 409.0197.
:2:1; [α20d] = −2.3 (c 0.3; H2O). 1H NMR (400 MHz, CD3OD) δ 4.54 (dd, 2H, J = 47.3, 5.8 Hz, H-6a, H-6b), 4.54 (d, J = 9.7 Hz, 1H, H-1), 3.89 (dd, J = 3.3, 0.8 Hz, 1H, H-4), 3.84–3.75 (m, 1H; H-5), 3.65 (at, J = 9.7 Hz, 1H, H-2), 3.47 (dd, J = 9.2, 3.4 Hz, 1H, H-3), 2.09 (s, 3H; SeCH3); 13C NMR (101 MHz, CD3OD) δ 83.8 (d, J = 167.6 Hz, C-6), 81.9 (C-1), 79.7 (d, J = 21.2 Hz, C-5), 75.7 (C-3), 71.7 (C-2), 70.3 (d, J = 6.6 Hz, C-4), 1.7 (SeCH3). 19F NMR (376 MHz, CD3OD) δ −231.58 (ddd, J = 47.3, 14.0 Hz); HRMS (ESI+): m/z calcd for C7H13FO5Se: 282.9861 [M + Na]+; found: 282.9869.
:1, v/v) was added. The mixture was allowed to reach room temperature and left stirring for 22 hours, then it was neutralized by adding acetic acid (1 mL), left stirring for additional 10 minutes, and concentrated in vacuo. The residue was taken up in EtOAc (30 mL), sequentially washed with water (2 × 20 mL), satd. aq. NaHCO3 solution (2 × 20 mL), brine (1 × 20 mL), dried over MgSO4, filtered, and concentrated in vacuo. The crude product was dissolved in anhydrous MeOH (3 mL) and a freshly prepared methanolic solution of NaOMe 1 M (100 μL) was added. The mixture was left stirring at room temperature 17 hours, then it was neutralized with Dowex 50WX80 cationic resin, filtered, and concentrated in vacuo. The crude was purified by flash chromatography (EtOAc/MeOH/H2O, 7:2:1, v/v) to give 3 (40 mg, 0.095 mmol, 24%) as a white amorphous solid. Rf 0.58 EtOAc/MeOH/H2O, 7:2:1; [α20d] = +7.25 (c 0.4; H2O). 1H NMR (400 MHz, D2O) δ 4.97 (d, J = 9.1 Hz, 1H, H-1), 4.50–4.25 (m, 2H, H-1′, H-2), 4.01–3.90 (m, 3H), 3.86–3.72 (m, 5H), 3.71–3.62 (m, 2H), 3.60–3.52 (m, 1H, H-2′), 2.17 (s, 3H; SeCH3); 13C NMR (101 MHz, D2O) δ 102.7 (C-1′), 90.5 (d, J = 183.4 Hz, C-2), 79.8, 77.4 (d, J = 8.1 Hz, C-4), 75.7 (d, J = 25.7 Hz, C-1), 75.3, 73.9 (d, J = 18.4 Hz, C-3), 72.4, 70.8 (C-2′), 68.5, 60.9, 59.9, 1.8 (SeCH3); 19F NMR (376 MHz, D2O) δ −188.19 (dd, J = 49.4, 15.4 Hz); HRMS (ESI+) m/z calcd for C13H23FO9Se: 445.0389 [M + Na]+; found: 445.0384.
63 (700 mg, 1.70 mmol) was dissolved in acetone (1.7 mL), then selenourea (209 mg, 1.70 mmol) was added and the mixture was heated to reflux for 1 hour. The formed white precipitate was filtered out, washed with acetone, and dried to give 13 (665 mg, 1.24 mmol, 73%) as a white powder. [α20d] = +11.5 (c 1.0, H2O) 1H NMR (500 MHz, D2O) δ 5.63 (ad, J = 3.2 Hz, 1H, H-4), 5.58–5.54 (m, 2H, H-1, H-2), 5.35 (dd, J = 8.2, 3.2 Hz, 1H, H-3), 4.42 (at, J = 6.0 Hz, 1H, H-5), 4.35–4.26 (m, 2H, H-6a, H-6b), 2.26 (s, 3H, OCOCH3), 2.18 (s, 3H, OCOCH3), 2.13 (s, 3H, OCOCH3), 2.08–2.05 (m, 3H, OCOCH3); 13C NMR (126 MHz, D2O) δ 173.4, 172.9, 172.8, 172.4 (4 OOCH3), 165.0 (SeC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 79.7 (C-1), 76.2 (C-5), 71.4 (C-3), 68.0 (C-4), 67.7 (C-2), 62.1 (C-6), 20.1, 20.0, 19.9 (4 OCOH3); HRMS (ESI+): m/z calcd for C15H23BrN2O9Se: 455.0569 [M − Br]+; found: 455.0552.
:1 → 8:2, v/v) to give 19 (150 mg, 0.19 mmol, 90%) as a white foam. Rf = 0.60, toluene/EtOAc 7:3; [α20d] = −19.2 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.47–7.43 (m, 2H, HAr), 7.39–7.27 (m, 8H, HAr), 5.31 (dd, J = 3.5, 1.1 Hz, 1H, H-4′), 5.21 (at, J = 10.1 Hz, 1H, H-2′), 4.92–4.77 (m, 4H, H-3′, CH2Ph, H-1′), 4.66 (d, J = 11.9 Hz, 1H, CH
N), 79.7 (C-1), 76.2 (C-5), 71.4 (C-3), 68.0 (C-4), 67.7 (C-2), 62.1 (C-6), 20.1, 20.0, 19.9 (4 OCOH3); HRMS (ESI+): m/z calcd for C15H23BrN2O9Se: 455.0569 [M − Br]+; found: 455.0552.
:1 → 8:2, v/v) to give 19 (150 mg, 0.19 mmol, 90%) as a white foam. Rf = 0.60, toluene/EtOAc 7:3; [α20d] = −19.2 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.47–7.43 (m, 2H, HAr), 7.39–7.27 (m, 8H, HAr), 5.31 (dd, J = 3.5, 1.1 Hz, 1H, H-4′), 5.21 (at, J = 10.1 Hz, 1H, H-2′), 4.92–4.77 (m, 4H, H-3′, CH2Ph, H-1′), 4.66 (d, J = 11.9 Hz, 1H, CH![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) Ph), 4.53 (d, J = 11.9 Hz, 1H, CHPh), 4.47–4.28 (m, 2H, H-1, H-2), 4.08–3.87 (m, 4H, H-6a, H-6b, H-6′a, H-6′b), 3.71–3.65 (m, 1H, H-5), 3.65–3.59 (m, 1H, H-3), 3.57 (s, 3H, OCH3), 3.45 (atd, J = 6.5, 6.1, 1.2 Hz, 1H, H-5′), 3.27 (at, J = 11.1 Hz, 1H, H-4), 2.14 (s, 3H, OCOCH3), 2.00 (s, 3H, OCOCH3), 1.98–1.96 (m, 6H, 2 OCOCH3); 13C NMR (101 MHz, CDCl3) δ 170.34, 170.33, 170.1, 169.6 (4 OOCH3), 138.2 (CAr), 137.9 (CAr), 128.6, 128.5, 128.4, 128.3, 128.1, 127.9, 127.8 (10 CAr), 101.4 (d, J = 23.0 Hz, C-1), 94.2 (d, J = 188.1 Hz, C-2), 79.0 (d, J = 17.9 Hz, C-3), 77.8 (under CDCl3 peak, C-1′), 76.4 (C-5), 75.5 (C-5′), 74.2 (CH2Ph), 73.6 (CH2Ph), 71.60 (C-3′), 70.1 (C-6), 68.5 (C-2′), 67.4 (C-4′), 61.8 (C-6′), 57.0 (OCH3), 41.9 (d, J = 6.8 Hz, C-4), 20.9, 20.8, 20.74, 20.72 (4 OCOH3); 19F NMR (376 MHz, CDCl3) δ −192.30 (add, J = 52.7, 14.5 Hz, F-2); HRMS (ESI+): m/z calcd for C35H43FO13Se: 793.1751 [M + Na]+; found: 793.1728.
