
Protonophoric and mitochondrial uncoupling activity of aryl-carbamate substituted fatty acids†
Hugo
MacDermott-Opeskin‡
 a,
Callum
Clarke‡
b,
Xin
Wu
cd,
Ariane
Roseblade
b,
Edward
York
b,
Ethan
Pacchini
b,
Ritik
Roy
b,
Charles
Cranfield
e,
Philip A.
Gale
cf,
Megan L.
O'Mara
ag,
Michael
Murray
h and
Tristan
Rawling
*b
a,
Callum
Clarke‡
b,
Xin
Wu
cd,
Ariane
Roseblade
b,
Edward
York
b,
Ethan
Pacchini
b,
Ritik
Roy
b,
Charles
Cranfield
e,
Philip A.
Gale
cf,
Megan L.
O'Mara
ag,
Michael
Murray
h and
Tristan
Rawling
*b
aResearch School of Chemistry, College of Science, The Australian National University, Canberra, ACT 0200, Australia
bSchool of Mathematical and Physical Sciences, Faculty of Science, University of Technology Sydney, Sydney, NSW 2007, Australia. E-mail: Tristan.Rawling@uts.edu.au; Tel: +61-2-9514-7956
cSchool of Chemistry, The University of Sydney, NSW 2006, Australia
dSchool of Chemistry and Molecular Biosciences, The University of Queensland, St Lucia, QLD 4072, Australia
eSchool of Life Sciences, Faculty of Science, University of Technology Sydney, Sydney, NSW 2007, Australia
fThe University of Sydney Nano Institute (SydneyNano), The University of Sydney, NSW 2006, Australia
gAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, St Lucia, QLD 4072, Australia
hDiscipline of Pharmacology, School of Medical Sciences, The University of Sydney, NSW 2006, Australia
First published on 1st December 2022
Abstract
Aryl-urea substituted fatty acids are protonophores and mitochondrial uncouplers that utilise a urea-based synthetic anion transport moiety to carry out the protonophoric cycle. Herein we show that replacement of the urea group with carbamate, a functional group not previously reported to possess anion transport activity, produces analogues that retain the activity of their urea counterparts. Thus, the aryl-carbamate substituted fatty acids uncouple oxidative phosphorylation and inhibit ATP production by collapsing the mitochondrial proton gradient. Proton transport proceeds via self-assembly of the deprotonated aryl-carbamates into membrane permeable dimeric species, formed by intermolecular binding of the carboxylate group to the carbamate moiety. These results highlight the anion transport capacity of the carbamate functional group.
Introduction
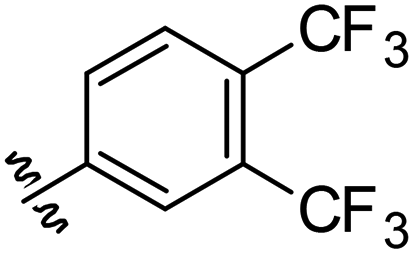
Synthetic anion transporters are molecules that facilitate the movement of anions across lipid membranes, a process that is hindered by the lipophilic core of lipid bilayers.1,2 Anion transport can occur through membrane channel formation or via a carrier mechanism in which the anion transporter binds an anion to form complexes that are capable of permeating lipid bilayers.3 Anion binding commonly occurs through non-covalent interactions between the anion and anion binding units in the transporter, which are often hydrogen bond donors.4 Urea and thiourea functional groups are among the most common functionalities employed in synthetic anion transporters because of their capacity to complex anions with high affinity through parallel hydrogen bonds.5 Other hydrogen bonding functional groups employed to date in synthetic anion transporters include squaramides,6 amides7 and various N-heterocycles.8–10We recently showed that incorporation of a urea-based anion transport moiety into a fatty acid produced a new class of atypical protonophores (termed aryl-ureas, Fig. 1) that act as mitochondrial uncouplers and inhibit cellular ATP production.11 Mitochondria produce ATP through oxidative phosphorylation, a process that couples nutrient oxidation to ATP synthesis through a transmembrane proton gradient. As nutrients are oxidised in the mitochondrial matrix, the energy released is used to pump protons from the mitochondrial matrix into the intermembrane space by a series of proteins embedded in the mitochondrial inner membrane (MIM), thereby establishing a proton gradient across the MIM. This gradient drives the flow of protons through the MIM embedded protein ATP-synthase, which converts ADP to ATP. Mitochondrial uncoupling can occur when protonophores, which are typically lipophilic weak acids, short circuit this mechanism by transporting protons across the MIM and into the matrix through the protonophoric cycle shown in Fig. 1a.12 In this typical protonophoric cycle the protonophore anion (A-, Fig. 1a) accepts a proton in the intermembrane space to form a neutral species (A-H) that readily crosses the MIM. Deprotonation of the protonophore in the more alkaline mitochondrial matrix result in the transport of one proton across the MIM and regenerates the anionic species (A−) that must permeate the lipophilic core of the MIM for continued cycling. Thus, the acidic groups in protonophores are most often conjugated to extended π-systems that delocalise charge to promote membrane permeability of the anionic protonophore and further cycling. Aryl-urea substituted fatty acids are of particular interest as they function through an atypical mechanism wherein the acidic group is not directly conjugated with a π-system (Fig. 1b). To permeate the MIM after deprotonation of the aryl-urea carboxylic acid group in the matrix, the aryl-ureas self-assemble into membrane permeable dimers, formed by intermolecular interaction of the fatty acid carboxylate group with the urea-based anion transport moiety (Fig. 1c).13,14 These dimers can return to the intermembrane space, where complex dissociation allows for continued cycling. Thus, the protonophoric and mitochondrial uncoupling activity of the aryl-ureas is directly linked to the carboxylate transport capacity of the synthetic anion transport group.
