
Controllable transformation of indoles using iodine(III) reagent†
Yinxiang
Jian‡
ac,
Peng
Liang‡
ab,
Xiaoyan
Li
ac,
Huawu
Shao
 *ac and
Xiaofeng
Ma
*ac
*ac and
Xiaofeng
Ma
*ac
aNatural Products Research Centre, Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu, 610041, People's Republic of China. E-mail: maxf@cib.ac.cn; shaohw@cib.ac.cn
bSchool of Chemical Engineering, Institute of Pharmaceutical Engineering Technology and Application, Key Laboratory of Green Chemistry of Sichuan Institutes of Higher Education, Sichuan University of Science & Engineering, Xueyuan Street 180, Huixing Road, Zigong, Sichuan 643000, People's Republic of China
cUniversity of Chinese Academy of Sciences, Beijing 100049, People's Republic of China
First published on 22nd November 2022
Abstract
Herein, an efficient and highly functional group-compatible procedure for controllable transformation of indoles by the combination of phenyliodine bis(trifluoroacetate) (PIFA) with n-Bu4NCl·H2O (TBAC) was exploited. Through controlling the amount of PIFA and TBAC from one to three equivalents, 3-chloro-indoles, 3-chloro-2-oxindoles, and 3,3-dichloro-2-oxindoles were obtained, respectively, in satisfactory to excellent yields. The advantages of the protocol include mild conditions, facile process with short reaction time, high yields, satisfactory functional group tolerance, and the use of PIFA, which is an air- and moisture-stable promoter. The mechanism studies showed that the reaction may proceed through a halonium ion species-mediated halogenation–elimination–halogenation stepwise process.
Introduction
Indole sub-units are prevalent in a myriad of natural products,1 marketed drugs,2 agrochemicals,3 and functional materials,4 and they are also useful building blocks in organic synthesis and fine chemistry. Therefore, the synthesis and functionalization of indoles are of considerable importance, and huge efforts have been devoted to these fields. Among multitudinous indole derivatives, 3-halo-indoles, 3-chloro-2-oxindoles, and 3,3-dichloro-2-oxindoles are three families that are prominent because of their wide presence in biologically active molecules5 (Scheme 1A) and broad application in the preparation of high-value heterocyclic molecules (Schemes 1B and C).6,7 Several methodologies have thus been developed for the synthesis of these frameworks.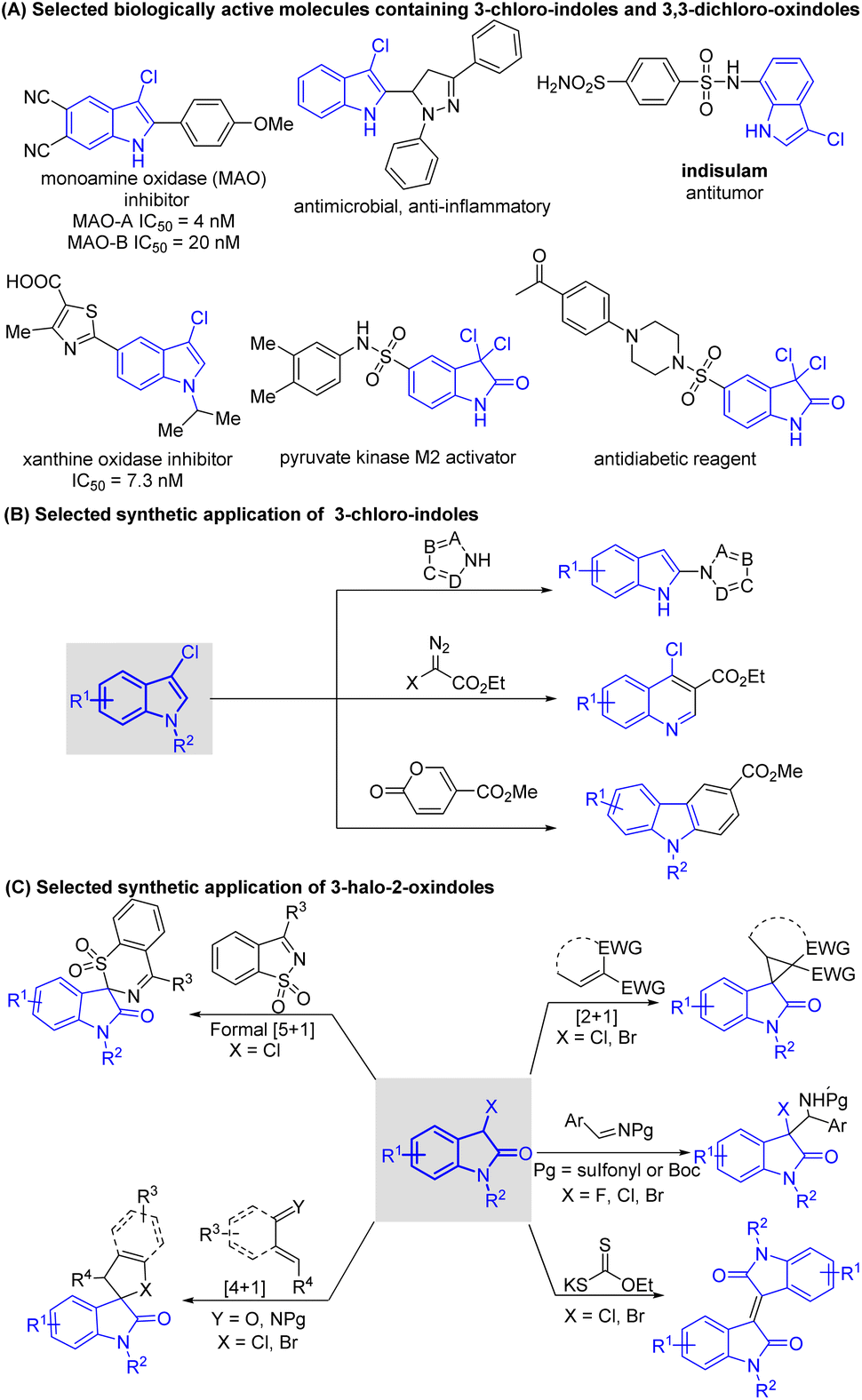 | ||
| Scheme 1 Biologically active molecules and synthetic applications of indole derivatives associated in this study. | ||
For the synthesis of 3-halo-indoles, the traditional procedures are based on the direct electrophilic halogenation of indoles using N-chlorosuccinimide (NCS)8a–d/N-bromosuccinimide (NBS)8e–g or Br2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9/I2,9d1,3-dichloro/dibromo/diiodo-5,5-dimethyl-2,4-imidazolidinedione (DCDMH)/DBDMH/DIDMH,10 SO2Cl2,11 and others.12 Moreover, in recent years, with the development of modern organic synthesis, many catalysed halogenation processes including electrochemical13 and photochemical8f reactions were developed, which realized greener and more sustainable halogenation procedures by improving the regioselectivity, averting overhalogenation, and minimizing the use of highly toxic reagents. The combination of hypervalent iodine reagents (such as (diacetoxyiodo)benzene (PIDA)) with halide anions is another much more attractive strategy because a wide range of 5-substituted indoles can be transformed to the corresponding 3-halo-indoles under very mild conditions with high efficiency (Schemes 2A and B).14
9/I2,9d1,3-dichloro/dibromo/diiodo-5,5-dimethyl-2,4-imidazolidinedione (DCDMH)/DBDMH/DIDMH,10 SO2Cl2,11 and others.12 Moreover, in recent years, with the development of modern organic synthesis, many catalysed halogenation processes including electrochemical13 and photochemical8f reactions were developed, which realized greener and more sustainable halogenation procedures by improving the regioselectivity, averting overhalogenation, and minimizing the use of highly toxic reagents. The combination of hypervalent iodine reagents (such as (diacetoxyiodo)benzene (PIDA)) with halide anions is another much more attractive strategy because a wide range of 5-substituted indoles can be transformed to the corresponding 3-halo-indoles under very mild conditions with high efficiency (Schemes 2A and B).14