:1, v/v) to give 26 (59 mg, 88 μmol, 91%) as a white foam. Rf = 0.39, toluene/acetone 9:1; [α20d] = −3.6 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.45 (dd, J = 3.4, 1.1 Hz, 1H, H-4′), 5.29 (ddd, J = 13.3, 11.5, 8.7 Hz, 1H, H-3), 5.18 (at, J = 9.9 Hz, 1H, H-2′), 5.10–4.99 (m, 2H, H-1′, H-3′), 4.59 (dd, J = 12.0, 2.1 Hz, 1H, H-6a), 4.51 (dd, J = 12.0, 4.9 Hz, 1H, H-6b), 4.44 (dd, J = 7.7, 2.6 Hz, 1H, H-1), 4.33–4.02 (m, 3H, H-2, H-6′a, H-6′b), 3.99–3.88 (m, 2H, H-5, H-5′), 3.56 (s, 3H, OCH3), 2.98 (at, J = 11.2 Hz, 1H, H-4), 2.17 (s, 3H, OCOCH3), 2.13 (s, 3H, OCOCH3), 2.09 (s, 3H, OCOCH3), 2.07 (s, 3H, OCOCH3), 2.03 (s, 3H, OCOCH3), 1.97 (s, 3H, OCOCH3); 13C NMR (101 MHz, CDCl3) δ 170.6, 170.30, 170.27, 169.94, 169.89, 169.86 (6 OOCH3), 101.4 (d, J = 22.3 Hz, C-1), 90.9 (d, J = 192.2 Hz, C-2), 77.3 (C-1′), 75.8 (C-5′), 73.8 (C-5), 71.6 (C-3′), 70.6 (d, J = 19.4 Hz, C-3), 67.9 (C-2′), 67.4 (C-4′), 64.5 (C-6), 62.4 (C-6′), 57.2 (OCH3), 41.2 (d, J = 4.9 Hz, C-4), 21.0, 20.9, 20.8, 20.74, 20.66 (6 OCOH3); 19F NMR (376 MHz, CDCl3) δ −196.30 (ddd, J = 51.0, 13.4, 2.6 Hz, F-2); HRMS (ESI+): m/z calcd for C25H35FO15Se: 697.1023 [M + Na]+; found: 697.1003.
:1, v/v) to afford 4 (16 mg, 38 μmol, 86%) as a white powder. Rf = 0.22, CH2Cl2/MeOH 9:1; [α20d] = −31.8 (c 1.0, MeOH); 1H NMR (400 MHz, CD3OD) δ 4.78 (d, J = 9.8 Hz, 1H, H-1′), 4.44 (dd, J = 7.7, 2.3 Hz, 1H, H-1), 4.10 (dd, J = 12.2, 2.0 Hz, 1H, H-6a), 3.98 (ddd, J = 51.5, 8.6, 7.7 Hz, 1H, H-2), 3.97 (dd, J = 12.2, 4.8 Hz, 1H, H-6b), 3.87 (dd, J = 3.4, 1.1 Hz, 1H, H-4′), 3.83–3.69 (m, 3H, H-3, H-6′a, H-2′), 3.69–3.61 (m, 2H, H-6′b, H-5), 3.56 (ddd, J = 7.4, 4.4, 1.1 Hz, 1H, H-5′), 3.53 (s, 3H, OCH3), 3.47 (dd, J = 9.2, 3.4 Hz, 1H, H-3′), 2.98 (at, J = 11.0 Hz, 1H, H-4); 13C NMR (101 MHz, CD3OD) δ 101.0 (d, J = 22.8 Hz, C-1), 92.9 (d, J = 187.4 Hz, C-2), 80.7 (C-5′), 80.4 (C-1′), 76.7 (C-5), 74.4 (C-3′), 72.3 (d, J = 18.4 Hz, C-3), 70.4 (C-2), 69.3 (C-4′), 62.3 (C-6), 61.4 (C-6′), 55.6 (OCH3), 41.9 (d, J = 5.7 Hz, C-4); 19F NMR (376 MHz, CD3OD) δ −197.77 (ddd, J = 51.5, 14.7, 2.3 Hz, F-2); HRMS (ESI+): m/z calcd for C13H23FO9Se: 445.0389 [M + Na]+; found: 445.0397.
Ph), 4.53 (d, J = 11.9 Hz, 1H, CHPh), 4.47–4.28 (m, 2H, H-1, H-2), 4.08–3.87 (m, 4H, H-6a, H-6b, H-6′a, H-6′b), 3.71–3.65 (m, 1H, H-5), 3.65–3.59 (m, 1H, H-3), 3.57 (s, 3H, OCH3), 3.45 (atd, J = 6.5, 6.1, 1.2 Hz, 1H, H-5′), 3.27 (at, J = 11.1 Hz, 1H, H-4), 2.14 (s, 3H, OCOCH3), 2.00 (s, 3H, OCOCH3), 1.98–1.96 (m, 6H, 2 OCOCH3); 13C NMR (101 MHz, CDCl3) δ 170.34, 170.33, 170.1, 169.6 (4 OOCH3), 138.2 (CAr), 137.9 (CAr), 128.6, 128.5, 128.4, 128.3, 128.1, 127.9, 127.8 (10 CAr), 101.4 (d, J = 23.0 Hz, C-1), 94.2 (d, J = 188.1 Hz, C-2), 79.0 (d, J = 17.9 Hz, C-3), 77.8 (under CDCl3 peak, C-1′), 76.4 (C-5), 75.5 (C-5′), 74.2 (CH2Ph), 73.6 (CH2Ph), 71.60 (C-3′), 70.1 (C-6), 68.5 (C-2′), 67.4 (C-4′), 61.8 (C-6′), 57.0 (OCH3), 41.9 (d, J = 6.8 Hz, C-4), 20.9, 20.8, 20.74, 20.72 (4 OCOH3); 19F NMR (376 MHz, CDCl3) δ −192.30 (add, J = 52.7, 14.5 Hz, F-2); HRMS (ESI+): m/z calcd for C35H43FO13Se: 793.1751 [M + Na]+; found: 793.1728.