 | ||
| Fig. 1 Mitochondrial uncoupling mechanisms of weak acid protonophores and aryl urea substituted fatty acids. Panel (a): proton transport across the mitochondrial inner membrane (MIM) in the protonophoric cycle. The protonophore (A−) is protonated in the intermembrane space, generating a neutral species that freely permeates the MIM. Proton transport occurs when the protonophore is deprotonated in the relatively alkaline matrix. The rate determining step in the cycle is permeation of the anionic species across the MIM, which allows the cycle to continue; panel (b): chemical structure of the aryl urea substituted fatty acids; panel (c): protonophoric cycle of the aryl urea substituted fatty acids. MIM permeation occurs via self-assembly into membrane permeable dimers formed by carboxylate binding to a urea-based anion transport group. | ||
In this paper we replaced the anion transport group in the aryl-urea scaffold with carbamate groups, and studied the anion-binding, protonophoric and mitochondrial uncoupling activities of these compounds using a range of chemical, biological and computational techniques. Carbamates are structurally related to amides, for which anion binding has been extensively explored using a variety of scaffolds.15–17 Additionally, some amide-based scaffolds have been demonstrated to possess anion transport activity.18–21 The carbamate functional group has been used in anion binding studies22,23 but has, to the best of our knowledge, not been assessed for anion transport activity. The carbamates were shown to depolarise mitochondria with similar potency to their aryl-urea counterparts. Using experimental and computational approaches we show that the carbamate analogues, like the aryl-ureas, self-associate into anionic dimers that can permeate the MIM. Together these results establish the anion transport capacity of the carbamate functional group for the first time.
Results and discussion
Compound design and synthesis
The protonophoric and mitochondrial uncoupling activity of the aryl-ureas is dependent on the anion transport capacity of the aryl-urea group. Substitution of the aromatic ring with lipophilic electron withdrawing groups was demonstrated to be critical to activity.11 Electron withdrawing groups increase the acidity and carboxylate affinity of the urea N–H groups to promote dimer formation, and lipophilic substituents increased the membrane permeability of the dimers. Thus, aryl-carbamate substituted fatty acids C1–C6 were substituted with lipophilic electron withdrawing groups.C1–C6 were synthesised in 3 steps as shown in Scheme 1. In the first step the carboxylic acid group in 1 was benzyl-ester protected by reaction with benzyl bromide with caesium carbonate as base. Carbamates 3–8 were prepared from 2 in a two-step, one-pot reaction. Appropriately substituted anilines were first reacted with N,N-carbonyldiimidazole to form intermediate N-carbamoylimidazoles. These intermediates serve as masked isocyanates,24 which were then reacted with 2 to form carbamates 3–8. Removal of the benzyl-protecting groups in 3–8 by palladium-catalysed hydrogenation yielded carbamates C1–C6.