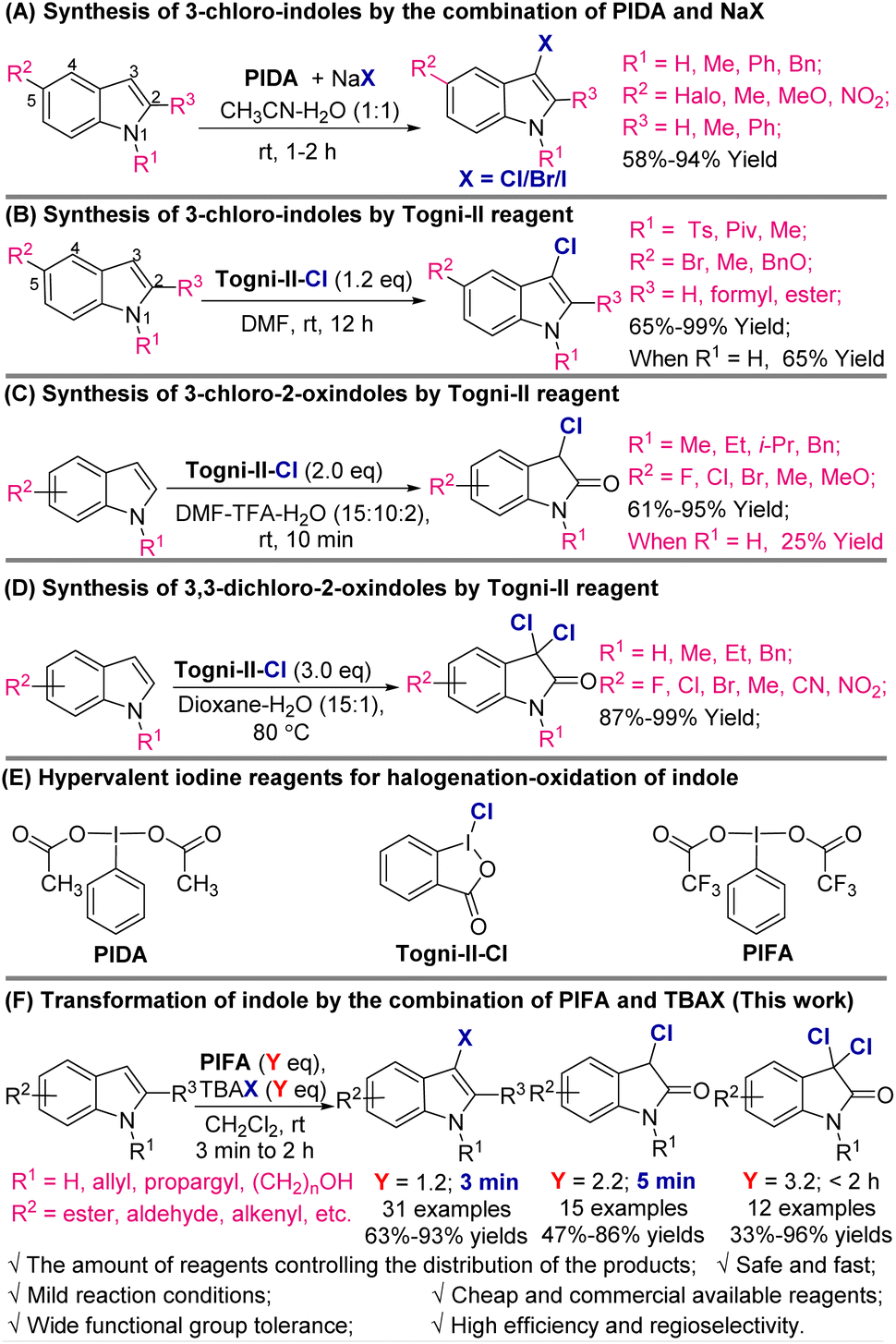 | ||
| Scheme 2 Synthesis of 3-chloro-indoles, 3-chloro-2-oxindoles, and 3,3-dichloro-2-oxindoles promoted by hypervalent iodine reagents. | ||
While they have been broadly employed in the construction of spirocyclic and 3,3-di-substituted oxindole scaffolds due to their bifunctional properties, the synthesis of 3-chloro-2-oxindoles is highly dependent on the FeCl3-prompted cyclization of β-nitrostyrene derivatives.15 The other procedures always involved multi-step transformation with narrow substrate scope and low yields. In 2018, Prathima and co-worker disclosed that in the presence of oxone and NaCl, indole-3-carboxaldehydes can be smoothly converted into 3-chloro-2-oxindoles. In 14 tested samples, products were produced in 79–92% yields, although it was a two-step process whereby it was necessary for the indole-3-carboxaldehydes to be prepared from indoles.16
In 2019, Yu's group developed an elegantly direct procedure for the synthesis of 3-chloro-2-oxindoles from indoles mediated by a hypervalent iodine reagent [1-chloro-1,2-benziodoxol-3-(1H)-one] (Togni-II-Cl) (Scheme 2C).17 The N-alkyl-substituted indoles and electron-donating groups or halide-substituted indoles were converted into 3-chloro-2-oxindoles in 61–95% yields, while simple 1H-indole offered the product in only 25% yield. Also, the reaction was performed in DMF using TFA as the co-solvent, which may result in the formation of unwanted byproducts when acid-sensitive groups are involved.
Lastly, 3,3-dichloro-2-oxindoles are extremely important oxindole derivatives that have wide biological activity, including anti-diabetic and anti-cancer activity. The synthesis of 3,3-dichloro-2-oxindoles from inexpensive and simple feedstocks remains rare. The first systematic study was from Yu's group.17 During their synthesis of 3-chloro-2-oxindoles from indoles, they found that when the amount of hypervalent iodine reagent was increased to three equivalents, they were able to isolate the corresponding 3,3-dichloro-2-oxindoles in 87% to 99% yields (Scheme 2D). This process seems more effective than the one used for the synthesis of 3-chloro-2-oxindoles because the N-unprotected indoles can be converted into the products in excellent yields, and some functional groups including halides, CN, and NO2 are well tolerated under the reaction conditions.
Despite these great successes, transformation of indoles to 3-halo-indoles, 3-chloro-2-oxindoles, and 3,3-dichloro-2-oxindoles in a controllable manner with satisfactory functional group tolerance and high selectivity remains a challenge. For example, halogenation of indoles is currently the most extensively studied among three transformations. However, most halogenation methodologies use C2-8a,e,f,14g or C5-substituted9d,14f indoles (among the most reactive positions after the C3-position1a,9b,9c), or indoles with electron-withdrawing groups protecting the N-atom,8c,12,14e to improve regioselectivity and minimize overhalogenation, while general protocols for the synthesis of 3-chloro-2-oxindoles and 3,3-dichloro-2-oxindoles from indoles, especially from 1-unsubstituted indoles, remain sparse.17 In these contexts, the systematic study of indole halogenation and oxidation reactions is thus particularly interesting and necessary.
In pursuit of our ongoing efforts on oxidative transformation of aromatic compounds,18 we previously described the hypervalent iodine reagent-mediated direct synthesis of 2-oxindoles from indoles.18b As the continuation of this work, herein, we report the results of our investigation of selective synthesis of 3-halo-indoles, 3-chloro-2-oxindoles, and 3,3-dichloro-2-oxindoles by the combination of phenyliodine bis(trifluoroacetate) (PIFA) and n-Bu4NX (TBAX, X = Cl, Br, I). By controlling the amount of PIFA-TBAC, the corresponding monohalogenation, chloridion–oxidation, and dichloridion–oxidation products were produced in satisfactory to excellent yield (Scheme 2F).