:1, v/v) to give 26 (59 mg, 88 μmol, 91%) as a white foam. Rf = 0.39, toluene/acetone 9:1; [α20d] = −3.6 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.45 (dd, J = 3.4, 1.1 Hz, 1H, H-4′), 5.29 (ddd, J = 13.3, 11.5, 8.7 Hz, 1H, H-3), 5.18 (at, J = 9.9 Hz, 1H, H-2′), 5.10–4.99 (m, 2H, H-1′, H-3′), 4.59 (dd, J = 12.0, 2.1 Hz, 1H, H-6a), 4.51 (dd, J = 12.0, 4.9 Hz, 1H, H-6b), 4.44 (dd, J = 7.7, 2.6 Hz, 1H, H-1), 4.33–4.02 (m, 3H, H-2, H-6′a, H-6′b), 3.99–3.88 (m, 2H, H-5, H-5′), 3.56 (s, 3H, OCH3), 2.98 (at, J = 11.2 Hz, 1H, H-4), 2.17 (s, 3H, OCOCH3), 2.13 (s, 3H, OCOCH3), 2.09 (s, 3H, OCOCH3), 2.07 (s, 3H, OCOCH3), 2.03 (s, 3H, OCOCH3), 1.97 (s, 3H, OCOCH3); 13C NMR (101 MHz, CDCl3) δ 170.6, 170.30, 170.27, 169.94, 169.89, 169.86 (6 OOCH3), 101.4 (d, J = 22.3 Hz, C-1), 90.9 (d, J = 192.2 Hz, C-2), 77.3 (C-1′), 75.8 (C-5′), 73.8 (C-5), 71.6 (C-3′), 70.6 (d, J = 19.4 Hz, C-3), 67.9 (C-2′), 67.4 (C-4′), 64.5 (C-6), 62.4 (C-6′), 57.2 (OCH3), 41.2 (d, J = 4.9 Hz, C-4), 21.0, 20.9, 20.8, 20.74, 20.66 (6 OCOH3); 19F NMR (376 MHz, CDCl3) δ −196.30 (ddd, J = 51.0, 13.4, 2.6 Hz, F-2); HRMS (ESI+): m/z calcd for C25H35FO15Se: 697.1023 [M + Na]+; found: 697.1003.
:1, v/v) to afford 4 (16 mg, 38 μmol, 86%) as a white powder. Rf = 0.22, CH2Cl2/MeOH 9:1; [α20d] = −31.8 (c 1.0, MeOH); 1H NMR (400 MHz, CD3OD) δ 4.78 (d, J = 9.8 Hz, 1H, H-1′), 4.44 (dd, J = 7.7, 2.3 Hz, 1H, H-1), 4.10 (dd, J = 12.2, 2.0 Hz, 1H, H-6a), 3.98 (ddd, J = 51.5, 8.6, 7.7 Hz, 1H, H-2), 3.97 (dd, J = 12.2, 4.8 Hz, 1H, H-6b), 3.87 (dd, J = 3.4, 1.1 Hz, 1H, H-4′), 3.83–3.69 (m, 3H, H-3, H-6′a, H-2′), 3.69–3.61 (m, 2H, H-6′b, H-5), 3.56 (ddd, J = 7.4, 4.4, 1.1 Hz, 1H, H-5′), 3.53 (s, 3H, OCH3), 3.47 (dd, J = 9.2, 3.4 Hz, 1H, H-3′), 2.98 (at, J = 11.0 Hz, 1H, H-4); 13C NMR (101 MHz, CD3OD) δ 101.0 (d, J = 22.8 Hz, C-1), 92.9 (d, J = 187.4 Hz, C-2), 80.7 (C-5′), 80.4 (C-1′), 76.7 (C-5), 74.4 (C-3′), 72.3 (d, J = 18.4 Hz, C-3), 70.4 (C-2), 69.3 (C-4′), 62.3 (C-6), 61.4 (C-6′), 55.6 (OCH3), 41.9 (d, J = 5.7 Hz, C-4); 19F NMR (376 MHz, CD3OD) δ −197.77 (ddd, J = 51.5, 14.7, 2.3 Hz, F-2); HRMS (ESI+): m/z calcd for C13H23FO9Se: 445.0389 [M + Na]+; found: 445.0397.
Procedure for α
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :β = 3:7.
Acetone (920 μL) was added to a vial containing 28 (340 mg, 916 μmol) and selenourea (113 mg, 916 μmol). The reaction mixture was heated to 65 °C for 3 hours before adding Et2O (5 mL). Collection of the white precipitate formed gave 29 (385 mg, 779 μmol, 87%) as an amorphous solid (α/β = 3:7). Rf = 0.71, MeCN/H2O 85:15; 1H NMR (500 MHz, D2O) δ 6.99 (dd, J = 5.6, 1.8 Hz, 1H, H-1α), δ 5.71 (dd, J = 10.0, 3.4 Hz, 1H, H-1β), 5.66 (td, J = 3.2, 1.2 Hz, 1H, H-4α), 5.64 (td, J = 3.2, 1.0 Hz, 1H, H-4β), 5.49–5.32 (m, 2H, H-2α, H-3α), 5.41 (ddd, J = 13.5, 9.2, 3.4 Hz, 1H, H-3β), 5.12 (ddd, J = 49.1, 9.8, 9.2 Hz, 1H, H-2β), 4.82–4.80 (m, 1H, H-5α), 4.43 (ddd, J = 6.5, 5.4, 1.1 Hz, 1H, H-5β), 4.36–4.25 (m, 4H, H-6aα, H-6bα, H-6aβ, H-6bβ), 2.25 (s, 3H, OCOCH3α), 2.24 (s, 3H, OCOCH3β), 2.14 (s, 6H, 2× OCOCH3β), 2.13 (s, 3H, OCOCH3α), 2.12 (s, 3H, OCOCH3α); 13C NMR (126 MHz, D2O) δ 173.4 (OOCH3β), 173.3 (OOCH3α), 172.9 (OOCH3α), 172.8 (OOCH3β), 172.6 (OOCH3β), 172.5 (OOCH3α), 164.20 (SeCNα), 164.24 (SeCNβ), 86.7 (d, J = 186.4 Hz, C-2β), 85.3 (d, J = 187.3 Hz, C-2α), 83.0 (d, J = 24.6 Hz, C-1α), 78.5 (d, J = 26.9 Hz, C-1β), 76.1 (C-5β), 71.7 (d, J = 19.1 Hz, C-3β), 70.7 (C-5α), 69.9 (d, J = 19.0 Hz, C-3α), 68.5 (d, J = 8.7 Hz, C-4β), 68.0 (d, J = 8.6 Hz, C-4α), 62.0 (C-6α), 20.1 (OCOH3β), 20.0 (OCOH3α), 20.03 (OCOH3α), 20.00 (OCOH3β), 19.9 (OCOH3β), 19.8 (OCOH3α); 19F NMR (470 MHz, D2O) δ −194.73 (dd, J = 48.1, 14.3 Hz, F-2α), −196.45 (dd, J = 48.5, 12.1 Hz, F-2β). HRMS (ESI+): m/z calcd for C13H20BrFN2O7Se: 415.0415 [M − Br]+; found: 415.0416.