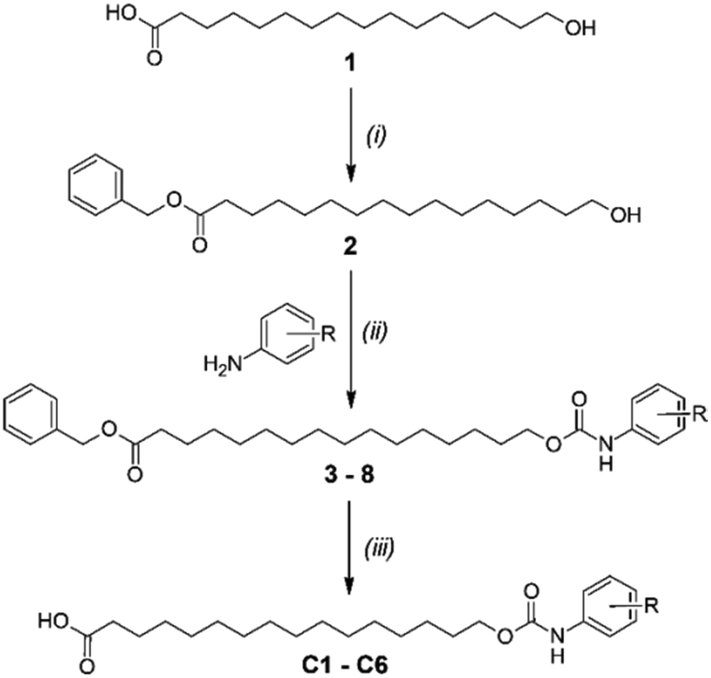 | ||
| Scheme 1 Synthesis of carbamates C1–C6. Reagents and conditions: (i) anhydrous DMF, Cs2CO3, benzyl bromide, 60 °C, 18 h; (ii) N,N-carbonyldiimidazole, anhydrous dichloromethane, rt, 2 h, then rt, 18 h; (iii) anhydrous tetrahydrofuran, H2, palladium/charcoal, rt, 18 h. | ||
Effects of C1–C6 on mitochondrial function in MDA-MB-231 cells
Like typical protonophores, the aryl-ureas can uncouple oxidative phosphorylation and inhibit ATP production by collapsing the proton gradient across the MIM. We therefore assessed the protonophoric and mitochondrial uncoupling activity of carbamates C1–C6 in MDA-MB-231 breast cancer cells. Effects on the proton gradient across the MIM were measured using the JC-1 assay. JC-1 is a fluorescent dye that forms aggregates in the matrix of polarised mitochondria that fluoresce red. In response to depolarisation of the MIM JC-1 disaggregates to monomers that diffuse in the cell cytosol and fluoresce green. The JC-1 IC50 was defined as the concentration required to shift the red: green fluorescence ratio by 50% of control cells. One hour treatments were used in JC-1 assays to observe the direct effects of the carbamates and minimise potential interference from other cellular processes such as apoptosis that can affect the proton gradient across the MIM. As shown in Table 1 (see Fig. S1† for dose–response plots) carbamates C1–C6 effectively depolarised mitochondria and shifted the JC-1 red: green fluorescence ratio with IC50 concentrations between 3–9 μM. The para-substituted analogue C4 was the least potent in the series (IC50 = 8.3 ± 1.1 μM), while the remaining analogues had similar IC50 concentrations of ∼4–5 μM. Interestingly, the JC-1 IC50 concentrations observed for the carbamates were very similar to those determined for the corresponding aryl-ureas U1–U6 recorded under identical experimental conditions (see Table 1).11 For example, the IC50 for the 4-Cl, 3-CF3-substituted aryl-urea U1 was 4.5 ± 1.1 μM11 while that for its carbamate counterpart C1 was 4.2 ± 0.1 μM.



The enzymatic activity of ATP-synthase is dependent upon the proton gradient across the MIM; thus depolarisation of the MIM should inhibit cellular ATP production. We therefore assessed the ability of C1–C6 to inhibit ATP synthesis in MDA-MB-231 cells. It was anticipated that ATP production would be impaired at concentrations above the JC-1 IC50, where significant proton transport and collapse of the proton gradient across the MIM occurs. As expected, carbamates C1–C6 inhibited ATP production at concentrations above their JC-1 IC50 values, (10 and 20 μM, Fig. 2a). Consistent with the JC-1 data, C4 was the least active of the carbamates and decreased ATP formation to 46.7 ± 6.5% of control values at 20 μM. To confirm that the reductions in intracellular ATP mediated by C1–C6 were not due to cell death we measured LDH release, which is a cytosolic enzyme that is released by cells during death. C1–C6 did not significantly increase LDH release relative to control (Fig. S2†), which suggests that the observed decreases in ATP were due to mitochondrial uncoupling and not cell death.
