
Cytotoxic alkaloids from the twigs and leaves of Cephalotaxus sinensis†
Fuxin
Zhang
,
Tao
Yang
,
Kailing
Yang
,
Ruixi
Zhou
,
Yu
Zhang
,
Wenwen
Chen
,
Zhetong
Liu
,
Guanqun
Zhan
 * and
Zengjun
Guo
*
* and
Zengjun
Guo
*
School of Pharmacy, Health Science Center, Xi'an Jiaotong University, Xi'an 710061, P. R. China. E-mail: zhanguanqun@mail.xjtu.edu.cn; guozj@mail.xjtu.edu.cn
First published on 28th November 2022
Abstract
Twelve new Cephalotaxus alkaloids (1–12) and nine known analogues (13–21) were isolated and identified from the twigs and leaves of Cephalotaxus sinensis. The structures of the new compounds (1–12) were elucidated by extensive spectroscopic analysis and single-crystal X-ray diffraction analysis. Cephalosine H (8) is the third example of an alkaloid containing the cephalolancine skeleton. Cephalosines J and K (10 and 11) are the rare natural Δ(2)1-alkene-6-hydroxyl homoerythrina-type alkaloids isolated from the Cephalotaxus genus. The racemization of cephalotaxine-type alkaloids is discussed. Alkaloids 6, 7, 11, 16, 18 and 19 exhibited broad and potent cytotoxicities against five human cancer cell lines, with IC50 values ranging from 0.053 to 10.720 μM, highlighting these compounds as promising leads for the development of new antitumor agents.
Introduction
The Cephalotaxus genus, the sole representative of the Cephalotaxaceae family, is widespread in Southern and Eastern Asia and consists of about 12 species and varieties.1 Among them, only six species and four varieties are native to China scattered in Sichuan, Shaanxi, Yunnan, Guangxi, Hainan, and Taiwan provinces.2,3 Over the past decades, scientists have obtained diverse and attractive secondary metabolites from the genus Cephalotaxus, including alkaloids, terpenoids, flavonoids, and phenylpropanoids.4,5 Notably, alkaloids, especially cephalotaxine-type alkaloids, have attracted considerable attention from medicinal chemists and pharmacologists owing to their potent antitumor activity and their unique pentacyclic ring structure with a side chain bearing an oxygen-containing chiral carbon center.6 Harringtonine (HT) and homoharringtonine (HHT), two representative cephalotaxine-type alkaloids with remarkable antileukemia activity, have been clinically used to treat chronic myeloid and acute non-lymphocytic leukemia in China since 1990.3,7Cephalotaxus sinensis (Rehder & E.H.Wilson) H.L.Li, a major species found in China and rich in resources, is used as a traditional folk medicine in China to treat inflammation, dyspepsia, ascariasis, and cough.8,9 Previous phytochemistry investigations of C. sinensis, concerning its characteristic Cephalotaxus alkaloids, are limited,10,11 reminding us that there may be more new alkaloids with significant antitumor activity waiting to be discovered.
In the present study, the twigs and leaves of C. sinensis collected from Qinling Mountains have been phytochemically investigated, leading to the isolation of twenty-one compounds (Fig. 1), including seven new cephalotaxine-type alkaloids (1–7) containing two pairs of enantiomers (1 and 2), one new cephalolancine-type alkaloid (8), four new homoerythrina-type alkaloids (9–12), and nine known analogues (13–21). Herein, the isolation, structure determination, and cytotoxicity and SARS-CoV-2 3CLpro inhibitory activities of compounds 1–21 are described.
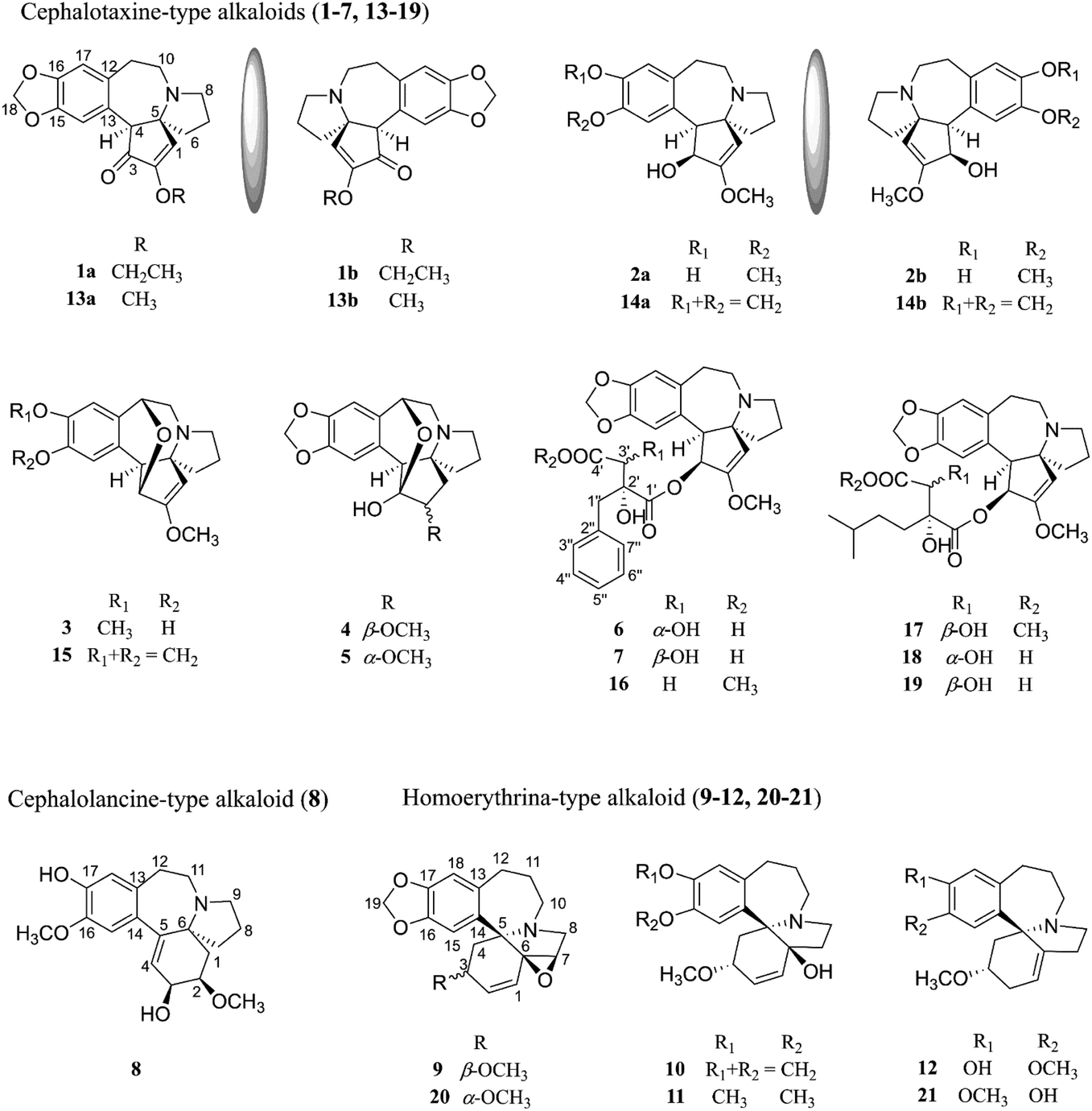 | ||
| Fig. 1 Chemical structures of the isolated compounds (1–21) from C. sinensis. | ||
Results and discussion
Structural elucidation
Cephalosine A (1), a colorless crystal, has the molecular formula of C19H21NO4 based on the [M + H]+ HRESIMS ion at m/z 328.1551 (calcd for C19H22NO4, 328.1549) and 13C NMR data. The NMR data of 1 (Tables 1 and 2) were similar to those of the co-isolated compound cephalotaxinone (17),12 except for the appearance of an oxyethyl group (δH 4.02, 2H, dq, J = 7.0, 2.8 Hz; 1.39, 3H, t, J = 7.0 Hz; δC 67.1; 14.7) in 1, replacing the oxymethyl group in 17. The HMBC correlations from H-19 (δH 4.02) to C-2 (δC 158.7) suggested that the oxyethyl group was attached to C-2. To further determine the stereochemistry of the chiral centers of alkaloid 1, a Ga Kα single-crystal X-ray diffraction analysis was performed. However, the results suggested the existence of a pair of enantiomers of 1. Subsequently, the enantiomers 1a and 1b (about 1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were further purified using a chiral column of Daicel Chiralpak IC (Fig. 2a) and confirmed using the opposite optical rotation and ECD data (Fig. 3a). Fortunately, the crystal of 1b was finally obtained and the absolute configuration of 1b was further established as 4R,5R by Ga Kα single-crystal X-ray diffraction analysis (Fig. 4). Thus, the absolute configurations of 1a and 1b were defined as 4S,5S and 4R,5R, respectively.
