
Selective reduction of nitroarenes using Ru/C and CaH2†
Ramiro
Robles-Henríquez
a,
Tomás
Chávez-Vega
a,
Sebastián
Gallardo-Fuentes
b,
Lucas
Lodeiro
 a,
Susan
Lühr
*a and
Marcelo
Vilches-Herrera
*a
a,
Susan
Lühr
*a and
Marcelo
Vilches-Herrera
*a
aChemistry Department, Faculty of Science, University of Chile, Las Palmeras 3425, Ñuñoa, Santiago, Chile. E-mail: susanluhr@uchile.cl; luis.vilches@u.uchile.cl
bInstituto de Química, Facultad de Ciencias, Pontificia Universidad Católica de Valparaíso, Avenida Universidad 330, Curauma, Valparaíso, Chile
First published on 28th November 2022
Abstract
Herein, we report an efficient and highly selective method for the reduction of aromatic, heteroaromatic and halonitro compounds using the readily available and cost-effective Ru/C as a catalyst along with unconventional CaH2 as a source of hydride. In most cases the corresponding anilines can be obtained by simple filtration without further purification. The use of 2-MeTHF and the simple operational work-up constitute a valid alternative to previous methodologies.
Introduction
The reduction of nitro compounds to the corresponding amines has been a matter of investigation since nitrobenzene was reduced to aniline using stoichiometric amounts of sodium sulfide more than a century ago.1 Since then, nitro compounds have been extensively used as starting materials for the synthesis of primary amines.2 The relevance of this transformation lies in the importance of amines in the synthesis of pharmaceuticals, agrochemicals and dyes, as well as intermediates or building blocks for the fine chemicals industry.3 Among the several methodologies developed for this pivotal transformation, including homogeneous catalysis,4 metal-free reductions,5 water–gas shift reaction,6 and the use of nanoparticles,7 the development of heterogeneous catalytic systems constitutes one of the most important approaches especially on an industrial scale,8 probably because of the simple work-up procedure, which consists of filtration and concentration to isolate the product. Typical heterogeneous catalysts are noble and non-noble metals such as palladium, platinum, ruthenium, iron, copper, manganese, nickel etc. These catalysts are used in the presence of molecular hydrogen (H2) or a hydrogen donor in catalytic (transfer) hydrogenation.9 Another approach is the use of hydride species that under aqueous or protic solvent conditions hydrolyze to give H2. Remarkably, despite the great advances in the field and the excellent contributions from different research groups, the main issue related to the use of heterogeneous catalysts is the chemoselective reduction of nitro groups in the presence of other reducible functionalities or nitro compounds containing heteroatoms.10 Thus, the development of new and highly chemoselective methodologies is of great relevance.11 Likewise, dehalogenation of halonitroaromatic compounds is commonly observed as a side reaction during the reduction reaction.12Among the available hydrides, aluminum hydride (LiAlH4) is a very reactive non-selective nitro reducing agent able to reduce other functionalities such as amides, nitriles and aldehydes among others, resulting in an obvious limitation for a more extended use.13 Moreover, LiAlH4 is a highly corrosive and flammable solid, and a tedious work-up is associated with its use. The presence of water results in the release of hydrogen, which can be ignited by the heat from the exothermic reaction.14 In contrast, boron hydrides and silanes have been pointed out as non-reactive reagents.15 On the other hand, sodium borohydride (NaBH4) is probably the most widely used reagent for this transformation,16 since its reactivity is enhanced by supported metal-based catalysts such as nickel, copper, and cobalt, and also palladium and platinum on carbon.17
Interestingly, calcium hydride (CaH2), which is normally used as a drying agent,18 has been scarcely exploited as a reducing agent. Only few reports exist about its utility as a hydride source for reduction reactions, despite its being air-stable, inexpensive, and easy to handle and by-products, such as calcium hydroxide (Ca(OH)2), derived from its use are non-toxic and can be separated by simple filtration.19 In 2005, the group of Okamoto reported the reduction of ketones and imines with the system CaH2/ZnX2.20 Two years later, in 2007, the same group expanded this work with the system ZnX2/CaH2/R3SiCl for the reduction of a variety of carbonyl compounds.21 In 2015 the group of Métay and Lemaire reported the reductive alkylation and reductive amination of carbonyl compounds using CaH2 but this time in combination with supported heterogeneous catalysts of Pd/C and Pt/C respectively.22 Based on these reports and as part of our continuing interest in the reduction of organic functionalities,23 we decided to explore the suitability of this hydride for the reduction of nitroarenes. Herein we report the highly selective reduction of aromatic and heteroaromatic nitro compounds using Ru/C and CaH2.
Results and discussion
In our seminal attempts, we studied the reduction of p-nitrophenol as a benchmark substrate. p-Nitrophenol is a toxic compound;24 nevertheless, it is a key intermediate in the synthesis of paracetamol (N-acetyl-4-aminophenol),25 one of the most prescribed drugs in the world, but also in the synthesis of dyes and agrochemicals.26 Using CaH2 and tetrahydrofuran (THF) as solvent, several supported heterogeneous catalysts were screened (Table 1).|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Conversion (%) | 1a (%) | Yield (%) |
| a Reaction conditions: 0.5 mmol substrate, [M] (5 mol%) and CaH2 (10 mmol) in THF (4.0 mL) at 100 °C for 20 hours in a sealed tube. Conversions, yields and ratios were determined by GC using decane as internal standard. | ||||