 | ||
| Fig. 2 Effects of carbamates C1–C6 on mitochondrial function in MDA-MB-231 breast cancer cells. (a) ATP formation by MDA-MB-231 cells after 48 h treatments with test compounds. Data represents mean ± SEM from three independent experiments. Difference from DMSO treated control: (‡) P < 0.001, (†) P < 0.01, (*) P < 0.05. (b) Oxygen consumption rates (OCR) in MDA-MB-231 cells after sequential addition of the ATP-synthase inhibitor oligomycin (1 μM), protonophore FCCP (1 μM) or test compound (C3, 20 μM), and then the ETC complex inhibitors rotenone/antimycin A (1 μM). | ||
The mitochondrial effects observed in JC-1 and ATP assays suggested that carbamates C1–C6 possess uncoupling activity similar to that of the aryl-ureas. To confirm this the uncoupling activity of carbamate C3 was assessed using the Seahorse Mito Stress test, which directly measures the oxygen consumption rate (OCR) of cells. In the assay MDA-MB-231 cells were treated with the ATP-synthase inhibitor oligomycin, which results in a decrease in OCR. Addition of an uncoupler under these conditions causes an increase in OCR. As shown in Fig. 2b, addition of the established protonophore FCCP to oligomycin-treated cells lead to a rapid increase in OCR, consistent with its uncoupling activity. Under the same conditions C3 also increased OCR, albeit with a slower rate of onset than FCCP. The differences in kinetics may result from differential subcellular localisation, with the more lipophilic C3 (clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P = 7.85, Table S3†) initially distributing to the plasma membrane before the mitochondria (FCCP clogP = 3.7112). Regardless, the observed increase in OCR confirms the uncoupling activity of C3.
P = 7.85, Table S3†) initially distributing to the plasma membrane before the mitochondria (FCCP clogP = 3.7112). Regardless, the observed increase in OCR confirms the uncoupling activity of C3.
Anion binding, self-association and proton transport studies
We next undertook mechanistic studies to determine if the carbamates elicited their protonophoric and uncoupling effects by the same mechanism as the aryl-ureas. Analogous aryl-ureas possess uncoupling activity because they self-associate into MIM-permeable dimers following deprotonation in the matrix that enable their return to the intermembrane space and allow for continued protonophoric cycling (Fig. 1). The aryl-urea group functions as an anion transporter by binding to the carboxylate group, which masks the negative charge and promotes movement of the carboxylate through the hydrophobic core of the lipid bilayer.To experimentally assess the anion affinity of the carbamate motif in comparison to the urea motif, we determined bromide (Br−) binding constants of a benzyl ester of C3 (C3-Bz) and a methyl ester of U3 (U3-Me) by 1H NMR titrations in CDCl3 (Fig. S3–S5†). The Br− affinity of C3-Bz (7.2 M−1) is 580-fold weaker than that of U3-Me (4200 M−1), attributed to the lack of a second NH hydrogen bonding site in the carbamate motif. Attempts to determine the AcO− binding constants of the two esters were unsuccessful due to AcO− induced ester hydrolysis.
To assess the capacity of the aryl-carbamates to self-associate, concentration-dependent 1H NMR experiments were performed for deprotonated form of carbamate C3 in CDCl3, using 1 equivalent of tetrabutylammonium hydroxide (TBAOH) for deprotonation of the carboxylic acid group. C3-TBAOH was found to self-associate in CDCl3 as evident by the pronounced upfield shifts of two aromatic CH peaks from 7.69 and 7.55 ppm to 7.87 and 7.84 ppm, respectively, by increasing the concentration of C3-TBAOH from 10 μM to 5 mM (Fig. S5†). The translational diffusion coefficient of C3-TBAOH (5 mM) was found to be (1.22 ± 0.01) × 10−9 m2 s−1, which is comparable to that of its urea analogue (U3-TBAOH, Table S1†). To further support aggregation of C3, we prepared a non-aggregating amide analogue of C3 in which the N–H binding unit is isolated from the aromatic ring (A3, Fig. S7†). No aggregation of A3 was observed in concentration-dependant studies (Fig. S7†) and the translational diffusion coefficient of A3-TBAOH (5 mM) was (1.51 ± 0.03) × 10−9 m2 s−1 (Table S1†), which is significantly faster than that of C3-TBAOH and U3-TBAOH and suggests that the dimer was likely the dominant aggregated species of these compounds. The slightly faster diffusion of C3-TBAOH compared with U3-TBAOH suggest that the aggregation of C3-TBAOH is less complete than U3-TBAOH, consistent with the weaker anion binding affinity of the carbamate motif.
Dimer formation was also evident in molecular dynamics (MD) simulations of C3 in a DOPC bilayer (see ESI† for methodology). Both the carboxylic acid (C3) and carboxylate (C3-D) protomers were examined, as well as systems containing a mix of protomers. All simulated compounds readily embedded themselves in the upper leaflet of the DOPC bilayer. Carboxylic acid C3 was observed to readily flip between leaflets during the simulation, while carboxylate C3-D was confined to the upper leaflet by its anionic tail. Formation of same protomer and cross protomer (Fig. 3) dimeric species was readily observed in the DOPC membrane environment, indicating the possible formation of head to tail dimeric species in the MIM. Although these dimeric species were not seen to transit between leaflets on the simulation timescale, it should be noted that in our MD simulations the bilayer lacked the charge gradient that drives anion permeation across the MIM in polarised mitochondria.
