Enhanced molecular binding affinity toward aromatic dications by anthracene-derived crown ethers in water†
Received
4th November 2022
, Accepted 5th December 2022
First published on 6th December 2022
Abstract
The pursuit of high molecular binding affinity using conventional crown ethers in water remains a challenging task in the field of supramolecular chemistry and may hold great promise in the creation of advanced biocompatible nanoconstructs. In this work, the molecular binding strength toward a series of structurally relevant cationic guests has been greatly enhanced by tetrasulfonated 1,5-dianthracenyl-42-crown-10 and as investigated by means of 1H NMR, UV–vis, and fluorescence spectroscopy, the host–guest association constants can reach up to 108 M−1 order of magnitude in aqueous solution. X-ray crystal diffraction analysis further demonstrates that the aromatic dication can be tightly encapsulated in the ring of anthracene-derived crown ether via multiple π-stacking and electrostatic interactions. Meanwhile, the obtained association constants are remarkably higher than the ones in the cases of the known benzene- and naphthalene-derived sulfonated crown ethers, substantiating that the appropriate extension of π-conjugation in the molecular skeleton of crown ether is a feasible method in attaining a highly affiliative host–guest complex. Taken together, our results indicate that the anthracene-based sulfonated crown ether can be developed as a new family of water-soluble macrocyclic receptors in the fabrication of functional nanoarchitectures.
Introduction
Progress in the field of supramolecular chemistry has been driven by the need for rationally designed synthetic receptors for applications in self-assembly processes,1 molecular machines,2 and functional materials,3 especially in aqueous environments.4 In this context, a variety of macrocyclic receptors, including crown ethers,5 cyclodextrins,6 calixarenes,7 and cucurbiturils,8 have drawn considerable attention in recent decades owing to their strong binding affinities toward a wide range of molecules and organic cations. Moreover, considering water is a unique, ubiquitous solvent in many chemical and biological processes, water-soluble macrocyclic receptors are of particular interest and are widely used as molecular sensors,9 amphiphilic assemblies,10 and hydrogels.11 Among the diverse water-soluble receptors, crown ethers exhibit particularly impressive performance because they are typically used in organic solution and lose most of their binding capability in an aqueous environment.12 Therefore, many efforts have been made in developing water-soluble crown ethers possessing high molecular binding strength. In the past years, several research groups,13 including ours,14 have made progress in enhancing the binding abilities of aromatic crown ethers in aqueous solution by introducing negatively charged functional groups. In particular, those aromatic crown ethers modified with sulfonate groups show exceptionally high affinities toward a variety of cationic guests, owing to the involvement of electrostatic and π-stacking interactions and the size-matching effect within the host–guest complexes.15
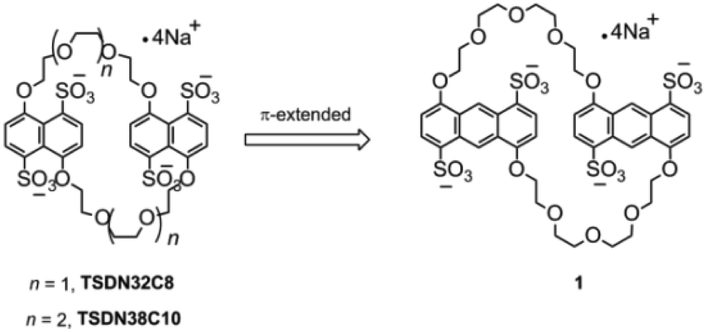
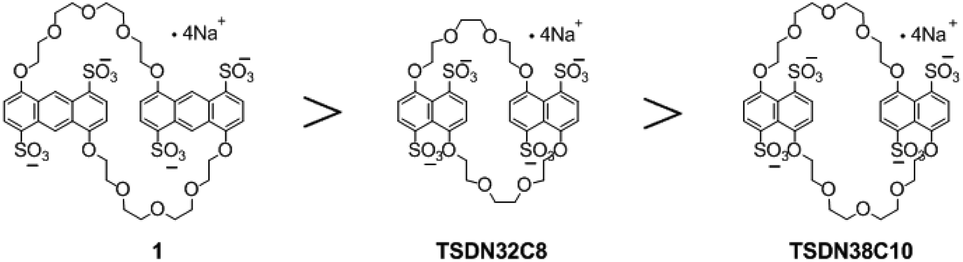
Crown ethers with an aromatic skeleton that can form pseudorotaxane-like complexes have been widely studied in recent years,16 and the introduction of sulfonate groups into the aromatic backbone of these crown ethers eventually results in the generation of negatively charged hosts with excellent water solubility and can strongly bind and recognize π-electron-deficient guests such as pyridinium ions and aromatic imides.13–15 The association constants (Ka) for these water-soluble complexes mainly range from 102 to 107 M−1, depending on the extension of the π-stacking area and charge numbers. Therefore, sulfonated crown ethers are suitable candidates for constructing tailor-made supramolecular assemblies with specific functions.17 For instance, we previously demonstrated that tetrasulfonated bis(m-phenylene)-26-crown-8 (TS26C8),14a tetrasulfonated dinaphtho-38-crown-10 (TS38C10),14b and tetrasulfonated dinaphtho-32-crown-8 (TS32C8)14b have different abilities of binding and recognizing paraquat, with Ka values of 103, 105, and 107 M−1, respectively. This variability is clearly due to TS26C8 with much smaller m-phenylene rings and less extensive π-conjugation than the other two naphthalene-based crown ethers.