:β = 3:7.
Acetone (920 μL) was added to a vial containing 28 (340 mg, 916 μmol) and selenourea (113 mg, 916 μmol). The reaction mixture was heated to 65 °C for 3 hours before adding Et2O (5 mL). Collection of the white precipitate formed gave 29 (385 mg, 779 μmol, 87%) as an amorphous solid (α/β = 3:7). Rf = 0.71, MeCN/H2O 85:15; 1H NMR (500 MHz, D2O) δ 6.99 (dd, J = 5.6, 1.8 Hz, 1H, H-1α), δ 5.71 (dd, J = 10.0, 3.4 Hz, 1H, H-1β), 5.66 (td, J = 3.2, 1.2 Hz, 1H, H-4α), 5.64 (td, J = 3.2, 1.0 Hz, 1H, H-4β), 5.49–5.32 (m, 2H, H-2α, H-3α), 5.41 (ddd, J = 13.5, 9.2, 3.4 Hz, 1H, H-3β), 5.12 (ddd, J = 49.1, 9.8, 9.2 Hz, 1H, H-2β), 4.82–4.80 (m, 1H, H-5α), 4.43 (ddd, J = 6.5, 5.4, 1.1 Hz, 1H, H-5β), 4.36–4.25 (m, 4H, H-6aα, H-6bα, H-6aβ, H-6bβ), 2.25 (s, 3H, OCOCH3α), 2.24 (s, 3H, OCOCH3β), 2.14 (s, 6H, 2× OCOCH3β), 2.13 (s, 3H, OCOCH3α), 2.12 (s, 3H, OCOCH3α); 13C NMR (126 MHz, D2O) δ 173.4 (OOCH3β), 173.3 (OOCH3α), 172.9 (OOCH3α), 172.8 (OOCH3β), 172.6 (OOCH3β), 172.5 (OOCH3α), 164.20 (SeCNα), 164.24 (SeCNβ), 86.7 (d, J = 186.4 Hz, C-2β), 85.3 (d, J = 187.3 Hz, C-2α), 83.0 (d, J = 24.6 Hz, C-1α), 78.5 (d, J = 26.9 Hz, C-1β), 76.1 (C-5β), 71.7 (d, J = 19.1 Hz, C-3β), 70.7 (C-5α), 69.9 (d, J = 19.0 Hz, C-3α), 68.5 (d, J = 8.7 Hz, C-4β), 68.0 (d, J = 8.6 Hz, C-4α), 62.0 (C-6α), 20.1 (OCOH3β), 20.0 (OCOH3α), 20.03 (OCOH3α), 20.00 (OCOH3β), 19.9 (OCOH3β), 19.8 (OCOH3α); 19F NMR (470 MHz, D2O) δ −194.73 (dd, J = 48.1, 14.3 Hz, F-2α), −196.45 (dd, J = 48.5, 12.1 Hz, F-2β). HRMS (ESI+): m/z calcd for C13H20BrFN2O7Se: 415.0415 [M − Br]+; found: 415.0416.
Procedure for α
:β = 1:99.
Acetone (1 mL) was added to a vial containing 28 (367 mg, 989 μmol) and selenourea (122 mg, 989 μmol). The reaction mixture was heated to 65 °C for 3 hours, and was left for 90 minutes at room temperature, before collection of the white precipitate formed to give 29β (234 mg, 474 μmol, 48%, 96% purity) as an amorphous solid.
Procedure for α
:β = 2:98.
Acetone (1.22 mL) was added to a vial containing 28 (367 mg, 989 μmol) and selenourea (122 mg, 989 μmol). The reaction mixture was heated to 65 °C for 3 hours, and was left for 3 days at room temperature, before filtration to give 29β (218 mg, 440 μmol, 44%, 98% purity) as a white crystalline solid. [α20d] = +12.5 (c 1.0; H2O); m.p. (°C): 168–168.5.
:2 → 1:1, v/v) gave 32 (198 mg, 515 μmol, 66%,) as an anomeric mix. Purification by flash column chromatography (cyclohexane/Et2O, 6:4, v/v) gave 32α (45 mg, 117 μmol, 15%) and 32β (73 mg, 190 μmol 24%) as transparent oils. 32α: Rf = 0.18, cyclohexane/Et2O 6:4; [α20d] = +222 (c 1.0, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 5.89 (dd, J = 5.8, 2.2 Hz, 1H, H-1), 5.48 (td, J = 3.1, 1.2 Hz, 1H, H-4), 5.25 (ddd, J = 12.2, 10.0, 3.5 Hz, 1H, H-3), 4.95 (ddd, J = 51.2, 9.9, 5.7 Hz, 1H, H-2), 4.54 (td, J = 6.4, 1.3 Hz, 1H, H-5), 4.12 (app. dd, J = 6.6, 1.7 Hz, 2H, H-6a, H-6b), 2.14 (s, 3H, OCOCH3), 2.05 (s, 3H, OCOCH3), 2.04 (s, 3H, OCOCH3), 2.02 (s, 3H, SeCH3); 13C NMR (126 MHz, CDCl3) δ 170.5, 170.1, 170.0 (3 OOCH3), 85.9 (d, J = 187.8 Hz, C-2), 79.2 (d, J = 25.1 Hz, C-1), 70.0 (d, J = 18.8 Hz, C-3), 68.6 (C-5), 68.4 (d, J = 8.3 Hz, C-4), 61.7 (C-6), 20.82, 20.75, 20.7 (3 OCOH3); 19F NMR (470 MHz, CDCl3) δ −196.01 (ddt, J = 51.0, 12.1, 2.5 Hz, F-2); HRMS (ESI+): m/z calcd for C13H19FO7Se: 409.0173 [M + Na]+; found: 409.0171. 32β: Rf = 0.14, cyclohexane/Et2O 6:4; [α20d] = +17.1 (c 2.0, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 5.53–5.40 (m, 1H, H-4), 5.09 (ddd, J = 13.0, 9.3, 3.5 Hz, 1H, H-3), 4.76 (dd, J = 9.8, 2.8 Hz, 1H, H-1), 4.60 (dt, J = 49.6, 9.6 Hz, 1H, H-2), 4.12 (dd, J = 11.4, 6.8 Hz, 1H, H-6a), 4.07 (dd, J = 11.3, 6.5 Hz, 1H, H-6b), 3.93 (td, J = 6.8, 1.2 Hz, 1H, H-5), 2.15 (s, 3H, SeCH3), 2.11 (s, 3H, OCOCH3), 2.03 (s, 3H, OCOCH3), 2.02 (s, 3H, OCOCH3); 13C NMR (126 MHz, CDCl3) δ 170.4, 170.1, 170.0 (3 OOCH3), 87.3 (d, J = 186.1 Hz, C-2), 76.3 (d, J = 26.0 Hz, C-1), 75.6 (C-5), 71.9 (d, J = 19.8 Hz, C-3), 68.1 (d, J = 8.4 Hz, C-4), 61.4 (C-6), 20.74, 20.69, 20.6 (3 OCOH3), 2.74 (SeCH3); 19F NMR (470 MHz, CDCl3) δ −196.28 (ddt, J = 49.6, 13.2, 2.9 Hz, F-2); HRMS (ESI+): m/z calcd for C13H19FO7Se: 409.0173 [M + Na]+; found: 409.0171.