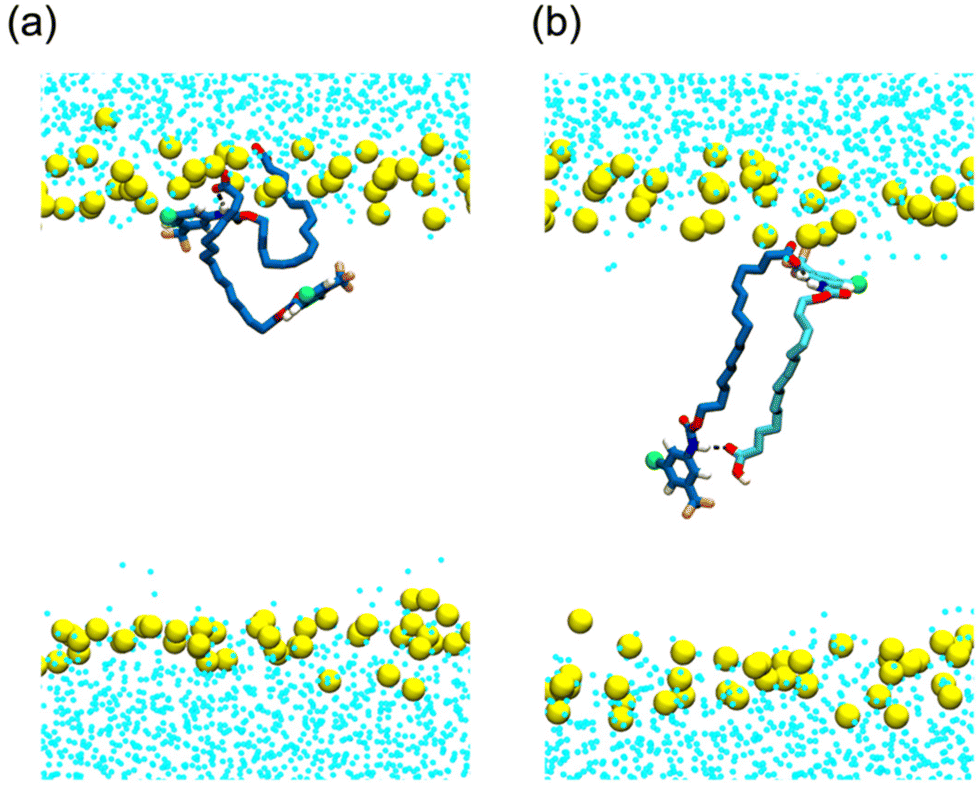 | ||
| Fig. 3 Snapshots from molecular dynamics simulations of C3, and the carboxylate equivalent C3-D in a DOPC bilayer. Formation of dimeric assemblies is evident in systems containing (a) C3-D, shown in blue liquorice representation, and (b) C3 and C3-D, shown in blue and cyan liquorice representation respectively. Water is shown as blue dots and the lipid headgroup phosphates are shown as yellow spheres. | ||
To further understand the capacity of the carbamates to carry out the carboxylate binding step, double hybrid DFT calculations were employed. Calculations at the DSD-PBEP86/aug-cc-pVTZ//M06-2X-D3(0)/6-311+G(d) level of theory25,26 were used to examine the binding energetics of compounds C1–C6 complexed with a model carboxylate (see ESI† for methodology). In addition to C1–C6 the corresponding aryl-urea analogues (U1–U6, Table S3†) were also examined for comparative purposes. Gas phase binding enthalpies revealed that tail-truncated aryl-urea analogues (U1–U6) bound a model carboxylate (propanoate) with greater affinity (−161.70 kJ mol−1 average, Table S2†) compared to carbamates C1–C6 (−107.92 kJ mol−1 average, Table S2†). Trends in gas phase affinities were reflected in implicit solvent with the aryl-urea > aryl-carbamate ordering preserved in both water and a non-polar solvent n-pentadecane (Table S2†). Unfavourable binding enthalpies in water (8.79, and 9.97 kJ mol−1 average for U and C series respectively) indicate facile dissociation of the carboxylate and anion binding motif in water. Additionally, favourable binding enthalpies in n-pentadecane (−95.74 and −56.70 kJ mol−1 average for U and C series, respectively) suggest strong association of the carboxylate and anion binding motif in a low dielectric medium, such as the interior of the MIM. Trends in binding enthalpies can be rationalised through examination of carboxylate coordination geometries across the aryl-urea and aryl-carbamate series exemplified by C3 and the corresponding aryl-urea (U3) in Fig. 4. As expected,5,27U3 donates two parallel N–H hydrogen bonds to the carboxylate moiety resulting in a bidentate coordination mode (Fig. 4a). C3 also donates two hydrogen bonds to the carboxylate moiety, one from the carbamate N–H and the second from a polarised C–H fragment of the adjacent phenyl group (Fig. 4b). Similar C–H hydrogen bonds have been observed in complexes of squaramide-based anion receptors with chloride.28 Aryl C–H hydrogen bonds are generally weaker than N–H hydrogen bonds, accounting for the lower propanoate binding enthalpies of aryl-carbamates compared to aryl-ureas.29
 | ||
| Fig. 4 Geometries of 3-chloro-5-triflouromethyl-substituted urea (U3), panel (a) and carbamate (C3, panel b) analogues complexed to propanoate. The HOMO of the resulting anionic complex is shown in panels (c) and (d) respectively. Geometries were optimized at the M06-2X-D3(0)/6-311+G(d) level of theory. Molecular orbital plots were constructed from a DSD-PBEP86/aug-cc-pVTZ single-point and contoured at the 0.003 isovalue. | ||
Finally, to confirm that the carbamates in their anionic form permeate bilayers as dimeric species we measured the concentration dependant protonophoric activity of C1–C6 using a tethered bilayer lipid membranes (tBLMs)-based assay we developed to measure proton transport in a cell free system.11 This system is comprised of a model lipid bilayer between gold electrodes, and proton transport across the membrane is detected as an increase in membrane conductance, which is measured by electrical impedance spectroscopy. A directly proportional relationship between bilayer