
The regioselective Wacker oxidation of internal allylamines: synthesis of functionalized and challenging β-amino ketones†‡
Mateusz
Garbacz
 and
Sebastian
Stecko
*
and
Sebastian
Stecko
*
Institute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: sebastian.stecko@icho.edu.pl
First published on 3rd November 2022
Abstract
A convenient and general protocol for the palladium-catalysed oxidation of internal allylamine derivatives to β-amino ketones is reported. The transformation occurs at room temperature and shows a wide substrate scope as well as high functional group and N-protecting group tolerance. We also describe potential applications of the method, e.g., the synthesis of bioactive molecules or simple transformations of selected β-amino ketones into other interesting building blocks.
Introduction
β-Amino ketones (β-AKs) are ubiquitous compounds in molecular science.1–4 While their structural motif is relatively rare in natural products and pharmaceuticals, many synthetic and medicinal chemists have been utilizing β-AKs as starting materials in the total synthesis of bioactive molecules.1,5–7 The bifunctional scaffold of β-AKs has been also found to be a versatile precursor for the asymmetric synthesis of anti- or syn-1,3-amino alcohols,6,8–10syn- and anti-1,3-diamines11,12 and α-ketoaziridines.13–15The high synthetic value of β-AKs stimulated the development of a plethora of strategies for their preparation, including diastereo- and enantioselective protocols (Scheme 1).1–4,6 Notably, the synthesis of non-racemic β-AKs is mostly dominated by the formation of the C2–C3 bond via the asymmetric Mannich reaction.6 Clearly, this reaction involving the addition of an enolizable carbonyl equivalent to the imine is a powerful tool to access these bifunctional compounds. However, it still has several drawbacks including its reliance on mostly pre-activated imines, a problematic control of selectivity in the case of ketoimines, and the limited pool of carbonyl partners. Furthermore, the asymmetric induction poses a challenge for the Mannich reaction. The diastereoselective mode requires pre-functionalized imines (with a chiral auxiliary or additional chelating centres). The enantioselective variant typically relies on organocatalytic approaches that have a strongly substrate-dependent efficiency. Direct formation of the N–C3 bond through 1,4-addition of a nitrogen nucleophile to the β-carbon vinylogously attached to an electron-withdrawing group (aza-Michael reaction) is another possible strategy for the construction of β-AKs. However, the catalytic, asymmetric aza-Michael reaction still poses a major challenge in organic synthesis. Most examples of this reaction involve acrylate substrates whose carboxylic group serves as a platform to install a chiral auxiliary group (diastereoselective approach) or an achiral functionality to achieve chelation with a chiral Lewis acid catalyst (enantioselective approach).9,16–23 Unfortunately, this asymmetric induction strategy lacks the functionality to install the chiral auxiliary group which makes it unsuitable for enones, which therefore require an enantioselective organocatalytic approach.6
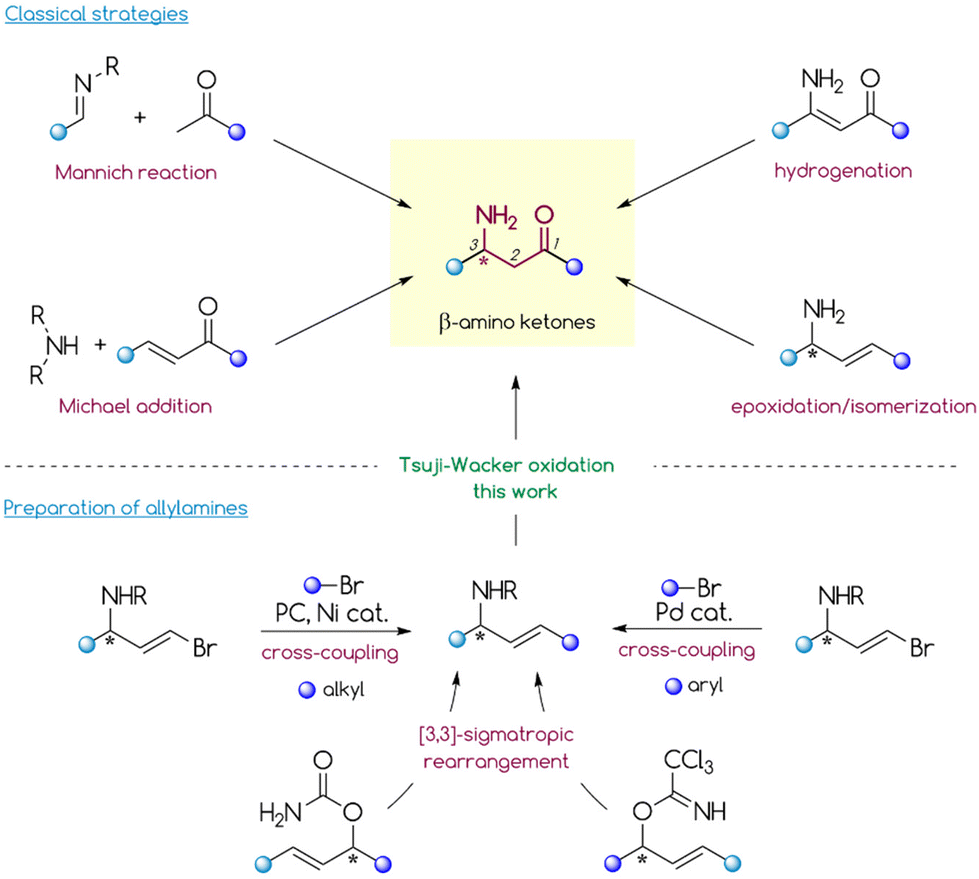 | ||
| Scheme 1 Strategies for preparing β-amino ketones and allylamines. | ||
Asymmetric Rh-catalysed hydrogenation of β-ketoenamides is another possible, albeit less explored, strategy to prepare β-AKs.24–28 This approach provides optically pure β-amino ketones in high yields with good to excellent enantioselectivity; however, it is also highly substrate-sensitive due to the specific requirements for coordination of bulky Rh-complexes to a double bond.
Other rarely explored strategies for preparing β-AKs rely on transformations of allylamines and propargylamines. For example, Coates,29 Che,30,31 and others32 successfully prepared β-AKs through an epoxidation/isomerization sequence that starts from the corresponding amines; however, the regioselectivity of this strategy is strongly substrate structure dependent. Pu's group33 and others34 reported the preparation of β-AKs via regioselective Au-catalysed hydration of propargylamines (including chiral), maintaining a high enantiomeric purity. Unfortunately, this process requires a donor group (e.g., tosyl or phosphinoyl) attached to the nitrogen atom to stabilize the intermediate Au-alkyne complex and achieve high regioselectivity and efficiency.
In contrast, another poorly explored approach involves the direct oxidation of internal allylamines via the anti-Markovnikov selective Tsuji–Wacker reaction. In general, the Tsuji–Wacker oxidation of terminal olefins to methyl ketones has a plethora of applications in the synthesis of fine chemicals, natural products, and pharmaceuticals.35–37 In comparison, analogous oxidation of internal alkenes appears to be rather rare, likely due to poor regioselectivity in the case of unbiased substrates. Moreover, their reaction rate is slower than that for terminal olefins and the process requires the presence of additional coordinating groups, although the course of the transformation remains substrate-dependent.35–37 Bearing this in mind, allylamines should be suitable substrates since the amine group may serve as a useful platform for installing directing groups to enhance reactivity and control the regioselectivity of the oxidation. Given the high potential of allylamines as β-AK precursors, it is surprising how few studies have examined the oxidation of allylamine derivatives (mostly N-Phth protected) with an internal double bond. For example, Feringa and co-workers38 reported the oxidation of allylic phthalimide to selectively deliver the corresponding methyl ketone. The same regioselectivity was observed by Sigman and co-workers39 when using the Pd(quinox)Cl2 complex and a large excess (12 eq.) of t-BuOOH as a co-oxidant. Notably, neither of the two studies used non-racemic starting materials. More recently, Kaneda and co-workers40 presented an elegant oxygen-coupled, copper-free Wacker oxidation of internal olefins. Although this protocol showed good substrate scope, it required high oxygen pressures (3–9 bar) and specialised equipment (autoclave), which limits its application in laboratory research.
The advantage of the last mentioned strategy is the availability of (optically pure) allylamines. Recently, we presented convenient protocols for the preparation of aliphatic allylamines either by [3,3]-sigmatropic rearrangement of allylcarbamates or via photoredox/Ni-catalysed cross-coupling with alkyl bromides.41,42 Notably, these protocols provide a simple, divergent, and reliable synthesis of non-racemic allylamines from readily available starting materials. It should be emphasized that the preparation of optically pure β-AKs with a certain substitution pattern, particularly at the nitrogen-bound C-stereocentre (e.g., Me/H, alkyl/alkyl, and aryl/aryl), is a challenging task to accomplish intermolecularly, for instance through Mannich or aza-Michael reactions, due to the poor stereodifferentiation of a pro-chiral functionality with highly similar substituents. Importantly, this drawback can be overcome by applying strategies for rapid generation of enantioenriched allylamines as developed by our group. In this study, we developed a general and convenient protocol for the direct functionalization of enantioenriched allylamines, particularly via Pd-catalysed oxidation, to access β-AKs with a broad scope of possible starting materials and the ultimate chemoselectivity profile, which remains an unsolved problem with the aforementioned traditional methods.
Results and discussion
As a first step, we subjected allylamine 1, with an internal double bond, to the Tsuji–Wacker oxidation under the conditions reported by Grubbs and co-workers for simple internal olefins.43 This choice was motivated by the mild reaction conditions (rt, atmospheric pressure) and the simple catalytic system. The initial oxidation reaction was carried out in the presence of Pd(OAc)2 (10 mol%), 1 eq. of benzoquinone (BQ), and HBF4 in the DMA–MeCN–H2O (3.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.5:1 v/v) solvent mixture at rt, yielding β-amino ketone 2 in 75% yield after 6 h as a sole regioisomer (Scheme 2). The inspection of the 1H NMR spectra of the crude reaction mixture did not reveal the presence of the alternative α-amino ketone 3. Further optimization studies showed that the reaction yield can be enhanced by replacing Pd(OAc)2 with Pd(TFA)2 (Scheme 2b, entries 1 vs. 3). According to Grubbs’ report, the presence of DMA was crucial to suppress side double bond chain-walking isomerization. During the optimization studies, we found that it was not necessary to add DMA to substrate 1 for the process to proceed efficiently and with complete regioselectivity. This observation led to a simplified solvent system that only required the MeCN–H2O mixture in subsequent experiments. As a result, the reaction rate was significantly increased, shortening the reaction time from 6 h to just 30 min. Eliminating DMA also improved the yield of product 2 (91% vs. 75%, Scheme 2b, entries 1 vs. 3) and greatly simplified its isolation and purification. We believe that the NHCbz group present in 1 plays a similar role to that of DMA. Due to an extra coordination of a β-Pd intermediate, it eliminates possible side isomerization processes and controls regioselectivity.
:3.5:1 v/v) solvent mixture at rt, yielding β-amino ketone 2 in 75% yield after 6 h as a sole regioisomer (Scheme 2). The inspection of the 1H NMR spectra of the crude reaction mixture did not reveal the presence of the alternative α-amino ketone 3. Further optimization studies showed that the reaction yield can be enhanced by replacing Pd(OAc)2 with Pd(TFA)2 (Scheme 2b, entries 1 vs. 3). According to Grubbs’ report, the presence of DMA was crucial to suppress side double bond chain-walking isomerization. During the optimization studies, we found that it was not necessary to add DMA to substrate 1 for the process to proceed efficiently and with complete regioselectivity. This observation led to a simplified solvent system that only required the MeCN–H2O mixture in subsequent experiments. As a result, the reaction rate was significantly increased, shortening the reaction time from 6 h to just 30 min. Eliminating DMA also improved the yield of product 2 (91% vs. 75%, Scheme 2b, entries 1 vs. 3) and greatly simplified its isolation and purification. We believe that the NHCbz group present in 1 plays a similar role to that of DMA. Due to an extra coordination of a β-Pd intermediate, it eliminates possible side isomerization processes and controls regioselectivity.
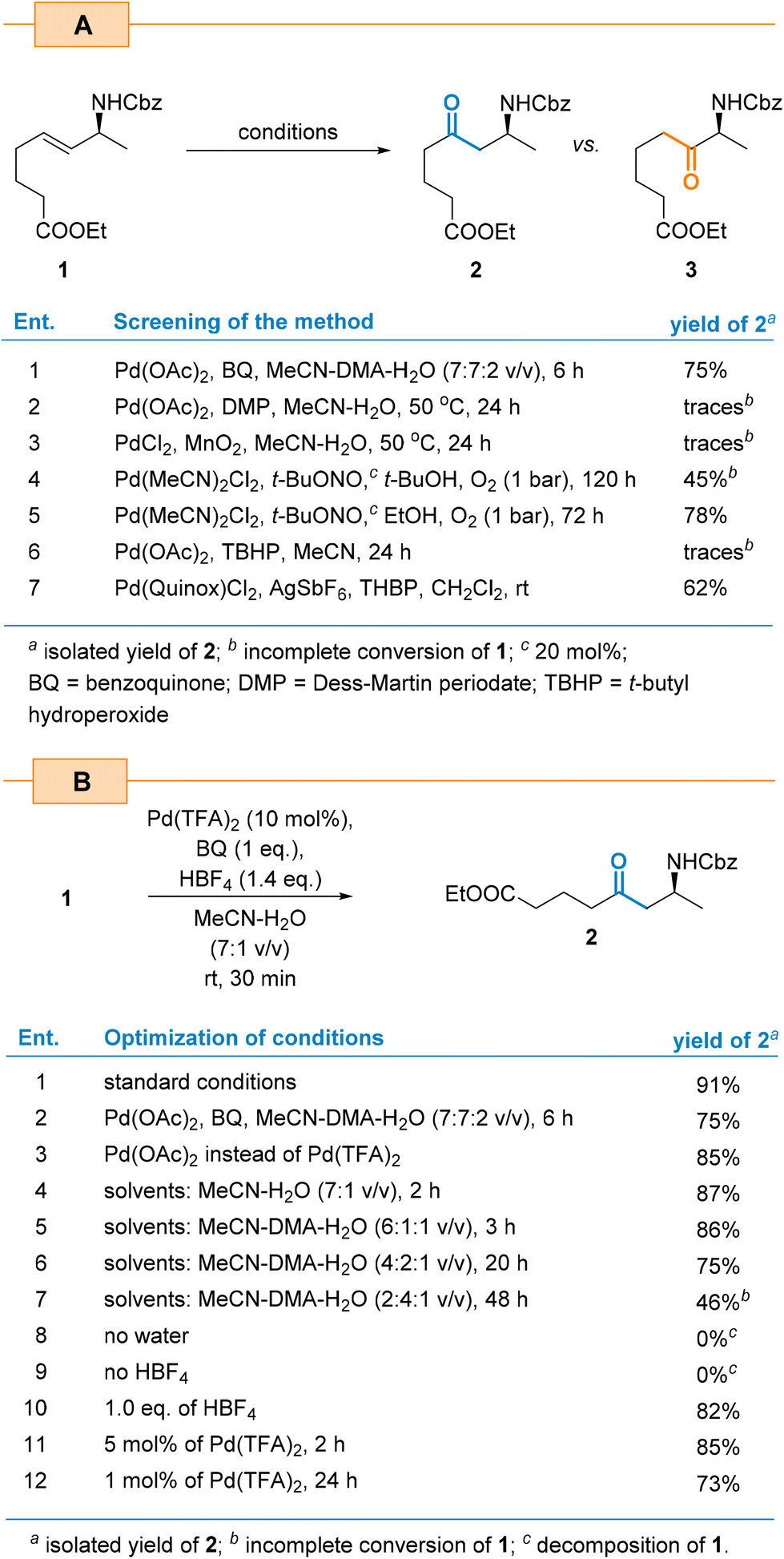 | ||
| Scheme 2 Optimization of the Wacker oxidation of allylamine 1. | ||
The change of the catalyst loading from the initial 10 mol% to 5 mol% decreased the yield from 91% to 85% (Scheme 2b, entries 1 vs. 11). At the same time, a fourfold extension of the reaction time was required to achieve full conversion of the starting material. Decreasing the catalyst amount further to 1 mol% increased the reaction time to achieve complete conversion of the starting material to 24 h and reduced the yield of product 2 to 73% (entry 12). We found that BQ was the best co-oxidant as the Dess–Martin reagent,44 MnO245 and TBHP46 yielded only traces of the product.
Independently, we also evaluated the aerobic conditions of the nitrite-Wacker oxidation reported by Kang and co-workers.47,48 The advantage of this method is that it uses oxygen as a terminal oxidant instead of stoichiometric BQ. Thus, in the presence of t-butyl nitrite (20 mol%) as a redox co-catalyst and t-BuOH as a solvent, amino ketone 2 was obtained in 45% yield only after 120 h (Scheme 2a, entry 4). Changing the solvent to EtOH enhanced the reaction rate and resulted in a 78% yield of product 2 after 72 h (entry 5). Hence, this approach resulted in a lower yield which suggests that the bulkier nucleophiles such as t-BuOH decreased the reaction rate. Interestingly, both cases exhibited the same regioselectivity which stands in contrast to Kang's findings.47 As demonstrated, the nitrite-Wacker oxidation of unbiased terminal alkenes with the Pd/t-BuONO system in EtOH leads to the Markovnikov product (ketone) whereas the anti-Markovnikov product (aldehyde in the case of terminal alkene) was formed when more hindered t-BuOH was applied.47 In the case of 1, regioselectivity was controlled solely by the directing group. The same regioselectivity was observed in the case of an oxidation of 1 in the presence of the Pd(Quinox)Cl2 complex and an excess of TBHP according to Sigman's protocol;39 however, the yield of the expected product was only 62% (Scheme 2a, entry 7).
Having identified the optimal conditions, we then directed our focus on the oxidation scope. Notably, although the nitrite-Wacker conditions described above delivered the model product in a lower yield, we decided to include this method in our scope studies, as it has the advantage of requiring oxygen rather than BQ.
The first class of substrates to be evaluated were allylamines bearing various N-protecting groups (Scheme 3). The oxidation proceeded efficiently for N-carbamate (2, 5) and N-amide type (7) protecting groups. The preparation of 4via oxidation of the corresponding N-Boc allylamine, using our standard conditions, failed. Plausibly, HBF4, required to facilitate the process, removed the Boc group to provide the corresponding free allylamine which was unreactive under these specific conditions. To overcome this limitation, we decided to replace the Pd(TFA)2/HBF4 catalytic system with a [Pd(MeCN)4(BF4)2] complex that, in fact, is generated in situ under our standard conditions. Now, with the acid having been removed from the reaction mixture, product 4 was obtained in an 85% yield. In comparison, the oxidation of the same substrate under the nitrite-Wacker conditions only resulted in a 61% yield of the desired product 4.
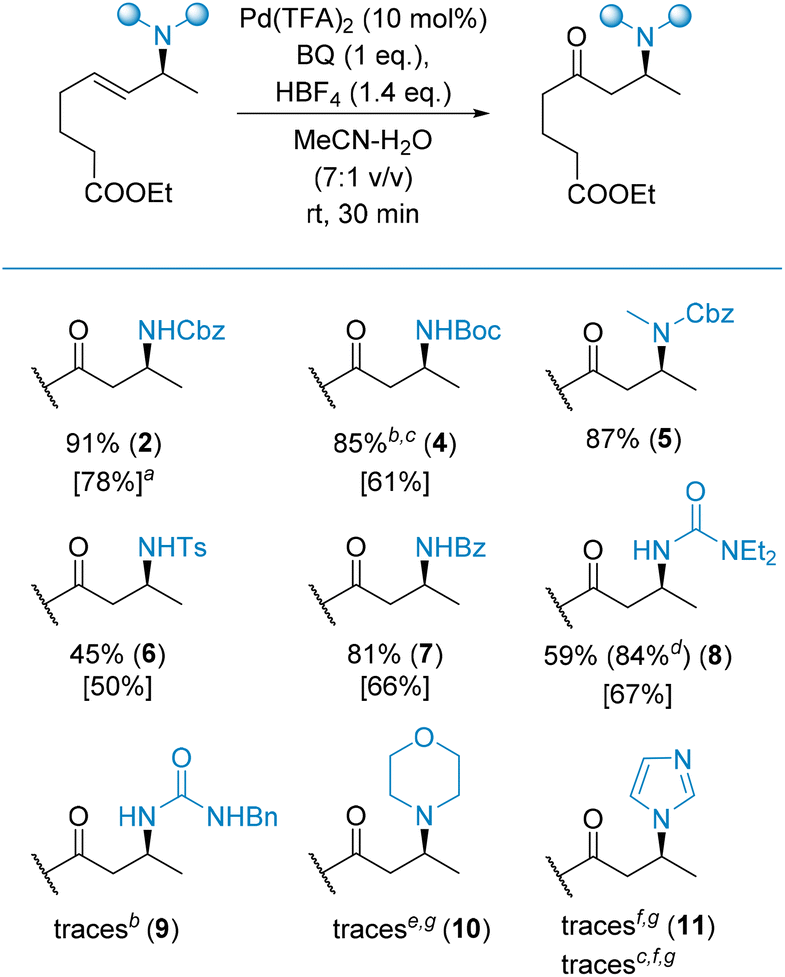 | ||
| Scheme 3 Effect of N-protection on the Tsuji–Wacker and nitrite-Wacker oxidation of allylamines. Standard conditions: Pd(TFA)2 (10 mol%), BQ (1 eq.), HBF4 (1.4 eq.), MeCN–H2O (7:1 v/v), rt, 30 min. aIn brackets, the yields of the nitrite-Wacker oxidation: Pd(MeCN)2Cl2 (5 mol%), t-BuONO (20 mol%), EtOH, O2 (1 atm), rt, 96 h; bdecomposition of the starting material under the standard conditions; creaction conditions: [Pd(MeCN)4(BF4)2] (7 mol%) instead of Pd(TFA)2/HBF4, rt, 30 min; daccording to the NMR of the crude reaction mixture with an internal standard; eincomplete conversion of the starting material; fdecomposition of the starting material; g4 eq. of HBF4 was used. | ||
Interestingly, N-Ts allylamine was oxidized too, delivering product 6 in a 45% yield, although along with several by-products, which we were unable to identify unambiguously. The oxidation of allyl systems with a urea moiety was more complicated. To be efficient, the distal nitrogen atom had to be completely protected. This resulted in a 59% yield of product 8 while other products (e.g., 9) were obtained in trace amounts only, along with a mixture of side products possibly arising from Pd-assisted intramolecular cyclization leading to imidazolidinone derivatives. Furthermore, the oxidation of N-allyl morpholine failed (yielding only traces of the desired product 10 along with side products), indicating that free amines are unsuitable substrates for the investigated Wacker reaction. The oxidation of N-allyl imidazole yielded only traces of the expected product 11 (under the standard conditions in the presence of an excess of HBF4 and in the presence of [Pd(MeCN)4(BF4)2]) again with side products, indicating that heterocyclic scaffolds bearing a lone electron pair are also unsuitable for the reaction conditions. Moreover, no starting material was recovered which suggests its either complete, but unproductive, conversion and/or decomposition.
During the next stage of scope studies, we examined the oxidation efficiency of four types of allylamines A–D (Scheme 4). These substrates were prepared by cross-coupling non-racemic 3-bromo allylamine derivatives with their corresponding alkyl bromides. For most cases, oxidation proceeded efficiently with complete regioselectivity (Scheme 4). Steric effects on oxidation were negligible; thus 1° (e.g., 12a, 13a, 13b), 2° (e.g., 12b, 12o–q, 13c, 13h, 13i, 13m–o), and 3° (e.g., 12c, 13d–f) alkyl groups were well tolerated regardless of the side ( or
or  or both, Scheme 4a and b) on which the allylamine scaffold is located (Scheme 4c). The analysis of the crude reaction mixtures of 12g–j did not reveal any presence of δ-AKs that could have resulted from the isomerization of the double bond toward the aryl ring. The reaction proceeded efficiently and regioselectively, also when the N-Cbz moiety was a part of a cyclic system (e.g., 13j or 14d) or for a cyclic allylamine that provided product 13k. Furthermore, the oxidation of α-tertiary allylamines proceeded smoothly and provided the corresponding β-AKs (14e–h) with a tetrasubstituted carbon stereocentre (Scheme 4d). Notably, the enantioselective synthesis of these systems, mainly via the Mannich reaction, is highly challenging due to the steric hindrance of ketoimines and their ability to exist as E/Z mixtures which significantly complicates stereocontrol of the addition reaction. The same holds true for type B non-racemic allylamines; their preparation through either the Mannich reaction (due to limited stability of imines) or conjugate addition to crotonyl enones (due to steric factors) is less effective and problematic. However, the proposed approach is able to address and overcome these issues. Additionally, our approach provides straightforward access to structurally diverse non-racemic allylamines through sigmatropic rearrangements.41,42 This is illustrated in Scheme 4d for type D α-tertiary allylamines; the preparation of such enantiopure systems bearing two aryl or alkyl groups that differ only slightly (
or both, Scheme 4a and b) on which the allylamine scaffold is located (Scheme 4c). The analysis of the crude reaction mixtures of 12g–j did not reveal any presence of δ-AKs that could have resulted from the isomerization of the double bond toward the aryl ring. The reaction proceeded efficiently and regioselectively, also when the N-Cbz moiety was a part of a cyclic system (e.g., 13j or 14d) or for a cyclic allylamine that provided product 13k. Furthermore, the oxidation of α-tertiary allylamines proceeded smoothly and provided the corresponding β-AKs (14e–h) with a tetrasubstituted carbon stereocentre (Scheme 4d). Notably, the enantioselective synthesis of these systems, mainly via the Mannich reaction, is highly challenging due to the steric hindrance of ketoimines and their ability to exist as E/Z mixtures which significantly complicates stereocontrol of the addition reaction. The same holds true for type B non-racemic allylamines; their preparation through either the Mannich reaction (due to limited stability of imines) or conjugate addition to crotonyl enones (due to steric factors) is less effective and problematic. However, the proposed approach is able to address and overcome these issues. Additionally, our approach provides straightforward access to structurally diverse non-racemic allylamines through sigmatropic rearrangements.41,42 This is illustrated in Scheme 4d for type D α-tertiary allylamines; the preparation of such enantiopure systems bearing two aryl or alkyl groups that differ only slightly ( and
and  in Scheme 4d) is highly challenging or impossible to achieve through any other known methods.49
in Scheme 4d) is highly challenging or impossible to achieve through any other known methods.49
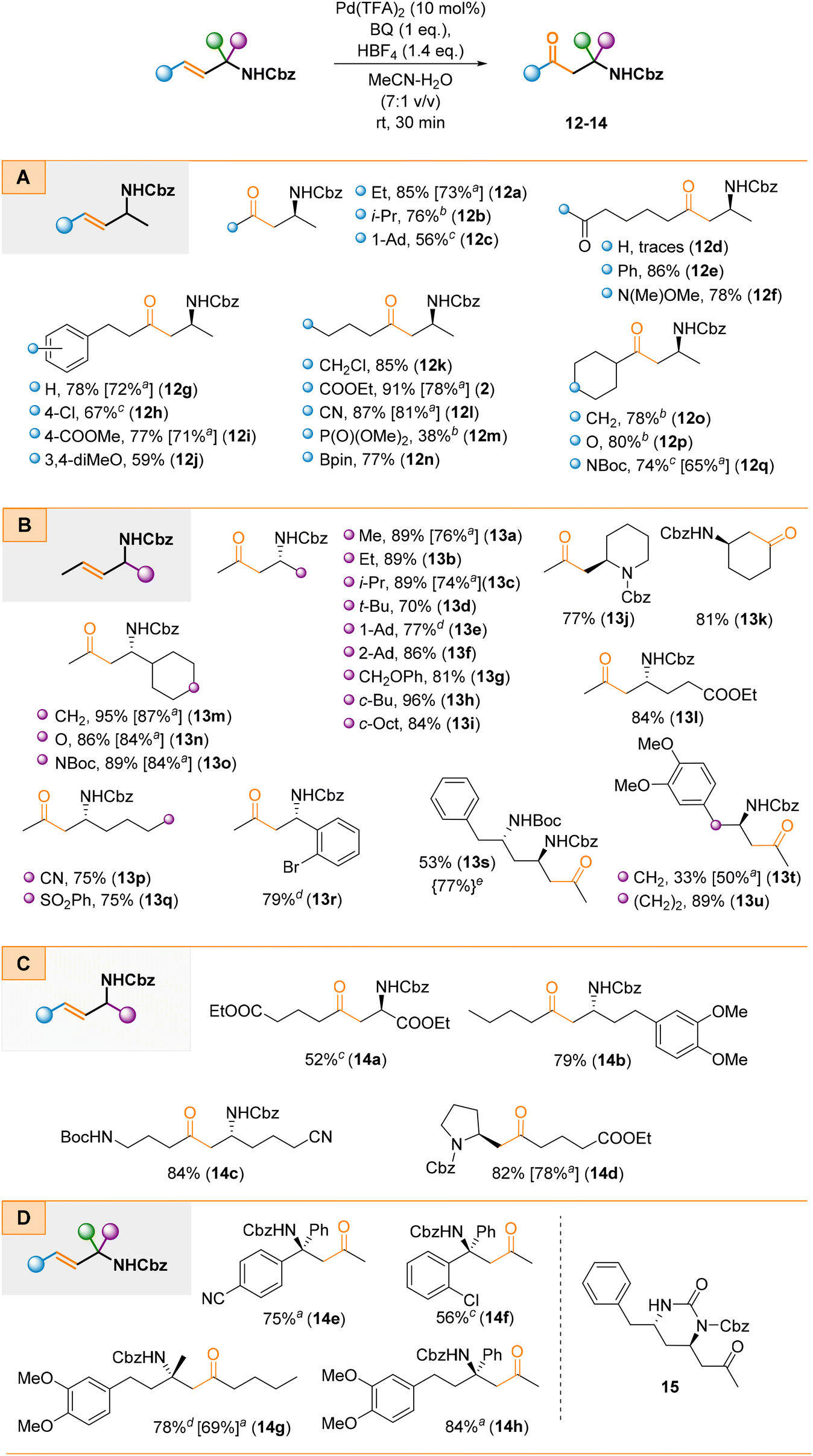 | ||
| Scheme 4 Scope studies on the Wacker oxidation of aliphatic allylamine derivatives. Standard conditions: Pd(TFA)2 (10 mol%), BQ (1 eq.), HBF4 (1.4 eq.), MeCN–H2O (7:1 v/v), rt, 30–90 min; ain brackets, the yields [%] of the nitrite-Wacker oxidation; Pd(MeCN)2Cl2 (5 mol%), t-BuONO (20 mol%), EtOH, O2 (1 atm), rt, 96 h; breaction time: 24 h; creaction time: 120 h; dreaction time: 8 h; ein brackets, the yield {%} of the reaction catalyzed by [Pd(MeCN)4(BF4)2] (7 mol%) which was used instead of Pd(TFA)2/HBF4, rt, 60 min. 1-Ad = 1-adamantyl; 2-Ad = 2-adamantyl; c-Bu = cyclobutyl; c-Oct = cyclooctyl. | ||
Furthermore, besides complete regioselectivity, the second key advantage of the proposed method is its high functional group tolerance. Typical electrophilic functionalities, such as ester (e.g., 2, 13l, 14a), nitrile (e.g., 12l, 13p, 14c), sulfonyl (13q), phosphonate (12m), ketone (12e), and Weinreb amide (12f) groups, were well tolerated. Unfortunately, the oxidation failed for a substrate bearing an aldehyde group. While the starting material was consumed, we only obtained traces of the expected product 12d along with multiple unidentified side products plausibly arisen via acid promoted addition/condensation reactions. These reaction conditions were also suitable for substrates containing additional, protected hydroxyl groups (13g). Interestingly and in contrast to the preparation of amino ketone 4 (see Scheme 3), the additional NHBoc group present outside the allylic system survived the standard reaction conditions and the corresponding diamino ketones (e.g., 12q, 13o, 13s, and 14c) were obtained in good yields. The lower yield for compound 13s (53%) resulted from partial subsequent intramolecular cyclization under the reaction conditions leading to tetrahydropyrimidin-2(1H)-one derivative 15. We were able to minimise the formation of this side product by replacing the standard catalytic system with the [Pd(MeCN)4(BF4)2] complex to suppress unwanted acid mediated cyclization. With this replacement product 13s was obtained in a 77% yield. Furthermore, a boronic acid ester moiety was compatible with the reaction conditions, which facilitated the synthesis of amino boronic acid derivatives such as compound 12n. Moreover, allylamines with an additional halide group attached to either the alkyl chains or the aromatic rings were suitable substrates and provided the expected β-AKs (e.g., 12k, 13r). These halo-derivatives are interesting building blocks, particularly as substrates for cyclization reactions to enable construction of heterocyclic scaffolds. In 13t an electron-rich aromatic ring was placed in close proximity to the allylic system which resulted in a significantly lower yield (33%). In fact, interception of proximal nucleophiles (including C-nucleophiles) by PdII species is a known reaction and has found numerous applications in the synthesis of carbo- or heterocyclic scaffolds.50–53 However, we were able to isolate 13t albeit along with multiple side-products (arising from Wacker-type cyclisation and possibly several consecutive reactions), which we were unable to isolate in an analytically pure form. Interestingly, in the case of 13u, which is a homolog of 13t, the desired β-amino ketone was isolated in an 89% yield. Plausibly, the increased distance between the nucleophilic electron-rich aromatic ring and the resulting PdII species made cyclisation impossible.
The oxidation of selected allylamines under the nitrite-Wacker conditions also delivered the desired products, although in slightly lower yields (Scheme 4, yields in square parenthesis). Under these conditions oxidation was also functional group tolerant. A significant drawback of this protocol is the long reaction time with oxidation requiring at least 96 h to achieve complete conversion of the starting material (Scheme 4).
Cinnamylamine derivatives also afforded the desired products in high yields as outlined in Scheme 5. As previously, high functional group tolerance was observed. Cinnamylamines bearing electron-neutral and electron-donating groups in the phenyl ring provided β-amino ketones only (16a–g). The same regioselectivity was observed in the case of halogen substituted analogues (16h, 16i). The presence of electron-withdrawing groups disturbs the electron density of a double bond, significantly decreasing the rate of the oxidation of such cinnamylamines (increasing reaction times from 3 h to 72 h). The disturbance of the electron density of alkenes also has an effect on the regioselectivity of the process and results in the formation of β-amino ketones along with α-amino regioisomers (16j–16q). For example, the presence of COOMe at the para position of phenyl provided regioisomeric amino ketones at a ratio of 90:10 (β-16j:α-16j). With the same group at the meta position, we only obtained β-regioisomer (16k). When COOMe was at the ortho position, we obtained a 65:35 mixture of isomers (β-16l/α-16l) along with compound 17, resulting from intramolecular transesterification of an enole intermediate. In the presence of a nitro group, an almost equimolar mixture of regioisomers was obtained (β-16q/α-16q). Notably, we were able to oxidize an allylamine bearing an additional aldehyde functionality attached to the benzene ring; thus, the products β-16n/α-16n were isolated in a moderate yield of 56%. In comparison, for the analogous reaction of an aliphatic system, only traces of the product 12d were noticed.
 | ||
| Scheme 5 The Tsuji–Wacker oxidation of cinnamylamine derivatives. Standard conditions: Pd(TFA)2 (10 mol%), BQ (1 eq.), HBF4 (1.4 eq.), MeCN–H2O (7:1 v/v), rt, 30–90 min; *overall yield of both regioisomers; areaction time: 8 h; breaction time: 24 h; creaction time: 30 h (after 24 h, additional amounts of the Pd catalyst (5 mol%) and BQ (0.5 eq.) were added to ensure the complete conversion of the starting material); dinseparable mixture; ersm 42%; fdecomposition of the substrate/product was observed; glow conversion of the starting material; nd = product was not detected. | ||
The oxidation of allylamines containing heterocyclic rings was more complicated. Saturated heterocyclic groups (e.g., tetrahydro-(2H)-pyran and N-protected piperidine) did not suppress the oxidation process which resulted in high yields of the expected products (e.g., 12p, 12q, 13n, 13o; cf., Scheme 4). On the other hand, amino ketone 18a was obtained in an average yield via the oxidation of the allylamine-bearing indol-5-yl group. When the allylamine moiety was attached to the electron-rich part of the indole ring (position 2 or 3), the complete conversion of the starting material was observed but only traces of the desired products 18d and 18g along with multiple unidentified side products were isolated. Arguably, this is a similar case to 13u, where a proximal nucleophilic centre (or group) could be intercepted the PdII species. The same course was observed for benzofuran analogues (18c and 18e). Interestingly, product 18b with a benzothiophen-3-yl ring was isolated in a 77% yield, whereas the analogue reaction of the closely related substrate with a benzothiophen-2-yl ring (product 18f) failed.
The presence of basic heterocyclic rings suppressed the oxidation and the synthesis of amino ketones 18g and 18h failed. This could be the result of deactivation of the Pd catalyst by the nitrogen lone pairs in combination with effects from the protonation of the heterocyclic nitrogen atom to provide a cationic moiety which acts as a strong electron-withdrawing group, thereby dramatically decreasing the reaction rate.
Furthermore, we synthesised products 19 which are challenging using the standard methods such as the Mannich reaction due to the specific electronic features of the required asymmetrical ketones. For example, deprotonation of β-ketoester or benzyl ketones proceeds at the most acidic position, and thus addition to the corresponding imine will not lead to amino ketones 19a or 19b but to isomeric products (Scheme 6). In order to obtain products 19a and 19b, a second deprotonation with a strong base is required, under the conditions promoting subsequent addition under kinetic control. In contrast, the Tsuji–Wacker oxidation of readily available allylamine 20a proceeded smoothly under mild conditions to provide amino ketone 19a solely.
 | ||
| Scheme 6 Synthesis of challenging β-amino ketones via Wacker oxidation. | ||
The oxidation of allylamines bearing an additional ester (20b) or ether (20c) function is more complicated. As these groups may also coordinate to Pd species, and they can compete with the NHCbz group and disturb the regioselectivity of the reaction as confirmed by the oxidation of 20b, since β-19b was obtained along with its α-regioisomer (β:α ratio of 65:35). In comparison, the closely related products 2 and 14a were obtained with complete regioselectivity since the distal location of the ester group does not allow for extra coordination to bonded Pd species. In the case of 20c, the competitive effect was stronger and resulted in the formation of an almost equimolar mixture of amino ketones β/α-19c.
Although this proposed approach has a great potential and utility for the synthesis of β-amino ketones, it has serious inherent limitations regarding the use of stoichiometric benzoquinone for large-scale applications. Unfortunately, the alternative nitrite-Wacker process, although highly attractive due to the use of oxygen as a final oxidant, has much lower efficiency. Moreover, oxidation with complete conversion of all starting material requires several days.
Although the catalytic aerobic oxidation of Pd(0) with oxygen is both an environmentally friendly and economical method for constructing complex organic molecules, this approach is unfavourable in terms of its kinetics and often requires high pressures.54,55 Already disclosed approach to overcome this limitation involves the use of stoichiometric oxidants (e.g., BQ) which have its own drawbacks such as unwanted waste and other by-products. An alternative strategy for the oxidation of palladium with oxygen requires a multi-stage relay as illustrated in Scheme 7A. This is redox cascade involves the participation of an electron transfer mediator (ETM) to improve the efficiency of the oxygen oxidation of hydroquinone and the subsequent oxidation of Pd(0) species.56 We decided to focus on two common ETM agents, Fe(pc) (pc: phthalocyanine)43,57 and Co(salophen),58,59 and evaluate their efficiency for the Wacker oxidation of allylamines.
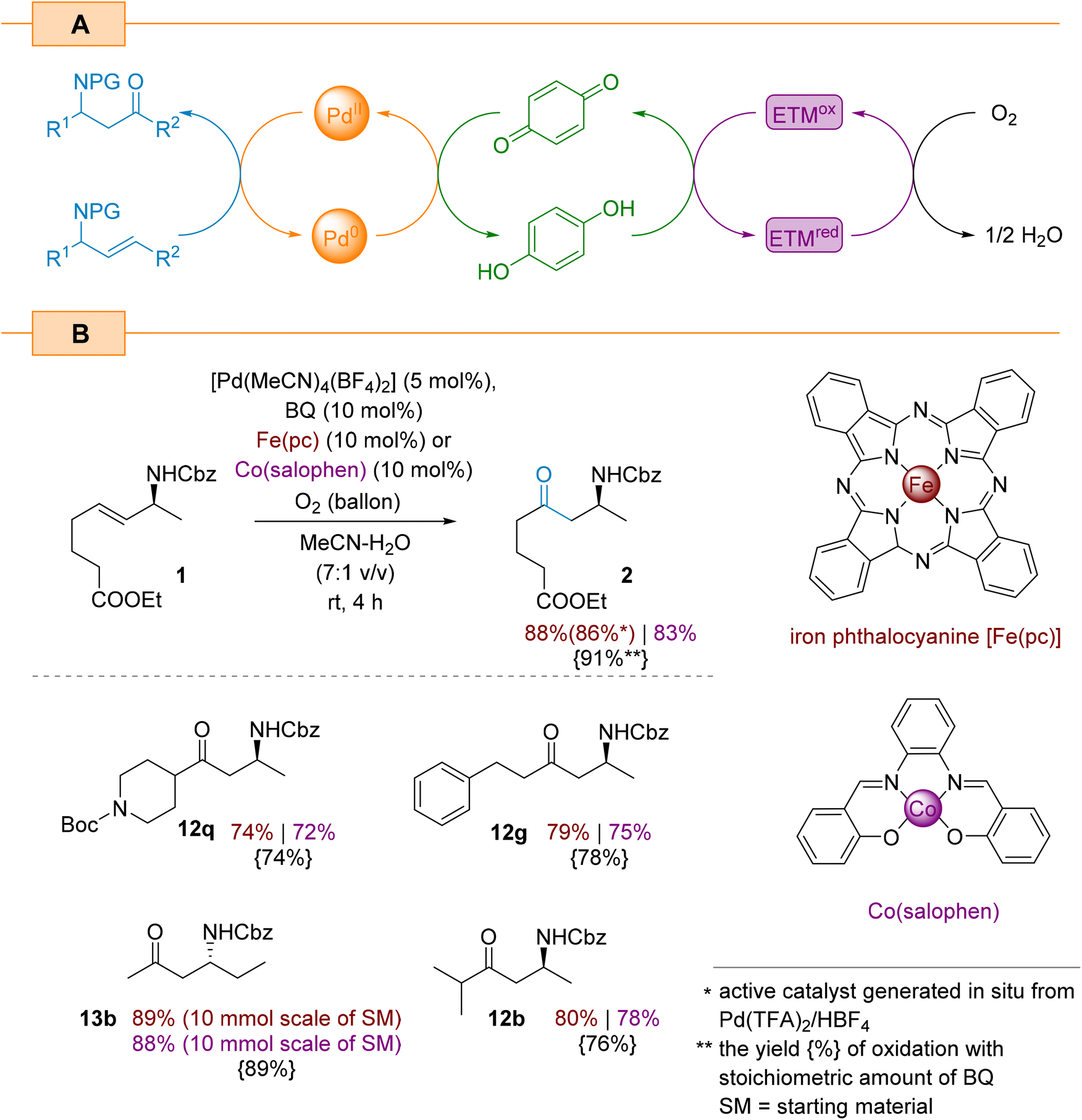 | ||
| Scheme 7 Catalytic aerobic the Wacker oxidation of internal allylamines. | ||
The control experiments with model allylamine 1 showed that the reaction using both ETMs (10 mol%) and benzoquinone (10 mol%) resulted in product 2 the yields of 88% and 83% when using Fe(pc) and Co(salophen) as ETMs, respectively. In both the cases, [Pd(MeCN)4(BF4)2] was used as the catalyst. We conducted further experiments on the Fe(pc)-aided oxidation which showed that the [Pd(MeCN)4(BF4)2] complex can be also generated in situ from Pd(TFA)2 and HBF4, and provides the target amino ketone 2 in a comparable yield (86%). When we applied both presented triple catalytic systems to oxidise selected substrates from Scheme 4, we again obtained comparable yields (Scheme 7B). As illustrated in Scheme 7B, the iron-based redox catalyst appears to be slightly more efficient than the cobalt-based one. Moreover, using 13b, we found that both catalytic protocols are scalable and deliver comparable isolated yields (scale: 10 mmol of starting material).
To further emphasize the utility of the β-amino ketones produced by the reported methods, amino ketone 13r was subjected to further transformations (Scheme 8). Addition of a Grignard reagent to ketone 13r provided a mixture of diastereoisomeric amino alcohols 21. Olefination of 13r with trimethyl phosphonoacetate provided the corresponding α,β-unsaturated ester 22 with a 62% yield. The reduction of ketone 13r with L-selectride provided a mixture of 1,3-amino alcohols 23 in a ratio of 2:1 (anti/syn), while a reduction with NaBH4 in the presence of CeCl3 provided amino alcohols 23 in the opposite stereoselectivity (4:1, syn/anti). A reagent namely NaBH4 in the presence of Lewis acid (e.g. Ce, Zn, Mn, or Ni salt) delivers an external hydride via a six-membered chelate involving the keto and amino groups (Scheme 8, TS-B),8 while anti selectivity plausibly results from intramolecular hydride delivery via a TS-A transition state.8 Lower selectivity in the last case plausibly reflects the difficulty of L-selectride to efficiently coordinate to the carbonyl group with the nitrogen atom in these compounds and results in only slight differentiation of both the Re and Si faces of the carbonyl.8
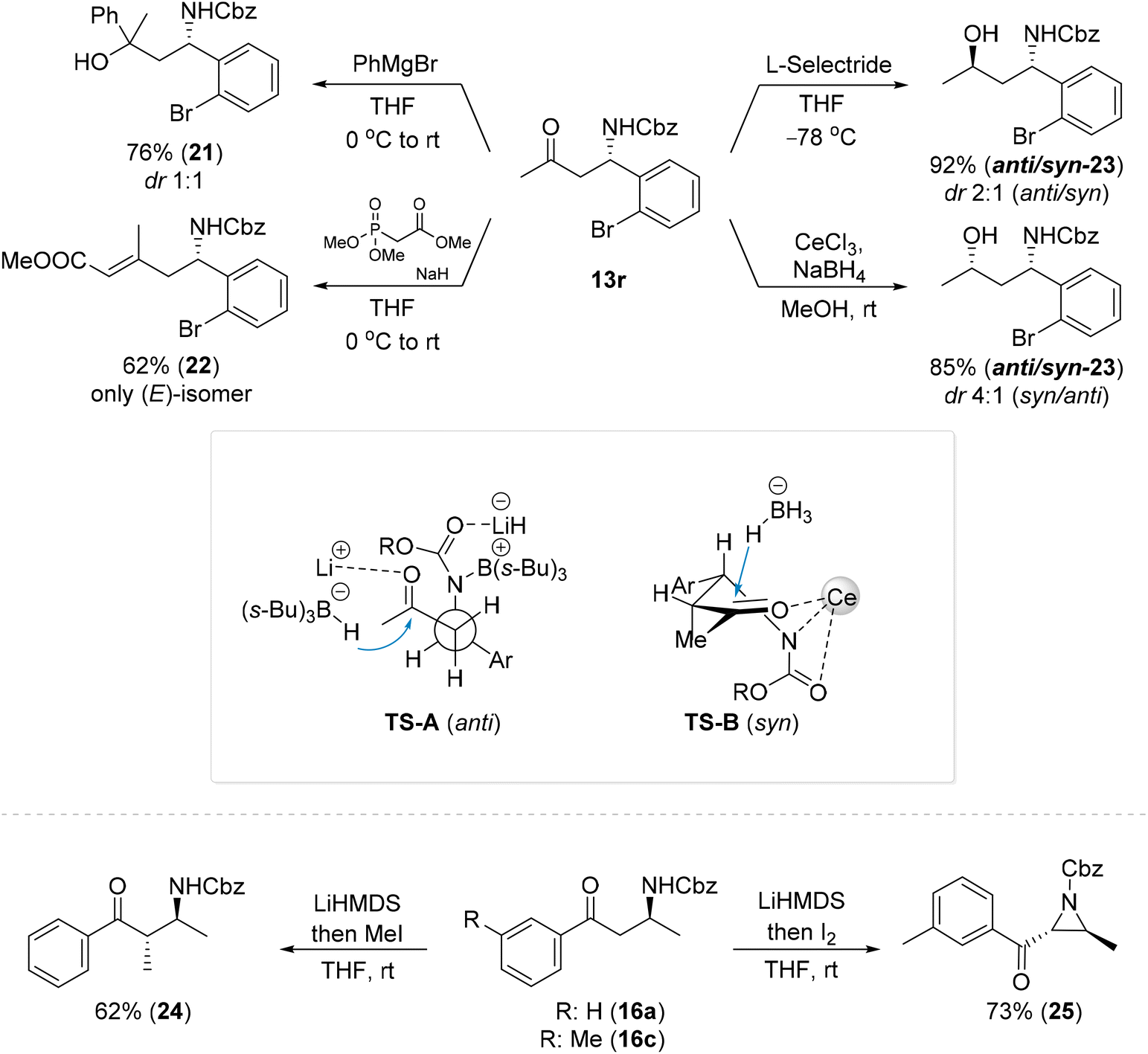 | ||
| Scheme 8 Selected transformations of β-amino ketones. | ||
Alkylation of 16a with MeI provided compound 24 as a sole isomer.60 The deprotonation of 16c followed by iodination and intramolecular cyclization led to a 73% yield of ketoaziridine 25, which is an interesting synthetic platform that can be regioselectively functionalized in various manners.13,61–66
To further demonstrate the utility of the reported oxidation protocol, we applied it to the synthesis of chromanylamine 30 (Scheme 9), which is a structural motif in a series of potent CFTC regulator correctors (e.g., 2667) for the treatment of cystic fibrosis, multiorgan disease of the lungs, sinuses, pancreas, and gastrointestinal tract, caused by a dysfunction or deficiency of the cystic fibrosis transmembrane conductance (CFTC) regulator protein. The Tsuji–Wacker oxidation of allylamine derivative 27 proved to be challenging due to the deactivating effect of the electron-poor ring. Nevertheless, it provided a β-amino ketone 28 a yield of 51% after 5 days. Removal of the Ac group under basic conditions furnished a free alcohol which, after subsequent cyclization, provided hemiacetal 29 in a yield of 85%. Its reduction under the conditions reported by Tunge and co-workers68 (by treatment with Et3SiH in the presence of BF3·Et2O) resulted in chromanylamine 30 as a single isomer. In contrast to the original protocol, we found that the reaction had to be performed at −30 °C (instead of −78 °C). A relative configuration of 30 was confirmed through NMR analysis. Compound 30 can serve as a direct precursor for preparing CFTC regulator correctors such as 26.
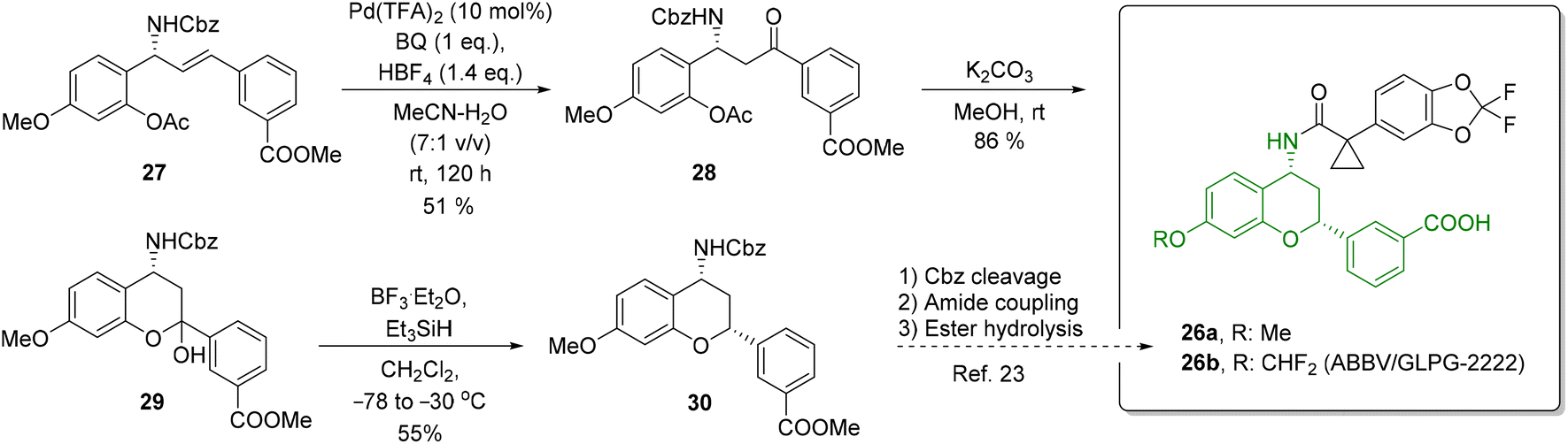 | ||
| Scheme 9 Synthesis of chromanylamine 30, a key structural scaffold of type 26 CFTC regulator correctors. | ||
Conclusions
In this work we could show that the Wacker oxidation provides a facile route for the preparation of functionalized β-amino ketones from a wide variety of structurally diverse allylamines. The approach is highly reliable and versatile which highlights its great potential for immediate applications in target-oriented syntheses and as a source for other synthetically useful building blocks. Some of the most prominent characteristics of this process are its high regioselectivity and its wide functional group tolerance. Moreover, various N-protecting groups, including carbamate, amide, sulfamide, or carbamoyl group, are well tolerated. In addition, we demonstrated the applicability of this procedure for the synthesis of bioactive compounds and further transformations of the resulting β-amino ketone derivatives. Importantly, we could demonstrate the catalytic aerobic variant of the reported Wacker oxidation using oxygen as a terminal oxidant in combination with Fe- or Co-based electron transfer mediators as redox catalysts for the hydroquinone re-oxidation. We were able to successfully demonstrate the scalability of this protocol, which, together with its simplicity, generality, and ability to provide access to amino ketones that are inaccessible to other strategies, should make this approach highly useful and enable others to find many applications for it across different chemical sciences.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support by the National Science Centre of Poland (research grant OPUS 2016/23/B/ST5/03322). Editorial assistance, in the form of language editing and correction, was provided by XpertScientific Editing and Consulting Services.References
- N. H. Nghi, H. B. Andrew and S. E. Brad, Curr. Org. Chem., 2014, 18, 260–289 CrossRef.
- A. Ricci, Amino Group Chemistry: From Synthesis to the Life Sciences, Wiley, 2008 Search PubMed.
- M. Braun, Modern Enolate Chemistry: From Preparation to Applications in Asymmetric Synthesis, Wiley, 2015 Search PubMed.
- D. Enders, K. E. Jaeger and G. Helmchen, Asymmetric Synthesis with Chemical and Biological Methods, Wiley, 2007 Search PubMed.
- A. S. Skwarecki, M. Schielmann, D. Martynow, M. Kawczyński, A. Wiśniewska, M. J. Milewska and S. Milewski, J. Pept. Sci., 2018, 24, e3060 CrossRef.
- S. Chacko, M. Kalita and R. Ramapanicker, Tetrahedron: Asymmetry, 2015, 26, 623–631 CrossRef CAS.
- M. R. Monaco, P. Renzi, D. M. Scarpino Schietroma and M. Bella, Org. Lett., 2011, 13, 4546–4549 CrossRef CAS PubMed.
- F. A. Davis, P. M. Gaspari, B. M. Nolt and P. Xu, J. Org. Chem., 2008, 73, 9619–9626 CrossRef CAS PubMed.
- J. Barluenga, B. Olano and S. Fustero, J. Org. Chem., 1985, 50, 4052–4056 CrossRef CAS.
- J. Barluenga, E. Aguilar, S. Fustero, B. Olano and A. L. Viado, J. Org. Chem., 1992, 57, 1219–1223 CrossRef CAS.
- M. Brodney, K. Gagnon, K.-N. Hu, A. Medek, P. Rose, Y. Shi, T. J. Senter and T. Wang, WO2020/131807 A1, 2020.
- U. Sorensen and A. Mete, WO2019/38315 A1, 2019.
- G. Luppi and C. Tomasini, Synlett, 2003, 797–800 CAS.
- C. Tomasini and A. Vecchione, Org. Lett., 1999, 1, 2153–2156 CrossRef CAS.
- H. Wang, J. Shi, J. Tan, W. Xu, S. Zhang and K. Xu, Org. Lett., 2019, 21, 9430–9433 CrossRef CAS.
- J. Humbrías-Martín, M. C. Pérez-Aguilar, R. Mas-Ballesté, A. Dentoni Litta, A. Lattanzi, G. Della Sala, J. A. Fernández-Salas and J. Alemán, Adv. Synth. Catal., 2019, 361, 4790–4796 CrossRef.
- T. Das, S. Mohapatra, N. P. Mishra, S. Nayak and B. P. Raiguru, ChemistrySelect, 2021, 6, 3745–3781 CrossRef CAS.
- Y. Zhang and W. Wang, Catal. Sci. Technol., 2012, 2, 42–53 RSC.
- K. Zheng, X. Liu and X. Feng, Chem. Rev., 2018, 118, 7586–7656 CrossRef CAS.
- J. Wang, P. Li, P. Y. Choy, A. S. C. Chan and F. Y. Kwong, ChemCatChem, 2012, 4, 917–925 CrossRef CAS.
- D. Enders, C. Wang and J. X. Liebich, Chem. – Eur. J., 2009, 15, 11058–11076 CrossRef CAS.
- J. Wang, P. Li, P. Y. Choy, A. S. C. Chan and F. Y. Kwong, ChemCatChem, 2012, 4, 1020–1020 CrossRef.
- S. Avidan-Shlomovich, H. Ghosh and A. M. Szpilman, ACS Catal., 2015, 5, 336–342 CrossRef CAS.
- H. Geng, K. Huang, T. Sun, W. Li, X. Zhang, L. Zhou, W. Wu and X. Zhang, J. Org. Chem., 2011, 76, 332–334 CrossRef CAS PubMed.
- J. S. Zhou, S. Guo, X. Zhao and Y. R. Chi, Chem. Commun., 2021, 57, 11501–11504 RSC.
- Q. Llopis, G. Guillamot, P. Phansavath and V. Ratovelomanana-Vidal, Org. Lett., 2017, 19, 6428–6431 CrossRef CAS PubMed.
- E. Cristóbal-Lecina, P. Etayo, S. Doran, M. Revés, P. Martín-Gago, A. Grabulosa, A. R. Costantino, A. Vidal-Ferran, A. Riera and X. Verdaguer, Adv. Synth. Catal., 2014, 356, 795–804 CrossRef.
- T. Imamoto, K. Tamura, Z. Zhang, Y. Horiuchi, M. Sugiya, K. Yoshida, A. Yanagisawa and I. D. Gridnev, J. Am. Chem. Soc., 2012, 134, 1754–1769 CrossRef CAS.
- J. R. Lamb, M. Mulzer, A. M. LaPointe and G. W. Coates, J. Am. Chem. Soc., 2015, 137, 15049–15054 CrossRef CAS PubMed.
- G. Jiang, J. Chen, H.-Y. Thu, J.-S. Huang, N. Zhu and C.-M. Che, Angew. Chem., Int. Ed., 2008, 47, 6638–6642 CrossRef CAS PubMed.
- J. Chen and C.-M. Che, Angew. Chem., Int. Ed., 2004, 43, 4950–4954 CrossRef CAS.
- A. D. Chowdhury, R. Ray and G. K. Lahiri, Chem. Commun., 2012, 48, 5497–5499 RSC.
- J. Ying and L. Pu, J. Org. Chem., 2016, 81, 8135–8141 CrossRef CAS PubMed.
- J. Ying, H. Wang, X. Qi, J.-B. Peng and X.-F. Wu, Organometallics, 2018, 37, 2837–2841 CrossRef CAS.
- S. E. Mann, L. Benhamou and T. D. Sheppard, Synthesis, 2015, 47, 3079–3117 CrossRef CAS.
- R. A. Fernandes, A. K. Jha and P. Kumar, Catal. Sci. Technol., 2020, 10, 7448–7470 RSC.
- B. W. Michel, L. D. Steffens and M. S. Sigman, in Organic Reactions, 2014, pp. 75–414, DOI:10.1002/0471264180.or084.02.
- B. Weiner, A. Baeza, T. Jerphagnon and B. L. Feringa, J. Am. Chem. Soc., 2009, 131, 9473–9474 CrossRef CAS.
- R. J. DeLuca, J. L. Edwards, L. D. Steffens, B. W. Michel, X. Qiao, C. Zhu, S. P. Cook and M. S. Sigman, J. Org. Chem., 2013, 78, 1682–1686 CrossRef CAS PubMed.
- T. Mitsudome, K. Mizumoto, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2010, 49, 1238–1240 CrossRef CAS PubMed.
- M. Garbacz and S. Stecko, Adv. Synth. Catal., 2020, 362, 3213–3222 CrossRef CAS.
- M. Garbacz and S. Stecko, Org. Biomol. Chem., 2021, 19, 8578–8585 RSC.
- B. Morandi, Z. K. Wickens and R. H. Grubbs, Angew. Chem., Int. Ed., 2013, 52, 2944–2948 CrossRef CAS.
- D. A. Chaudhari and R. A. Fernandes, J. Org. Chem., 2016, 81, 2113–2121 CrossRef CAS.
- R. A. Fernandes, G. V. Ramakrishna and V. Bethi, Org. Biomol. Chem., 2020, 18, 6115–6125 RSC.
- J. Zhao, L. Liu, S. Xiang, Q. Liu and H. Chen, Org. Biomol. Chem., 2015, 13, 5613–5616 RSC.
- K.-F. Hu, X.-S. Ning, J.-P. Qu and Y.-B. Kang, J. Org. Chem., 2018, 83, 11327–11332 CrossRef CAS.
- X.-S. Ning, M.-M. Wang, C.-Z. Yao, X.-M. Chen and Y.-B. Kang, Org. Lett., 2016, 18, 2700–2703 CrossRef CAS.
- In the case of the Mannich reaction, it is highly difficult to achieve enantio-differentiation for ketoimines bearing barely the same groups at the imines’ carbon atom. 1,4-Addition to β,β-disubstituted Micheal acceptors is unfavorable and slow due to steric factors and reversibility.
- Y. Deng, A. K. Å. Persson and J.-E. Bäckvall, Chem. – Eur. J., 2012, 18, 11498–11523 CrossRef CAS PubMed.
- H. A. Döndaş, M. d. G. Retamosa and J. M. Sansano, Organometallics, 2019, 38, 1828–1867 CrossRef.
- Y. Ping, Y. Li, J. Zhu and W. Kong, Angew. Chem., Int. Ed., 2019, 58, 1562–1573 CrossRef CAS PubMed.
- R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981–3019 CrossRef CAS.
- J. Liu, A. Guðmundsson and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2021, 60, 15686–15704 CrossRef CAS PubMed.
- J. Piera and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2008, 47, 3506–3523 CrossRef CAS PubMed.
- S. Manna, W.-J. Kong and J.-E. Bäckvall, in Advances in Catalysis, ed. M. Diéguez and O. Pàmies, Academic Press, 2021, vol. 69, pp. 1–57 Search PubMed.
- C. J. Pereira Monteiro, M. A. Ferreira Faustino, M. d. G. Pinho Morgado Silva Neves, M. M. Quialheiro Simões and E. Sanjust, Catalysts, 2021, 11, 122 CrossRef.
- C. W. Anson, S. Ghosh, S. Hammes-Schiffer and S. S. Stahl, J. Am. Chem. Soc., 2016, 138, 4186–4193 CrossRef CAS.
- C. C. Pattillo, I. I. Strambeanu, P. Calleja, N. A. Vermeulen, T. Mizuno and M. C. White, J. Am. Chem. Soc., 2016, 138, 1265–1272 CrossRef CAS.
- S. Gair, R. F. W. Jackson and P. A. Brown, Tetrahedron Lett., 1997, 38, 3059–3062 CrossRef CAS.
- O. Yasuyuki, K. Kiichi and M. Teruaki, Bull. Chem. Soc. Jpn., 2005, 78, 1309–1333 CrossRef.
- K. O'Brien, K. ó. Proinsias and F. Kelleher, Tetrahedron, 2014, 70, 5082–5092 CrossRef.
- B. Schweitzer-Chaput, M. Keita, T. Milcent, S. Ongeri and B. Crousse, Tetrahedron, 2012, 68, 7028–7034 CrossRef CAS.
- T. K. Chakraborty, G. Animesh and R. T. Venugopal, Chem. Lett., 2003, 32, 82–83 CrossRef CAS.
- S. Hanessian and L.-D. Cantin, Tetrahedron Lett., 2000, 41, 787–790 CrossRef CAS.
- X.-F. Tan, F.-G. Zhang and J.-A. Ma, Beilstein J. Org. Chem., 2020, 16, 638–644 CrossRef CAS PubMed.
- X. Wang, B. Liu, X. Searle, C. Yeung, A. Bogdan, S. Greszler, A. Singh, Y. Fan, A. M. Swensen, T. Vortherms, C. Balut, Y. Jia, K. Desino, W. Gao, H. Yong, C. Tse and P. Kym, J. Med. Chem., 2018, 61, 1436–1449 CrossRef CAS PubMed.
- K. Li, K. Vanka, W. H. Thompson and J. A. Tunge, Org. Lett., 2006, 8, 4711–4714 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01843h |
| ‡ This paper is dedicated to Prof. Marek Chmielewski on his 80th birth anniversary. |
| This journal is © The Royal Society of Chemistry 2023 |