| 1 | Pt/C | 99 | <1 | 69 |
| 2 | Pd/C | 99 | <1 | 60 |
| 3 | Rh/C | 95 | 5 | 39 |
| 4 | Ru/C | 99 | 1 | 99 |
| 5 | Pt/Al2O3 | 95 | 5 | 81 |
| 6 | Pd/Al2O3 | 98 | 2 | 50 |
| 7 | Rh/Al2O3 | 99 | <1 | 62 |
| 8 | Ru/Al2O3 | 21 | 78 | 12 |
| 9 | Pt/SiO2 | 23 | 76 | 13 |
| 10 | Pd/SiO2 | 43 | 56 | 4 |
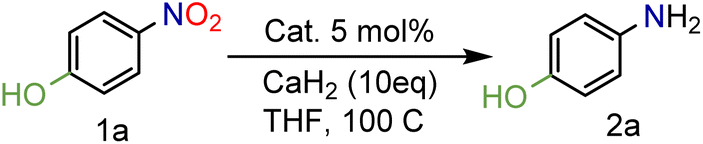
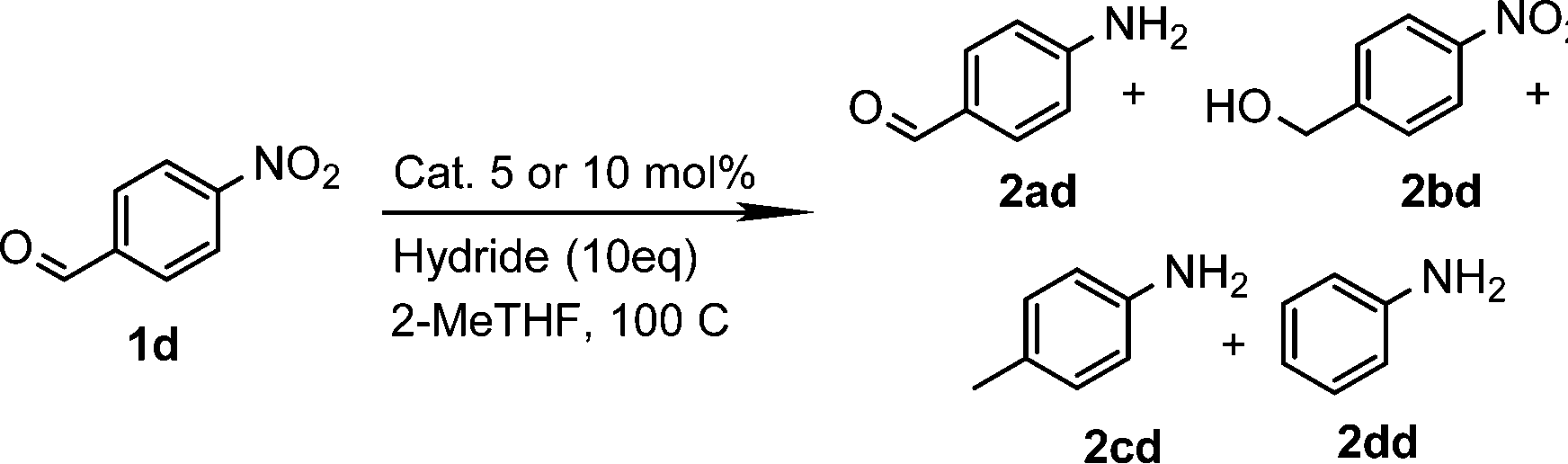
From these data, we observed that heterogeneous catalysts supported on carbon and alumina were the best systems in terms of conversion and yields (entries 1–7), except for Ru/Al (entry 8) with similar values to those for catalysts supported on silica (entries 9 and 10). On the other hand, it is well known that the type of metal employed in the reaction is the most important parameter regarding the activity and selectivity. Thus, it is accepted that, at least in the hydrogenation of nitrobenzenes, the activity of unsupported and carbon-supported metals is as follows: Pt > Pd > Rh > Ir > Os > Ru.27 In contrast to this accepted tendency, the highest activity of carbon-supported catalysts was found to be for Ru on carbon, with 99% conversion and 99% yield (entry 4). However, for alumina-supported catalysts, the tendency was as expected with Pt on alumina giving a significant yield of 81% (entry 5). Although ruthenium belongs to the platinum series, it is undoubtedly the most cost-effective metal amongst the precious metals. No less important is the fact that the order of selectivity in the reduction of halonitroarenes is Ru > Pt > Rh > Pd, which could be of great interest for the reduction of these substrates.28 Encouraged by this result, and as part of our interest in the development of sustainable methodologies,29 we decided to explore the use of 2-methyltetrahydrofuran (2-MeTHF) as an environmentally friendly alternative solvent30 to the more widely used THF and in line with the principles of Green Chemistry.31 Considering that solvents are the main source of waste in chemical processes,32 the introduction of eco-friendly alternatives has become an area of intense investigation.33 In this context, 2-MeTHF can be obtained from biomass resources and is abiotically degraded by sunlight and air.34 To our delight excellent conversion and yield were also obtained; therefore we selected it as a reaction medium for this reaction, although an additional screening of solvents was performed. In toluene and ethanol, the reaction did not work. In N-methylpyrrolidone and acetonitrile, 75% yield of the corresponding amine was obtained, whereas in DMF the yield was 90%. Thus, the reaction is still active in other solvents but affording low yields. The use of DMF is not attractive since it is toxic, cannot be as easily removed as THF or 2-MeTHF, and a more tedious work-up is necessary. Control experiments without catalyst or without CaH2 were performed. In addition, although great efforts have been developed to understand the nature of hydrides due to their capacity for hydrogen storage,35 less explored has been how different sources of hydrides can influence a particular reaction. Hence, comparative experiments using NaBH4 as an alternative hydride source with the carbon-supported catalysts were also performed (Table 2). It is important to note that the reduction of the substrate in question by NaBH4 has been reported to be thermodynamically feasible but kinetically restricted in the absence of a catalyst.36 As expected, no reaction occurred without catalyst (entry 2) or without CaH2 (entry 3). Under these conditions, the use of NaBH4 was less effective than using CaH2 in the synthesis of p-aminophenol when used in combination with Pd/C or Ru/C (entries 5 and 7) achieving only 1% and 72% yields respectively. Interestingly, the reduction of this substrate was reported by Neilson, Wood and Wylie in 1962 with 69% yield when the reaction was carried out with Pd/C at room temperature.37 The use of the expensive Pt/C (entry 4) showed better results than observed with CaH2 (Table 1, entry 1) but similar to those obtained with Ru/C and CaH2 (entry 1), with 98% conversion and yield. These results suggested to us that CaH2 could play an important role in the reduction. Therefore, in order to gain insights into its role in the reaction, an additional comparative experiment was performed by selecting a nitroarene containing a functionality reactive in the presence of NaBH4. In this context, NaBH4 is well known to afford the reduction of aromatic aldehydes to the corresponding benzyl alcohols in a reaction that proceeds without catalyst at room temperature.38 Under those conditions the nitro group remains unaffected.39 However, this can be transformed into the desired amine by modifying the properties of NaBH4 in the presence of a catalyst (e.g. Pd/C). Thus, it is accepted that the surface of the catalyst is crucial for the activation of the hydrogen.40 With this in mind, p-nitrobenzaldehyde was the substrate of choice (Table 3). From these data, we confirmed that the hydride source is a determinant for the outcome of the reaction. By using NaBH4, a mixture of products with a different distribution ratio was observed (entries 1–4), whereas the use of CaH2 clearly afforded more selective reactions (entries 5–8). To our delight, the chemoselective reduction of the nitro group (2ad, entry 8) was observed with Ru/C with 99% yield and conversion. On the other hand, interesting is the formation of p-methylaniline (2cd) as the main product using Pd/C or Rh/C with 92% and 70% yield respectively (entries 5 and 7). In turn, the reduction of the nitro group along with a decarbonylation reaction was observed with Pt/C with 52% yield (entry 6).