:1) were further purified using a chiral column of Daicel Chiralpak IC (Fig. 2a) and confirmed using the opposite optical rotation and ECD data (Fig. 3a). Fortunately, the crystal of 1b was finally obtained and the absolute configuration of 1b was further established as 4R,5R by Ga Kα single-crystal X-ray diffraction analysis (Fig. 4). Thus, the absolute configurations of 1a and 1b were defined as 4S,5S and 4R,5R, respectively.
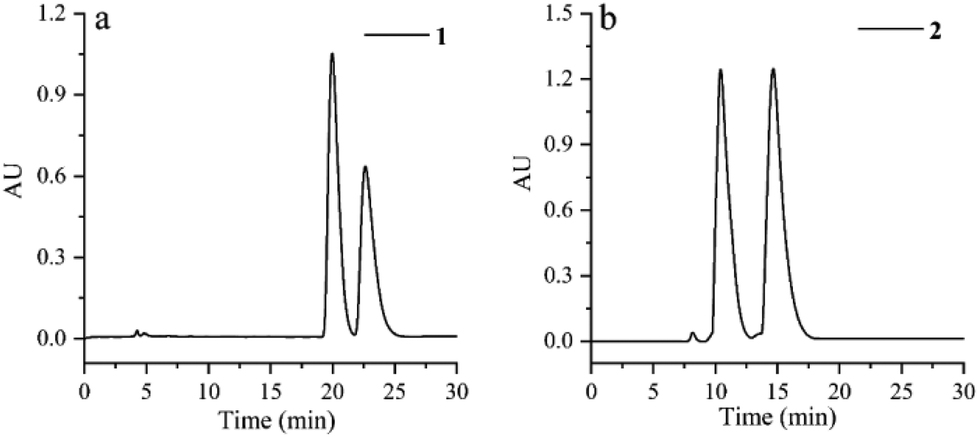 | ||
| Fig. 2 HPLC analysis for racemization of compounds 1 and 2 using a Daicel Chiralpak IC column. (a) MeOH/H2O, 80:20, 0.8 mL min−1, tR 20.0 min for 1a and tR 22.6 min for 1b; (b) MeOH/H2O/NH3·H2O, 78:22:0.03, 0.8 mL min−1, tR 10.4 min for 2b and tR 14.6 min for 2a. | ||
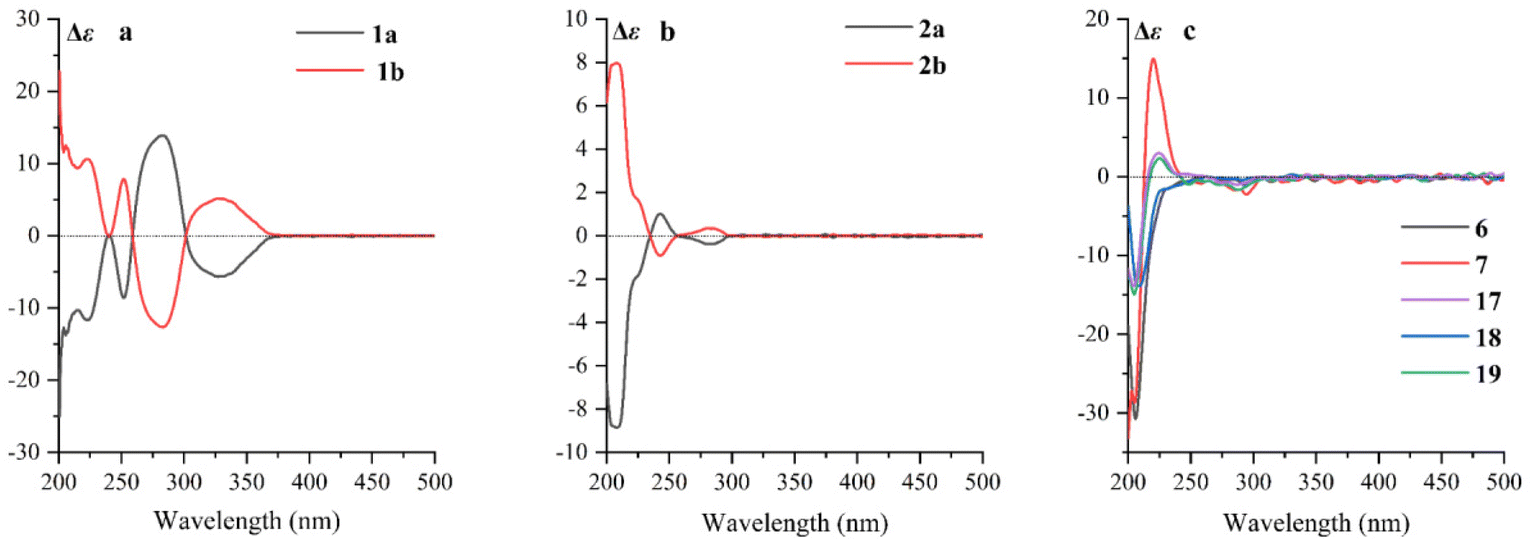 | ||
| Fig. 3 (a) ECD spectra of 1a and 1b; (b) ECD spectra of 2a and 2b; (c) ECD spectra of 6, 7, and 17–19. | ||
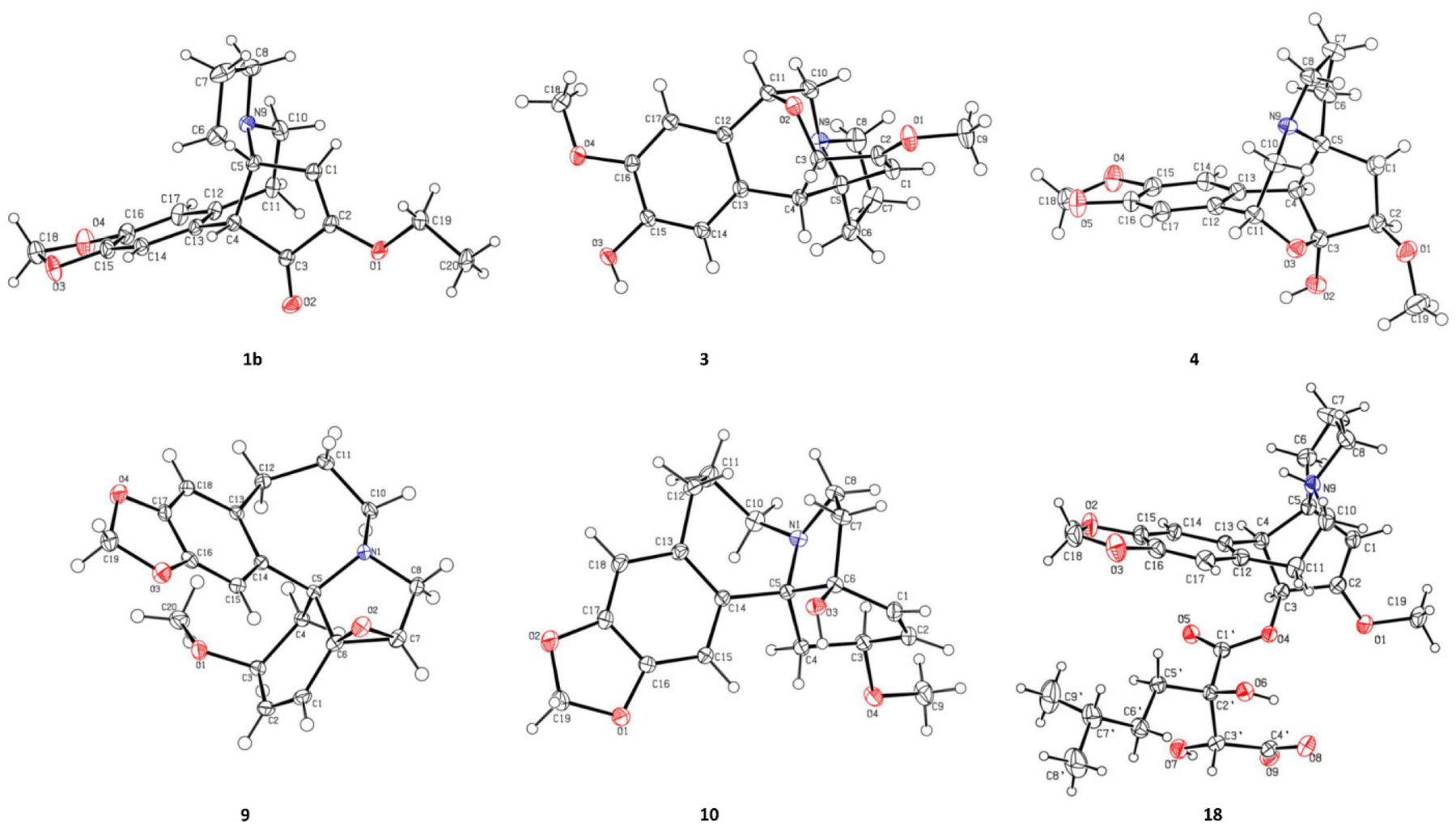 | ||
| Fig. 4 ORTEP drawing of compounds 1b, 3, 4, 9, 10, and 18. | ||
| No. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1 | 6.68, s | 5.01, s | 4.70, br s | 1.91, dd (14.0, 9.0) | 2.26, dd (15.4, 6.2) |
| 1.78, dd (14.0, 9.0) | 1.53, br d (15.4) | ||||
| 2 | 3.86, t (9.0) | 3.54, d (6.2) | |||
| 3 | 4.70, d (9.2) | 3.99, dd (5.3, 0.5) | |||
| 4 | 3.67, s | 3.64, d (9.2) | 3.2 d (5.3) | 3.12 s | 3.34, overlap |
| 6 | 2.05, ddd (12.5, 12.5, 8.8) | 1.98, ddd (12.2, 10.8, 9.0) | 1.79, 2H, overlap | 1.73, m | 1.73, 2H, m |
| 1.99, ddd (12.5, 8.8, 3.2) | 1.86, ddd (12.2, 9.0, 3.0) | 1.62, m | |||
| 7 | 1.94, m | 1.79, m | 1.79, overlap | 1.64, 2H, m | 1.68, 2H, m |
| 1.79, m | 1.70, m | 1.71, m | |||
| 8 | 2.95, ddd (9.5, 9.5, 3.4) | 2.90, ddd (9.5, 9.5, 3.5) | 2.53, ddd (9.2, 8.5, 2.8) | 2.65, ddd (9.0, 8.5, 3.4) | 2.67, dd (9.0, 8.5, 3.5) |
| 2.75, ddd (9.5, 9.5, 7.5) | 2.62, overlap | 2.44, q (9.2) | 2.32, dq (9.0, 2.2) | 2.33, dq (9.0, 2.2) | |
| 10 | 2.85, ddd (11.8, 11.8, 7.8) | 2.84, ddd (11.5, 11.5, 7.8) | 2.78, 2H, overlap | 2.80, dd (12.5, 4.0) | 2.77, dd (12.6, 4.0) |
| 2.55, dd (11.8, 8.0) | 2.62, overlap | 2.73, d (12.5) | 2.64, d (12.6) | ||
| 11 | 2.47, dd (15.0, 7.8) | 3.36, ddd (14.5, 11.5, 7.8) | 4.76, dd (4.0, 2.0) | 4.92, br d (4.0) | 4.86, overlap |
| 2.37, ddd (15.0, 11.8, 8.0) | 2.28, dd (14.5, 7.0) | ||||
| 14 | 6.75, s | 6.73, s | 6.71, s | 6.75, s | 6.75, s |
| 17 | 6.67, s | 6.61, s | 6.80, s | 6.78, s | 6.76, s |
| 18 | 5.91, 2H, brs | 5.94, d (1.1) | 5.93, d (1.0) | ||
| 5.92, d (1.1) | 5.92, d (1.0) | ||||
| 19 | 4.02, 2H, dq (7.0, 2.8) | ||||
| 20 | 1.39, 3H, t (7.0) | ||||
| 2-OCH3 | 3.70, 3H, s | 3.71, 3H, s | 3.50, 3H, s | 3.42, 3H, s | |
| 15-OCH3 | 3.83, 3H, s | ||||
| 16-OCH3 | 3.85, 3H, s |
| No. | 1 | 2 | 3 | 4 | 5 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 126.8 | 98.0 | 97.6 | 31.7 | 32.4 | 39.4 | 130.2 | 135.1 | 135.1 | 117.8 |
| 2 | 158.7 | 163.3 | 167.3 | 85.9 | 86.4 | 79.2 | 132.2 | 130.8 | 130.7 | 33.4 |
| 3 | 203.8 | 74.7 | 78.3 | 104.8 | 106.9 | 66.5 | 74.4 | 76.5 | 76.6 | 76.0 |