conductance and protonophore concentration indicates that the membrane permeant species is the monomeric anion, while bilayer conductance increases with the square of the protonophore concentration when an anionic dimer is the permeant species.30 To perform these studies we utilised tethered bilayer lipid membranes (tBLMs) formed from 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), a major lipid component of the MIM.31 As shown in Fig. S7a,†C1–C6 produced an increase in bilayer conductance that was proportional to the square of carbamate concentration, which indicates that the carbamates in their anionic form permeate the membrane as dimers. The magnitude of the conductance increases produced by C1–C6 were similar (Fig. S7b†), which is consistent with the JC-1 data, where each carbamate dissipated the proton gradient across the MIM with IC50 concentrations between 3.9–8.3 μM (see Table 1).
Taken together, these data provide evidence that carbamates C1–C6 uncouple mitochondria and transport protons across lipid bilayers via the dimerisation mechanism first identified for the aryl-ureas. Since mitochondrial uncoupling activity in these scaffolds is directly linked to the carboxylate transport capacity of the synthetic anion transport group, these data show, for the first time, that the carbamate functional group can act as synthetic anion transport motif. While urea based scaffolds can donate two parallel hydrogen bonds to the transported carboxylate moiety, the carbamates examined herein only provide a single traditional hydrogen bond to the transported carboxylate group along with an aryl C–H hydrogen bond. As a result, the carbamates bind carboxylate with weaker overall affinity as demonstrated via anion binding studies and DFT calculations. Carbamate based receptors are expected to possess greater hydrogen bond donor capacity, and therefore anion affinity, than their amide counterparts due to increased Brønstead acidity of the N–H moiety. However, several studies have revealed a delicate interplay of conformational effects in the anion binding capacity of urea and amide containing anion binding motifs,18,19,32,33 and is of particular importance as the carbamates studied in this work lack a large scaffold or multiple coordinating groups that can pre-organise the anion binding site.
One surprising observation from the JC-1 assays is that carbamates C1–C6 depolarise mitochondria with equivalent potencies to their urea counterparts. Although cell-based data is influenced by cellular processes (metabolism etc.), the result is unexpected because anion affinity has been identified as an important determinant of anion transport capacity,34 and the experimental and computational data showed that the ureas bind to carboxylate with significantly higher affinity. Very high anion affinity can adversely impact anion transport by hindering anion release, therefore it is possible slower anion release by U1–U6 results in equivalent anion transport activity to C1–C6. However, this is unlikely as hindered anion release has only been observed to affect anion transporters that bind through four or more hydrogen bonds.35 In addition to anion affinity, lipophilicity35–38 (expressed as logP) and molecular size also affect anion transporter activity.35 Molecular size is unlikely to explain the relative activities of the carbamates and ureas as both scaffolds have similar size. Carbamates C1–C6 are more lipophilic than their urea counterparts U1–U6 (see calculated logP values in Table SC in ESI†), and although the difference is small (∼0.7) it may contribute to the surprising activity of C1–C6 relative to U1–U6.