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Conversion (%) | Yield (%) |
| a Reaction conditions: 0.5 mmol substrate, [M] (5 mol%). b CaH2 (10 mmol) in 2-MeTHF (4.0 mL) at 100 °C for 20 hours in a sealed tube. c NaBH4 (5 mmol) in 2-MeTHF (4.0 mL) at 100 °C for 20 hours in a sealed tube. Conversions, yields and ratios were determined by GC. | |||
| 1b | Ru/C | 99 | 99 |
| 2b | — | 0 | 0 |
| 3b | Ru/C | 0 | 0 |
| 4c | Pt/C | 98 | 98 |
| 5c | Pd/C | 60 | <1 |
| 6c | Rh/C | 99 | 48 |
| 7c | Ru/C | 72 | 72 |
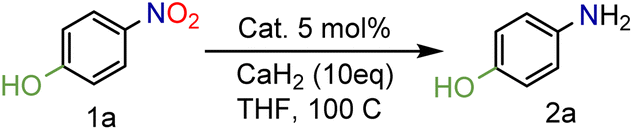
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
|||
|---|---|---|---|
| Entry | Cat. | Conv. (%) | Selectivity: 2ad/2bd/2cd/2dd |
| a Reaction conditions: 0.5 mmol substrate, [M] (5 mol%). b NaBH4 (5 mmol). c CaH2 (10 mmol), 2-MeTHF (4.0 mL), 100 °C, 20 hours. Conversions, yields and ratios were determined by GC. | |||
| 1b | Pd/C | 90 | 1/1/4/55 |
| 2b | Pt/C | 53 | 3/9/11/23 |
| 3b | Rh/C | 52 | 2/1/25/11 |
| 4b | Ru/C | 62 | 1/27/13/6 |
| 5c | Pd/C | 99 | 0/0/92/8 |
| 6c | Pt/C | 99 | 0/0/48/52 |
| 7c | Rh/C | 99 | 0/0/70/7 |
| 8c | Ru/C | 99 | 99/0/0/0 |
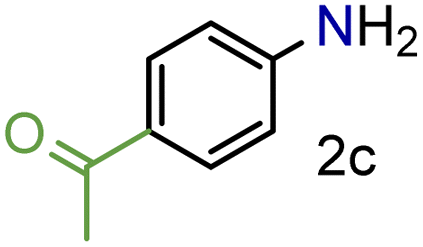
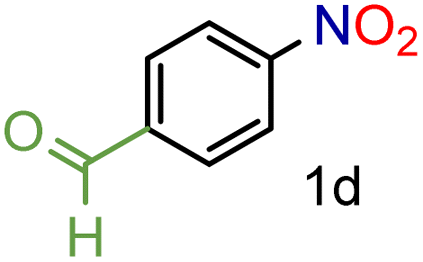
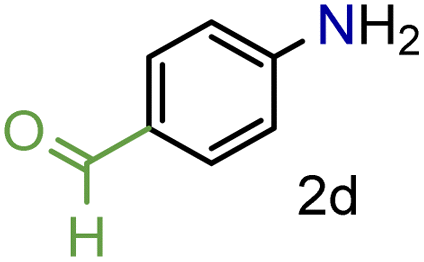
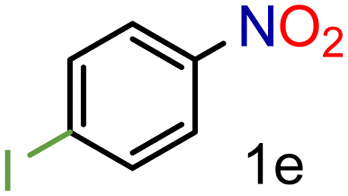
Encouraged by these results we decided to prove the scope and generality of the methodology. Several nitroarenes containing different functionalities were screened (Table 4). The products were identified using the corresponding commercially available anilines or those prepared according to literature reports. To validate the method as a green methodology, the reaction media were filtered off, concentrated under vacuum and the 1H NMR spectra were recorded without further purification (ESI†).
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Conv./sel (%) | GC yieldb (%) |
| a Reaction conditions: 0.5 mmol substrate, [M] (5 mol%) and CaH2 (10 mmol) in 2-MeTHF (4.0 mL) at 100 °C for 20 hours in a sealed tube. b Values in parentheses refer to isolated yields. c The reaction was scaled up 5 times. | ||||
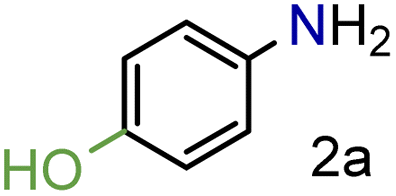
| 1c |
|
|
99/99 | 99 (96) |
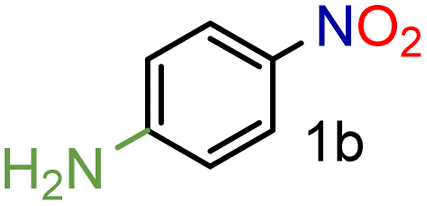
| 2 |
|
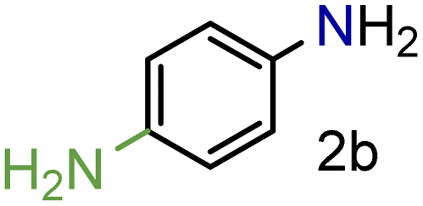
|
99/99 | 99 (97) |
| 3 |
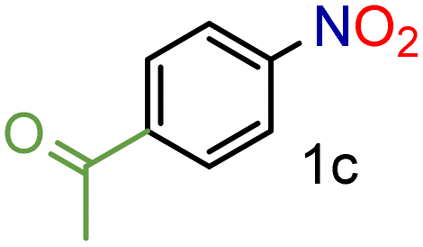
|
|
99/99 | 99 (97) |
| 4 |
|
|
98/99 | 98 (96) |
| 5 |
|
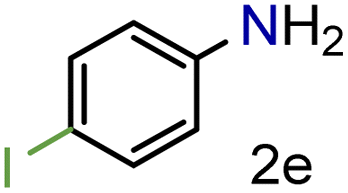
|
92/99 | 92 (90) |
| 6 |
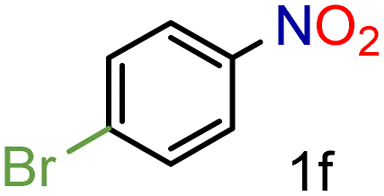
|
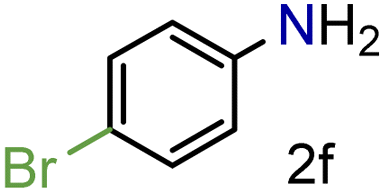
|
82/99 | 82 (77) |
| 7 |
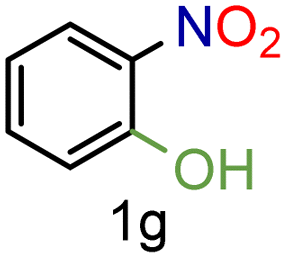
|
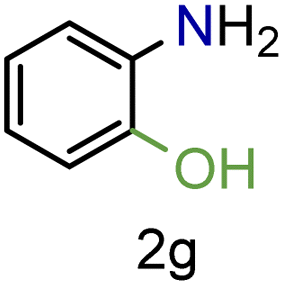
|
99/99 | 99 (96) |
| 8 |
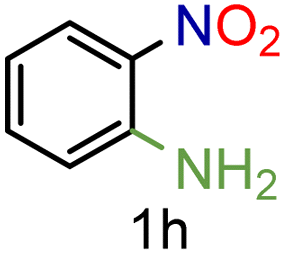
|
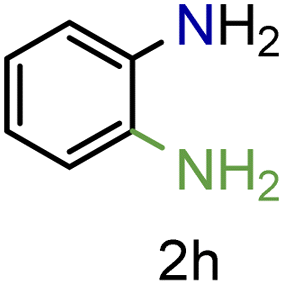
|
99/99 | 99 (95) |
| 9 |
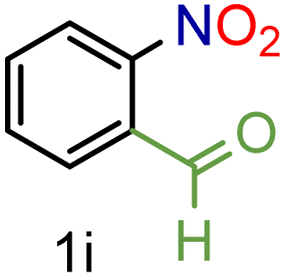
|
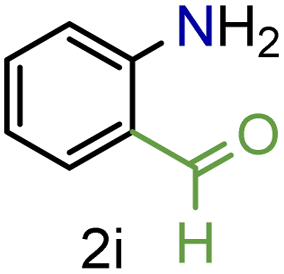
|
88/93 | 88 (80) |
| 10 |
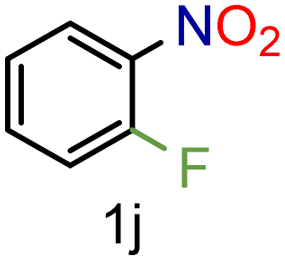