| 4 | 60.9 | 58.8 | 50.0 | 55.8 | 54.6 | 130.9 | 27.8 | 37.4 | 37.2 | 39.2 |
| 5 | 67.0 | 72.5 | 75.5 | 66.4 | 67.9 | 147.8 | 66.1 | 71.5 | 71.2 | 70.5 |
| 6 | 39.5 | 44.0 | 34.5 | 39.1 | 39.6 | 69.5 | 70.0 | 80.2 | 80.2 | 144.3 |
| 7 | 20.8 | 20.9 | 22.4 | 20.9 | 20.9 | 39.3 | 61.2 | 38.4 | 38.4 | 28.6 |
| 8 | 53.8 | 54.5 | 50.3 | 50.6 | 50.9 | 25.5 | 55.8 | 48.2 | 48.2 | 47.5 |
| 9 | 50.1 | |||||||||
| 10 | 48.8 | 49.8 | 53.5 | 55.2 | 55.5 | 50.4 | 43.3 | 43.5 | 51.2 | |
| 11 | 32.0 | 32.0 | 78.9 | 77.0 | 77.0 | 46.5 | 30.0 | 29.1 | 29.1 | 24.5 |
| 12 | 132.0 | 134.7 | 130.5 | 132.1 | 131.9 | 32.5 | 36.8 | 34.1 | 33.8 | 37.8 |
| 13 | 130.4 | 128.6 | 128.2 | 127.5 | 128.3 | 130.1 | 136.5 | 135.3 | 134.9 | 136.3 |
| 14 | 113.7 | 118.1 | 116.9 | 110.8 | 110.8 | 132.0 | 135.5 | 139.4 | 138.5 | 132.1 |
| 15 | 148.7 | 147.1 | 148.6 | 148.4 | 148.2 | 115.2 | 112.1 | 112.4 | 117.2 | 117.1 |
| 16 | 147.8 | 146.6 | 148.5 | 148.7 | 148.7 | 147.6 | 146.4 | 146.8 | 147.6 | 146.3 |
| 17 | 111.2 | 118.2 | 109.9 | 106.9 | 106.9 | 147.8 | 147.2 | 147.2 | 148.6 | 146.9 |
| 18 | 102.4 | 102.3 | 102.3 | 117.1 | 112.4 | 111.6 | 115.9 | 120.2 | ||
| 19 | 67.1 | 102.2 | 102.2 | |||||||
| 20 | 14.7 | |||||||||
| 2-OCH3 | 57.6 | 57.8 | 58.8 | 58.1 | 57.3 | |||||
| 3-OCH3 | 57.0 | 56.6 | 56.7 | 56.3 | ||||||
| 15-OCH3 | 56.9 | |||||||||
| 16-OCH3 | 56.8 | 56.5 | 56.8 | 56.8 | ||||||
| 17-OCH3 | 56.5 |
Cephalosine B (2) has a molecular formula of C18H23NO4, as determined using the [M + H]+ ion at m/z 318.1704 (calcd for C18H24NO4, 318.1705) in the HRESIMS and 13C NMR data. Analysis of its NMR data (Tables 1 and 2) revealed that compound 2 corresponded well to 14,13 except for the presence of resonances for an oxymethyl group (δH 3.83, 3H, s; δC 56.9) in 2 and the absence of resonances for a methylenedioxy moiety in 14 (δH 5.86, 1H, d, J = 1.2 Hz; δH 5.85, 1H, d, J = 1.2 Hz; δC 102.1). The methoxy group was located at C-15 as supported by the HMBC correlation from 15-OCH3 (δH 3.83) to C-15 (δC 147.1) along with the NOESY correlation from 15-OCH3 (δH 3.83) to H-14 (δH 6.73). The relative configurations of 2 were established by analysis of the proton–proton coupling constants and the NOESY spectrum. The correlations of H-1 (δH 5.01) with H-6β (δH 1.86) and of H-4 (δH 3.64) with H-6α (δH 1.98)/H-3 (δH 4.70)/H-14 (δH 6.73) combined with the same coupling constant between H-3 and H-4 (J3,4 = 9.2 Hz) with those of co-isolated cephalotaxine-type alkaloids (16–19) suggested that the cephalotaxine core in 2 has the same relative configurations as those of 16–19.3,6 However, the optical rotation data of 2 were much smaller than those of the analogues (16–19) suggesting that 2 may be a pair of enantiomers (2a and 2b), and was purified and confirmed in the same way as 1 (Fig. 2b). Considering the similar ECD spectra of 2a and cephalofortine C, and the opposite ECD spectra of 2b and cephalofortine C,14 the absolute configurations of 2a and 2b were defined to be 3S,4S,5R and 3R,4R,5S, respectively.