:05; [α20d] = +28 (c 1.0; H2O); 1H NMR (400 MHz, D2O) δ 4.90 (dd, J = 10.0, 2.1 Hz, 1H, H-1), 4.56 (dt, J = 49.8, 9.5 Hz, 1H, H-2), 4.09 (t, J = 3.5 Hz, 1H, H-4), 3.96 (ddd, J = 14.6, 9.1, 3.5 Hz, 1H, H-3), 3.85–3.68 (m, 3H, H-5, H-6a, H-6b), 2.19 (s, 3H, SeCH3); 13C NMR (101 MHz, D2O) δ 90.2 (d, J = 179.2 Hz, C-2), 80.2 (C-5), 76.4 (d, J = 26.0 Hz, C-1), 72.0 (d, J = 18.3 Hz, C-3), 69.4 (d, J = 9.2 Hz, C-4), 60.8 (C-6), 1.9 (SeCH3); 19F NMR (376 MHz, D2O) δ −196.07 (ddt, J = 49.8, 14.7, 2.7 Hz, F-2); HRMS (ESI+): m/z calcd for C7H13FO4Se: 282.9856 [M + Na]+; found: 282.9859.
:05; [α20d] = +269 (c 1.0; H2O); 1H NMR (400 MHz, D2O) δ 5.96 (dd, J = 5.7, 2.1 Hz, 1H, H-1), 4.88 (ddd, J = 51.5, 9.7, 5.8 Hz, 1H, H-2), 4.25 (ddd, J = 6.6, 5.1, 1.1 Hz, 1H, H-5), 4.07 (td, J = 3.5, 1.1 Hz, 1H, H-4), 4.01 (ddd, J = 13.5, 9.8, 3.5 Hz, 1H, H-3), 3.81–3.77 (m, 2H, H-6a, H-6b), 2.07 (s, 3H, SeCH3); 13C NMR (101 MHz, D2O) δ 89.2 (d, J = 180.9 Hz, C-2), 79.6 (d, J = 24.6 Hz, C-1), 73.1 (C-5), 69.6 (d, J = 16.8 Hz, C-3), 69.4 (d, J = 9.2 Hz, C-4), 60.8 (C-6), 2.3 (Se–CH3); 19F NMR (376 MHz, D2O) δ −195.48 (ddt, J = 51.1, 13.3, 2.7 Hz, F-2); HRMS (ESI+): m/z calcd for C7H13FO4Se: 282.9856 [M + Na]+; found: 282.9856.
64 (284 mg, 886 μmol) in pyridine (4.4 mL) at 0 °C. The reaction mixture was left to reach room temperature. After 2 hours CH2Cl2 (50 mL) was added, and the mixture was washed with satd. aq. NaHCO3/ice (50 mL). The organic phase was dried before removal of solvents under reduced pressure to give crude 3162 (361 mg, 90% crude weight, 92% pure by NMR). Triethylamine (90 μL, 666 μmol) was added to a stirred solution of 31 (216 mg, 476 μmol) and 29α/β (165 mg, 333 μmol) in anhydrous CH3CN (5.6 mL) at room temperature, and the mixture was stirred for 90 minutes before removal of solvents under reduced pressure. Purification by flash column chromatography (cyclohexane/EtOAc, 1:1, v/v) gave 33 (137 mg, 203 μmol, 61%, α/β = 0.28:1) as a transparent oil. Purification by flash column chromatography (cyclohexane/Et2O, 6:4 → 4:6, v/v) gave 33α (15 mg, 22 μmol, 7%) as a transparent oil, and 33β (33 mg, 49 μmol, 15%) as a transparent amorphous solid. 33α: Rf = 0.43, cyclohexane/EtOAc 1:1; [α20d] = +70.4 (c 0.1, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 6.01 (dd, J = 5.5, 1.6 Hz, 1H, H-1′), 5.45 (td, J = 3.3, 1.3 Hz, 1H, H-4′), 5.21 (dd, J = 11.3, 9.4 Hz, 1H, H-3), 5.04–4.84 (m, 2H, H-3′, H-2′), 4.84 (dd, J = 9.4, 8.0 Hz, 1H, H-2), 4.69 (dd, J = 11.9, 2.2 Hz, 1H, H-6b), 4.41 (td, J = 6.3, 1.3 Hz, 1H, H-5′), 4.40 (d, J = 7.9 Hz, 1H, H-1), 4.32 (dd, J = 12.0, 5.9 Hz, 1H, H-6a), 4.14 (dd, J = 11.4, 6.5 Hz, 1H, H-6b′), 4.09 (dd, J = 11.4, 6.6 Hz, 1H, H-6a′), 3.76 (ddd, J = 11.2, 5.8, 2.2 Hz, 1H, H-5), 3.48 (s, 3H, OCH3), 3.10 (t, J = 11.3 Hz, 1H, H-4), 2.13 (s, 3H, OCOCH3), 2.09 (s, 3H, OCOCH3), 2.06 (s, 3H, OCOCH3), 2.05 (s, 3H, OCOCH3), 2.03 (s, 3H, OCOCH3), 2.02 (s, 3H, OCOCH3); 13C NMR (101 MHz, CDCl3) δ 170.6, 170.5, 170.0, 169.93, 169.89, 169.8 (6 OOCH3), 101.5 (C-1), 84.9 (d, J 191.2 Hz, C-2′), 83.3 (d, J = 24.7 Hz, C-1′), 74.4 (C-3), 73.4 (C-5), 72.9 (C-2), 69.7 (C-5′), 69.6 (C-3′), 67.9 (d, J = 6.9 Hz, C-4′), 64.7 (C-6), 61.1 (C-6′), 57.1 (OCH3), 42.5 (C-4), 21.0, 20.9, 20.8, 20.70, 20.68, 20.66 (6 OCOH3); 19F NMR (376 MHz, CDCl3) δ −192.76–−193.15 (m, F-2′); HRMS (ESI+): m/z calcd for C25H35FO15Se: 697.1020 [M + Na]+; found: 697.1018. 33β: Rf = 0.42, cyclohexane/EtOAc 1:1; [α20d] = −16.4 (c 1.0, CH2Cl2); 1H-NMR (400 MHz, CDCl3) δ 5.47 (ddd, J = 3.7, 2.7, 1.1 Hz, 1H, H-4′), 5.17 (dd, J = 11.5, 9.3 Hz, 1H, H-3), 5.08 (ddd, J = 13.0, 9.4, 3.5 Hz, 1H, H-3′), 4.93 (dd, J = 9.7, 2.8 Hz, 1H, H-1′), 4.90 (dd, J = 9.3, 8.0 Hz, 1H, H-2), 4.71 (dd, J = 12.1, 2.1 Hz, 1H, H-6b), 4.59 (dt, J = 49.4, 9.6 Hz, 1H, H-2′), 4.39 (d, J = 7.9 Hz, 1H, H-1), 4.35 (dd, J = 12.1, 6.0 Hz, 1H, H-6a), 4.11 (m, 1H, H-6a′), 4.09 (dd, J = 12.1, 6.0 Hz, 1H, H-6b′), 3.97 (td, J = 6.4, 1.2 Hz, 1H, H-5′), 3.83 (ddd, J = 11.2, 6.0, 2.1 Hz, 1H, H-5), 3.49 (s, 3H, OCH3), 3.06 (t, J = 11.3 Hz, 1H, H-4), 2.17 (s, 3H, OCOCH3), 2.08 (s, 3H, OCOCH3), 2.07 (s, 3H, OCOCH3), 2.06 (s, 3H, OCOCH3), 2.05 (s, 3H, OCOCH3), 2.04 (s, 3H, OCOCH3); 13C-NMR (101 MHz, CDCl3) δ 170.7, 170.4, 170.3, 170.1, 169.9, 169.8 (6 OOCH3), 101.5 (C-1), 87.0 (d, J 187.2 Hz, C-2′), 76.1 (d, J 26.2 Hz, C-1′), 76.0 (C-5′), 74.1 (C-5), 72.9 (C-2), 71.7 (d, J 19.7 Hz, C-3′), 71.7 (C-3), 68.0 (d, J 8.2 Hz, C-4′), 64.8 (C-6), 61.7 (C-6′), 57.0 (OCH3, 40.5 (C-4), 21.2, 21.0, 20.9, 20.8, 20.73, 20.69 (6 OCOH3); 19F-NMR (376 MHz, CDCl3) δ −195.79 (ddt, J = 49.4, 13.1, 2.7 Hz, F-2′); HRMS (ESI+): m/z calcd for C25H35FO15Se: 697.1020 [M + Na]+; found: 697.1017.