|
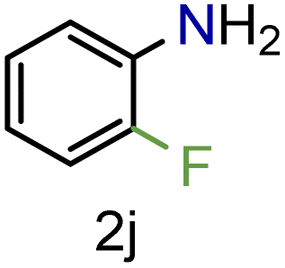
|
99/99 | 99 (91) |
| 11 |
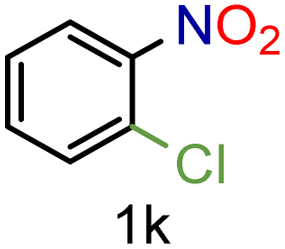
|
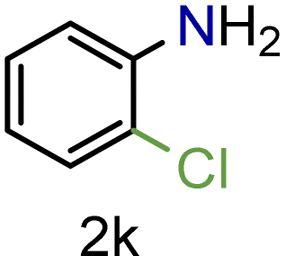
|
70/99a | 70 (55) |
| 12 |
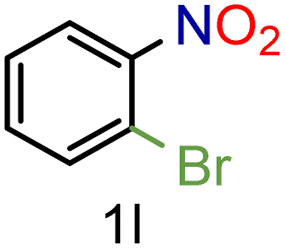
|
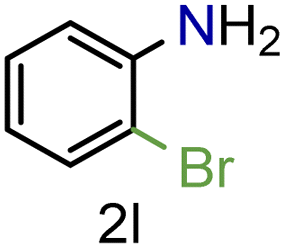
|
73/99 | 73 (71) |
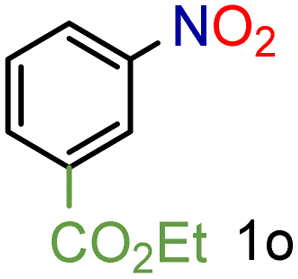
| 13 |
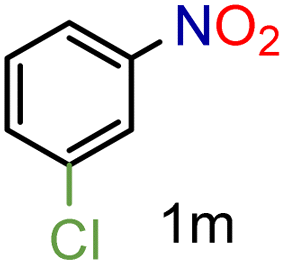
|
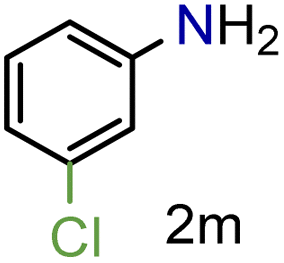
|
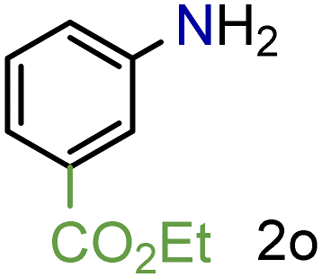
97/99b | 97 (97) |
| 14 |
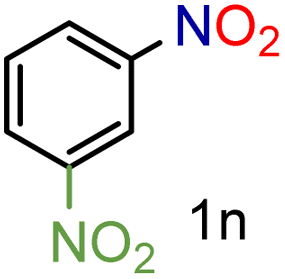
|
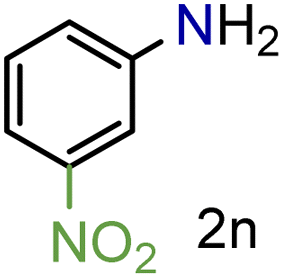
|
79/99 | 79 (76) |
| 15 |
|
|
97/99 | 97 (91) |
| 16 |
|
|
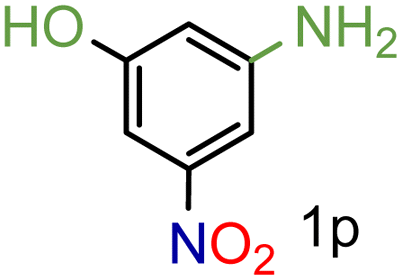
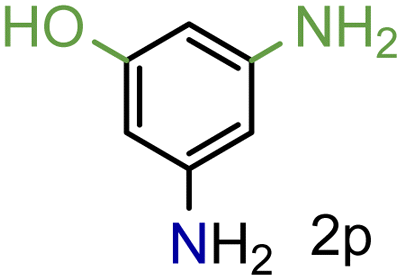
99/99 | 99 (98) |
| 17 |
|
|
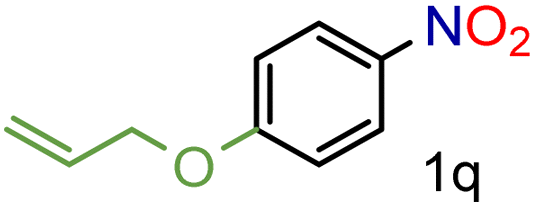
72/99 | 72 (71) |
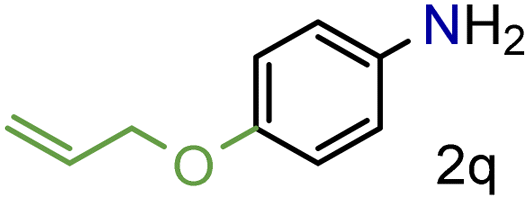
| 18 |
|
|
99/99 | 99 (99) |
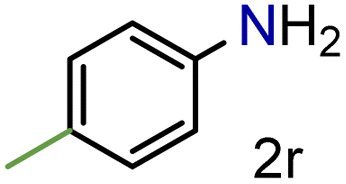
| 19 |
|
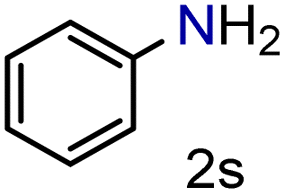
|
99/99 | 99 (99) |
| 20 |
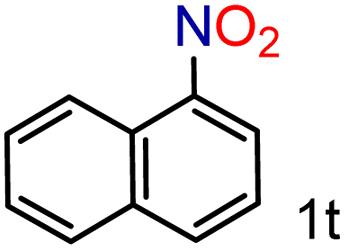
|
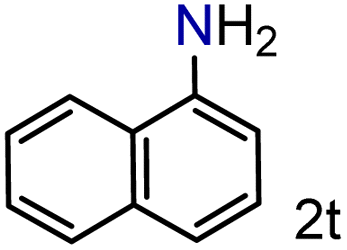
|
99/99 | 99 (92) |
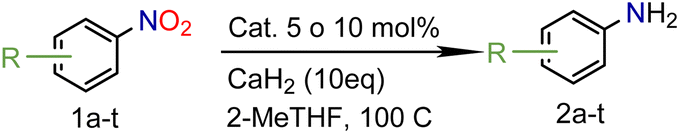
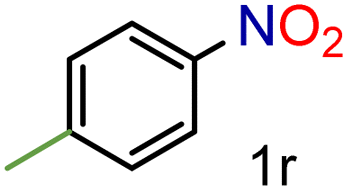
To our delight conversions and yields up to 99%, calculated on the basis of GC analysis, were obtained. The 1H NMR analysis of the crude products of the reactions showed the efficiency of the reduction and the catalytic system. The corresponding anilines were easily obtained in most cases without the necessity of column chromatography or other purification technique which constitutes a great advantage of the process. Remarkably, excellent selectivity was observed for all substrates containing other reducible functional groups (entries 3, 4, 9, 15 and 17). This is particularly relevant considering the selectivity problems associated with the use of heterogeneous catalysts.10 Aldehydes and ketones at 2- and 4-positions relative to the nitro group (entries 3, 4 and 9) remained unaffected during the reduction process and were obtained with good (87%) to excellent yields (99%). The corresponding 1H NMR analysis confirmed the presence of the formyl and carbonyl groups respectively. In the case of the ester group, it was obtained with 97% yield (entry 15) and a substrate containing an ether linkage with a double bond was well tolerated (entry 17) with 72% yield. The case of halonitroarenes (entries 5, 6, 10–13) represents one of the major achievements of this methodology. These compounds have been classified as very challenging substrates because one of the major issues frequently observed with their reduction is the undesired hydrodehalogenation.12 The C–X bond is a useful entity especially for cross-coupling reactions; therefore the presence of the halogen is relevant in molecules with nitro groups that need to be reduced. The position and nature of the halogen have also been investigated. Thus, the selectivity for the obtention of haloanilines