Compared to the procedures in Scheme 2A–D, the properties of this process include (i) the first examples for conversion of indoles into 3-halo-indoles, 3-chloro-2-oxindoles, and 3,3-dichloro-2-oxindoles by simply controlling the amount of reagents for halogenation; (ii) the most general protocol for regioselective 3-chlorination of indoles developed to date, with a variety of solvents that can be employed; (iii) likely reaction through halogenation–elimination–halogenation stepwise-delivered diversity of products with oxidation-sensitive groups such as aldehyde, alkenyl, alkynyl, and alcohol tolerated; and (iv) rapid reactions for 3-halo-indoles and 3-chloro-oxindoles that finish in less than 5 minutes.
Results and discussion
To optimize the halogenation reaction of indoles mediated by hypervalent iodine reagent, we initially studied the influence of solvents and the chloro source (Table 1) using 1H-indole (1a) as a model substrate. Identification of the reaction parameters resulted in rapid determination of the optimum conditions for the synthesis of 3-chloro-1H-indole (Table 1, entry 3). That is, when using PIFA as the oxidant and n-Bu4NCl·H2O (TBAC) as the chloro source, the product of monochlorination (2a) can be isolated in 93% yield. Moreover, the rapid reaction finishes in less than 3 minutes. Clearly, the type of chloro source is crucial to the transformation because neither NaCl nor NH4Cl produced any desired product (Table 1, entries 1 and 2).|
|
|||||
|---|---|---|---|---|---|
| Entry | Oxidant | Cl source | Solvent | T (°C) | Yieldb (%) |
| a General conditions: 1a (1.0 equiv.), oxidant (1.2 equiv.), Cl source (1.2 equiv.). b Isolated yield. | |||||
| 1 | PIFA | NaCl | CH2Cl2 | rt | N.R. |
| 2 | PIFA | NH4Cl | CH2Cl2 | rt | N.R. |
| 3 | PIFA | TBAC | CH 2 Cl 2 | rt | 93 |
| 4 | PIFA | TBAC | DCE | rt | 73 |
| 5 | PIFA | TBAC | Toluene | rt | 53 |
| 6 | PIFA | TBAC | THF | rt | 56 |
| 7 | PIFA | TBAC | 1,4-Dioxane | rt | 47 |
| 8 | PIFA | TBAC | CH3CN | rt | 67 |
| 9 | PIFA | TBAC | Et2O | rt | 73 |
| 10 | PIFA | TBAC | Acetone | rt | 64 |
| 11 | PIFA | TBAC | CHCl3 | rt | 60 |
| 12 | PIFA | TBAC | EtOAc | rt | 48 |
| 13 | PIDA | TBAC | CH2Cl2 | rt | Complex |
| 14 | PIFA | TBAC | CH2Cl2 | 0 | 90 |
| 15 | PIFA | TBAC | CH2Cl2 | 40 | 89 |

The reaction can also be performed in a wide range of other solvents including 1,2-dichloroethane (DCE), toluene, tetrahydrofuran (THF), 1,4-dioxane, CH3CN, ether (Et2O), acetone, CHCl3, and ethyl acetate. In all of these solvents, the 3-chloro-indole product (2a) was isolated in 47% to 73% yield (Table 1, entries 4–12). Changing the oxidant to PIDA resulted in the formation of a complex mixture (Table 1, entry 13). When the reaction was carried out at 0 °C and 40 °C, respectively, the product can still be formed at approximately 90% yield (Table 1, entries 14 and 15).
After determining the optimal reaction conditions, we proceeded to investigate the substrate scope. As shown in Table 2, we found that the reaction exhibits remarkable functional group tolerance, and it is compatible with numerous varieties of substituents on the phenyl ring and N-substituent. The substrates with an electron-donating group on the N-atom, such as Me, allyl, and Bn, were smoothly chloridized at the 3-position in satisfactory to excellent yields with high regioselectivity (Table 2, 2b–2d).
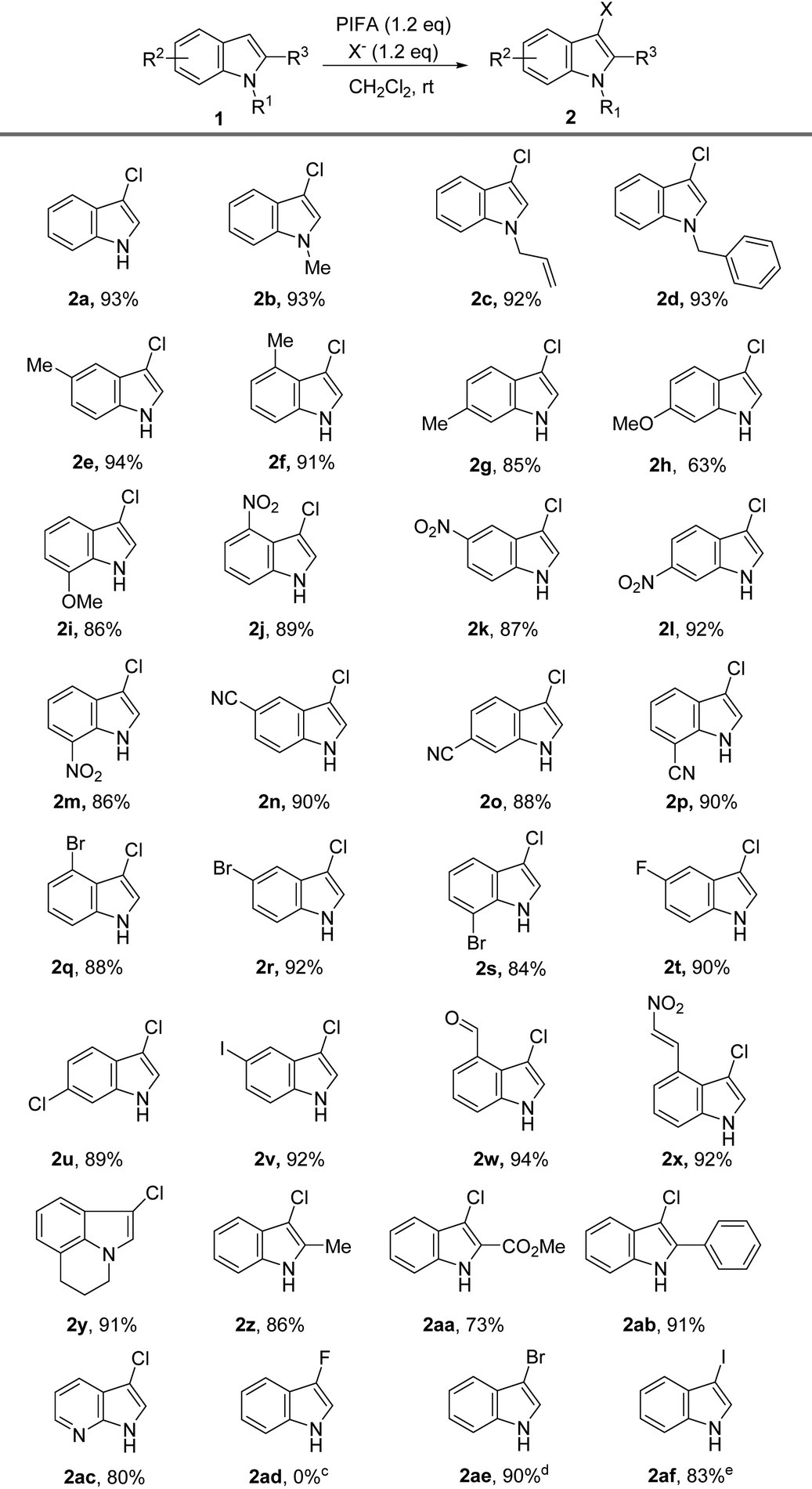
The electron-donating (such as Me, MeO) and electron-withdrawing substituents (such as NO2, CN, F, Cl, Br, I) at the 4-, 5-, 6-, or 7-position of the phenyl ring were compatible with the chlorination reaction. In all the tested cases, the corresponding 3-chloro-indoles were provided in satisfactory to excellent yields, and all the reactions were finished in less than 3 minutes (Table 2, 2e–2v). The substrates substituted with a double bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), aldehyde, or β-nitrostyrene were all compatible under the reaction conditions, achieving the products in 92% to 94% yields (Table 2, 2c, 2w, 2x), which further demonstrated the efficiency and highly functional group tolerance of our approach for the synthesis of 3-chloro-indoles.