Compound 3, a colorless crystal, has a molecular formula of C18H21NO4 as established using its HRESIMS (m/z 316.1551 [M + H]+, calcd 316.1549) and 13C NMR data. The NMR data of 3 (Tables 1 and 2) closely resembled those of 15,15 except for a methoxy group (δH 3.85, 3H, s; δC 56.8) in 3 instead of a methylenedioxy moiety (δH 5.92, 1H, s; δH 5.91, 1H, s; δC 102.3) in 15. These were verified from the HMBC correlation between the methoxy group (δH 3.85) and C-16 (148.5) along with the NOESY correlations observed from the methoxy group (δH 3.85) to H-17 (δH 6.80). Finally, the single crystals of 3 were obtained from a MeOH/H2O (20:1) solution. The absolute configuration of each stereogenic center in 3 was defined to be 3S,4S,5R,11R by Ga Kα single-crystal X-ray crystallographic data analysis with a Flack parameter of 0.11 (5) (Fig. 4).
The molecular formula of cephalosine D (4) was determined to be C18H21NO5 on the basis of HRESIMS (m/z 332.1504 [M + H]+, calcd 332.1498) and 13C NMR data. The 1D NMR data (Tables 1 and 2) of 4 closely resembled those of cephalotine B,16 with the main difference being the presence of a methoxy group (δH 3.50, 3H, s; δC 58.8) in 4 replacing the C-2 hydroxyl group in cephalotine B, which was confirmed by the HMBC correlation from the methoxy group to C-2 (δC 85.9). Thus, 4 was an etherified derivative of cephalotine B. Finally, the absolute configuration of 4 was defined as 2R,3S,4S,5S,11R based on single-crystal X-ray diffraction analysis (Fig. 4).
Compound 5 has the same molecular formula of C18H21NO5 as that of 4, determined using the same [M + H]+ ion at m/z 332.1486 (calcd for C18H22NO5, 332.1498) in the HRESIMS and 13C NMR data. The NMR data of 5 showed high similarities to those of 4 (Tables 1 and 2), with the only difference being the signals vicinal to C-2, indicating the β-orientation of H-2. The β-orientation of H-2 was determined based on the coupling constant between H-1 and H-2 in 5 (J1,2 = 6.2 Hz),16 differing from that of 4 (J1,2 = 9.0 Hz) with an α-orientation of H-2. The NOESY cross-peaks from 2-OCH3 (δH 3.42) to H-1α (δH 1.53) and H-6α (δH 1.73) further supported the elucidation. Thus, the structure of cephalosine E (5) was determined as shown.
Compounds 6 and 7 share the same molecular formula, C29H31NO9, as determined using the negative-ion peak at m/z [M − H]− 536.1932 and 536.1933, respectively (calcd for C29H30NO9, 536.1926). The 1H and 13C NMR data of compounds 6 and 7 (Table 3) were similar to those of the co-isolated compounds 18 and 19, respectively, with the differences being restricted to the substituents on the C-2′ position. The signals in 1H and 13C NMR spectra established the presence of a phenmethyl moiety in 6 (δH 7.21, m, 2H, H-3′′/H-7′′; 7.20, m, 2H, H-4′′/H-6′′; 7.19, m, 1H, H-5′′; 2.82, d, J = 14.1 Hz, 1H, H-1a′′; 2.73, d, J = 14.1 Hz, 1H, H-1b′′; δC 137.0, C-2′′; 131.7, C-3′′/C-7′′; 128.9, C-4′′/C-6′′; 127.7, C-5′′; 40.9, C-1′′) and 7 (δH 7.22, m, 2H, H-3′′/H-7′′; 7.21, m, 2H, H-4′′/H-6′′; 7.18, m, 1H, H-5′′; 3.07, d, J = 13.8 Hz, 1H, H-1a′′; 2.80, d, J = 13.8 Hz, 1H, H-1b′′; δC 136.9, C-2′′; 132.0, C-3′′/C-7′′, 128.9, C-4′′/C-6′′; 127.8, C-5′′; 42.4, C-1′′), which was further confirmed by the HMBC correlations from H-1′′ to C-2′′/C-3′′/C-7′′. The connectivity of the phenmethyl moiety at C-2′ in 6 and 7 was established on the basis of HMBC cross-peaks from H-1′′ to C-1′/C-2′/C-3′. Therefore, the relative configurations of 6 and 7 were deduced to be the same as those of 18 and 19, respectively. The absolute configurations of 6 and 7 were determined based on similar ECD curves as those of 18 and 19, respectively (Fig. 3c). The structures of 18 and 19 were almost identical except for the configuration at C-3′, while the ECD curve of 18 differed from 19 with only a minor difference at around 225 nm, a negative Cotton effect in 18 and a positive Cotton effect in 19, indicating that the Cotton effect at around 225 nm was a characteristic of the configuration at C-3′ in cephalotaxine esters. The ECD spectrum of 18, whose absolute configuration was determined to be 3S,4S,5R,2′R,3′R by Ga Kα single-crystal X-ray diffraction analysis (Fig. 4), was in good agreement with a previous report,3 further supporting the deduction. Therefore, the absolute configurations of 6 and 7 were determined to be the same as those of 18 (3S,4S,5R,2′R,3′R) and 19 (3S,4S,5R,2′R,3′S), respectively, based on their similar ECD data (Fig. 3c).
| No. | 6 | 7 | ||
|---|---|---|---|---|
| δ H | δ C | δ H | δ C | |
| 1 | 5.23, s | 101.2 | 5.23, s | 101.2 |
| 2 | 159.9 | 160.0 | ||
| 3 | 5.81, d (9.7) | 76.7 | 5.92, d (9.8) | 76.5 |
| 4 | 3.88, d (9.7) | 56.9 | 3.88, d (9.8) | 56.5 |
| 5 | 72.3 | 72.1 | ||
| 6 | 2.00, ddd (12.5, 10.5, 9.0) | 44.1 | 2.02, ddd (12.5, 10.5, 9.0) | 44.0 |
| 1.94, ddd (12.5, 9.0, 3.0) | 1.94, ddd (12.5, 9.0, 3.0) | |||
| 7 | 1.83, m | 21.0 | 1.83, m | 20.9 |
| 1.74, m | 1.74, m | |||
| 8 | 2.96, ddd (9.5, 9.5, 3.8) | 54.8 | 2.96, ddd (9.5, 9.5, 3.8) | 54.7 |
| 2.65, dd (9.5, 7.0) | 2.65, dd (9.5, 7.0) | |||
| 10 | 2.91, ddd (11.8, 11.8, 7.0) | 49.9 | 2.87, ddd (11.8, 11.8, 7.0) | 49.7 |
| 2.69, dd (11.8, 8.0) | 2.63, dd (11.8, 8.0) | |||
| 11 | 3.36, ddd (14.2, 11.8, 8.0) | 32.6 | 3.15, ddd (14.1, 11.8, 8.0) | 32.2 |
| 2.55, dd (14.2, 7.0) | 2.39, dd (14.1, 7.0) | |||
| 12 | 134.7 | 134.9 | ||
| 13 | 129.8 | 130.1 | ||
| 14 | 6.63, s | 114.4 | 6.60, s | 114.6 |
| 15 | 148.5 | 148.4 | ||
| 16 | 147.7 | 147.3 | ||
| 17 | 6.74, s | 111.2 | 6.69, s | 111.1 |
| 18 | 5.89, d (1.2) | 102.2 | 5.90, d (1.2) | 102.3 |
| 5.77, d (1.2) | 5.79, d (1.2) | |||
| 1′ | 173.1 | 173.5 | ||
| 2′ | 81.6 | 81.1 | ||
| 3′ | 3.17, s | 76.3 | 3.26, s | 75.7 |
| 4′ | 173.0 | 173.1 | ||
| 1′′ | 2.82, d (14.1) | 40.9 | 3.07, d (13.8) | 42.4 |
| 2.73, d (14.1) | 2.80, d (13.8) | |||
| 2′′ | 137.0 | 136.9 | ||
| 3′′ | 7.21, m | 131.7 | 7.22, m | 132.0 |
| 4′′ | 7.20, m | 128.9 | 7.21, m | 128.9 |
| 5′′ | 7.19, m | 127.7 | 7.18, m | 127.8 |
| 6′′ | 7.20, m | 128.9 | 7.21, m | 128.9 |
| 7′′ | 7.21, m | 131.7 | 7.22, m | 132.0 |
| 2-OCH3 | 3.63, 3H, s | 57.9 | 3.72, 3H, s | 57.8 |
The determination of the absolute configuration at C-3′ in cephalotaxine-type alkaloids has been an attractive and challenging work for a long time. Two reliable methods, chemical methods and CD experiments, were developed to solve the problem. The chemical methods including Mosher's method and chemical transformations were applied for the determination of the absolute configuration, such as cephalezomines C and D,17 cephastigiamide A,18 and cephaloverines E and F;3 however this method is limited by the amount of the compounds. The CD spectrum of the molybdate complex of the diacid derived from the hydrolysis of isoharringtonine was first used to determine the absolute configuration of its side chain;19 then this method was applied for 3′S-hydroxyneoharringtonine,20 while the presence of other vicinal diols or carboxyl groups restrict the application of this method. In this study, we observed that the Cotton effect at around 225 nm in the ECD spectra is a characteristic for determining the absolute configuration at C-3′ in cephalotaxine esters (excludes nitrogen oxides). It should be a simple and convenient method, especially for compounds with low molecular weight.