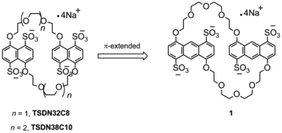
The pursuit of highly affinitive macrocyclic hosts has long been a focus area in supramolecular chemistry research.18 Although the sulfonated naphtho crown ethers TS38C10 and TS32C8 are useful hosts in the supramolecular assembly process, developing more sulfonated crown ethers with stronger binding ability is still of fundamental importance in supramolecular chemistry. We reasoned that we could increase the affinity of this type of macrocyclic host by replacing the aromatic units of the previously reported sulfonated crown ethers with anthracene units to increase the π-conjugation (Fig. 1) and thus increase the π-stacking interactions between the π-electron-deficient guests and the planes of the aromatic rings. Herein, as part of our ongoing work on developing sulfonated crown ethers with specific host–guest binding characteristics,14 we report the synthesis of a new anthracene-derived tetrasulfonated crown ether containing two 4,8-disulfonate-1,5-anthracyl units linked by two tetraethylene glycol chains, which is named tetrasulfonated 1,5-dianthracenyl-42-crown-10 (1). In addition, we evaluated the abilities of this macrocycle to bind seven typical cationic guest molecules (G1–G7, Scheme 1) in aqueous media. We found that 1 could bind tightly to the guests with two cations and the obtained Ka values were in the range of 103–108 M−1, mainly depending on the extension of π-conjugation in these guest molecules. According to these results, the anthracene-based crown ether 1 is a more powerful host than the known tetrasulfonated benzo- and naphtho-crown ethers. Our findings are also expected to encourage the synthesis of other water-soluble crown ethers with larger aromatic cores and may facilitate the development of more advanced functional supramolecular assemblies based on water-soluble macrocycles.19
 |
| | Fig. 1 Structures of previously reported dinaphtho-tetrasulfonated crown ethers and tetrasulfonated crown ethers bearing aromatic moieties with the more extended anthracene unit. | |
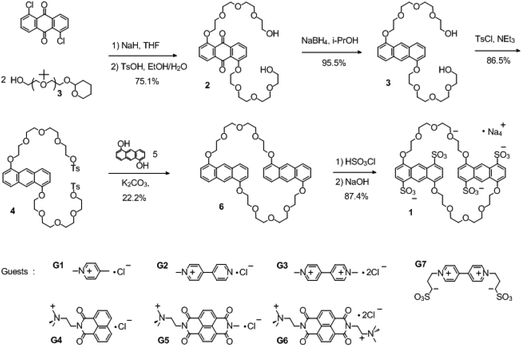 |
| | Scheme 1 Synthesis of 1 and molecular structures of the guests. | |