C), aldehyde, or β-nitrostyrene were all compatible under the reaction conditions, achieving the products in 92% to 94% yields (Table 2, 2c, 2w, 2x), which further demonstrated the efficiency and highly functional group tolerance of our approach for the synthesis of 3-chloro-indoles.
The tricyclic indole is also a suitable substrate for the reaction, and gave the corresponding 3-chlorination indole product 2y in 91% yield. We examined the influence of the substituted group on the 2-position, because the 2-substituted 3-chloro-indoles are broadly used as bioactive molecules, as shown in Scheme 1A. Obviously, there were negligible effects on the efficiency of the reaction by the substitutes in the 2-position of indole, and the tested three substrates offered the products in satisfactory to excellent yields (2z, 2aa, 2ab). 7-Azaindole was also converted into a 3-chlorination product (2ac) in 80% yield. To further investigate the scope of the reaction, we studied the influence of the different halogen sources, including n-Bu4NF·6H2O (TBAF), n-Bu4NBr (TBAB), and n-Bu4NI (TBAI). Even though no desired product (2ad) was detected when TBAF was employed, the halogen atom from TBAB and TBAI was smoothly transformed into indole, and the corresponding 3-bromo-1H-indole 2ae and 3-iodo-1H-indole192af were provided in 90% and 83% yield, respectively (Table 2). Because 3-iodo-indoles are extremely important building blocks for the preparation of 3-substituted indole derivatives that are prevalent in numerous biologically active molecules,19e our protocol would therefore provide a novel and useful strategy for the synthesis of 3-iodo-indoles.
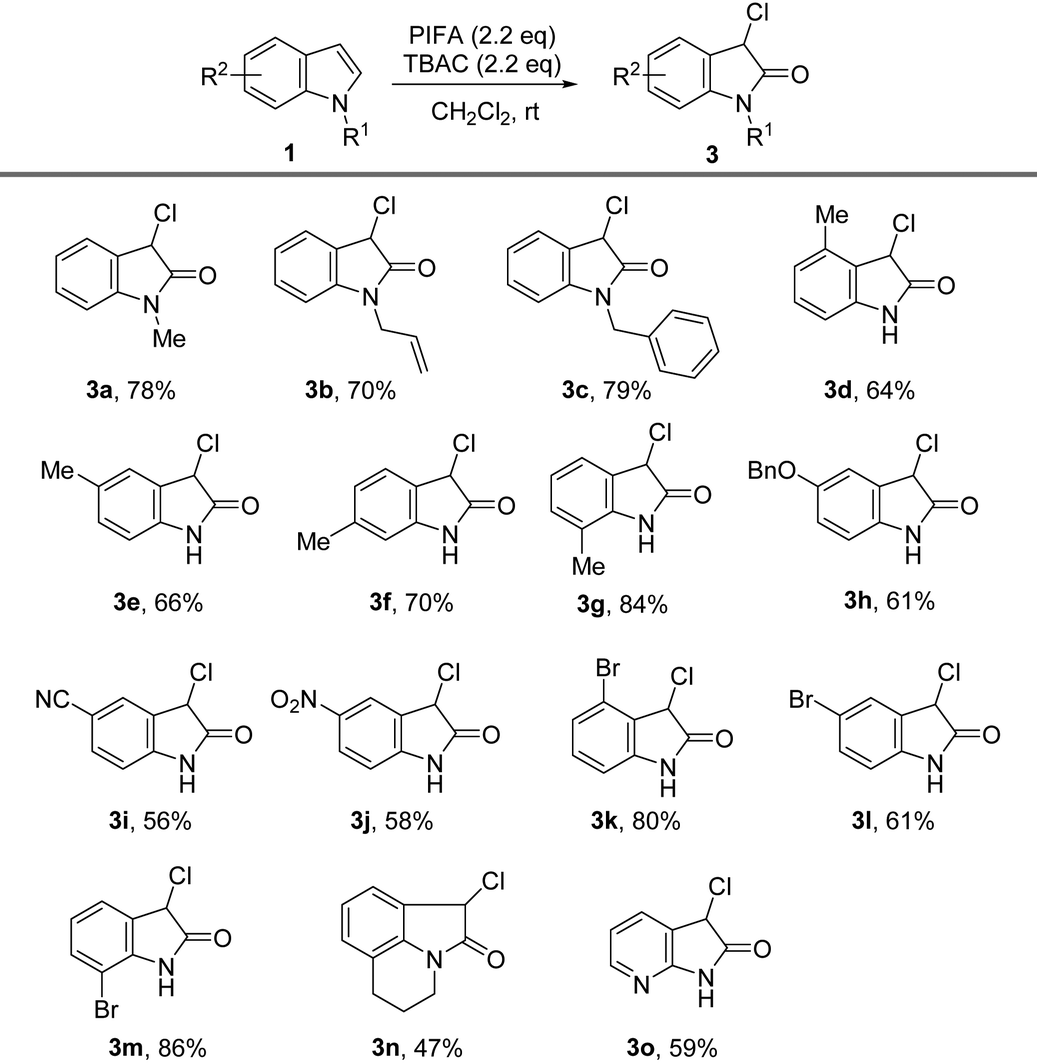
We previously showed that the combination of PIFA and TBAC oxidized 3-substituted indoles to corresponding 2-oxindoles. The efficient halogenation and oxidation of various indoles under similar conditions fascinated us, and thus, we further explored the possibility of synthesizing 3-chloro-2-oxindoles directly from indoles. Interestingly, by simply increasing the amount of PIFA and TBAC to 2.2 equiv., we were able to isolate 3-chloro-2-oxindole from indole in 83% yield without altering any other reaction conditions,18b and the reaction was as efficient as the process for the synthesis of 3-chlorination indole, with both reactions finishing in less than 5 min.
Herein, we further investigated the substrate scope for the direct synthesis of 3-chloro-2-oxindoles. As illustrated in Table 3, varieties of 3-chloro-2-oxindoles were successfully obtained in moderate to satisfactory yields directly from indoles. The electron-donating substituents such as Me, Bn, and allyl on the N-atom of indole reacted well under the standard reaction conditions, offering the corresponding products in 70%–79% yields (Table 3, 3a–3c). The electron-donating groups at the 4-, 5-, 6-, and 7-position of indoles are also suitable for the oxidation reaction, and the corresponding 3-chloro-2-oxindole products were achieved in moderate to satisfactory yields (Table 3, 3d–3h). Indoles with electron-withdrawing groups, including NO2, CN, Cl, and Br at different positions of the phenyl ring, were also smoothly chlorinated and oxidized to 3-chloro-2-oxindoles in up to 86% yield (Table 3, 3i–3m). The reaction also tolerated substrates with tricyclic-containing indoles, despite producing the product (3n) with a diminished yield (47%). Finally, 3-chloro-7-azaoxindole 3o was successfully obtained in approximately 60% yield with this chlorination–oxidation protocol.