Cephalosine H (8) was obtained as a white amorphous powder with a molecular formula of C19H25NO4 from its HRESIMS (m/z 332.1865 [M + H]+, calcd for C19H26NO4,332.1862) and 13C NMR data. The NMR data of 8 (Tables 2 and 4) were similar to those of cephalounei A,21 except for the presence of an additional methoxy group (δH 3.81, 3H, s; δC 56.5) in 8 and the absence of a methylenedioxy moiety (δH 5.89, 1H, s; 5.88, 1H, s; δC 102.4) in cephalounei A. The additional methoxy group was located at C-16 based on the HMBC correlation from the methoxy group (δH 3.81) to C-16 (δC 147.6) and the NOESY correlation from the methoxy group (δH 3.81) to H-15 (δH 6.65). Furthermore, 2D NMR experiments including HSQC, HMBC, 1H–1H COSY, and NOESY established the structure of 8 (Fig. 5). The absolute configuration of 8 was elucidated to be the same as that of cephalounei A based on their similar ECD and optical rotation data.21 Thus, the absolute configuration of 8 was assigned to be 2R,3S,6S, and named cephalosine H.
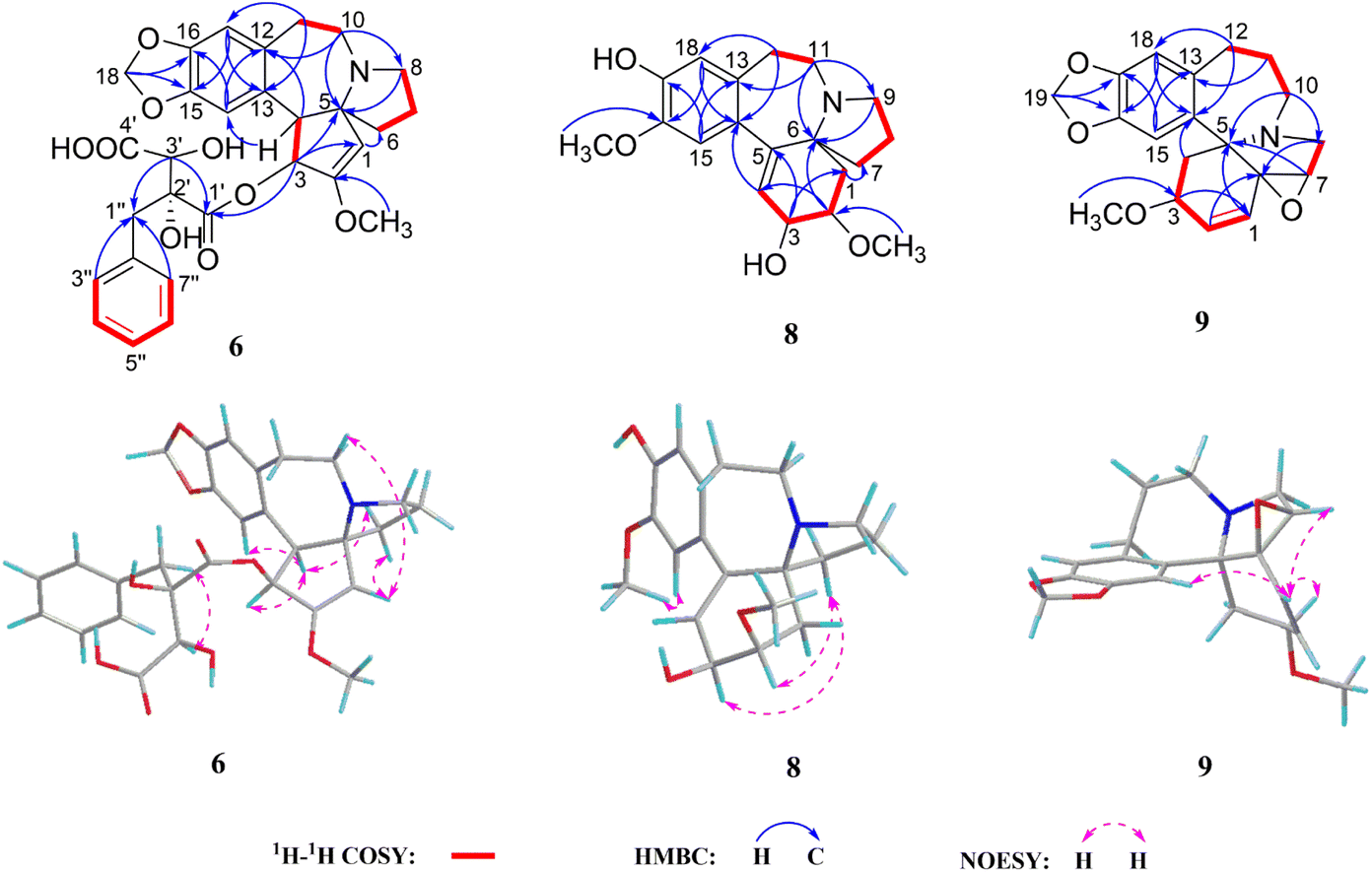 | ||
| Fig. 5 1H–1H COSY, key HMBC, and NOESY correlations of 6, 8, and 9. | ||
| No. | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|
| 1 | 1.99, dd (13.5, 8.5) | 5.89, br d (10.0) | 5.70, dd (10.2, 1.7) | 5.72, dd (10.3, 2.0) | 5.58, br s |
| 1.77, overlap | |||||
| 2 | 3.65, ddd (8.5, 3.5, 2.5) | 6.07, dd (10.0, 4.5) | 5.90, ddd (10.2, 1.7, 1.7) | 5.94, ddd (10.3, 2.0, 2.0) | 2.64, m |
| 1.99, m | |||||
| 3 | 4.36, t (3.8) | 3.92, t (4.5) | 3.79, m | 3.81, ddd (9.0, 5.0, 2.0) | 3.29, m |
| 4 | 5.69, d (3.8) | 3.25, d (14.5) | 2.15, dd (13.2, 5.2) | 2.19, dd (13.3, 5.0) | 2.78, dd (11.5, 3.2) |
| 1.75, dd (14.5, 4.5) | 2.08, dd (13.2, 9.0) | 2.11, dd (13.3, 9.0) | 1.49, t (11.5) | ||
| 7 | 1.77, overlap | 3.61, br s | 1.94, ddd (12.0, 9.0, 9.0) | 1.94, ddd (12.1, 9.0, 9.0) | 2.46, m |
| 1.68, m | 1.70, ddd (12.0, 6.5, 1.2) | 1.71, ddd (12.1, 6.3, 1.2) | 2.31, m | ||
| 8 | 1.75, m | 3.22, d (12.0) | 3.06, ddd (9.0, 9.0, 1.2) | 3.09, ddd (9.0, 9.0, 1.2) | 2.74, ddd (9.0, 8.9, 7.0) |
| 1.62, m | 2.75, d (12.0) | 2.89, overlap | 2.91, overlap | 2.70, overlap | |
| 9 | 2.85, ddd (9.2, 6.7, 2.7) | ||||
| 2.80, ddd (9.2, 9.0, 6.4) | |||||
| 10 | 3.12, ddd (13.0, 12.5, 2.0) | 3.00, dd (15.0, 8.3) | 3.02, dd (15.0, 8.0) | 3.45, ddd (14.6, 12.9, 2.0) | |
| 2.81, ddd (13.0, 3.5, 3.5) | 2.89, overlap | 2.91, overlap | 3.14, ddd (14.6, 4.4, 3.2) | ||
| 11 | 3.41, ddd (15.0, 12.5, 4.0) | 1.83, m | 2.12, m | 2.16, m | 1.83, m |
| 2.96, ddd (15.0, 5.4, 3.0) | 1.69, m | 1.79, m | 1.82, m | 1.57, m | |
| 12 | 3.16, ddd (17.7, 12.5, 5.4) | 3.35, t (15.0) | 2.93, ddd (14.5, 12.2, 4.0) | 2.98, ddd (14.5, 12.3, 4.3) | 3.11, ddd (15.2, 12.5, 1.1) |
| 2.76, ddd (17.7, 4.0, 3.0) | 2.70, dd (15.0, 4.5) | 2.51, ddd (14.5, 4.0, 4.0) | 2.55, ddd (14.5, 4.3, 4.3) | 2.70, overlap | |
| 15 | 6.65, s | 6.76, s | 6.87, s | 6.97, s | 6.73, s |
| 18 | 6.52, s | 6.59, s | 6.54, s | 6.65, s | 6.62, s |
| 19 | 5.85, 2H, s | 5.85, 2H, s | |||
| 2-OCH3 | 3.46, 3H, s | ||||
| 3-OCH3 | 3.05, 3H, s | 3.30, 3H, s | 3.32, 3H, s | 3.21, 3H, s | |
| 16-OCH3 | 3.81, 3H, s | 3.78, 3H, s | 3.73, 3H, s | ||
| 17-OCH3 | 3.80, 3H, s |
Cephalosine I (9) has a molecular formula of C19H21NO4 as deduced from its positive ion peak at m/z 328.1551 [M + H]+ (calcd for 328.1543, C19H22NO4). The 1D NMR data (Tables 2 and 4) of 9 showed high similarity to those of the co-isolated compound fortunine (20),22 except for the signals vicinal to C-3, indicating 9 was a C-3 epimer of 20. The absolute configuration of 9 was defined as 3S,5R,6S,7R by Ga Kα X-ray crystallographic data analysis (Fig. 4).