:8, v/v) gave 33β (132 mg, 20 μmol, 56%) as a white amorphous solid.
:15; [α20d] = −22.2 (c 1.0; H2O); 1H NMR (400 MHz, D2O) δ 5.11 (dd, J = 10.0, 2.2 Hz, 1H, H-1′), 4.55 (dt, J = 50.0, 9.5 Hz, 1H, H-2′), 4.38 (d, J = 8.1 Hz, 1H, H-1), 4.20 (dd, J = 12.5, 2.1 Hz, 1H, H-6b), 4.07 (t, J = 3.3 Hz, 1H, H-4′), 4.00–3.91 (m, 2H, H-6a, H-3′), 3.83–3.78 (m, 1H, H-5), 3.79–3.70 (m, 3H, H-5′, H-6a′, H-6b′), 3.65 (dd, J = 10.9, 8.9 Hz, 1H, H-3), 3.58 (s, 3H, OCH3), 3.31 (t, J = 8.4 Hz, 1H, H-2), 3.02 (t, J = 11.1 Hz, 1H, H-4); 13C NMR (101 MHz, D2O) δ 102.9 (C-1), 90.7 (d, J = 180.6 Hz, C-2′), 80.3 (C-5′), 76.4 (C-5), 76.0 (d, J = 25.9 Hz, C-1′), 74.4 (C-2), 73.2 (C-3), 72.1 (d, J = 18.3 Hz, C-3′), 69.3 (d, J = 9.2 Hz, C-4′), 62.1 (C-6), 61.0 (C-6′), 57.0 (OCH3), 43.4 (C-4); 19F NMR (376 MHz, D2O) δ −195.71 (ddt, J = 49.9, 14.9, 2.8 Hz, F-2′). HRMS (ESI+): m/z calcd for C13H23FO9Se: 445.0384 [M + Na]+; found: 445.0383.
:15; [α20d] = +224 (c 1.0; H2O); 1H NMR (500 MHz, D2O) δ 6.27 (dd, J = 5.9, 1.8 Hz, 1H, H-1′), 4.86 (ddd, J = 51.7, 10.2, 6.0 Hz, 1H, H-2′), 4.38 (d, J = 8.0 Hz, 1H, H-1), 4.25 (ddd, J = 6.7, 5.1, 1.2 Hz, 1H, H-5′), 4.18 (dd, J = 12.2, 2.2 Hz, 1H, H-6b), 4.08 (td, J = 3.4, 1.1 Hz, 1H, H-4′), 3.96 (ddd, J = 12.6, 10, 3.5 Hz, 1H, H-3′), 3.92 (dd, J = 12.3, 6.0 Hz, 1H, H-6a), 3.84–3.69 (m, 4H, H-3, H-5, H-6a′, H-6b′), 3.58 (s, 3H, OCH3), 3.26 (dd, J = 9.1, 8.0 Hz, 1H, H-2), 2.91 (t, J = 11.0 Hz, 1H, H-4); 13C NMR (126 MHz, D2O) δ 102.9 (C-1), 88.5 (d, J 183.2 Hz, C-2′), 80.4 (d, J 24.7 Hz, C-1′), 75.8 (C-3), 75.4 (C-5), 74.4 (C-2), 73.7 (C-5′), 69.5 (d, J 17.1 Hz, C-3′), 69.3 (d, J 9.3 Hz, C-4′), 62.4 (C-6), 60.7 (C-6′), 57.0 (OCH3), 42.6 (C-4); 19F NMR (376 MHz, D2O) δ −193.87 (app. dd, J = 50.8, 12.3 Hz, F-2′). HRMS (ESI+): m/z calcd for C13H23FO9Se: 445.0384 [M + Na]+; found: 445.0386.
Conclusions
In summary, a set of 19F and 77Se substituted saccharides (1–5) has been synthesized. While selenoglycosides 1 and 3 were prepared by reacting the corresponding glycosyl bromides with methyl diselenide under reducing conditions, compounds 2, 4, and 5 were synthesized via the formation of the corresponding isoselenouronium salts as selenyl transfer reagents. The proposed approach for the synthesis of pseudo-lactosides 4 and 5 allowed for the efficient introduction of a selenium atom at the interglycosidic linkage and a fluorine at C-2, either on the D-glucose or the D-galactose moiety. In the case of the preparation of pseudo-lactoside 5β and selenogalactoside 2α/β, the 2′-F substituent complicated the synthesis, yielding an α/β mixture in the formation of the selenourea salt 29. However, the almost pure β-form could be obtained by fractional precipitation directly from the reaction mixture and the following displacement reactions were found to be stereospecific and with no anomerisation occurring. To the best of our knowledge, the synthesised compounds represent the first example of a set of small carbohydrates functionalised with both selenium and fluorine, constituting valuable tools for structural elucidations of protein–carbohydrate interactions exploiting the complementary reporting abilities of the two unnatural substitutions.Author contributions
Conceptualization: S. O.; synthesis and characterization: C. R., D. B., A. S. I.; writing original draft: C. R.; manuscript review & editing: S. O., C. R., D. B.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from European Commission's GLYCOPHARM ITN project (contract no. 317297) and Science Foundation Ireland (grants no. 13/IA/1959 and 16/RC/3889).References
- G. F. Audette, L. T. J. Delbaere and J. Xiang, Curr. Protein Pept. Sci., 2003, 4, 11–20 CrossRef CAS PubMed.