At this stage, we were curious what would happen if we further increased the amount of PIFA and TBAC. Much to our surprise, when 5-chloro-indole was treated with 3.2 equiv. of PIFA combined with same amount of TBAC, we observed a clear conversion after stirring the reaction mixture at room temperature for 1.5 hours. The major product was proved to be 3,3,5-trichloro-2-oxindole (4a), and the yield for this transformation was 86%. 5-Bromo-indole also delivered the product 4b in up to 95% yield. We then focused our attention on the functional group tolerance of this oxidation–dichlorination protocol. Some representative examples are collected in Table 4. Indoles with electron-withdrawing groups (including NO2, CN, and ester) substituted on the phenyl ring were all smoothly converted into dichloridized oxindole products in up to 96% yield (4c–4e). Indole substrates with electron-donating groups (such as Me) on the phenyl ring also generated the corresponding product (4f) in 60% yield under the standard conditions.
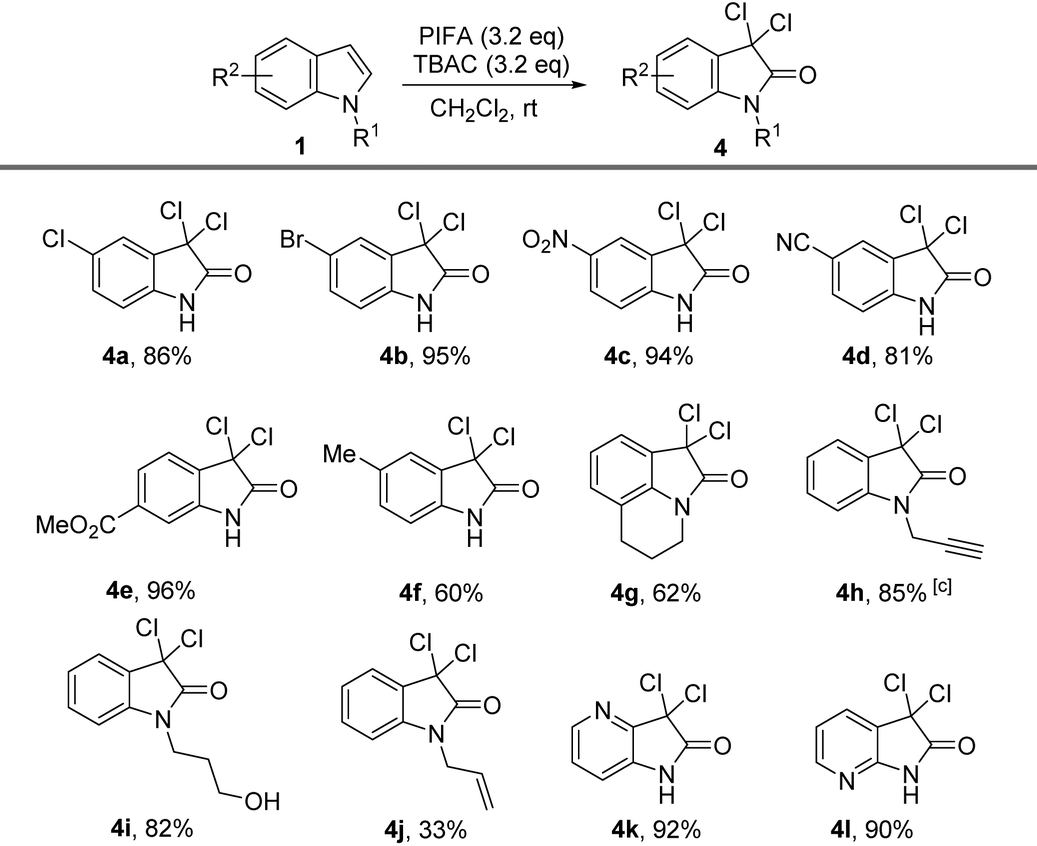
The high yield under the very mild reaction conditions encouraged us to further extend this protocol to tricyclic and N-substituted indoles. The functional groups at N-atoms such as alkynyl and free hydroxyl groups were all compatible under the reaction conditions, offering the products (4h, 4i) in 85% and 82% yield, respectively. N-Allyl-substituted groups were also successfully transformed into the 3,3-dichloro-2-oxindole 4j despite in the reduced yield. Moreover, azaindoles including 4-azaindole and 7-azaindole were smoothly converted into 3,3-dichloro-2-oxindoles (4k, 4l) in excellent yields.
To clarify the mechanism of the halogenation and oxidation processes, some control experiments were carried out (Scheme 3). It was found that the 3-chlorination product was formed in 71% and 90% yield when 1.0 equiv. of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and 2,6-di-tert-butyl-4-methylphenol (BHT), respectively, was added to the reaction mixture, which may rule out the possibility of a radical process (Scheme 3, Eqn. 1). The lower yield from TEMPO possibly occurred because it reacted with PIFA, inhibiting the formation of active halonium ion.20 Additionally, treatment of 5-bromo-3-chloroindole with 1.2 equiv. of PIFA and TBAC smoothly formed 3l in 53% yield (Scheme 3, Eqn. 2). When 3l was further reacted with PIFA and TBAC (1.2 equiv.), 4b was isolated as the only product in 69% yield (Scheme 3, Eqn. 3, Conditions A), which suggested that direct chlorination of 3-chloro-2-oxindole to form 3,3-dichloro-2-oxindole may take place in our case. We have previously proved that when PIFA was stirred with TBAC, it formed the highly active intermediate 5 (vide infra), which may trigger the subsequent chlorination and oxidation.18b Moreover, treatment of 3l with 5 or HOCl (in situ formed from the reaction of NaOCl and HClaq) produced 4b in 64% and 69% yield, respectively (Scheme 3, Eqn. 3, Conditions B and C). These control experiments suggest that intermediate 5 and HOCl may be involved in all three transformations.
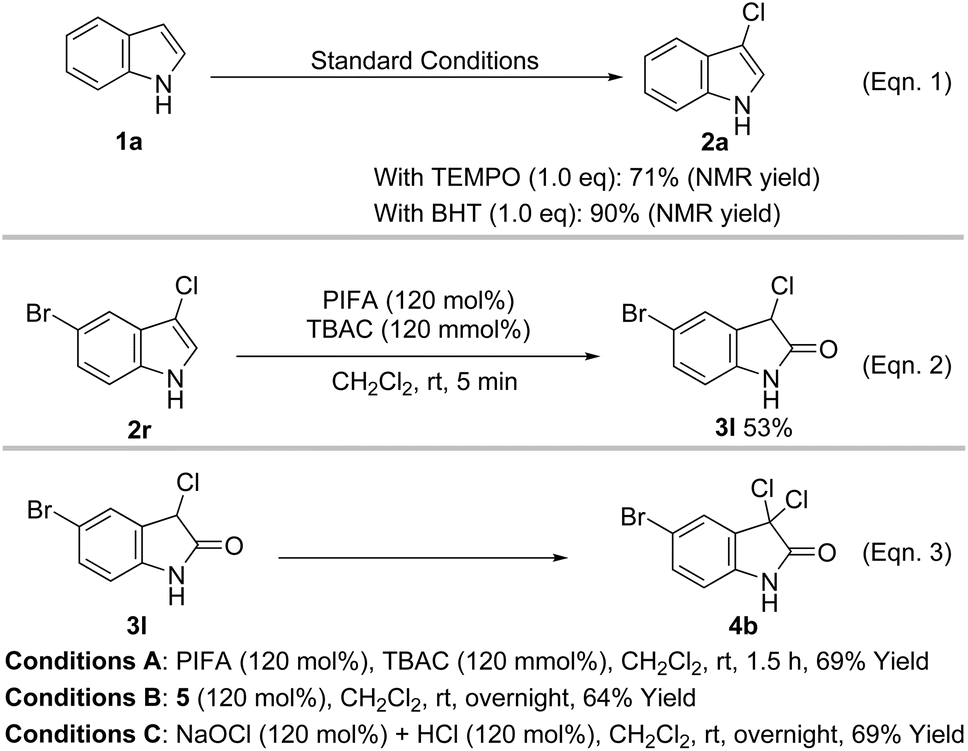 | ||
| Scheme 3 Control experiments. | ||
On the basis of the above and the previous experiments, a plausible mechanism for the transformations is proposed in Scheme 4. The reaction between TBAC and PIFA generates a new and highly active hypervalent iodine intermediate 5, which is followed by reductive elimination to form Cl–OH species.18b Electrophilic chlorination of indole by 5 or Cl–OH produces intermediate 6,1a which then undergoes aromatization to form 3-chloro-indole product 2e.