Cephalosine J (10), existing as colorless crystals, possesses the molecular formula of C19H23NO4 as determined using the HRESIMS (m/z 330.1710 [M + H]+, calcd for C19H24NO4, 330.1705) and 13C NMR data. The NMR data of 10 resembled those of 3-epi-schellhammericine,23 except for the significant downfield shifts of C-6 (δC 80.2), C-7 (δC 38.4), C-1 (δC 135.1), and C-5 (δC 71.5) in 10, suggesting that a hydroxyl group was attached to C-6 in 10. To determine the configuration of the hydroxyl group at the quaternary carbon C-6, the crystals of 10 were obtained from a MeOH/H2O (20:1) solution. Therefore, the absolute configuration of 10 was determined to be 3R,5R,6S by the Ga Kα X-ray crystallographic data analysis with a Flack parameter of 0.06 (3) (Fig. 4).
Cephalosine K (11) possesses a molecular formula of C20H27NO4 as established using the HRESIMS peak at m/z 346.2021 [M + H]+ (calcd for 346.2013, C20H28NO4). The 1H and 13C NMR data of 11 (Tables 2 and 4) resembled those of 10 except for the absence of a methylenedioxy moiety in 10 and the presence of two additional methoxy groups (δH 3.78, 3H, s; δC 56.8 and δH 3.80, 3H, s; δC 56.5) in 11. The HMBC correlation from the methoxy group (δH 3.78) to C-16 (δC 147.6) together with the key NOESY correlation of the methoxy group (δH 3.78) and H-15 (δH 6.97) in 11 indicate that the later methoxy group was attached at C-16. The other methoxy group (δH 3.80, s; δC 56.5) attached to C-17 was determined by the same procedure. Hence, the absolute configuration of 11 was determined to be the same as that of 10 and verified using the similar ECD spectra of these two compounds.
Though the planar structures of these two compounds has been reported as reduction products of comosimine and 3-epi-wilsonine, respectively, the absolute configurations of the hydroxyl at C-6 in 10 and 11 has not been determined.24–26 In this work, the absolute configuration of the hydroxyl group at C-6 in homoerythrina-type alkaloids 10 and 11 was determined by single X-ray crystallographic data analysis.
Cephalosine L (12) has the molecular formula C19H25NO3 based on the HRESIMS ion at m/z 316.1917 [M + H]+ (calcd for 316.1913, C19H26NO3) and 13C NMR. The NMR data of compound 12 closely resembled those of 21,27 except for slight differences in the chemical shifts of the aromatic ring, suggesting a methoxy to be attached to C-16 in 12, instead of C-17 in 21. The HMBC correlation from 16-OCH3 (δH 3.73) to C-16 (δC 146.3), along with the key NOESY correlation from 16-OCH3 (δH 3.73) to H-15 (δH 6.73) in 12 indicated the connection of the methoxy group with C-16. Therefore, the structure of 12 was defined as shown in Fig. 1 and named cephalosine L.
The known alkaloids 13–21 were identified as cephalotaxinone (13),12 cephalotaxine (14),13 3-deoxy-3,11-epoxy-cephalotaxine (15),15 neoharringtonine (16),20 isoharringtonine (17),28 cephaloverine E (18),3 5′-des-O-methylisoharringtonine (19),29 fortunine (20),30 and taxodine (21),27 respectively, via comparison of the reported spectroscopic data.
Racemization of cephalotaxine alkaloids
The racemization of cephalotaxine-type alkaloids has puzzled researchers for a long time. In 1969, Abraham et al. found that the racemization occurred in the process of crystallization of cephalotaxine methiodide.31 Subsequently, Huang et al. reported cephalotaxine, isolated from Cephalotaxus fortune, containing 15% racemate in 1980.32 In 2015, Zhou et al. confirmed that the racemization of cephalotaxine was not generated in the process of salt formation and crystallization.33 However, it is still not clear whether the racemization of cephalotaxine alkaloids is generated by natural or artificial means, especially in the process of extraction and isolation. Thus, we simulated the extraction and isolation process of compounds 2 and 14 to study the racemization of cephalotaxine-type alkaloids.As shown in Fig. 6 and Fig. S1,† (−) cephalotaxine (14a) and (−) cephalosine B (2a) were not observed to be converted into their enantiomers after being heated at 75 °C in 95% ethanol for 9 hours, suggesting that the racemization was not generated in the process of extraction. These two compounds were then dispersed in hydrochloric acid solution (pH = 2) for 24 h, followed by placing them in an alkaline solution (pH = 9) for 24 h. Interestingly, racemization was not observed under these reaction conditions. Moreover, racemization did not occur during silica gel column chromatography and HPLC analysis. Considering these results, we suggest that the racemization of cephalotaxine-type alkaloids is not generated in the process of extraction and isolation but it exists in nature.