has been ordered as F > Cl > Br > I.41 On the other hand, ortho- and para-substituted halonitroarenes have been pointed out to be more susceptible to the dehalogenation reaction than meta derivatives. From our results and in contrast to those studies, no dehalogenation was observed with 4-iodonitrobenzene, one of the most demanding and difficult substrates due to the easy hydrogenolysis of the C–I bond (entry 5, 92%). Similar result was obtained with 4-bromonitrobenzene (entry 6, 82%). ortho-Halo-substituted substrates were reduced without dehalogenation with moderate yields as in the case of 2-chloronitrobenzene (entry 11) but with good (73%) to excellent yields (99%) in the case of 2-bromo- and 2-fluoronitrobenzene respectively. Notably, in the reduction of 2-chloronitrobenzene, carried out at 10 mol% of catalyst loading, full conversion was obtained, nevertheless with a considerable amount of the dehalogenated product (41%). Therefore 5 mol% of the catalyst was used. Considering that a common strategy to suppress dehalogenation is the use of additives of different nature, metallic, acidic or basic, this methodology outperformed those methods reported in the literature since no additives are required. The selective partial reduction of 1,3-dinitrobenzene (entry 14) has already been observed with Ru/C but using molecular hydrogen at a pressure of 15 bar.42 The mono reduction was achieved with 79% yield along with 16% of m-phenylenediamine. Unfortunately, our attempts to obtain the latter by increasing the amount of CaH2 failed. The method was also efficient for the reduction of polyaromatic systems, such as 1-nitronaphthalene, which was reduced with excellent yield (entry 20). Considering the safety issues related to the use of H2, the facile procedure of this methodology constitutes another excellent alternative to the tedious and hazardous methods reported in the literature.43
Because several molecules include the presence of aromatic heterocycles containing the nitro functionality, we also tested a small library of substituted nitropyridines to expand the scope of the methodology (Table 5). Moreover, the selective reduction of nitro compounds in the presence of electronically activated heteroaryl halides remains challenging since these substrates are more susceptible to hydrodehalogenation.44
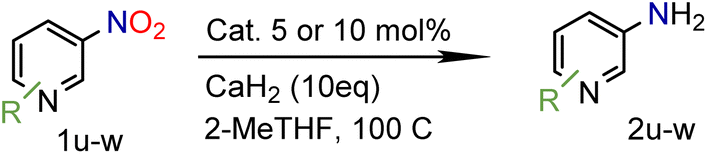
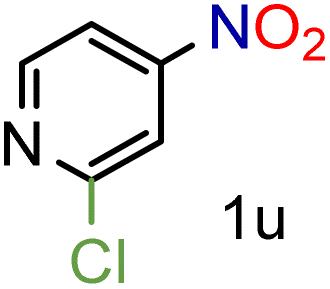
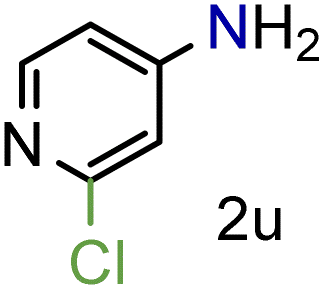
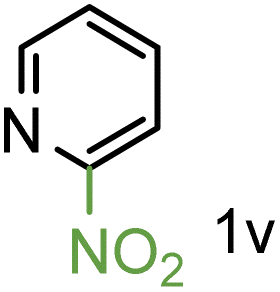
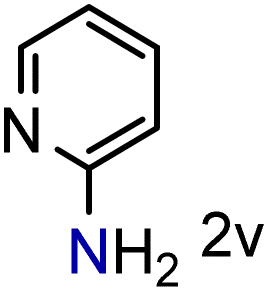
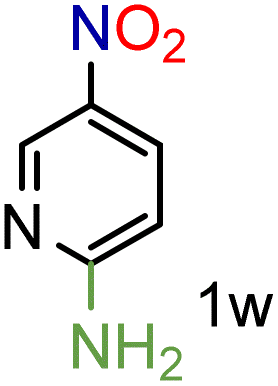
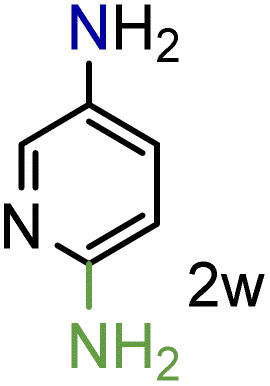
We were pleased to observe that these substrates could also be reduced with high yields and conversions (entries 1–3). Remarkably, the reduction of 2-chloronitropyridine occurs without dehalogenation (entry 1). 1H NMR analysis of the crude products of the reactions showed that no purification was necessary (ESI†).