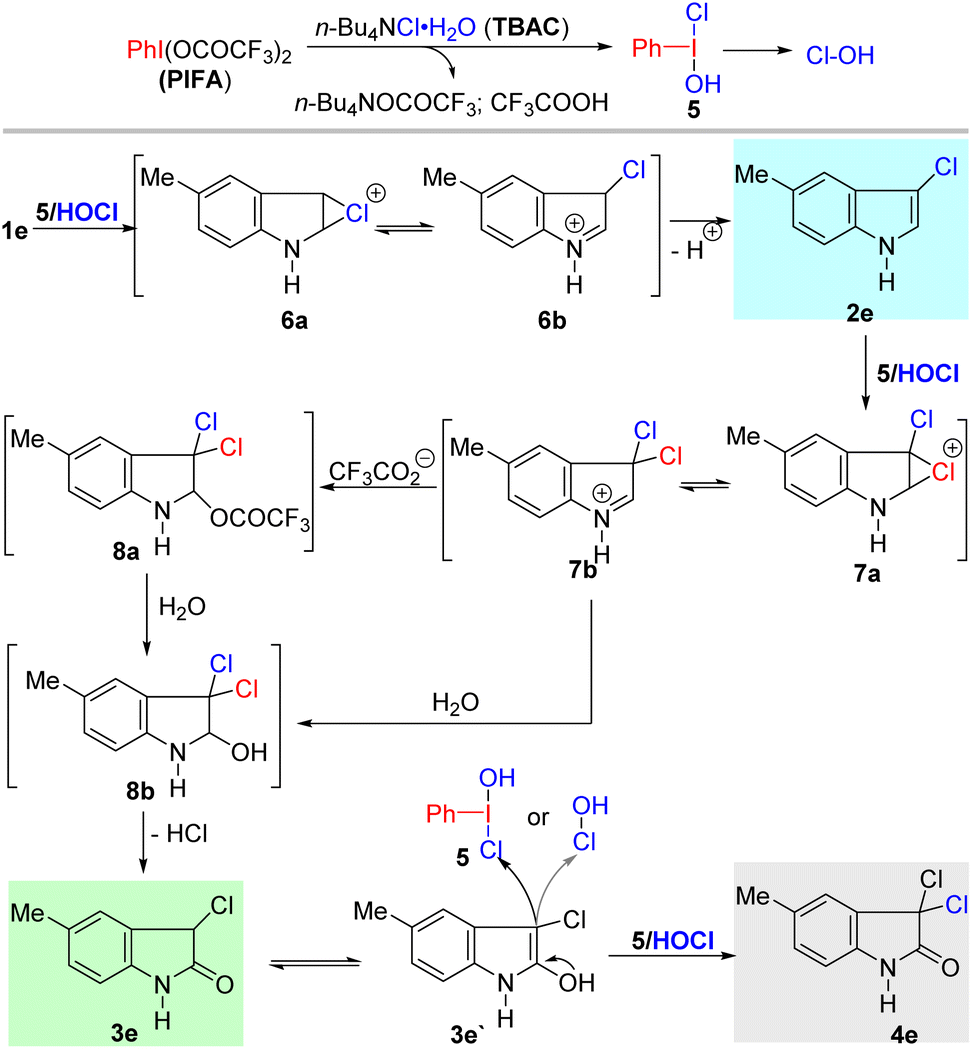 | ||
| Scheme 4 Plausible reaction mechanism for the PIFA-mediated transformation of indoles. | ||
Subsequently, 2e participates in the second chlorination by 5 or Cl–OH to provide intermediate 7, which is attacked by trifluoroacetate anion or H2O to form intermediate 8a and 8b, respectively. In the presence of H2O, intermediate 8a can also participate in hydrolysis reactions to produce 8b.21 Elimination of HCl from 8b provides 3-chloro-2-oxindole 3e. Lastly, chlorination of 3e by 5 or Cl–OH can offer the 3,3-dichloro-2-oxindole 4e. This is different from the Togni-II-Cl-promoted dichlorination–oxidation process reported by Yu's group, in which they proved that the oxidation of 8b can directly form 4e.17
The highly efficient, mild reaction conditions and controllable selectivity of our protocols were very useful for the synthesis of 3-chloro-indoles, 3-chloro-2-oxindoles, and 3,3-dichloro-2-oxindoles so that a wide range of indole-based biologically active molecules could be constructed (Scheme 5). For example, when indole (9) was treated with PIFA-TBAC under the standard conditions for the dichloridation–oxidation, the product 3,3-dichloro-2-oxindole (10) that would be used as an anticancer reagent17 was formed in 79% yield (Scheme 5A). The application of our methodology to the formal synthesis of ropinirole hydrochloride, an FDA-approved drug for the treatment of Parkinson's disease, is also shown in Scheme 5B. Chloridation–oxidation of alcohol 11 formed 3-chloro-2-oxindole 12 in 51% yield. Catalytic hydrogenation of 12 in the presence of H2 generated 2-oxindole 13 in 90% yield. Conversion of 13 to ropinirole hydrochloride (14) has been previously achieved after two-step simple transformations.22
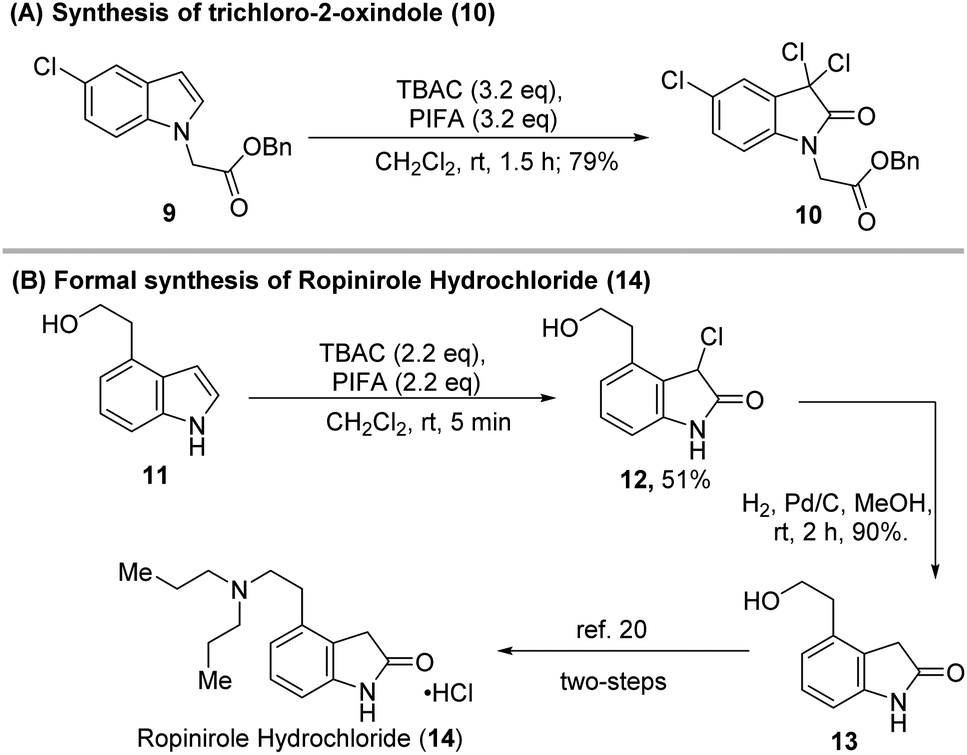 | ||
| Scheme 5 Applications of the PIFA-mediated transformation of indoles. | ||
Conclusions
We developed a mild and controllable protocol for the synthesis of 3-halo-indoles, 3-chloro-2-oxindoles, and 3,3-dichloro-2-oxindoles from indoles that was promoted by the combination of PIFA and TBAC. It is expected that the protocol will be widely used because of several advantages, including the use of inexpensive reagents, broad substrate scope, wide functional group compatibility, controllable distribution of products, and satisfactory isolated yields. Moreover, it was found that the strategy can also be used to realize the synthesis of 3-iodo indoles and 3-bromo indoles by replacing TBAC with TBAI or TBAB.Mechanism studies revealed that the reactions may undergo halogenation–elimination (formal oxidation)–halogenation stepwise, which, in turn, will lead to the formation of different products (3-chloro-indoles, 3-chloro-2-oxindoles, and 3,3-dichloro-2-oxindoles) by controlling the amount of PIFA-TBAC. The application of the protocol was demonstrated by the synthesis of biologically active molecules, including ropinirole hydrochloride, an FDA-approved drug that is used for the treatment of Parkinson's disease.