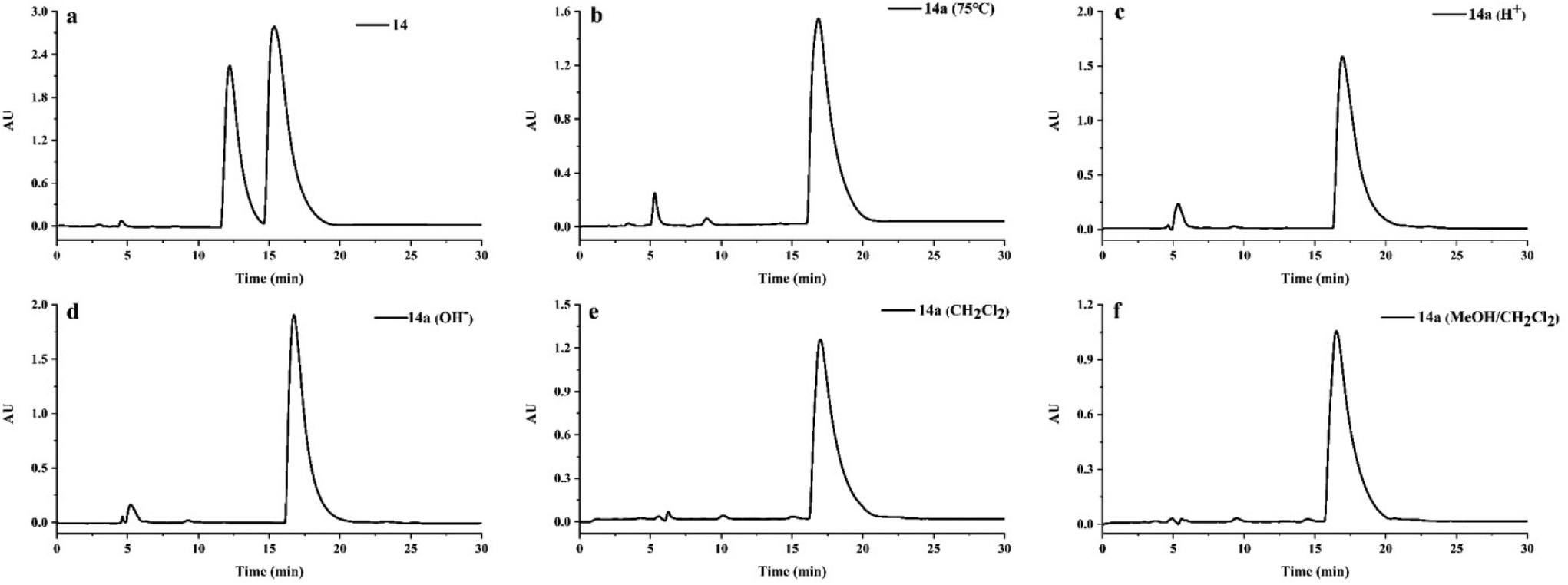 | ||
| Fig. 6 HPLC analysis for racemization of compound 14a using a Daicel Chiralpak IC column (MeOH/H2O/NH3·H2O, 78:22:0.03, 0.8 mL min−1). Reaction conditions: (a) a natural mixture of compound 14; (b) heating in 95% ethanol at 75 °C for 9 hours; (c) dispersing in hydrochloric acid solution (pH = 2) for 24 h; (d) keeping in alkaline solution (pH = 9) for 24 h; (e) keeping in dichloromethane solution for 24 h; (f) keeping in the mixed solutions (CH2Cl2:MeOH:Et2NH = 5:1:0.02) for 24 h. | ||
Cytotoxic activities of the isolated alkaloids
Since two representative cephalotaxine-type alkaloids HHT and HT exhibit remarkable anti-cancer activities and have been clinically used to treat chronic myeloid and acute non-lymphocytic leukemia in China since 1990,3,7 all isolated alkaloids (except for 17) were preliminarily evaluated for their cytotoxic effects against five human cancer cell lines (human promyelocytic leukemia cell line HL-60, human lung cancer cell line A-549, human hepatocarcinoma cell line SMMC-7721, human breast cancer cell line MDA-MB-231, and human colon cancer cell line SW480). As shown in Table 5, new alkaloids 6, 7, and 11, as well as known compounds 16 and 18–19 exhibited broad and potent cytotoxicity against all the tested cancer cell lines, with IC50 values ranging from 0.053 to 10.720 μM. Among them, the cephalotaxine ester 16 exhibited the most potent activities with IC50 values ranging from 0.053 to 0.151 μM. Compound 16 was even more cytotoxic to SMMC-7721 than the positive controls HHT, cisplatin and paclitaxel. Further comparison of the bioactivities of the alkaloids with different configurations of the hydroxyl group at C-3′ (6–7 and 18–19) implied that the compounds with α-OH at C-3′ showed stronger cytotoxicity against HL-60 than against other cancer cells (A-549, SMMC-7721, MDA-MB-231, and SW480 cell lines), whereas the compounds with β-OH at C-3′ showed stronger cytotoxicity against the cancer cells (A-549, SMMC-7721, MDA-MB-231, and SW480 cell lines) than HL-60.| Compounds | IC50b (μM) |
||||
|---|---|---|---|---|---|
| HL-60 | A549 | SMMC-7721 | MDA-MB-231 | SW480 | |
| a Compound 17 was not detected, other compounds, IC50 > 40 μM. b Values were expressed as means ± SD (n = 3). c Positive control. | |||||
| 1a | >40 | 33.750 ± 0.630 | 17.050 ± 0.160 | 26.340 ± 0.830 | 29.960 ± 1.340 |
| 6 | 0.400 ± 0.005 | 0.840 ± 0.011 | 1.066 ± 0.086 | 1.773 ± 0.043 | 3.904 ± 0.233 |
| 7 | 2.236 ± 0.583 | 0.727 ± 0.020 | 0.945 ± 0.014 | 0.878 ± 0.057 | 1.452 ± 0.037 |
| 11 | 8.428 ± 1.210 | 5.556 ± 0.318 | 6.272 ± 0.198 | 10.320 ± 0.540 | 10.720 ± 0.930 |
| 16 | 0.133 ± 0.005 | 0.053 ± 0.007 | 0.054 ± 0.001 | 0.151 ± 0.008 | 0.133 ± 0.005 |
| 18 | 0.053 ± 0.011 | 1.663 ± 0.303 | 3.030 ± 0.073 | 3.347 ± 0.239 | 4.942 ± 0.184 |
| 19 | 0.604 ± 0.087 | 0.188 ± 0.163 | 0.180 ± 0.009 | 0.494 ± 0.042 | 0.438 ± 0.050 |
| HHTc | 0.027 ± 0.002 | 0.029 ± 0.002 | 0.120 ± 0.006 | 0.077 ± 0.005 | 0.066 ± 0.003 |
| Cisplatinc | 4.156 ± 0.189 | 9.104 ± 0.300 | 4.732 ± 0.543 | 14.030 ± 0.750 | 26.340 ± 0.830 |
| Paclitaxelc | <0.008 | <0.008 | 0.123 ± 0.008 | <0.008 | <0.008 |
SARS-CoV-2 3CLpro inhibitory activities of compounds 1–21
HHT was reported to inhibit SARS-CoV-2 in vitro with an IC50 value of 2.10 μM,34 indicating that other cephalotaxine-type alkaloids may exhibit potential anti-SARS-CoV-2 activity. 3CLpro has a pivotal role in mediating the replication and transcription of coronaviruses, making it an attractive drug target for SARS-CoV-2.35 Thus, all isolated compounds were evaluated for their SARS-CoV-2 3CLpro inhibitory activities. Unfortunately, none of them showed significant activities at 40 μM.Conclusions
In summary, twelve new Cephalotaxus alkaloids (1–12) were isolated and identified from the twigs and leaves of C. sinensis. The Cotton effect at around 225 nm in the ECD spectra has been first reported as a characteristic for determining the absolute configuration at C-3′ in cephalotaxine esters. The racemization of cephalotaxine-type alkaloids is evidenced to be natural. The cytotoxicity assay revealed that the cephalotaxine alkaloids (6, 7, 16, 18, and 19) and one homoerythrina-type alkaloid (11) exhibited broad and potent cytotoxicity against five human cancer cell lines (HL-60, A-549, SMMC-7721, MDA-MB-231, and SW480 cell lines), with IC50 values ranging from 0.053 to 10.720 μM. Among them, the cephalotaxine ester 16 exhibited the most potent activities with IC50 values of 0.053–0.151 μM. The cytotoxicity of 16 against SMMC-7721 was even better than those of the positive controls HHT, cisplatin, and paclitaxel. This study not only enriches the chemical diversity of Cephalotaxus alkaloids in Cephalotaxaceae plants but also contributes to the further development of Cephalotaxus alkaloids as a source of potential anti-tumor agents.Author contributions
G. Zhan and Z. Guo designed the study and modified the manuscript; F. Zhang conducted the main experimental procedures, analyzed the data, and wrote the manuscript; T. Yang and K. Yang contributed to the isolation; R. Zhou and Y. Zhang contributed to checking the experimental data; W. Chen and Z. Liu contributed to checking the references.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 32270412 and 31800286), the China Postdoctoral Science Foundation (No. 2021M702636), the Inheritance and Innovation of Traditional Chinese Medicine & Key Scientific Research on the Development of “Qin Medicine” of Shaanxi Administration of Traditional Chinese Medicine (2021-02-ZZ-018), and the Key R&D Plan of Shaanxi Province, China (No. 2020ZDLSF05-05). The Shaanxi Plant Extracts Engineering Technology Research Center and “Qiyao” Resources and Anti-tumor Key Laboratory of Shaanxi Administration of Traditional Chinese Medicine also supported this work. We are grateful to the staff, Gang Chang and Lu Bai, at the Instrumental Analysis Centre of Xi'an Jiaotong University for NMR and HRESIMS tests.References
- H. Abdelkafi and B. Nay, Natural products from Cephalotaxus sp.: chemical diversity and synthetic aspects, Nat. Prod. Rep., 2012, 29, 845–869 RSC.
- D. C. Hao, X. D. Hou, X. J. Gu, P. G. Xiao and G. B. Ge, Ethnopharmacology, chemodiversity, and bioactivity of Cephalotaxus medicinal plants, Chin. J. Nat. Med., 2021, 19, 321–338 CAS.
- G. L. Ma, N. Guo, X. L. Wang, J. Li, Z. X. Jin, Y. Han, S. Z. Dong, J. Xiong and J. F. Hu, Cytotoxic secondary metabolites from the vulnerable conifer Cephalotaxus oliveri and its associated endophytic fungus Alternaria alternate Y-4-2, Bioorg. Chem., 2020, 105, 104445 CrossRef CAS.