Based on our experimental findings a computational study was carried out to gain more insight about the differences observed with the different catalysts addressing the question as to how metal surfaces activate the nitro group of aromatic substrates. Considering this as a binding phenomenon, the adsorption energies of the model substrate p-nitrophenol on Ru(0001), Rh(111), Pd(111), and Pt(111) surfaces were obtained with the aid of DFT calculations (see Computational Details). The corresponding adsorption energies (ΔEbind) of the resulting binding geometries were calculated as follows:
| ΔEbind = Eadsorbate − (Esurface + Esubstrate) | (1) |
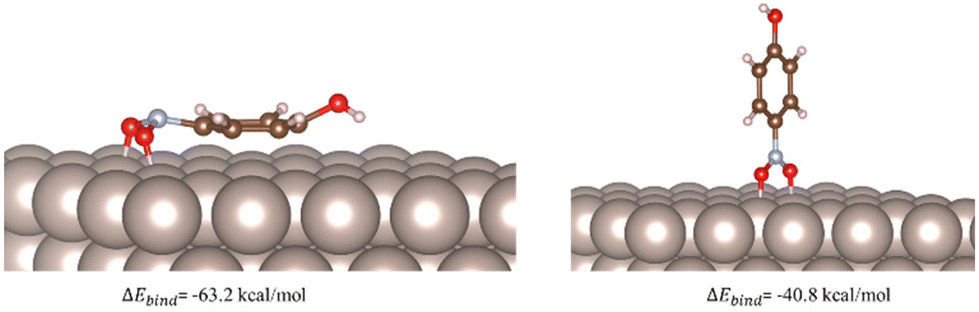 | ||
| Fig. 1 Computed binding geometries and adsorption energies for the physisorption of p-nitrophenol on the Ru(0001) surface. | ||
In order to understand the factors behind the observed chemoselectivity, we also computed the adsorption energies of p-nitrobenzaldehyde on the Ru(0001) surface. As expected, for the low-lying adsorbate geometry (ΔEbind = −78.2 kcal mol−1), the substrate binds to the metal surface with the aromatic ring oriented parallel to the surface. Again, this binding mode makes both the N–O bonds longer than in the isolated substrate (1.35 Å and 1.39 Å respectively) and the improper COON dihedral angle deviating 30.0° from planarity, whereas the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond is oriented nearly parallel to the surface with an improper OCHC dihedral angle of 8.5° (Fig. 2).
O bond is oriented nearly parallel to the surface with an improper OCHC dihedral angle of 8.5° (Fig. 2).
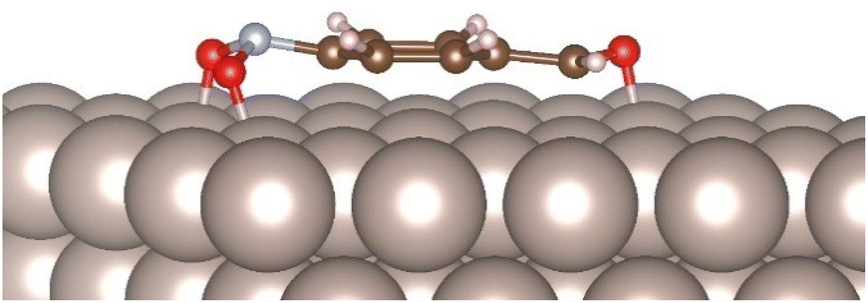 | ||
| Fig. 2 Computed low-lying binding geometry for the physisorption of p-nitrobenzaldehyde on Ru(0001) surface. | ||
As a result of this less geometrical deformation of the carbonyl functionality, the C–O bond elongation is less pronounced (from 1.22 Å to 1.30 Å) than that observed with the nitro group, highlighting that the adsorbed nitro functionality is more activated for the bond-forming and bond-cleavage processes and in consequence selectively reduced. However, elucidating the detailed mechanism of this transformation, including the role of the hydride source in the reaction, requires further theoretical and experimental studies beyond of the scope of the present work.
Experimental
General procedure
The corresponding nitro (hetero)arene (0.5 mmol) and Ru/C (5%; 5–10 mol%) were weighed into a 15 mL sealed tube and 4 mL of 2-MeTHF was added to the tube followed by CaH2 (10 equiv.). The reaction mixture was allowed to stir at 100 °C for 20 h. After cooling, the reaction was then filtered through a millipore syringe filter (Durapore (PVDF) with graduated multilayer glass prefilter, 25 mm diameter, 0.45 μm). 0.5 μL of the filtrated reaction was added to a vial containing additional 0.5 μL of 2-MeTHF and injected into a GC. The remaining solution was concentrated under vacuum and the corresponding 1H NMR spectra were recorded.Computational details
All calculations were performed with CP2K 7.145 using periodic boundary conditions. The wavefunctions and the electronic density were represented and extrapolated by the hybrid Gaussian and plane wave (GPW) method within the QuickStep module.46 Energy and force evaluations were performed using the PBE exchange–correlation functional (PBE96),47 with the Becke–Johnson damped Grimme's correction scheme DFT-D3(BJ) to account for dispersion interactions,48 with a cutoff radius of 32.0 Å and three-body terms. The molecular orbital occupation numbers were smeared with the Fermi–Dirac distribution at an electronic temperature of 300 K. The valence electron (Kohn–Sham) wavefunctions of all elements were expanded using the DZVP-MOLOPT gaussian basis set (-SR version for metallic elements),49 with the corresponding pseudopotential from the GTH library.50 Explicit valence shells included are: H(1s), C(2s2p), N(2s2p), O(2s2p), Br(4s4p), Ru(4s4p4d5s), which correspond to 1, 4, 5, 6, 7 and 16 valence electrons, respectively. The density was represented with a plane wave energy cutoff of 750 Ry, and a multigrid of five levels with a relative cutoff of 60 Ry. We used a convergence criterion of 10−7 for SCF cycles. Metallic ruthenium was optimized by means of simultaneous relaxation of cell and geometry over a two-atom hexagonal primitive cell [MP-Ru], maintaining cell symmetry and angles, at 0,1 kbar and using a Γ-centered Monkhorst–Pack k-point grid of 11 × 11 × 6. The ruthenium slab model was constructed for the (0001) surface with eight-atom layer thickness by means of geometrical relaxation with a Γ-centered Monkhorst–Pack k-point grid of 11 × 11 × 1. Finally, adsorption of organic molecules (through different functional groups of the molecules) on ruthenium was carried out with a 6 × 6 expanded Ru(0001) surface with a single Γ-centered k-point, constraining the position of the last three atomic layers of ruthenium.Conclusions
We have developed a new and simple synthetic methodology for the reduction of aromatic and heteroaromatic nitro compounds using Ru/C and CaH2 as hydride source. Our findings showed the key role of CaH2 which acts in a synergistic manner with the heterogeneous catalyst allowing one to reach high conversions and selectivities of nitro compounds containing other reducible functionalities. In addition, halonitroarenes can also be reduced without dehalogenation. In this context the reported method overcomes one of the main problems associated with the use of heterogeneous catalysts. The reaction proceeds without the use of additives in 2-MeTHF as an environmentally friendly solvent. In addition, the corresponding anilines can be obtained by simple filtration without the necessity of further purification. Because of this operational simplicity, the process aims to be an alternative to other reported methods.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Powered@NLHPC: This research was partially supported by the supercomputing infrastructure of the NLHPC (ECM-02). S. G.-F. acknowledges financial support by the ANID project FONDECYT Iniciación No. 11221216. We acknowledge the FONDECYT Regular Project No. 1210708.References
- H. K. Porter, in Organic Reactions, John Wiley & Sons, Hoboken,NJ, 2011, p. 455 Search PubMed.