Author contributions
Y. J., P. L., and X. L. performed the experiments and analyzed the results. X. M. and H. S. conceived the idea and guided the project. P. L. and X. M. drafted the manuscript with feedback from all co-authors. All authors have approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank the National Natural Science Foundation of China (22001246 and 21772192); the start-up grant from Chengdu Institute of Biology, Chinese Academy of Sciences; the Biological Resources Program (KFJ-BRP-008) from the Chinese Academy of Sciences; and the Thousand Talents Program of Sichuan Province, for the financial support.References
- (a) R. J. Sundberg, Top. Heterocycl. Chem., 2010, 26, 47–115 CrossRef CAS; (b) M. Lounasmaa and A. Tolvanen, Nat. Prod. Rep., 2000, 17, 175–191 RSC; (c) M. Somei and F. Yamada, Nat. Prod. Rep., 2004, 21, 278–311 RSC; (d) J. S. Cannon and L. E. Overman, Angew. Chem., Int. Ed., 2012, 51, 4288–4311 CrossRef CAS PubMed; (e) H. Achenbach and K. Biemann, J. Am. Chem. Soc., 1965, 87, 4944–4950 CrossRef CAS; (f) N. K. Garg, R. Sarpong and B. M. Stoltz, J. Am. Chem. Soc., 2002, 124, 13179–13184 CrossRef CAS PubMed.
- (a) Z. Yousuf, A. K. Richards, A. N. Dwyer, B. Linclaua and D. C. Harrowven, Org. Biomol. Chem., 2015, 13, 10532–10539 RSC; (b) R. Silvestri, G. D. Martino, G. L. Regina, M. Artico, S. Massa, L. Vargiu, M. Mura, A. G. Loi, T. Marceddu and P. L. Colla, J. Med. Chem., 2003, 46, 2482–2493 CrossRef CAS PubMed; (c) P. Marchand, M. L. Borgne, M. Palzer, G. L. Bauta and R. W. Hartmann, Bioorg. Med. Chem. Lett., 2003, 13, 1553–1555 CrossRef CAS PubMed.
- (a) A. Andreani and M. Rambaldi, J. Heterocycl. Chem., 1988, 25, 1519–1523 CrossRef CAS; (b) H. Firouzabadi, N. Iranpoor, M. Jafarpour and A. Ghaderi, J. Mol. Catal. A: Chem., 2006, 253, 249–251 CrossRef CAS; (c) G.-G. Liu, H. Zhao, Y.-B. Lan, B. Wu, X.-F. Huang, J. Chen, J.-C. Tao and X.-W. Wang, Tetrahedron, 2012, 68, 3843–3850 CrossRef CAS.
- (a) C. Li, M. Liu, N. G. Pschirer, M. Baumgarten and K. Mullen, Chem. Rev., 2010, 110, 6817–6855 CrossRef CAS PubMed; (b) Q. Li, G. Yu, J. Huang, H. Liu, Z. Li, C. Ye, Y. Liu and J. Qin, Macromol. Rapid Commun., 2008, 29, 798–803 CrossRef CAS; (c) Q. Li, Z. Li, C. Ye and J. Qin, J. Phys. Chem. B, 2008, 112, 4928–4933 CrossRef CAS PubMed; (d) M. S. Park, D. H. Choi, B. S. Lee and J. Y. Lee, J. Mater. Chem., 2012, 22, 3099–3104 RSC.
- (a) Z. V. Chirkova, M. V. Kabanova, S. I. Filimonov, I. G. Abramov, A. Petzer, J. P. Petzer, S. I. Firgang and K. Y. Suponitsky, Bioorg. Med. Chem. Lett., 2015, 25, 1206–1211 CrossRef CAS PubMed; (b) S. Mostafa, M. K. El-Dean Adel, A. Mostafa and H. Reda, J. Heterocycl. Chem., 2018, 55, 1166–1175 CrossRef; (c) T. Owa, A. Yokoi, K. Yamazaki, K. Yoshimatsu, T. Yamori and T. Nagasu, J. Med. Chem., 2002, 45, 4913–4922 CrossRef CAS PubMed; (d) T. Han, M. Goralski, N. Gaskill, E. Capota, J. Kim, T. C. Ting, Y. Xie, N. S. Williams and D. Nijhawan, Science, 2017, 356, eaal3755 CrossRef PubMed; (e) T. Uehara, Y. Minoshima, K. Sagane, N. H. Sugi, K. O. Mitsuhashi, N. Yamamoto, H. Kamiyama, K. Takahashi, Y. Kotake, M. Uesugi, A. Yokoi, A. Inoue, T. Yoshida, M. Mabuchi, A. Tanaka and T. Owa, Nat. Chem. Biol., 2017, 13, 675–680 CrossRef CAS PubMed; (f) J. U. Song, S. P. Choi and T. H. Kim, Bioorg. Med. Chem. Lett., 2015, 25, 1254–1258 CrossRef CAS PubMed; (g) M. B. Boxer, M. Shen, D. S. Auld, C. J. Thomas and M. J. Walsh, WO2011137089A1, 2011; (h) J. Xiao, W. Westbroek, O. Motabar, W. A. Lea, X. Hu, A. Velayati, W. Zheng, N. Southall, A. M. Gustafson, E. Goldin, E. Sidransky, K. Liu, A. Simeonov, R. J. Tamargo, A. Ribes, L. Matalonga, M. Ferrer and J. J. Marugan, J. Med. Chem., 2012, 55, 7546–7559 CrossRef CAS PubMed.
- (a) M. Poirier, S. Goudreau, J. Poulin, J. Savoie and P. L. Beaulieu, Org. Lett., 2010, 12, 2334–2337 CrossRef CAS PubMed; (b) S. Peeters, L. N. Berntsen, P. Rongved and T. Bonge-Hansen, Beilstein J. Org. Chem., 2019, 15, 2156–2160 CrossRef CAS PubMed; (c) T. Guney, J. J. Lee and G. A. Kraus, Org. Lett., 2014, 16, 1124–1127 CrossRef CAS PubMed.
- (a) L. Liu, Y. Li, T. Huang, D. Kong and M. Wu, Beilstein J. Org. Chem., 2021, 17, 2321–2328 CrossRef CAS PubMed and references cited therein; (b) R.-L. Li, Q.-Y. Fang, M.-Y. Li, X.-S. Wang and L.-M. Zhao, Chem. Commun., 2021, 57, 11322–11325 RSC; (c) J. Meesin, N. Chotsaeng and C. Kuhakarn, Synlett, 2022, 1317–1322 CrossRef CAS.