- D. Solís, J. Jiménez-Barbero, M. Martín-Lomas and T. Díaz-Mauriño, Eur. J. Biochem., 1994, 223, 107–114 CrossRef PubMed.
- C. Sandstroem, B. Hakkarainen, E. Matei, A. Glinchert, M. Lahmann, S. Oscarson, L. Kenne, A. M. Gronenborn, C. Sandström, B. Hakkarainen, E. Matei, A. Glinchert, M. Lahmann, S. Oscarson, L. Kenne and A. M. Gronenborn, Biochemistry, 2008, 47, 3625–3635 CrossRef CAS PubMed.
- R. C. Saliba and N. L. Pohl, Curr. Opin. Chem. Biol., 2016, 34, 127–134 CrossRef CAS PubMed.
- I. P. Street, C. R. Armstrong and S. G. Withers, Biochemistry, 1986, 25, 6021–6027 CrossRef CAS PubMed.
- I. J. Goldstein, C. M. Reichert and A. Misaki, Ann. N. Y. Acad. Sci., 1974, 234, 283–296 CrossRef CAS PubMed.
- J. Xia, J. Xue, R. D. Locke, E. V. Chandrasekaran, T. Srikrishnan and K. L. Matta, J. Org. Chem., 2006, 71, 3696–3706 CrossRef CAS PubMed.
- D. J. Vocadlo and S. G. Withers, Carbohydr. Res., 2005, 340, 379–388 CrossRef CAS PubMed.
- L. Wang, Y. Liu, L. Wu and X. L. Sun, Biochim. Biophys. Acta, Proteins Proteomics, 2016, 1864, 143–153 CrossRef CAS PubMed.
- J. D. Belcher, C. Chen, J. Nguyen, F. Abdulla, P. Nguyen, M. Nguyen, N. M. Okeley, D. R. Benjamin, P. D. Senter and G. M. Vercellotti, PLoS One, 2015, 10, e0117772 CrossRef PubMed.
- T. Diercks, J. P. P. Ribeiro, F. J. Cañada, S. André, J. Jiménez-Barbero and H.-J. Gabius, Chem. – Eur. J., 2009, 15, 5666–5668 CrossRef CAS PubMed.
- E. Matei, S. André, A. Glinschert, A. S. Infantino, S. Oscarson, H.-J. Gabius and A. M. Gronenborn, Chem. – Eur. J., 2013, 19, 5364–5374 CrossRef CAS PubMed.
- M. Kurfiřt, M. Dračínský, L. Červenková Šťastná, P. Cuřínová, V. Hamala, M. Hovorková, P. Bojarová and J. Karban, Chem. – Eur. J., 2021, 27, 13040–13051 CrossRef PubMed.
- J. Ribeiro, T. Diercks, J. Jiménez-Barbero, S. André, H.-J. Gabius and F. J. Cañada, Biomolecules, 2015, 5, 3177–3192 CrossRef CAS PubMed.
- L. Unione, M. Alcalá, B. Echeverria, S. Serna, A. Ardá, A. Franconetti, F. J. Cañada, T. Diercks, N. Reichardt and J. Jiménez-Barbero, Chem. – Eur. J., 2017, 23, 3957–3965 CrossRef CAS PubMed.
- T. Diercks, A. S. Infantino, L. Unione, J. Jiménez-Barbero, S. Oscarson and H. J. Gabius, Chem. – Eur. J., 2018, 24, 15761–15765 CrossRef CAS PubMed.
- J. Daniel Martínez, P. Valverde, S. Delgado, C. Romanò, B. Linclau, N. C. N. C. Reichardt, S. Oscarson, A. Ardá, J. Jiménez-Barbero and F. Javier Cañada, Molecules, 2019, 24, E2337 CrossRef PubMed.
- V. Denavit, D. Lainé, C. Bouzriba, E. Shanina, É. Gillon, S. Fortin, C. Rademacher, A. Imberty and D. Giguère, Chem. – Eur. J., 2019, 25, 4478–4490 CrossRef CAS PubMed.
- J. D. Martínez, A. I. Manzano, E. Calviño, A. De Diego, B. Rodriguez De Francisco, C. Romanò, S. Oscarson, O. Millet, H. J. Gabius, J. Jiménez-Barbero and F. J. Cañada, J. Org. Chem., 2020, 85, 16072–16081 CrossRef PubMed.
- J. A. Garnett, Y. Liu, E. Leon, S. A. Allman, N. Friedrich, S. Saouros, S. Curry, D. Soldati-Favre, B. G. Davis, T. Feizi and S. Matthews, Protein Sci., 2009, 18, 1935–1947 CrossRef CAS PubMed.
- S. A. Allman, H. H. Jensen, B. Vijayakrishnan, J. A. Garnett, E. Leon, Y. Liu, D. C. Anthony, N. R. Sibson, T. Feizi, S. Matthews and B. G. Davis, ChemBioChem, 2009, 10, 2522–2529 CrossRef CAS PubMed.
- J. S. Andrews, B. D. Johnston and B. M. Pinto, Carbohydr. Res., 1998, 310, 27–33 CrossRef CAS.
- S. Mehta, J. Andrews, B. D. Johnston, B. Svensson and B. M. Pinto, J. Am. Chem. Soc., 2001, 117, 9783–9790 CrossRef.
- S. André, K. E. Kövér, H. J. Gabius and L. Szilágyi, Bioorg. Med. Chem. Lett., 2015, 25, 931–935 CrossRef PubMed.
- H. Kaltner, T. Szabó, F. Krisztina, S. André, S. Balla, J. C. Manning, L. Szilágyi and H.-J. Gabius, Bioorg. Med. Chem., 2017, 25, 3158–3170 CrossRef CAS PubMed.
- M. Raics, Á. K. Balogh, C. Kishor, I. Timári, F. J. Medrano, A. Romero, R. M. Go, H. Blanchard, L. Szilágyi, K. E. Kövér and K. Fehér, Int. J. Mol. Sci., 2022, 23, 2494 CrossRef CAS PubMed.
- L. Buts, R. Loris, E. De Genst, S. Oscarson, M. Lahmann, J. Messens, E. Brosens, L. Wyns, H. De Greve and J. Bouckaert, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2003, D59, 1012–1015 CrossRef CAS PubMed.