- N. Ono, The Nitro Group in Organic Synthesis, John Wiley & Sons, New York, NY, 2001 Search PubMed.
- (a) F. Ullmann, W. Gerhartz, Y. S. Yamamoto, F. T. Campbell, R. Pfefferkorn and J. F. Rounsaville, Ullmann's Encyclopedia of Industrial Chemistry, Verlag Chemie, Weinheim, Germany, 2012, vol. A2, pp. 647–718 Search PubMed; (b) P. N. Rylander, Catalytic Hydrogenation in Organic Syntheses, Academic Press, 1979 Search PubMed; (c) S. Nishimura, Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, John Wiley & Sons, 2001 Search PubMed; (d) R. M. Bullock, Science, 2013, 342, 1054–1055 CrossRef CAS PubMed; (e) A. M. Smith and R. Whyman, Chem. Rev., 2014, 114, 5477–5510 CrossRef CAS PubMed.
- W. G. Jia, M. X. Cheng, Q. T. Xu, L. L. Gao and G. Yuan, Polyhedron, 2018, 69–75 CrossRef CAS.
- M. Orlandi, D. Brenna, R. Harms, S. Jost and M. Benaglia, Org. Process Res. Dev., 2018, 22, 430–445 CrossRef CAS.
- (a) A. M. Tafesh and J. Weiguny, Chem. Rev., 1996, 96, 2035–2052 CrossRef CAS PubMed; (b) S. Cenini and F. Ragaini, Catalytic Reductive Carbonylation of Organic Nitro Compounds, Kluwer Academic Press, Dordrecht, 1996 Search PubMed; (c) L. He, L. C. Wang and H. Sun, Angew. Chem., Int. Ed., 2009, 48, 9538–9541 CrossRef CAS PubMed; (d) J. Huang, L. Yu and L. He, Green Chem., 2011, 13, 2672–2677 RSC; (e) K. Nomura, J. Mol. Catal. A: Chem., 1995, 95, 203 Search PubMed; (f) F. Ragaini and S. Cenini, J. Mol. Catal. A: Chem., 1996, 105, 145 CrossRef CAS; (g) L. Liu, B. Qiao, Z. Chen, J. Zhang and Y. Deng, Chem. Commun., 2009, 653 RSC; (h) F. A. Westerhaus, I. Sorribes, G. Wienhöfer, K. Junge and M. Beller, Synlett, 2015, 26, 313 CrossRef CAS.
- (a) L. Gregor, A. K. Reilly, T. A. Dickstein, S. Mazhar, S. Bram, D. G. Morgan, Y. Losovyj, M. Pink, B. D. Stein, V. G. Matveeva and L. M. Bronstein, ACS Omega, 2018, 3, 14717–14725 CrossRef CAS PubMed; (b) Y. Duan, X. Dong, T. Song, Z. Wang, J. Xiao, Y. Yuan and Y. Yang, ChemSusChem, 2019, 12, 4636 CrossRef CAS PubMed.
- D. Formenti, F. Ferretti, F. K. Scharnagl and M. Beller, Chem. Rev., 2019, 119, 2611–2680 CrossRef CAS PubMed.
- (a) X. Chen, X. Y. Zhou, H. Wu, Y. Z. Lei and J. H. Li, Synth. Commun., 2018, 48, 2475–2484 Search PubMed; (b) Q. Tang, Z. Yuan, S. Jin, K. Yao, H. Yang, Q. Chi and B. Liu, React. Chem. Eng., 2020, 5, 58 Search PubMed; (c) B. Zeynizadeh, F. Mohammad Aminzadeh and H. Mousavi, Res. Chem. Intermed., 2021, 47, 3289–3312 CrossRef CAS.
- (a) H. S. Wei, X. Y. Liu, A. Q. Wang, L. L. Zhang, B. T. Qiao, X. F. Yang, Y. Q. Huang, S. Miao, J. Y. Liu and T. Zhang, Nat. Commun., 2014, 5, 8 Search PubMed; (b) Y. J. Shu, H. C. Chan, L. F. Xie, Z. P. Shi, Y. Tang and Q. S. Gao, ChemCatChem, 2017, 9, 4199–4205 CrossRef CAS; (c) H. U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 Search PubMed.
- (a) Z. N. Hu, J. Liang, K. Ding, Y. Ai, Q. Liang and H. B. Sun, Appl. Catal., A, 2021, 626, 118339 Search PubMed; (b) S. Sun, M. Du, R. Zhao, X. Jv, P. Hu, Q. Zhang and B. Wang, Green Chem., 2020, 22, 4640–4644 RSC.
- M. Pietrowski, Curr. Org. Chem., 2012, 9, 470–487 CAS.
- R. F. Nystrom and W. G. Brown, J. Am. Chem. Soc., 1948, 70(11), 3738–3740 CrossRef CAS PubMed.
- (a) P. Carson and C. Mumford, Hazardous Chemicals Handbook, Butterworth-Heinemann, Oxford, 2nd edn, 2002, p. 232 Search PubMed; (b) G. Weiss, Hazardous Chemicals Data Book, Noyes Data Corporation, New York, 2nd edn, 1985, p. 639 Search PubMed.
- E. R. Burkhardt and K. Matos, Chem. Rev., 2006, 106, 2617 Search PubMed.
- B. M. Trost and I. Fleming, Comprehensive Organic Synthesis, Selectivity, Strategy and Efficiency in Modern Organic Chemistry, Pergamon, Oxford, 1991 Search PubMed.
- (a) B. Zeynizadeh and K. J. Zahmatkesh, Chin. Chem. Soc., 2003, 50, 267 CrossRef CAS; (b) M. Periasamy and M. J. Thirumalaikumar, Organomet. Chem., 2000, 609, 137 Search PubMed; (c) A. Rahman and S. B. Jonnalagadda, Catal. Lett., 2008, 123, 264 Search PubMed; (d) I. Pogorelić, M. Filipan-Litvić, S. Merkaš, G. Ljubić, I. Cepanec and M. J. Litvić, Mol. Catal. A: Chem., 2007, 274, 202 CrossRef; (e) T. Satoh, S. Suzuki, Y. Suzuki, Y. Miyaji and Z. Imai, Tetrahedron Lett., 1969, 10, 4555 CrossRef.
- R. E. Gawley and A. Davis, Calcium Hydride, Encyclopedia of Reagents for Organic Synthesis, 2001, DOI:10.1002/047084289X.rc005.
- K. Nagashima, H. Visbal and K. Hirao, Int. J. Appl. Ceram. Technol., 2016, 13, 265–268 CrossRef CAS.
- T. Aida, N. Kuboki, K. Kato, W. Uchikawa, C. Matsuno and S. Okamoto, Tetrahedron Lett., 2005, 46, 1667–1669 CrossRef CAS.
- A. Tsuhako, J. Q. He, M. Mihara, N. Saino and S. Okamoto, Tetrahedron Lett., 2007, 48, 9120–9123 CrossRef CAS.
- (a) C. Guyon, E. Da Silva, R. Lafon, E. Métay and M. Lemaire, RSC Adv., 2015, 5, 2292 RSC; (b) C. Guyon, M.-C. Duclos, M. Sutter, E. Métay and M. Lemaire, Org. Biomol. Chem., 2015, 13, 7067–7075 Search PubMed.
- (a) M. Vilches-Herrera, S. Gallardo-Fuentes, M. Aravena-Opitz, M. Yáñez-Sánchez, H. Jiao, J. Holz, A. Börner and S. Lühr, J. Org. Chem., 2020, 85, 9213–9218 Search PubMed; (b) M. Vilches-Herrera, S. Werkmeister, K. Junge, A. Börner and M. Beller, Catal. Sci. Technol., 2014, 4, 629–632 RSC.
- (a) M. Misra, S. R. Chowdhury and T. I. Lee, Appl. Catal., B, 2020, 272, 118991 CrossRef CAS; (b) Z. Wu, X. Yuan and H. Zhong, et al. , Sci. Rep., 2016, 6, 25638 CrossRef CAS PubMed.
- Z. Dong, X. Le, X. Li, W. Zhang, C. Dong and J. Ma, Appl. Catal., B, 2014, 129–135 Search PubMed.
- (a) X. Zhu, S. Liang, S. Chen, X. Liu and R. Li, Catal. Sci. Technol., 2020, 10, 8332–8338 RSC; (b) P. P. Ghimire, L. Zhang and U. A. Kinga, et al. , J. Mater. Chem. A, 2019, 7, 9618–9628 RSC; (c) I. Ibrahim, I. O. Ali and T. M. Salama, et al. , Appl. Catal., B, 2016, 181, 389–402 Search PubMed.
- S. Nishimura, Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, Wiley-Interscience, 2001 Search PubMed.
- X. Wang, M. Liang, J. Zhang and Y. Wang, Curr. Org. Chem., 2007, 11, 299–314 Search PubMed.
- (a) N. Lezana, A. Galdámez, S. Lühr and M. Vilches-Herrera, Green Chem., 2016, 18, 3712–3717 Search PubMed; (b) M. Concha, A. Cortínez, N. Lezana, M. Vilches-Herrera and S. Lühr, Catal. Sci. Technol., 2022, 12, 6833–6890 Search PubMed.
- V. Pace, P. Hoyos, L. Castoldi, P. Dominguez de María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369–1379 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521–527 RSC.
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC.
- R. Palkovitz, Angew. Chem., Int. Ed., 2010, 49, 4336–4338 CrossRef PubMed.
- Q. Wang, J. Guo and P. Chen, Joule, 2020, 4, 705–709 CrossRef.
- B. Baruah, G. J. Gabriel, M. J. Akbashev and M. E. Booher, Langmuir, 2013, 29, 4225–4234 CrossRef CAS PubMed.
- T. Neilson, H. C. S. Wood and A. G. Wylie, J. Chem. Soc., 1962, 371–372 Search PubMed.
- (a) A. Wolfson and C. Dlugy, Org. Commun., 2009, 2, 34–41 Search PubMed; (b) S. Y. Han, E. M. Shulman-Roskes, K. Misiura, L. W. Anderson, E. Szymajda, M. P. Gamcsik, Y. H. Chang and S. M. Ludeman, 34, 1994, 247–254 CAS.
- (a) S. Senaweera and J. D. Weaver, Chem. Commun., 2017, 53, 7545–7548 RSC; (b) J. Desroches, P. A. Champagne, Y. Benhassine and J.-F. Paquin, Org. Biomol. Chem., 2015, 13, 2243–2246 Search PubMed.
- W. B. Smith, J. Heterocycl. Chem., 1987, 24, 745–748 CrossRef CAS.
- (a) M. Pietrowski, Selective hydrogenation of unsaturated functional groups over heterogeneous ruthenium catalysts, in Ruthenium: Properties, Production and Applications, ed. D. B. Watson, Nova Science Pub Inc., 2011 Search PubMed; (b) R. L. Augustine, Heterogeneous Catalysis for the Synthetic Chemist, CRC Press, 1996, p. 672 Search PubMed; (c) J. R. Kosak, Catalysis in organic synthesis, Academic Press, 1980 Search PubMed; (d) H. U. Blaser, A. Schnyder, H. Steiner, F. Rössler and P. Baumeister, Selective Hydrogenation of Functionalized Hydrocarbons, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH, 2008, vol. 8 Search PubMed.
- J. Hou, Y. Ma, Y. Li, F. Guo and L. Lu, Chem. Lett., 2008, 37, 974–975 CrossRef CAS.
- (a) T. W. Graham Solomons and C. B. Fryhle, Organic Chemistry, John Wiley & Sons, New York, 7th edn, 2000, p. 701 Search PubMed; (b) W. Herbst and K. Hunger, Industrial Organic Pigments: Production, Properties, Applications, Wiley-VCH, Weinheim, 3rd rev. edn, 2004, p. 185 CrossRef; (c) S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, Cambridge, 2004, p. 77 Search PubMed.
- A. Kasparian, C. Savarin, A. Allgeier and Sh. Walker, J. Org. Chem., 2011, 76, 9841–9844 CrossRef CAS PubMed.
- (a) J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 Search PubMed; (b) T. D. Kühne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. Hossein, M. H. Bani-Hashemian, V. Weber, U. Borštnik, M. Taillefumier, A. Shoshana Jakobovits, A. Lazzaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Glöβ, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- (a) S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703 CrossRef CAS PubMed; (b) C. Hartwigsen, S. Goedecker and J. Hutter, Phys. Rev. B, 1998, 58, 3641 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01807a |
| This journal is © The Royal Society of Chemistry 2023 |