Another possible explanation for the equal activity of the ureas and carbamates is the capacity of both functional groups to delocalise the charged anion and produce complexes with increased lipophilicity and membrane permeability. Although the effect of charge delocalisation on anion transport has not been widely studied, molecular orbital analysis did suggest dispersal of the carboxylate charge across the extended π-system of the aryl-ureas promoted uncoupling activity.11 Examination of the molecular orbitals of carbamate C3 and urea U3 complexed to a model carboxylate moiety (propanoate) revealed that U3-propanoate and C3-propanoate HOMOs show delocalisation of electron density across the aryl π-system (Fig. 4c and d). The efficient delocalisation found in C3-propanoate is noteworthy given only one traditional hydrogen bond is present. Efficient charge transfer from the carboxylate moiety in the carbamate series is further demonstrated in Natural Population Analysis (NPA)39 calculations that indicate comparable delocalisation between the carbamide and urea series (Table S4,† average difference of 0.012 natural charge units). Thus, it is possible that charge delocalisation contributes to the similar levels of activity for the carbamate and urea series.
Conclusions
In this paper we report on the synthesis and mechanistic evaluation of a series of protonophoric mitochondrial uncouplers consisting of a long chain fatty acid linked with an aryl-carbamate anionophore. Uncoupling activity was established through cell-based assays, and mechanistic studies revealed that like their urea counterparts, the carbamate analogues self-assemble into membrane permeable anionic dimers that allow for protonophoric activity. Despite weaker carboxylate binding enthalpies, carbamates C1–C6 had similar uncoupling activity to their urea counterparts in cell-based assays, which may result from higher carbamate lipophilicity or similar capacity to delocalise charge of its anion guest. This study provides the first experimental evidence to establish the carbamate function group as a synthetic anion transport motif.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This study was supported by grants from the Australian National Health and Medical Research Council (1031686 and 1087248). P. A. G. thanks the Australian Research Council (DP200100453) for funding. X. W. thanks the Australian Research Council for a DECRA fellowship (DE220101000). X. W, M. M. and P. A. G. acknowledge and pay respect to the Gadigal people of the Eora Nation, the traditional owners of the land on which we research, teach and collaborate at the University of Sydney. This research/project was undertaken with the assistance of resources and services from the National Computational Infrastructure (NCI), which is supported by the Australian Government. We acknowledge the NMR Facility of Mark Wainwright Analytical Centre at the University of New South Wales, and the Centre for Advanced Imaging at the University of Queensland for NMR support.References
- A. Ebert, C. Hannesschlaeger, K.-U. Goss and P. Pohl, Biophys. J., 2018, 115, 1931–1941 CrossRef CAS.
- A. P. Davis, D. N. Sheppard and B. D. Smith, Chem. Soc. Rev., 2007, 36, 348–357 RSC.
- B. A. McNally, W. M. Leevy and B. D. Smith, Supramol. Chem., 2007, 19, 29–37 CrossRef CAS PubMed.
- L. Chen, S. N. Berry, X. Wu, E. N. W. Howe and P. A. Gale, Chem, 2020, 6, 61–141 CAS.
- V. B. Bregovic, N. Basaric and K. Mlinaric-Majerski, Coord. Chem. Rev., 2015, 295, 80–124 CrossRef.
- N. Busschaert, I. L. Kirby, S. Young, S. J. Coles, P. N. Horton, M. E. Light and P. A. Gale, Angew. Chem., Int. Ed., 2012, 51, 4426–4430 CrossRef CAS.
- K. M. Bak, K. Chabuda, H. Montes, R. Quesada and M. J. Chmielewski, Org. Biomol. Chem., 2018, 16, 5188–5196 RSC.
- J. Shang, W. Si, W. Zhao, Y. Che, J.-L. Hou and H. Jiang, Org. Lett., 2014, 16, 4008–4011 CrossRef CAS.
- J. T. Davis, P. A. Gale and R. Quesada, Chem. Soc. Rev., 2020, 49, 6056–6086 RSC.
- J. T. Davis, O. Okunola and R. Quesada, Chem. Soc. Rev., 2010, 39, 3843–3862 RSC.
- T. Rawling, H. MacDermott-Opeskin, A. Roseblade, C. Pazderka, C. Clarke, K. Bourget, X. Wu, W. Lewis, B. Noble, P. A. Gale, M. L. O'Mara, C. Cranfield and M. Murray, Chem. Sci., 2020, 11, 12677–12685 RSC.