- (a) S. Song, X. Li, J. Wei, W. Wang, Y. Zhang, L. Ai, Y. Zhu, X. Shi, X. Zhang and N. Jiao, Nat. Catal., 2020, 3, 107–115 CrossRef CAS; (b) S. M. Maddox, C. J. Nalbandian, D. E. Smith and J. L. Gustafson, Org. Lett., 2015, 17, 1042–1045 CrossRef CAS PubMed; (c) R. C. Samanta and H. Yamamoto, Chem. – Eur. J., 2015, 21, 11976–11979 CrossRef CAS PubMed; (d) K. A. Miller, T. R. Welch, T. J. Greshock, Y. Ding, D. H. Sherman and R. M. Williams, J. Org. Chem., 2008, 73, 3116–3119 CrossRef CAS PubMed; (e) Y. Nishii, M. Ikeda, Y. Hayashi, S. Kawauchi and M. Miura, J. Am. Chem. Soc., 2020, 142, 1621–1629 CrossRef CAS PubMed; (f) X. Jiang, J. Yang, F. Zhang, P. Yu, P. Yi, Y. Sun and Y. Wang, Adv. Synth. Catal., 2016, 358, 2678–2683 CrossRef CAS; (g) P. K. Chhattise, A. V. Ramaswamy and S. B. Waghmode, Tetrahedron Lett., 2008, 49, 189–194 CrossRef CAS.
- (a) P. Martin, Tetrahedron Lett., 1987, 28, 1645–1646 CrossRef CAS; (b) O. R. Suarez-Castillo, L. Beiza-Granados, M. Melendez-Rodriguez, A. Alvarez-Hernandez, M. S. Morales-Rios and P. Joseph-Nathan, J. Nat. Prod., 2006, 69, 1596–1600 CrossRef CAS PubMed; (c) Y. Liu and G. W. Gribble, J. Nat. Prod., 2002, 65, 748–749 CrossRef CAS PubMed; (d) V. Bocchi and G. Palla, Synthesis, 1982, 1096–1097 CrossRef CAS.
- (a) A. H. Sandtorv and H.-R. Bjorsvik, Adv. Synth. Catal., 2013, 355, 499–507 CrossRef CAS; (b) J. Yan, T. Ni and F. Yan, Tetrahedron Lett., 2015, 56, 1096–1098 CrossRef CAS.
- M. R. Brennan, K. L. Erickson, F. S. Szmalc, M. J. Tansey and J. M. Thornton, Heterocycles, 1986, 24, 2879–2885 CrossRef CAS.
- (a) R. A. Rodriguez, C.-M. Pan, Y. Yabe, Y. Kawamata, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2014, 136(19), 6908–6911 CrossRef CAS PubMed; (b) S. Song, X. Sun, X. Li, Y. Yuan and N. Jiao, Org. Lett., 2015, 17, 2886–2889 CrossRef CAS PubMed.
- L. Sun, X. Zhang, Z. Li, J. Ma, Z. Zeng and H. Jiang, Eur. J. Org. Chem., 2018, 4949–4952 CrossRef CAS.
- For selected important reviews on iodine(III) reagents: (a) L. A. Segura-Quezada, K. R. Torres-Carbajal, K. A. Juárez-Ornelas, A. J. Alonso-Castro, R. Ortiz-Alvarado, T. Dohi and C. R. Solorio-Alvarado, Org. Biomol. Chem., 2022, 20, 5009–5034 RSC and cited therein; (b) R. Budhwan, S. Yadav and S. Murarka, Org. Biomol. Chem., 2019, 17, 6326–6341 RSC; (c) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed; (d) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650–682 CrossRef CAS PubMed. For examples on hypervalent iodine reagents promoted halogenation of indoles. (e) S. C. Fosu, C. M. Hambira, A. D. Chen, J. R. Fuchs and D. A. Nagib, Chem, 2019, 5, 417–428 CrossRef CAS PubMed; (f) V. Himabindu, S. P. Parvathaneni and V. J. Rao, New J. Chem., 2018, 42, 18889–18893 RSC; (g) M. Wang, Y. Zhang, T. Wang, C. Wang, D. Xue and J. Xiao, Org. Lett., 2016, 18, 1976–1979 CrossRef CAS PubMed.
- (a) P. Demerseman, J. Guillaumel, J. M. Clavel and R. Royer, Tetrahedron Lett., 1978, 23, 2011–2012 CrossRef; (b) J. Guillaumel, P. Demerseman, J. M. Clavel and R. Royer, Tetrahedron, 1980, 36, 2459–2465 CrossRef CAS; (c) J. Guillaumel, P. Demerseman, J. M. Clavel and R. J. Royer, Heterocycl. Chem., 1980, 17, 1531–1536 CrossRef CAS.
- V. L. Reddy, P. S. Prathima, V. J. Rao and R. Bikshapathi, New J. Chem., 2018, 42, 20152–20155 RSC.
- X. Jiang, L. Yang, W. Yang, Y. Zhu, L. Fang and C. Yu, Org. Biomol. Chem., 2019, 17, 6920–6924 RSC.
- (a) A. Wang, Y.-Z. Liu, Z. Shen, Z. Qiao and X. Ma, Org. Lett., 2022, 24, 1454–1459 CrossRef CAS PubMed; (b) P. Liang, H. Zhao, T. Zhou, K. Zeng, W. Jiao, Y. Pan, Y. Liu, D. Fang, X. Ma and H. Shao, Adv. Synth. Catal., 2021, 363, 3532–3538 CrossRef CAS; (c) X. Ma, Y. Z. Liu, L. Du, J. W. Zhou and I. E. Markó, Nat. Commun., 2020, 11, 914 CrossRef CAS PubMed; (d) X. Ma, J. J. Farndon, T. A. Young and J. F. Bower, Angew. Chem., Int. Ed., 2017, 56, 14531–14535 CrossRef CAS PubMed.
- (a) M. G. Saulnier and G. W. Gribble, J. Org. Chem., 1983, 48, 2690–2695 CrossRef CAS; (b) J. Barluenga, M. Trincado, E. Rubio and J. M. Gonzalez, Angew. Chem., Int. Ed., 2003, 42, 2406–2409 CrossRef CAS PubMed; (c) Q. Huang and R. C. Larock, J. Org. Chem., 2003, 68, 7342–7349 CrossRef CAS PubMed; (d) M. Amjad and D. W. Knight, Tetrahedron Lett., 2004, 45, 539–541 CrossRef CAS; (e) D. Yue, T. Yao and R. C. Larock, J. Org. Chem., 2006, 71, 62–69 CrossRef CAS PubMed.
- (a) M. Nakajima, S. Nagasawa, K. Matsumoto, T. Kuribara, A. Muranaka, M. Uchiyama and T. Nemoto, Angew. Chem., Int. Ed., 2020, 59, 6847–6852 CrossRef CAS PubMed; (b) C. Xu, X. Song, J. Guo, S. Chen, J. Gao, J. Jiang, F. Gao, Y. Li and M. Wang, Org. Lett., 2018, 20, 3933–3937 CrossRef CAS PubMed; (c) Z. Xu, R. Jia, Z. Ma, S. Cao, L. Shen and H. Ji, Synlett, 2019, 1909–1913 CAS.
- (a) D.-D. Tu, L.-Y. Ma, X.-G. Tong, X. Deng and C.-F. Xia, Org. Lett., 2012, 14, 4830–4833 CrossRef CAS PubMed; (b) D. Kajiyama, T. Saitoh, S. Yamaguchi and S. Nishiyama, Synthesis, 2012, 1667–1671 CAS; (c) J. Loh and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 46–48 CrossRef PubMed.
- J. D. Hayler, S. L. B. Howie, R. G. Giles, A. Negus, P. W. Oxley, T. C. Walsgrove and M. Whiter, Org. Process Res. Dev., 1998, 2, 3–9 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01951e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |