
Targeting the tubulin C-terminal tail by charged small molecules†‡
Shuo
Li
 a,
Mattia
Mori
b,
Mingyan
Yang
a,
Soumia
Elfazazi
c,
Rafael
Hortigüela
c,
Peter
Chan
e,
Xinyue
Feng
d,
April
Risinger
e,
Zhiyou
Yang
d,
María Ángela
Oliva
c,
J.
Fernando Díaz
c and
Wei-Shuo
Fang
*a
a,
Mattia
Mori
b,
Mingyan
Yang
a,
Soumia
Elfazazi
c,
Rafael
Hortigüela
c,
Peter
Chan
e,
Xinyue
Feng
d,
April
Risinger
e,
Zhiyou
Yang
d,
María Ángela
Oliva
c,
J.
Fernando Díaz
c and
Wei-Shuo
Fang
*a
aState Key Laboratory of Bioactive Substances and Functions of Natural Medicines & MHC Key Laboratory of Biosynthesis of Natural Products, Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, 2A Nan Wei Road, Beijing 100050, China. E-mail: wfang@imm.ac.cn
bDepartment of Biotechnology, Chemistry and Pharmacy, University of Siena, via Aldo Moro 2, Siena 53100, Italy
cCentro de Investigaciones Biológicas Margarita Salas, Consejo Superior de Investigaciones Científicas, Ramiro de Maeztu 9, Madrid 28040, Spain
dCollege of Food Science and Technology, Guangdong Ocean University, Guangdong Provincial Key Laboratory of Aquatic Product Processing and Safety, Guangdong Province Engineering Laboratory for Marine Biological Products, Guangdong Provincial Engineering Technology Research Center of Seafood, Key Laboratory of Advanced Processing of Aquatic Product of Guangdong Higher Education Institution, Zhanjiang 524088, China
eDepartment of Pharmacology, University of Texas Health Science Center at San Antonio, San Antonio, Texas 78229, USA
First published on 28th November 2022
Abstract
The disordered tubulin C-terminal tail (CTT), which possesses a higher degree of heterogeneity, is the target for the interaction of many proteins and cellular components. Compared to the seven well-described binding sites of microtubule-targeting agents (MTAs) that localize on the globular tubulin core, tubulin CTT is far less explored. Therefore, tubulin CTT can be regarded as a novel site for the development of MTAs with distinct biochemical and cell biological properties. Here, we designed and synthesized linear and cyclic peptides containing multiple arginines (RRR), which are complementary to multiple acidic residues in tubulin CTT. Some of them showed moderate induction and promotion of tubulin polymerization. The most potent macrocyclic compound 1f was found to bind to tubulin CTT and thus exert its bioactivity. Such RRR containing compounds represent a starting point for the discovery of tubulin CTT-targeting agents with therapeutic potential.
Introduction
The microtubule (MT) cytoskeleton plays critical roles in multiple biological processes, including mitosis, intracellular signaling, trafficking, mobility and morphology. Apart from assembling into centrioles, axonemes and spindles, MTs also form extensive interactions with microtubule-associated proteins (MAPs) to efficiently regulate versatile MT functions. The α/β-tubulin heterodimer is the structural element that polymerizes into MTs. Human tubulins are encoded by multiple genes, consisting of at least six α-tubulin isotypes and seven β-tubulin isotypes.1 The spatial and temporal distribution of tubulin isotypes varies, and some isotypes are also closely related to drug resistance in cancer chemotherapy.2Over the past decades, hundreds of microtubule-targeting agents (MTAs) have been discovered, some of which are used in the treatment of cancers, gout3 and parasitic infection.4 Currently, at least seven MTA binding sites have been located within α- or β-tubulin or at their interface,5 with new sites continually being discovered.6,7 Taxane and vinca site MTAs have been widely used in clinical chemotherapy,5,8,9 but these suffer from toxicity and drug resistance, which limits their further applications. One form of clinically relevant drug resistance is due to the overexpression of certain tubulin isotypes (such as β-III), which has led to a pursuit of isotype selective MTAs but only with limited success to date.10–13 This is largely because the impacts of isotypic structural variations on MTA binding sites are poorly understood.
The tubulin C-terminal tail (CTT), defined by tens of C-terminal residues in both α- and β-tubulin subunits, varies more in the sequence between isotypes compared to the highly conserved tubulin core. Tubulin CTT is also subject to many types of post-translational modifications (tyrosination, glutamylation, glycylation, etc.), which imparts further structural complexity14 that can be interpreted by MAPs, motor proteins, plus-end tracking proteins, MT severing enzymes and other effectors to generate sophisticated biological readouts.15 Thus, MTAs that interact with tubulin CTT have the potential to target specific tubulin isotype(s) or to interfere with the interactions between MTs and their binding partners. Several peptide fragments, most of which are from internal peptides bound to MTs, have been discovered in a serendipitous way as tubulin CTT binders.16–19 However, the key structural elements that contribute to such binding remain to be illustrated.
Compared with the globular tubulin core, tubulin CTT lacks an ordered structure and is termed an intrinsically-disordered protein region (IDPR or IDR). Several in vitro and in silico studies indicated the intrinsic disorder properties of tubulin CTT.20–22 Targeting of an intrinsically-disordered protein (IDP) or IDPR is challenging because the vast majority of knowledge and principles that guide the interaction of ligands with proteins with defined structures may not be applied to the design of IDP binders. In practice, people choose to take a detour by targeting the ordered structure in the protein–protein interaction (PPI) complex, i.e., after the IDP is transited from the disordered state to the ordered state, such as in the p53–MDM2 interaction.23,24 However, such a transition for tubulin CTT remains to be illustrated. On the other hand, direct targeting of an IDP in its disordered state has also been attempted, such as for c-Myc, although only compounds with low micromolar binding efficacy resulted from this approach.25
To design a tubulin CTT binder, we chose to use the primary structure as our target and then introduced multiple charge centers in our compounds that are complementary to the abundant acidic residues (Asp, Glu) in tubulin CTT. We preferred this approach because the electrostatic interactions involving tubulin CTT are known to regulate the MT structure and function. The charge repulsion between adjacent tubulin CTTs destabilizes the MT lattice and influences MT dynamics, and the removal of tubulin CTT by subtilisin proteolysis lowers the critical concentration of tubulin polymerization and reduces the sensitivity of tubulin polymerization to ionic strength variation.26,27 Meanwhile, the clustered acidic residues within tubulin CTT may form discrete charge patches, providing an electrostatic recognition pattern for interaction with the basic grooves of tubulin CTT binding proteins. Here, we designed and synthesized a series of compounds containing triple arginines (RRR) in linear and cyclic forms, based on the study that tens of micromolar binding affinity is observed between polyarginine and polyglutamic acid.28 The binding of these compounds to tubulin CTT and its impact on tubulin polymerization were assessed.
Results and discussion
Synthesis of linear and cyclic RRR peptides
In order to balance the charged and hydrophilic RRR sequence, which is believed to have poor cell permeability, hydrophobic alkyl linkers were introduced to furnish macrocycles 1a–1f. The different length of linkers in the macrocycles also provided a way to fine-tune the rigidity of 1a–1f. The corresponding linear peptides 2a–2f were also synthesized as references to examine the relative influence of physicochemical properties and conformational rigidity. Furthermore, glycol linkers were incorporated to furnish macrocycles 3a–3d to explore the role of linkers with different chemotypes (Fig. 1).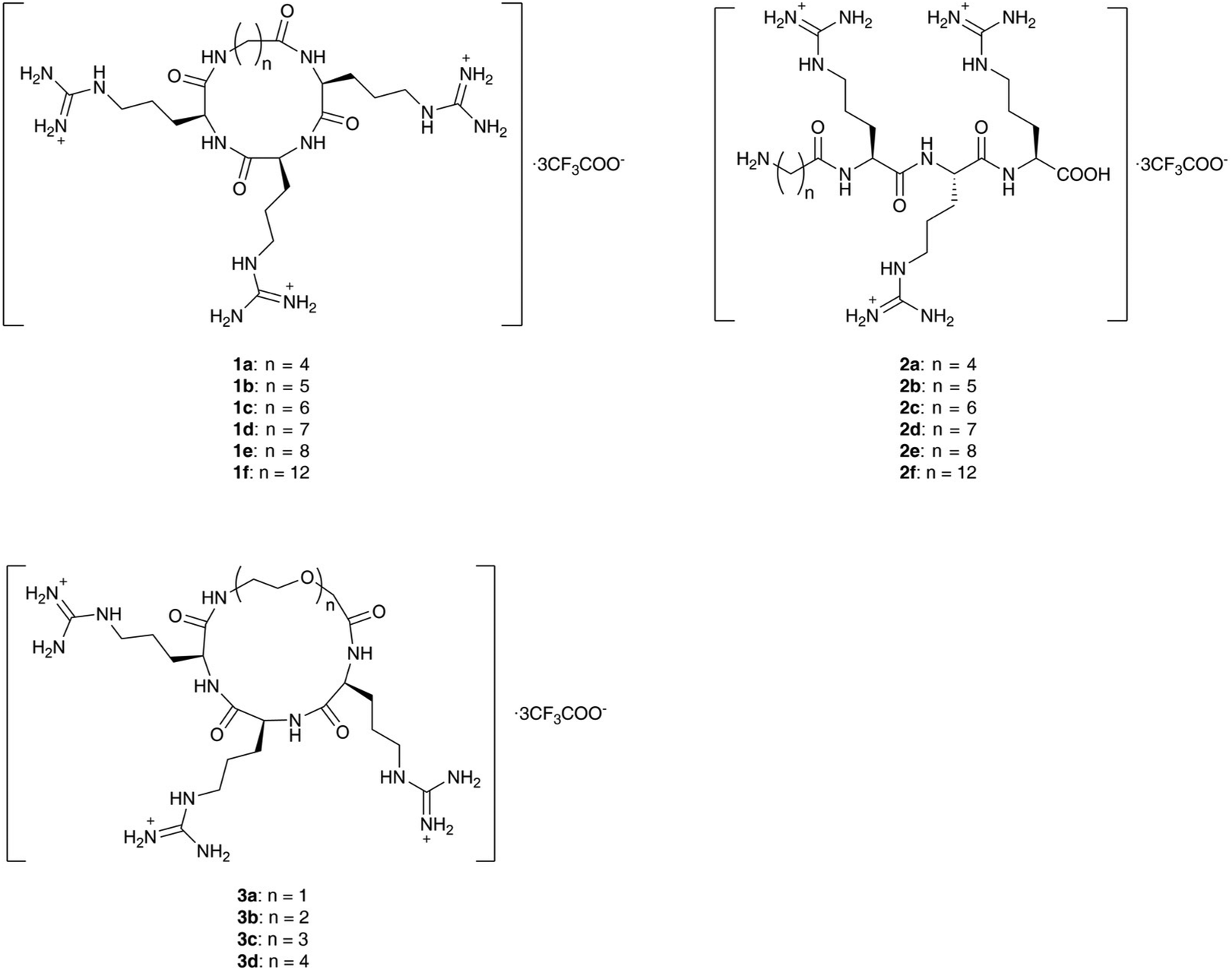 | ||
| Fig. 1 Structures of linear and cyclic peptides containing the RRR sequence to be synthesized. | ||
The alkyl linkers 4a–4f used in the synthesis of series 1 and 2 were prepared by N-protection of ω-amino acids or selective functionalization of dicarboxylic acids (Scheme S1‡). The glycol linkers 4g–4j used in the synthesis of series 3 were directly purchased (Scheme S1‡). Then peptide elongation and linker incorporation for all series 1–3 were performed on the acid-labile 2-chlorotrityl (2-CTC) resin29 by Fmoc-based solid phase synthesis.
For the synthesis of alkyl linked macrocycles 1a–1f, the linkers were initially attached to the N- or C-terminal of the RRR sequence. However, a poor yield was observed when such substrates were subjected to cyclization. Alternatively, the alkyl linkers were inserted into the RRR sequence to give the linear precursors 1a′–1f′ (Scheme 1), which were obtained after the treatment with 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP). The cyclization was realized in the solution phase by slowly adding the linear precursors 1a′–1f′ to a solution of coupling reagents to alleviate the competition of intermolecular oligomerization while maintaining a rapid reaction. Macrocycles in the protected form 1a′′–1f′′ were obtained in moderate to high yields and no oligomerized byproducts were observed. Finally, deprotection by a TFA-based cleavage cocktail afforded the desired macrocycles 1a–1f (Scheme 1).
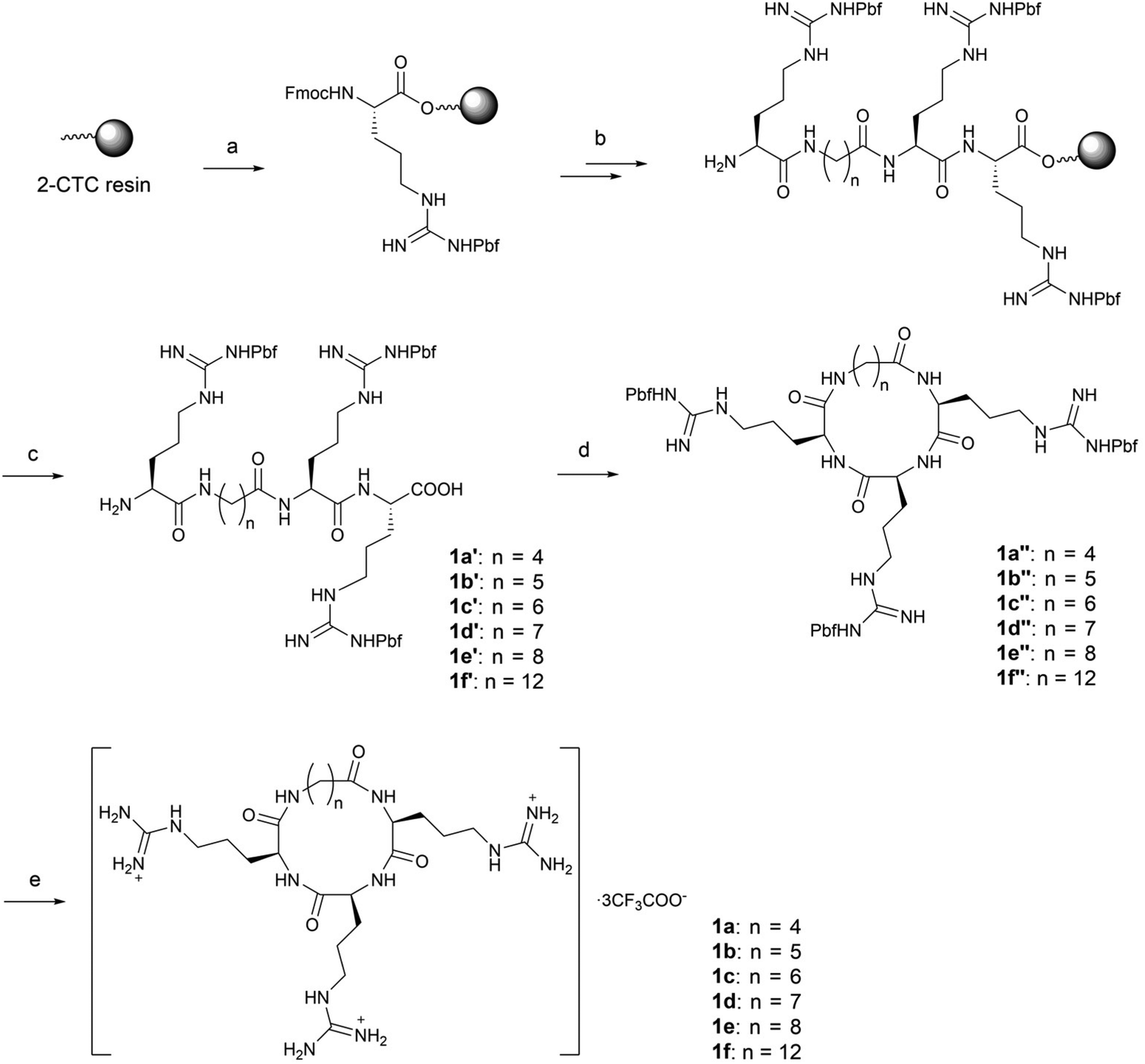 | ||
Scheme 1 Synthesis of compounds 1a–1f. Reagents and conditions: (a) Fmoc-Arg(Pbf)-OH, DIEA, DMF, r.t.; (b) standard solid-phase peptide synthesis procedure: (i) coupling: corresponding alkyl linkers 4a–4f, HBTU, HOBt, DIEA, DMF, r.t.; (ii) Fmoc deprotection: 20% piperidine/DMF, r.t.; (iii) Phth deprotection: 60% hydrazine/DMF, r.t.; (c) 20% HFIP/DCM, r.t.; (d) HATU, DIEA, DMF, 0 °C; (e) TFA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TIS:H2O = 95:2.5:2.5, r.t., 35–44% overall yield. :TIS:H2O = 95:2.5:2.5, r.t., 35–44% overall yield. | ||
The linear peptides bearing alkyl linkers, 2a–2f, were directly cleaved from the resin by the TFA-based cocktail after sequential conjugation of triple arginines (RRR) and the corresponding alkyl linkers (Scheme 2).
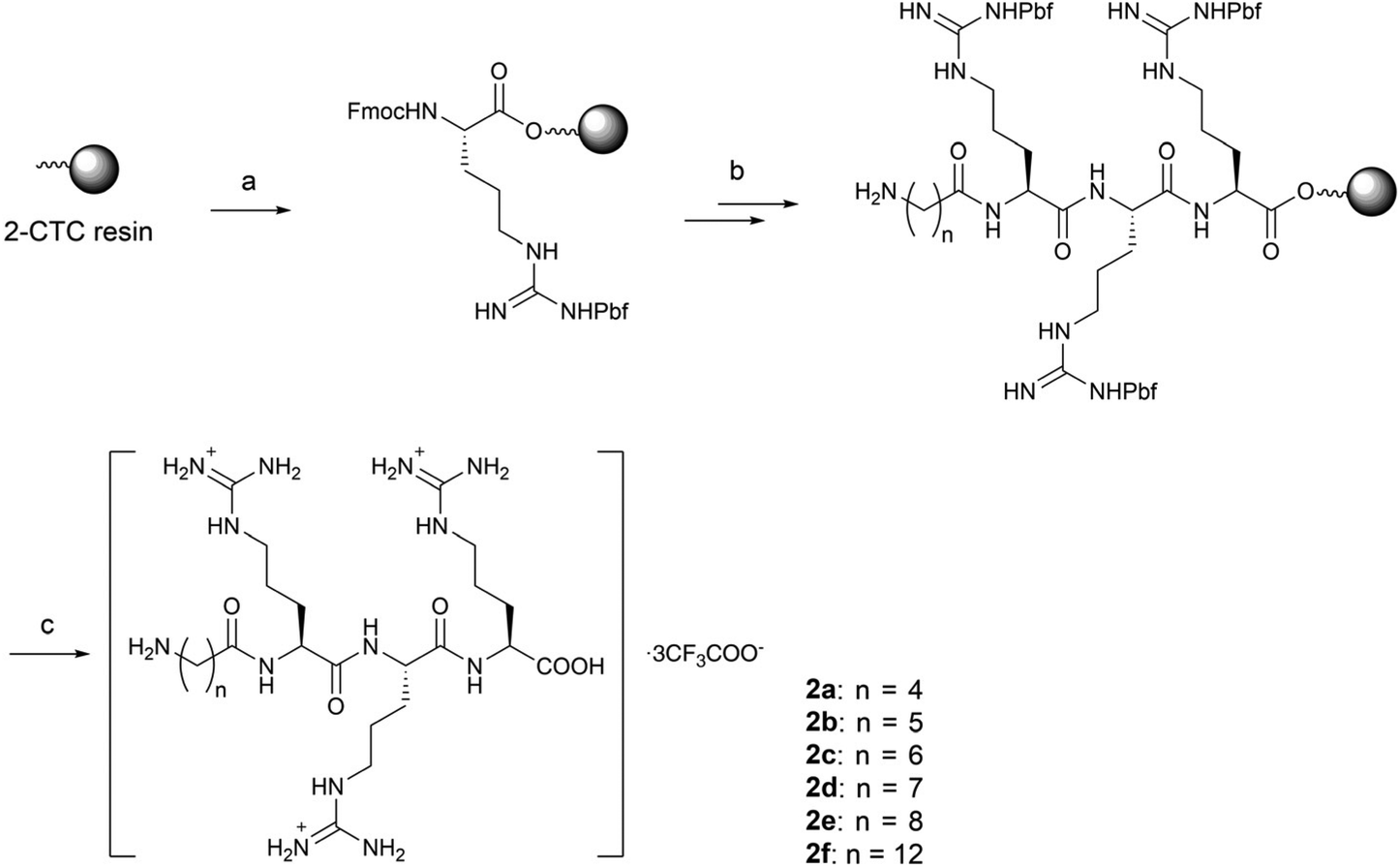 | ||
| Scheme 2 Synthesis of compounds 2a–2f. Reagents and conditions: (a) Fmoc-Arg(Pbf)-OH, DIEA, DMF, r.t.; (b) standard solid-phase peptide synthesis procedure: (i) coupling: corresponding alkyl linkers 4a–4f, HBTU, HOBt, DIEA, DMF, r.t.; (ii) Fmoc deprotection: 20% piperidine/DMF, r.t.; (iii) Phth deprotection: 60% hydrazine/DMF, r.t.; (c) TFA:TIS:H2O = 95:2.5:2.5, r.t., 17–50% overall yield. | ||
For glycol linked macrocycles 3a–3d, we introduced linkers containing one to four ethylene glycols (4–13 atoms length) in order to compare with the length of alkyl linkers (4–12 atoms length) in series 1. The synthesis of 3a–3d was realized in a similar way to that of macrocycles 1a–1f (Scheme 3).
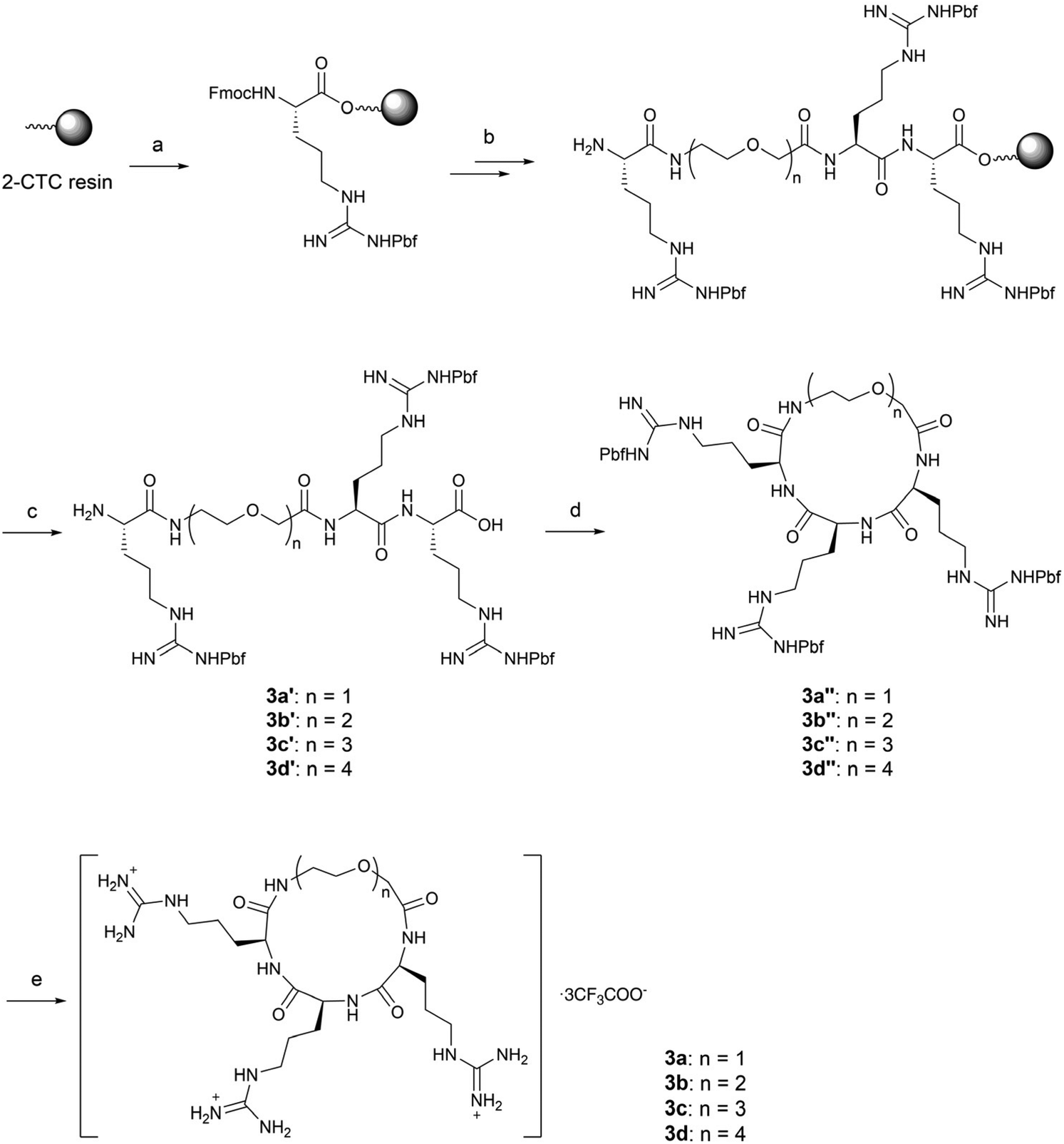 | ||
| Scheme 3 Synthesis of compounds 3a–3d. Reagents and conditions: (a) Fmoc-Arg(Pbf)-OH, DIEA, DMF, r.t.; (b) standard solid-phase peptide synthesis procedure: (i) coupling: corresponding glycol linkers 4g–4j, HBTU, HOBt, DIEA, DMF, r.t.; (ii) Fmoc deprotection: 20% piperidine/DMF, r.t.; (c) 20% HFIP/DCM, r.t.; (d) HATU, DIEA, DMF, 0 °C; (e) TFA:TIS:H2O = 95:2.5:2.5, r.t., 12–33% overall yield. | ||
Linear and cyclic RRR peptides promote tubulin polymerization and bind to the tubulin C-terminal tail
Next, we screened all the above linear and cyclic RRR peptides in a biochemical tubulin polymerization assay. The result showed that most of the series 1 compounds at 100 μM were able to promote tubulin polymerization comparable to that of paclitaxel at 10 μM, and series 2 and 3 had slightly weaker activity (Fig. 2A). This demonstrated that the RRR sequence was able to provide the basic requirement for promoting tubulin polymerization, possibly through interacting with tubulin CTT and shielding the acidic residues. | ||
| Fig. 2 Effect on tubulin polymerization of the linear and cyclic peptides containing the RRR sequence. A: biochemical tubulin polymerization assay of series 1–3. The final concentration of the compounds was 100 μM. B: biochemical tubulin polymerization assay of 1a–1f at multiple concentrations. The concentration of tubulin was 20 μM in all experiments. Paclitaxel (10 μM) and podophyllotoxin (10 μM) were used as references in all assays. | ||
The effects of series 1 on biochemical tubulin polymerization at different concentrations were also evaluated. Macrocycles 1 bearing longer alkyl linkers induced a greater extent of tubulin polymerization and a shortened nucleation phase, which was more apparent at higher concentrations (Fig. 2B). Below 30 μM, none of the macrocycles 1a–1f were able to promote tubulin polymerization (Fig. 2B). On the other hand, although the linear peptides 2a–2e and the glycol-linked macrocycles 3a–3d promoted tubulin polymerization at 100 μM, they showed poor correlation between the activity and the linker length. It is worth noting that 2f did not show any tubulin polymerization promotion ability (vs. DMSO), whereas its counterpart 1f is the most potent compound in series 1.
A closer view of the structure–activity relationship for three series of compounds with comparable length linkers, i.e., 1a/2a/3a, 1d/2d/3b and 1f/2f/3d, provides some more information. While the macrocycles 1a/1d showed comparable activity to their linear counterparts 2a/2d in the tubulin polymerization assay, 1f induced a much higher extent of tubulin polymerization and significantly shortened the nucleation phase compared its linear counterpart 2f (Fig. S2‡). This suggests the importance of a balance of flexibility and rigidity in the compounds to promote tubulin polymerization. The guanidine group of arginine, the assumed structural segment that contributes to biochemical activity, experiences more constraints in the macrocycles with shorter linkers which are not beneficial for tubulin polymerization, whereas arginines (or guanidine groups) constrained to some extent in the macrocycles with longer linkers are beneficial for tubulin polymerization.
The morphology of the polymerized MTs in the presence of 1f was also observed. The polymerized MTs were prepared with 1 mM of 1f. As expected, 1f significantly enhanced the turbidity of the system to about one order of magnitude higher than 50 μM paclitaxel did (data not shown). Highly increased turbidity is usually related to the formation of open sheets, stacking rings or helical arrangements, but in this case we found that 1f induced the formation of normal MTs (Fig. 3).
 | ||
| Fig. 3 Negative stain electron microscopy of microtubules assembled in the presence of 1f. | ||
We used a co-sedimentation assay to obtain a direct confirmation of the binding of 1f to MTs, and to do so we incubated 1f with crosslinked stabilized MTs or tubulin under assembly conditions, followed by centrifugation and SDS-PAGE analysis of pellets and supernatants. In the absence of MTs 1f remained fully soluble in the supernatant (96 ± 1%, n = 3), while in the presence of MTs it co-sedimented with both crosslinked stabilized MTs (ν = 0.9 ± 0.1) and native MTs (ν = 0.64 ± 0.05) (Fig. 4). It should be noted that only MTs sedimented under the centrifugation conditions (no tubulin dimers, tubulin aggregates or the peptide) and hence, the presence of the macrocycle 1f in the sedimentation suggested its interaction with MTs.
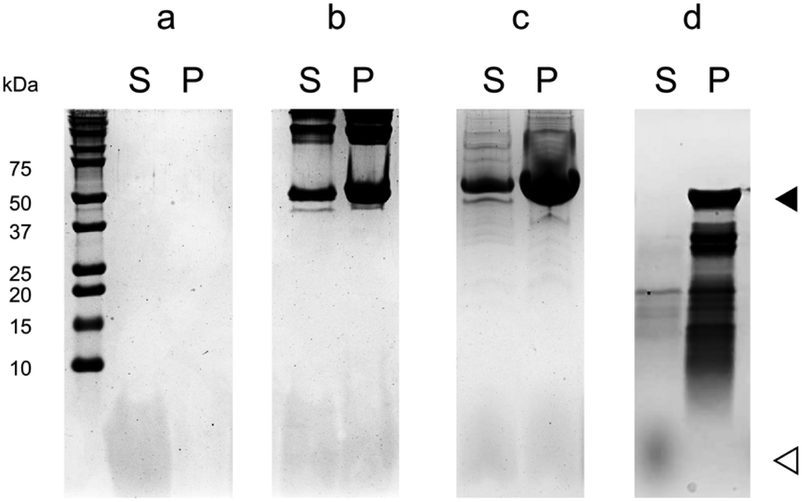 | ||
| Fig. 4 Co-sedimentation assay for the interaction between 1f and the microtubule. The protein weight is indicated by the marker in the left-most column. The filled triangle represents tubulin subunits and the hollowed triangle represents 1f. Compound 1f was incubated (a) without a protein, (b) with crosslinked stabilized MTs, (c) with native MTs, and (d) with MTs assembled from subtilisin-digested tubulin before subjecting to SDS-PAGE. S, supernatant; P, precipitate. | ||
In order to confirm the binding site, we analyzed the interaction of 1f with MTs assembled from subtilisin-digested tubulin, in which tubulin CTT was cleaved by this protease upon short periods of incubation (Fig. S1‡).30 Unlike in the case of crosslinked stabilized MTs or native MTs, 1f was not able to co-sediment with MTs assembled from subtilisin-digested tubulin (Fig. 4), suggesting that 1f directly binds to MTs through tubulin CTT.
Cyclic RRR peptides are capable of reversing axonal atrophy
The biological consequences of our compounds were assessed. We performed cellular assays and found that the incubation of 1f at as high as 10 mM with the SH-SY5Y cancer cell line did not inhibit cellular proliferation. Meanwhile, we found that 50 μM of 1f did not affect the proliferation or the appearance of cellular MTs in MDA-MB-231 or HCC1937 TNBC cell lines.We also investigated whether these compounds act against Aβ25–35-induced axonal atrophy. Aβ25–35 was dissolved in distilled water and incubated at 37 °C for 4 days for aggregation. Thereafter, mouse primary neurons were pretreated with 10 μM Aβ25–35 for 0.5 h and then co-treated with all compounds 1–3 for 3 days. Compared with the control, Aβ25–35 significantly decreased the axonal density, while compounds 1f, 3a, and 3c (10 μM) partially reversed Aβ25–35-induced axonal atrophy (Fig. 5).
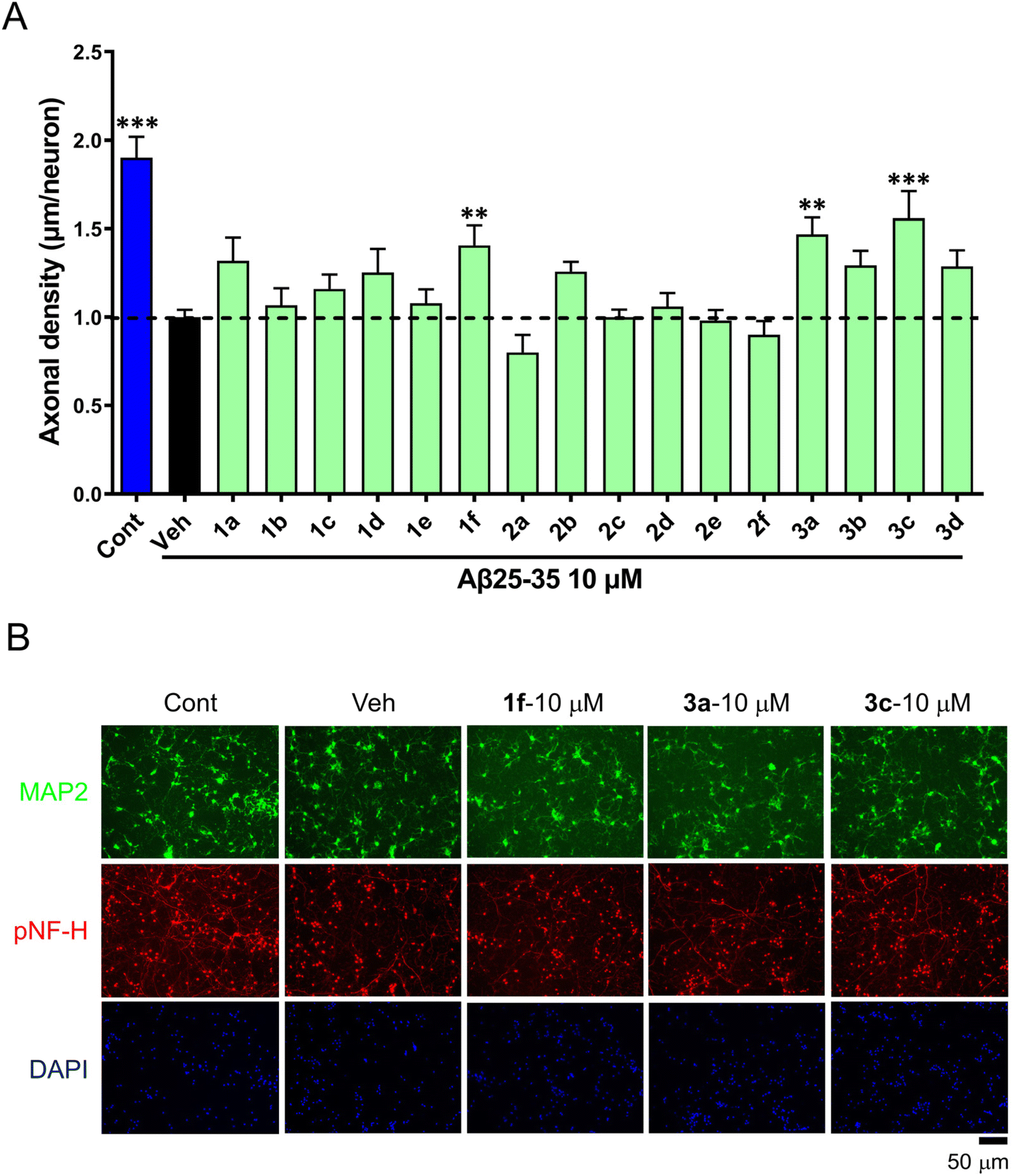 | ||
| Fig. 5 The effects of compounds against Aβ25–35-induced axonal atrophy in cultured primary cortical neurons. The cortical neurons were cultured for 3 days, pretreated with 10 μM Aβ25–35 for 0.5 h, and post co-cultured with compounds for 3 days. A: normalized statistical axonal density. The error bars represent the standard error of mean (n = 10–23). **p < 0.01, ***p < 0.001 vs. vehicle, one-way ANOVA post hoc Dunnett's test was performed. B: the representative photos of p-neurofilament heavy subunit (pNF-H)-positive neurites and MAP2-positive neurons. Cont, control; Veh, vehicle. | ||
Together, these data suggested that 1f was unable to disrupt the gross MT structure in cancer cells, but may function in mouse neurons, possibly through tubulin/MT modulation. However, the cellular bioavailability and/or target engagement should be further assessed.
Molecular dynamics simulation suggests efficient interaction between series 1 and the tubulin C-terminal tail
To gain insight into the potential recognition pattern between series 1 compounds and tubulin CTT, we performed molecular dynamics (MD) simulations. We initially examined the simulated binding between the compounds and the isolated peptide of βI-tubulin CTT (Ac-EEEEDFGEEAEEEA). As expected, the tubulin CTT peptide maintained a high degree of disorder during MD simulations and all tested compounds (1a, 1e and 1f) were predicted to bind to tubulin CTT. Theoretical affinity was estimated by the molecular mechanics Poisson–Boltzmann surface area (MM/PBSA) and generalized Born surface area (MM/GBSA) approaches, showing that 1a had a slightly higher interaction energy with tubulin CTT compared to 1e and 1f, with the latter two showing similar delta energies of binding values (Fig. 6A). The recognition was mostly driven by long-range electrostatic interactions, whereas hydrogen bonds established between arginines and acidic residues in tubulin CTT stabilized the complex (data not shown). No specific role of the alkyl linker in the interaction was observed.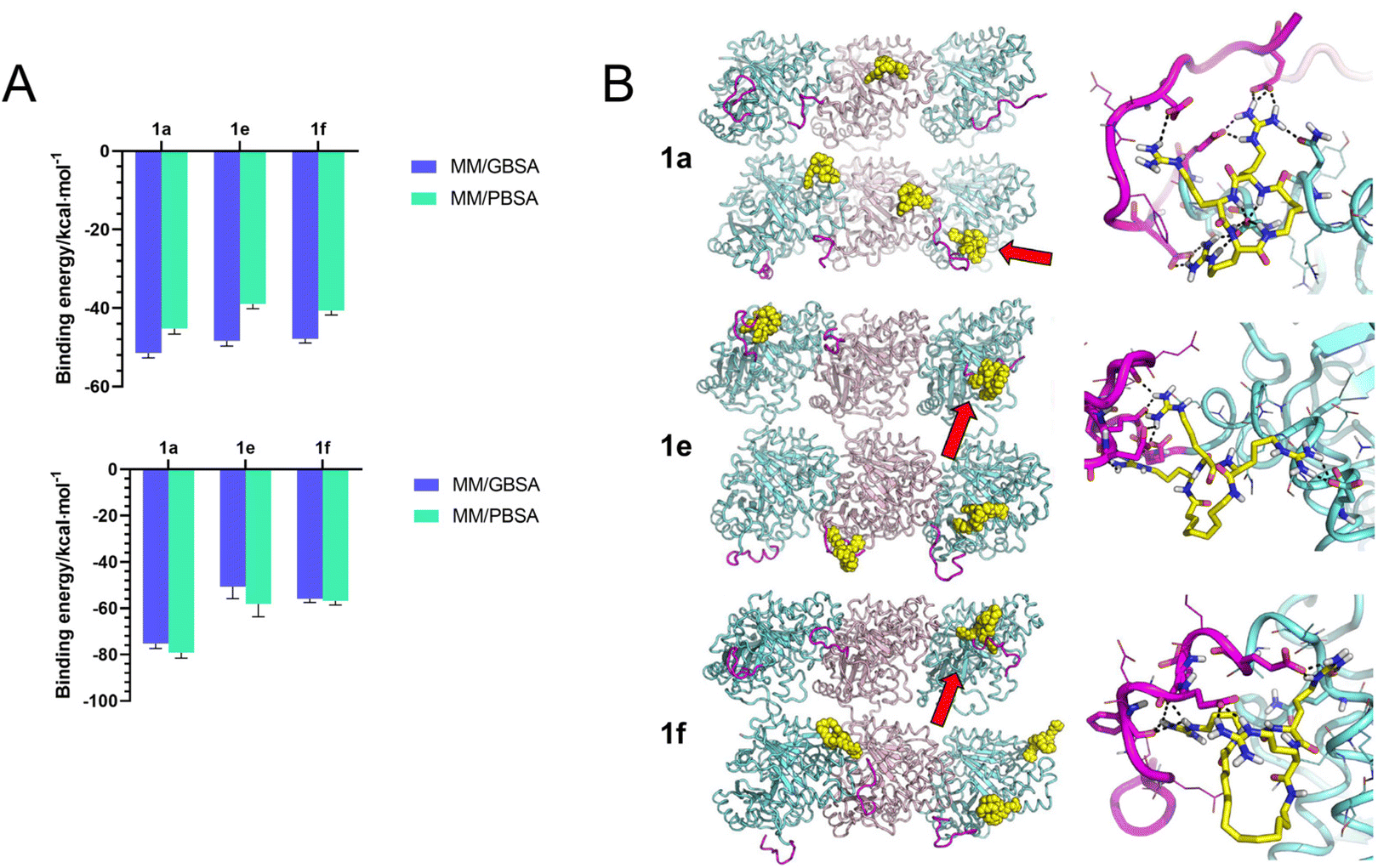 | ||
| Fig. 6 In silico prediction of the interaction between series 1 and the tubulin C-terminal tail. A: theoretical binding energy of 1a, 1e and 1f to the isolated peptide of βI-tubulin CTT (top) or tubulin CTT in the MT lattice (bottom). B: interaction between 1a, 1e and 1f with the MT lattice. The overview of the binding pattern is shown on the left. The interaction between compounds and tubulin CTT is indicated by red arrows and shown in detail on the right. Pink ribbon, α-tubulin. Cyan ribbon, β-tubulin. Purple ribbon, tubulin CTT. Yellow, compounds 1a, 1e and 1f. | ||
Next, we investigated whether 1a, 1e and 1f could bind to tubulin CTT in the context of a MT lattice. The MD trajectories of the MT lattice were first simulated in the absence of any compounds. The conformation of the tubulin core was stable in MD simulations, while the conformation of tubulin CTT significantly distorted to establish contacts with itself, with the solvent or with the adjacent tubulin cores, in agreement with the intrinsically disordered nature of tubulin CTT. Subsequently, unstrained MD trajectories were produced for each compound using the MT lattice structure. Like the binding with the isolated tubulin CTT peptide, 1a, 1e and 1f were predicted to be able to recognize tubulin CTT in the more complex system, and to decrease the overall disorder of this region, although the order of binding affinity in silico does not correspond to their potency to promote tubulin polymerization in vitro (Fig. 6A). The binding models revealed that the recognition was still mainly attributed to electrostatic interactions with no contribution from the alkyl linker (Fig. 6B). Interestingly, some compounds were predicted to bridge the tubulin CTT to interact with the tubulin core. The backbone NH of 1a established a peculiar hydrogen bond network with an aspartic acid on the tubulin core and 1e adopted an elongated conformation to contact the tubulin core while binding to tubulin CTT (Fig. 6B, right panels). Nevertheless, the specificity or affinity of series 1 for tubulin CTT may be questionable, as in most of the simulations no more than two of the four molecules reached the expected target, i.e., tubulin CTT. In summary, 1a, 1e and 1f were predicted to be capable of interacting with tubulin CTT in molecular simulations, but non-specific interactions with other parts of the tubulin structure were also possible.
Discussion
Inspired by the electrostatic interaction between tubulin CTT and its binding partners, linear and cyclic peptides containing three consecutive arginines (RRR) were designed. The selection of arginine rather than other natural basic amino acids (lysine or histidine) is supported by a study demonstrating an additional hydrogen bond between the guanidine group in arginine and the carboxylic group in glutamic acid, compared with lysine and histidine.28 Although structurally simple, our RRR macrocycles represented by 1f were able to promote tubulin polymerization through interacting with tubulin CTT, but no cellular activity consistent with MT disruption was observed in multiple cancer cell lines.The current binding models between tubulin CTT and its binding partners indicate that multivalent charge interactions are necessary to stabilize the complex.31–34 For example, multiple lysines and arginines are well-positioned in the basic grooves within the CAP-Gly domain of CLIP-170 and in the N-terminal and central domains of TTLL7 to facilitate their interactions with tubulin CTT.35,36 In a recent study, the conjugation of four decapeptides of KA7 (KAKAKAKAKAKAKA) to star-PEG (a dendrimer with four polyglycol arms) furnished a star-shaped polymer with nanomolar potency to MTs and some MAP-like properties,37 whereas KA7 itself did not show either MT binding or MAP-like properties. In this study, three consecutive arginines seem to be barely enough for the binding to tubulin CTT, as suggested by the co-sedimentation assay and MD simulations. These results raise the possibility of designing tubulin CTT binders with much lower molecular weight.
Apart from the multivalent interaction, the spatial arrangement of charge centers on tubulin CTT is also important for the recognition of certain binding partners, as found in tubulin tyrosine ligase (TTL) recognition of the terminal EE sequence in α-tubulin CTT,38 and inhibition of the MT severing enzymes spastin and katanin by the isolated β-tubulin CTT peptide with an appropriate sequence.39 In our study, the exploration of the charge center position was achieved by incorporating variable linkers into the macrocycles (series 1 and 3). In series 1, compounds bearing longer linkers are more potent in inducing tubulin polymerization and there is a decline of such activity when the linker becomes shorter. This may indicate that the cyclization with shorter linkers, especially in the case of 1a with a four-carbon linker, leads to inappropriate conformational constraints in the macrocycle, and suggests that the appropriate flexibility of the molecules will facilitate the binding to tubulin CTT.
In cellular assays, our compounds cannot inhibit the proliferation of cancer cells, but are able to reverse Aβ25–35-induced axonal atrophy in neurons. Poor cell permeability and low to medium affinity and/or efficacy of our compounds are proposed to account for the non-toxicity to cancer cells. Based on the activity in neurons, the compounds with glycol linkers seem more cell permeable than those with alkyl linkers. Nonetheless, the interaction of our cyclic peptides with membrane receptors to realize cellular effects can also not be excluded. Both cellular permeability and target engagement have to be assessed for these CTT binding cyclic peptides to explain the difference in bioactivity in cancer cells and neurons.
Conclusion
To the best of our knowledge, this work provides a new chemotype of MT stabilizer, and is possibly the first report on tubulin CTT binders with the molecular weight similar to marketed small molecule drugs. A more comprehensive insight into their recognition pattern by tubulin CTT will facilitate the design of new and more potent and/or selective tubulin CTT binders.Abbreviations
| 2-CTC | 2-Chlorotrityl chloride |
| CTT | C-terminal tail |
| DAPI | 4′,6-Diamidino-2-phenylindole |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCM | Dichloromethane |
| DEAD | Diethyl azodicarboxylate |
| DIEA | N,N-Diisopropylcarbodiimide |
| DMF | N,N-Dimethylformamide |
| Fmoc | Fluorenylmethyloxycarbonyl |
| HATU | O-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate |
| HBTU | O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate |
| HFIP | 1,1,1,3,3,3-Hexafluoro-2-propanol |
| HOBt | 1-Hydroxybenzotriazole |
| HPLC | High-performance liquid chromatography |
| IDP | Intrinsically-disordered protein |
| IDR | Intrinsically-disordered protein region |
| MAP | Microtubule-associated protein |
| MD | Molecular dynamics |
| MM/GBSA | Molecular mechanics/generalized Born surface area |
| MM/PBSA | Molecular mechanics/Poisson–Boltzmann surface area |
| MT | Microtubule |
| MTA | Microtubule-targeting agent |
| Pbf | (2,2,4,6,7-Pentamethyl-2,3-dihydrobenzo[b]furan-5-yl)sulfonyl |
| Phth | Phthalic |
| r.t. | Room temperature |
| Su | Succinimide |
| THF | Tetrahydrofuran |
| TFA | Trifluoroacetic acid |
| TIS | Triisopropylsilane |
| TTL | Tubulin tyrosine ligase. |
Author contributions
Compound synthesis and validation, S. Li and M. Yang; biochemical tubulin polymerization assay, S. Li; morphology of the microtubule, S. Elfazazi, R. Hortigüela, M. Á. Oliva and J. F. Díaz; co-sedimentation assay, S. Elfazazi, R. Hortigüela, M. Á. Oliva and J. F. Díaz; cell assay on cancer cell lines, P. Chan and A. Risinger; cell assay on neuronal cell lines, X. Feng and Z. Yang; modeling, M. Mori; manuscript writing, S. Li and W.-S. Fang; manuscript revision, S. Li, W.-S. Fang, J. F. Díaz, M. Mori, Z. Yang and A. Risinger; manuscript approval, all authors; supervision of the project, W.-S. Fang.Conflicts of interest
The authors declare no conflicts of financial interest.Acknowledgements
We thank Dr Norbert Sewald from Bielefeld University, Germany, for his generous advice on peptide synthesis. We also thank Ganadería Fernando Díaz for calf brain supply and Escuela Hongaresa de Esgrima de Pontevedra for its private donation to the project. This research was funded by the CAMS Innovation Fund for Medical Sciences (Grant No. 2016-I2M-1-010), State Key Lab Grant type C (Grant No. GTZC201709) (W.-S. F.), Ministerio de Ciencia e Innovación (Grant No. PID2021-123399OB-I00 /AEI/10.13039/501100011033) (M. Á. O.), Ministerio de Ciencia e Innovacion (Grant No. PID2019-104545RBI00 /AEI/10.13039/501100011033), European Commission-NextGenerationsEU (Regulation EU 2020/2094, through CSIC's Global Health Platform (PTI Salud Global)), Proyecto de Investigación en Neurociencia Fundacion Tatiana Pérez de Guzmán el Bueno 2020 (J. F. D.), the Voelcker Fund (A. R.), the Natural Science Foundation of Guangdong Province of China (Grant No. 2022A1515011419), and the Key Project in Higher Education of Guangdong, China (Grant No. 2022ZDZX2029) (Z. Y.).References
- R. F. Ludueña, in International Review of Cytology, Elsevier, 1998, vol. 178, pp. 207–275 Search PubMed.
- L. J. Leandro-García, et al. , Cytoskeleton, 2010, 67, 214–223 CrossRef.
- P. Richette, et al. , Ann. Rheum. Dis., 2017, 76, 29–42 CrossRef CAS.
- B. P. Chatterji, et al. , Expert Opin. Ther. Pat., 2011, 21, 167–186 CrossRef CAS.
- M. O. Steinmetz and A. E. Prota, Trends Cell Biol., 2018, 28, 776–792 CrossRef CAS PubMed.
- S. Matthew, et al. , Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2021847118 CrossRef CAS.
- J. Yang, et al. , Sci. Adv., 2021, 7, eabg4168 CrossRef CAS.
- Y.-N. Cao, et al. , Eur. J. Med. Chem., 2018, 143, 806–828 CrossRef CAS PubMed.
- E. Martino, et al. , Bioorg. Med. Chem. Lett., 2018, 28, 2816–2826 CrossRef PubMed.
- C. Ferlini, et al. , Cancer Res., 2005, 65, 2397–2405 CrossRef CAS.
- R. Matesanz, et al. , Bioorg. Med. Chem., 2014, 22, 5078–5090 CrossRef CAS PubMed.
- P. Cai, et al. , Cancer Lett., 2013, 341, 214–223 CrossRef CAS.
- A. Pepe, et al. , Bioorg. Med. Chem. Lett., 2009, 19, 3300–3304 CrossRef CAS PubMed.
- C. Janke and J. C. Bulinski, Nat. Rev. Mol. Cell Biol., 2011, 12, 773–786 CrossRef CAS.
- C. Janke, J. Cell Biol., 2014, 206, 461–472 CrossRef CAS.
- R. Tarazona, et al. , J. Immunol., 2000, 165, 6776–6782 CrossRef CAS PubMed.
- T. Sarkar, et al. , Proteins: Struct., Funct., Bioinf., 2001, 44, 262–269 CrossRef CAS.
- R. B. Maccioni, et al. , Eur. J. Biochem., 1986, 154, 427–435 CrossRef CAS.
- Y. Laurin, et al. , Biochemistry, 2015, 54, 3660–3669 CrossRef CAS.
- A. Vemu, et al. , J. Biol. Chem., 2016, 291, 12907–12915 CrossRef CAS PubMed.
- H. Freedman, et al. , Proteins: Struct., Funct., Bioinf., 2011, 79, 2968–2982 CrossRef CAS.
- Y. Luo, et al. , Nat. Commun., 2020, 11, 18 CrossRef CAS.
- L. T. Vassilev, et al. , Science, 2004, 303, 844–848 CrossRef CAS.
- H. Lee, et al. , J. Biol. Chem., 2000, 275, 29426–29432 CrossRef CAS PubMed.
- D. I. Hammoudeh, et al. , J. Am. Chem. Soc., 2009, 131, 7390–7401 CrossRef CAS.
- J. Wolff, et al. , Protein Sci., 1996, 5, 2020–2028 CrossRef CAS.
- L. Serrano, et al. , Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 5989–5993 CrossRef CAS.
- V. Petrauskas, et al. , J. Phys. Chem. B, 2015, 119, 12164–12171 CrossRef CAS PubMed.
- K. Barlos, et al. , Int. J. Pept. Protein Res., 1991, 37, 513–520 CAS.
- S. De la Vina, et al. , Biochemistry, 1988, 27, 5352–5365 CrossRef CAS PubMed.
- A. E. Prota, et al. , J. Cell Biol., 2013, 200, 259–270 CrossRef CAS PubMed.
- A. Szyk, et al. , Nat. Struct. Mol. Biol., 2011, 18, 1250–1258 CrossRef CAS.
- F. Li, et al. , Nat. Struct. Mol. Biol., 2019, 26, 583–591 CrossRef CAS PubMed.
- C. P. Garnham, et al. , Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6545–6550 CrossRef CAS.
- M. Mishima, et al. , Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10346–10351 CrossRef CAS.
- C. P. Garnham, et al. , Cell, 2015, 161, 1112–1123 CrossRef CAS.
- H. Drechsler, et al. , Mol. Biol. Cell, 2019, 30, 2953–2968 CrossRef CAS.
- M. Ruediger, et al. , Eur. J. Biochem., 1994, 220, 309–320 CrossRef CAS.
- A. Roll-Mecak and R. D. Vale, Nature, 2008, 451, 363–367 CrossRef CAS.
Footnotes |
| † This paper is dedicated to the late Professor Maurizio Botta, University of Siena. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01910h |
| This journal is © The Royal Society of Chemistry 2023 |