- E. S. Childress, S. J. Alexopoulos, K. L. Hoehn and W. L. Santos, J. Med. Chem., 2018, 61, 4641–4655 CrossRef CAS PubMed.
- X. Wu and P. A. Gale, J. Am. Chem. Soc., 2016, 138, 16508–16514 CrossRef CAS.
- E. N. W. Howe and P. A. Gale, J. Am. Chem. Soc., 2019, 141, 10654–10660 CrossRef CAS.
- U. Manna and G. Das, Coord. Chem. Rev., 2021, 427, 213547 CrossRef CAS.
- S. K. Dey, A. Basu, R. Chutia and G. Das, RSC Adv., 2016, 6, 26568–26589 RSC.
- A. Hennig, L. Fischer, G. Guichard and S. Matile, J. Am. Chem. Soc., 2009, 131, 16889–16895 CrossRef CAS.
- E. B. Park and K.-S. Jeong, Chem. Commun., 2015, 51, 9197–9200 RSC.
- P. V. Santacroce, J. T. Davis, M. E. Light, P. A. Gale, J. C. Iglesias-Sanchez, P. Prados and R. Quesada, J. Am. Chem. Soc., 2007, 129, 1886–1887 CrossRef CAS.
- L. Yuan, J. Shen, R. Ye, F. Chen and H. Zeng, Chem. Commun., 2019, 55, 4797–4800 RSC.
- O. A. Okunola, J. L. Seganish, K. J. Salimian, P. Y. Zavalij and J. T. Davis, Tetrahedron, 2007, 63, 10743–10750 CrossRef CAS.
- Y. Wang, E. Duran, D. Nacionales, A. Valencia, C. Wostenberg and E. R. Marinez, Tetrahedron Lett., 2008, 49, 6410–6412 CrossRef CAS.
- Y. Xia, B. Wu, S. Li, Z. Yang, Y. Liu and X.-J. Yang, Supramol. Chem., 2010, 22, 318–324 CrossRef CAS.
- T. Rawling, A. M. McDonagh, B. Tattam and M. Murray, Tetrahedron, 2012, 68, 6065–6070 CrossRef CAS.
- S. Kozuch and J. M. L. Martin, Phys. Chem. Chem. Phys., 2011, 13, 20104–20107 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889–3915 RSC.
- V. Amendola, G. Bergamaschi, M. Boiocchi, L. Fabbrizzi and M. Milani, Chem. – Eur. J., 2010, 16, 4368–4380 CrossRef CAS.
- L. M. Eytel, H. A. Fargher, M. M. Haley and D. W. Johnson, Chem. Commun., 2019, 55, 5195–5206 RSC.
- S. McLaughlin, J. Membr. Biol., 1972, 9, 361–372 CrossRef CAS.
- S. E. Horvath and G. Daum, Prog. Lipid Res., 2013, 52, 590–614 CrossRef CAS.
- S. J. Brooks, S. E. García-Garrido, M. E. Light, P. A. Cole and P. A. Gale, Chem. – Eur. J., 2007, 13, 3320–3329 CrossRef CAS.
- I. Sandler, F. A. Larik, N. Mallo, J. E. Beves and J. Ho, J. Org. Chem., 2020, 85, 8074–8084 CrossRef CAS PubMed.
- N. Busschaert, S. J. Bradberry, M. Wenzel, C. J. E. Haynes, J. R. Hiscock, I. L. Kirby, L. E. Karagiannidis, S. J. Moore, N. J. Wells, J. Herniman, G. J. Langley, P. N. Horton, M. E. Light, I. Marques, P. J. Costa, V. Felix, J. G. Frey and P. A. Gale, Chem. Sci., 2013, 4, 3036–3045 RSC.
- I. Marques, P. M. R. Costa, M. Q. Miranda, N. Busschaert, E. N. W. Howe, H. J. Clarke, C. J. E. Haynes, I. L. Kirby, A. M. Rodilla, R. Perez-Tomas, P. A. Gale and V. Felix, Phys. Chem. Chem. Phys., 2018, 20, 20796–20811 RSC.
- V. Saggiomo, S. Otto, I. Marques, V. Felix, T. Torroba and R. Quesada, Chem. Commun., 2012, 48, 5274–5276 RSC.
- M. J. Spooner and P. A. Gale, Chem. Commun., 2015, 51, 4883–4886 RSC.
- N. J. Knight, E. Hernando, C. J. E. Haynes, N. Busschaert, H. J. Clarke, K. Takimoto, M. Garcia-Valverde, J. G. Frey, R. Quesada and P. A. Gale, Chem. Sci., 2016, 7, 1600–1608 RSC.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob02049a |
| ‡ These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2023 |