- N. Kostlanova, E. P. Mitchell, H. Lortat-Jacob, S. Oscarson, M. Lahmann, N. Gilboa-Garber, G. Chambat, M. Wimmerova and A. Imberty, J. Biol. Chem., 2005, 280, 27839–27849 CrossRef CAS PubMed.
- T. Suzuki, H. Makyio, H. Ando, N. Komura, M. Menjo, Y. Yamada, A. Imamura, H. Ishida, S. Wakatsuki, R. Kato and M. Kiso, Bioorg. Med. Chem., 2014, 22, 2090–2101 CrossRef CAS PubMed.
- C. Hamark, J. Landström and G. Widmalm, Chem. – Eur. J., 2014, 20, 13905–13908 CrossRef CAS PubMed.
- I. Pérez-Victoria, O. Boutureira, T. D. W. Claridge and B. G. Davis, Chem. Commun., 2015, 51, 12208–12211 RSC.
- M. Raics, I. Timári, T. Diercks, L. Szilágyi, H. J. Gabius and K. E. Kövér, ChemBioChem, 2019, 20, 1688–1692 CrossRef CAS PubMed.
- T. Suzuki, C. Hayashi, N. Komura, R. Tamai, J. Uzawa, J. Ogawa, H. N. Tanaka, A. Imamura, H. Ishida, M. Kiso, Y. Yamaguchi and H. Ando, Org. Lett., 2019, 21, 6393–6396 CrossRef CAS PubMed.
- T. Diercks, F. J. Medrano, F. G. FitzGerald, D. Beckwith, M. J. Pedersen, M. Reihill, A. K. Ludwig, A. Romero, S. Oscarson, M. Cudic and H. J. Gabius, Chem. – Eur. J., 2021, 27, 316–325 CrossRef CAS PubMed.
- P. J. Card, J. Org. Chem., 1983, 48, 393–395 CrossRef CAS.
- K. Dax, M. Albert, J. Ortner and B. J. Paul, Carbohydr. Res., 2000, 327, 47–86 CrossRef CAS PubMed.
- Z. Witczak and S. Czernecki, in Advances in carbohydrate chemistry and biochemistry, Academic Press, 1998, vol. 53, pp. 143–195 Search PubMed.
- M. Salvadó, B. Amgarten, S. Castillón, G. J. L. Bernardes and O. Boutureira, Org. Lett., 2015, 17, 2836–2839 CrossRef PubMed.
- A. A. Kumar, T. Z. Illyés, K. E. Kövér and L. Szilágyi, Carbohydr. Res., 2012, 360, 8–18 CrossRef CAS PubMed.
- Y. Kawai, H. Ando, H. Ozeki, M. Koketsu and H. Ishihara, Org. Lett., 2005, 7, 4653–4656 CrossRef CAS PubMed.
- T. Manna and A. K. Misra, Org. Biomol. Chem., 2019, 17, 8902–8912 RSC.
- T. Suzuki, N. Komura, A. Imamura, H. Ando, H. Ishida and M. Kiso, Tetrahedron Lett., 2014, 55, 1920–1923 CrossRef CAS.
- A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Essentials of glycobiology, Cold Spring Harbor Laboratory Press, 2nd edn, 2009 Search PubMed.
- S. H. Barondes, D. N. W. Cooper, M. A. Gitt and H. Leffler, J. Biol. Chem., 1994, 269, 20807–20810 CrossRef CAS PubMed.
- L. Astorgues-Xerri, M. E. Riveiro, A. Tijeras-Raballand, M. Serova, C. Neuzillet, S. Albert, E. Raymond and S. Faivre, Cancer Treat. Rev., 2014, 40, 307–319 CrossRef CAS PubMed.
- A. B. Samal, H. J. Gabius and A. V. Timoshenko, Anticancer Res., 1995, 15, 361–367 CAS.
- F. Strino, J. H. Lii, C. A. K. Koppisetty, P. G. Nyholm and H. J. Gabius, J. Comput.-Aided Mol. Des., 2010, 24, 1009–1021 CrossRef CAS PubMed.
- H. Ippel, M. C. Miller, S. Vértesy, Y. Zhang, F. J. Cañada, D. Suylen, K. Umemoto, C. Romanò, T. Hackeng, G. Tai, H. Leffler, J. J. Kopitz, S. André, D. Kubler, J. Jiménez-Barbero, S. Oscarson, H.-J. Gabius and K. H. Mayo, Glycobiology, 2016, 6, 1–16 CrossRef.
- P. Traar, F. Belaj and K. A. Francesconi, Aust. J. Chem., 2004, 57, 1051–1053 CrossRef CAS.
- S. Wagner, C. Mersch and A. Hoffmann-Röder, Chem. – Eur. J., 2010, 16, 7319–7330 CrossRef CAS PubMed.
- D. Lafont, P. Boullanger, F. Carvalho and P. Vottero, Carbohydr. Res., 1997, 297, 117–126 CrossRef CAS.
- R. Miethchen, P. M. Collins and R. J. Ferrier, Monosaccharides: Their Chemistry and Their Roles in Natural Products, John Wiley & Sons, Ltd., 1995 Search PubMed.
- M. Albert, D. Karl and J. Ortner, Tetrahedron, 1998, 54, 4839–4848 CrossRef CAS.
- P. J. Garegg, H. Hultberg and S. Wallin, Carbohydr. Res., 1982, 108, 97–101 CrossRef.
- S. Czernecki and D. Randriamandimby, J. Carbohydr. Chem., 1996, 15, 183–190 CrossRef CAS.
- C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17–60 CrossRef CAS.
- M. Niemietz, L. Perkams, J. Hoffman, S. Eller, C. Unverzagt, Y. Ito, M. Niemietz, M. Pischl and C. Raps, Chem. Commun., 2011, 47, 10485–10487 RSC.
- A. J. Birch, J. Chem. Soc., 1944, 430–436 RSC.
- M. Nanami, H. Ando, Y. Kawai, M. Koketsu and H. Ishihara, Tetrahedron Lett., 2007, 48, 1113–1116 CrossRef CAS.
- J. I. Padrón and J. T. Vázquez, Tetrahedron: Asymmetry, 1995, 6, 857–858 CrossRef.
- J. St-Gelais, C. Leclerc and D. Giguère, Carbohydr. Res., 2022, 511, 1–9 CrossRef PubMed.
- M. J. Kiefel, B. Beisner, S. Bennett, I. D. Holmes and M. Von Itzstein, J. Med. Chem., 1996, 39, 1314–1320 CrossRef CAS PubMed.
- H. J. Berthold, S. Franke, J. Thiem and T. Schotten, J. Org. Chem., 2010, 75, 3859–3862 CrossRef CAS PubMed.
- P. Kováč, E. A. Sokoloski and C. P. J. Glaudemans, Carbohydr. Res., 1984, 128, 101–109 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob02299k |
| ‡ Present address: Department of Chemistry, Technical University of Denmark, Kemitorvet 207, Kgs. Lyngby, Denmark. |
| This journal is © The Royal Society of Chemistry 2023 |