- Y. Chang, F. C. Meng, R. Wang, C. Wang, X. Y. Lu and Q. W. Zhang, Chemistry, bioactivity, and the structure-activity relationship of cephalotaxine-type alkaloids from Cephalotaxus sp., in Studies in Natural Products Chemistry, Elsevier, Amsterdam, 2017, vol. 53, pp. 339–373 Search PubMed.
- C. Jiang, J. Xue, Y. Yuan, Y. Li, C. Zhao, Q. Jing, X. Zhang, M. Yang, T. Han, J. Bai, Z. Li, D. Li and H. Hua, Progress in structure, synthesis and biological activity of natural cephalotane diterpenoids, Phytochemistry, 2021, 192, 112939 CrossRef CAS.
- G. Z. Ma, P. F. Li, L. Liu, W. D. Z. Li and L. Chen, Diastereoselective synthesis of cephalotaxus esters via asymmetric mukaiyama aldol reaction, Org. Lett., 2017, 19, 2250–2253 CrossRef CAS PubMed.
- C. X. Zhao, B. Q. Li, Z. X. Shao, D. H. Li, Y. K. Jing, Z. L. Li and H. M. Hua, Cephasinenoside A, a new cephalotane diterpenoid glucoside from Cephalotaxus sinensis, Tetrahedron Lett., 2019, 60, 151154 CrossRef CAS.
- S. Ma, R. Jia, M. Guo, K. Qin and L. Zhang, Insecticidal activity of essential oil from Cephalotaxus sinensis and its main components against various agricultural pests, Ind. Crops Prod., 2020, 150, 112403 CrossRef CAS.
- Y. Y. Fan, J. B. Xu, H. C. Liu, L. S. Gan, J. Ding and J. M. Yue, Cephanolides A–J, cephalotane-type diterpenoids from Cephalotaxus sinensis, J. Nat. Prod., 2017, 80, 3159–3166 CrossRef CAS.
- S. Ma, X. Shi, H. Yan, Z. Ma and X. Zhang, Antiphytoviral activity of alkaloids from Cephalotaxus sinensis, Ind. Crops Prod., 2016, 94, 658–664 CrossRef CAS.
- Y. X. Cai, X. Y. Xie, Q. Q. Zhou, X. Huang and J. B. Xu, Two novel cephalotaxine-type alkaloids from Cephalotaxus sinensis, China J. Chin. Mater. Med., 2022, 47, 2994–2999 Search PubMed.
- D. Weisleder, R. G. Powell and C. R. Smith Jr., Carbon-13 nuclear magnetic resonance spectroscopy of cephalotaxus alkaloids, Org. Magn. Reson., 1980, 13, 114–115 CrossRef CAS.
- S. Sakamoto, T. Miyamoto, K. Usui, H. Tanaka and S. Morimoto, Sodium-periodate-mediated harringtonine derivatives and their antiproliferative activity against HL-60 acute leukemia cells, J. Nat. Prod., 2018, 81, 34–40 CrossRef CAS.
- L. Zhu, L. J. Gong, D. R. Zhu, J. M. Zhu, Y. Li, L. Y. Kong and J. G. Luo, Cephalotaxine-type alkaloids from the seeds of Cephalotaxus fortunei and their cytotoxic activities, Phytochemistry, 2021, 191, 112903 CrossRef CAS.
- R. G. Powell, R. V. Madrigal, C. R. Smith Jr. and K. L. Mikolajczak, Alkaloids of Cephalotaxus harringtonia var drupacea., 11-Hydroxycephalotaxine and drupacine, J. Org. Chem., 1974, 39, 676–680 CrossRef CAS.
- L. Ni, X. H. Zhong, J. Cai, M. F. Bao, B. J. Zhang, J. Wu and X. H. Cai, Five new alkaloids from Cephalotaxus lanceolata and C. fortunei var. alpina, Nat. Prod. Bioprospect., 2016, 6, 149–154 CrossRef CAS.
- H. Morita, M. Arisaka, N. Yoshida and J. Kobayashi, Cephalezomines A–F, potent cytotoxic alkaloids from Cephalotaxus harringtonia var. nana, Tetrahedron, 2000, 56, 2929–2934 CrossRef CAS.
- H. Morita, Y. Nagakura, T. Hosoya, W. Ekasari, A. Widyawaruyanti, K. Mori-Yasumoto, S. Sekita and Y. Hirasawa, Cephastigiamide A, and antiplasmodial activity of Cephalotaxus alkaloids from Cephalotaxus harringtonia Forma Fastigiata, Heterocycles, 2010, 81, 441–450 CrossRef CAS.
- S. Brandange, S. Josephson, S. Vallen and R. G. Powell, Absolute-configuration of 2,3-dihydroxy-2-isopentylbutanedioic acid, a component of alkaloid isoharringtonine, Acta Chem. Scand., Ser. B, 1974, 28, 1237–1248 CrossRef.
- I. Takano, I. Yasuda, M. Nishijima, Y. Yanagi, K. Takeya and H. Itokawa, Ester-type cephalotaxus alkaloids from Cephalotaxus harringtonia var. Drupacea, Phytochemistry, 1997, 44, 735–738 CrossRef CAS PubMed.
- K. K. Mei, G. K. Wang, H. P. Cai and Z. H. Luo, Cephalounei A, a new cephalotaxus alkaloid from the powdered stems of Cephalotaxus fortune Hook. F, Rec. Nat. Prod., 2019, 13, 506–511 CrossRef CAS.
- M. Qiu, B. Lu, X. Ma and R. Nie, Alkaloids from Cephalotaxus fortunei collected in Lijinag, Acta Bot. Yunnanica, 1997, 19, 97–99 CAS.
- V. L. Tran, L. N. Thi, T. T. T. Phuong, L. N. Thi, A. H. Ngoc, H. L. T. Thu, V. C. Tran, N. P. Thi, P. D. Thi and V. S. Tran, The alkaloidal constituents of Cephalotaxus mannii collected in Lam Dong Province, Vietnam, Chem. Nat. Compd., 2017, 53, 1122–1126 CrossRef.
- N. Langlois, B. Das, P. Potier and L. Lacombe, Plants from new caledonia. IV. alkaloids of Phelline comosa, Bull. Soc. Chim. Fr., 1970, 10, 3535–3543 Search PubMed.
- R. G. Powell, K. L. Mikolajczak, D. Weisleder and C. R. Smith Jr., Alkaloids of Cephalotaxus wilsoniana, Phytochemistry, 1972, 11, 3317–3320 CrossRef CAS.
- I. Bick, C. Ralph and S. Panichanun, Homoerythrina and related alkaloids, in Alkaloids: chemical and biological perspectives, Springer, New York, 1991, pp. 1–41 Search PubMed.
- R. G. Powell, Structures of homoerythrina alkaloids from Cephalotaxus harringtonia, Phytochemistry, 1972, 11, 1467–1472 CrossRef CAS.
- Y. N. Li, K. M. Wu and L. Huang, The synthesis of isoharringtonine and the isolation of isomers, Acta Pharm. Sin., 1984, 19, 582–589 CAS.
- I. Takano, I. Yasuda, M. Nishijima, Y. Hitotsuyanagi, K. Takeya and H. Itokawa, Alkaloids from Cephalotaxus harringtonia, Phytochemistry, 1996, 43, 299–303 CrossRef CAS.
- Z. Liu, Q. Du, K. Wang, L. Xiu and G. Song, Completed preparative separation of alkaloids from Cephaltaxus fortunine by step-pH-gradient high-speed counter-current chromatography, J. Chromatogr. A, 2009, 1216, 4663–4667 CrossRef CAS PubMed.
- D. J. Abraham, R. D. Rosenstein and E. L. McGandy, Single crystal X-ray structures of chemotherapeutic agents II, the structure of cephalotaxine methiodide, Tetrahedron Lett., 1969, 10, 4085–4086 CrossRef.
- W. K. Huang, Y. L. Li and X. F. Pan, The synthesis of deoxyharringtonine and the isolation of stereoisomers, Sci. China, Ser. A: Math., Phys., Astron., 1979, 12, 1171–1180 Search PubMed.
- Y. Q. Zhou, Studies on the total synthlesis and racemization mechanism of cephalotaxine, Ph.D. Dissertation, Nankai University, Tianjin, 2015 Search PubMed.
- K. T. Choy, A. Y. L. Wong, P. Kaewpreedee, S. F. Sia, D. Chen, K. P. Y. Hui, D. K. W. Chu, M. C. W. Chan, P. P. H. Cheung, X. Huang, M. Peiris and H. L. Yen, Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in Vitro, Antiviral Res., 2020, 178, 104786 CrossRef CAS.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, HRESIMS, UV, and ECD spectra. CCDC 2213948–2213954. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ob01980a |
| This journal is © The Royal Society of Chemistry 2023 |