Results and discussion
Synthesis of the hosts
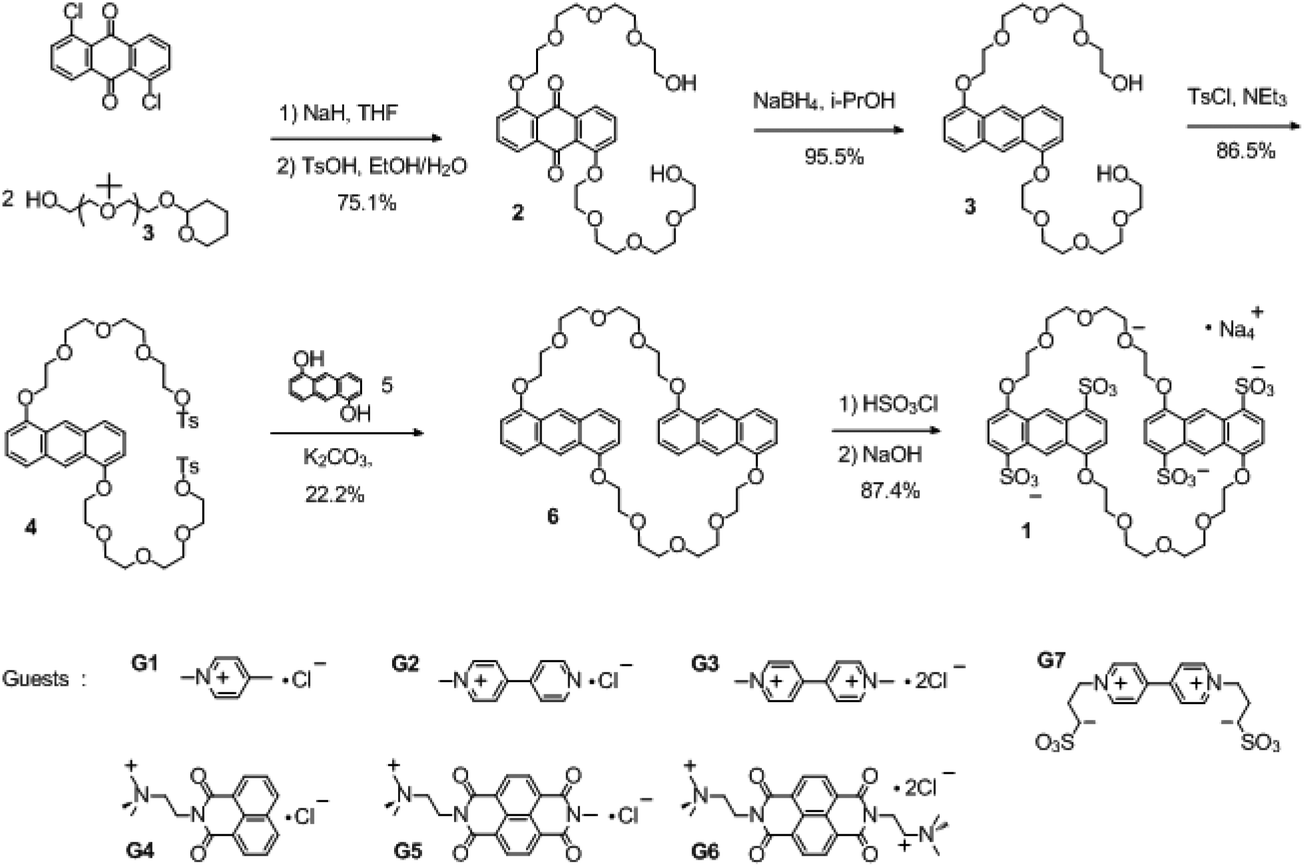
Tetrasulfonated anthracenyl crown ether 1 was synthesized by sulfonation of the corresponding neutral precursors 6 with chlorosulfonic acid (Scheme 1). The neutral precursor 6, namely, 1,5-dianthracenyl-42-crown-10, was synthesized starting from 1,5-dichloroanthraquinone and tetrahydropyran-protected tetraethylene glycol (2), as shown in Scheme 1. Briefly, a nucleophilic aromatic substitution reaction between 1,5-dichloroanthraquinone and tetrahydropyran-protected tetraethylene glycol (3 equiv.) gave tetraethylene glycol-modified anthraquinone 3. Reduction of 3 with NaBH4 in isopropanol followed by tosylation gave ditosylate 4, which was converted into neutral anthracenyl crown ether 6 by macrocyclization under high-dilution conditions. Then 6 was sulfonated with chlorosulfonic acid (4.1 equiv.) in dichloromethane at room temperature. Subsequent neutralization with sodium hydroxide and recrystallization in alcohol–water gave 1 as pure sodium salts, which showed good solubility (up to 30 mM) in water. It is noteworthy that only slightly more than 4 equiv. of chlorosulfonic acid was necessary to introduce four sulfonate groups onto crown ether 6, i.e., this reaction consumed less of the sulfonating reagent than our previously reported procedure for the preparation of tetrasulfonated benzo and naphtho crown ethers (each crown ether consumed at least 8 equiv. of chlorosulfonic acid).14 We attributed this difference to the higher reactivity of the 1,5-dioxyanthracene units relative to that of the other aromatic units.
Selection of guest molecules
In our previous work, we concluded that the strong host–guest binding abilities of tetrasulfonated benzo and naphtho crown ethers were mainly due to a combination of the electrostatic attraction, π-stacking interaction, and size-matching effect.15a Compared with these crown ethers, macrocycle 1 has a more-extended aromatic skeleton that can be expected to generate a broader area of π-overlap with cationic π-conjugated guests and provide more extensive π-stacking interaction. To confirm this expectation, six guests (G1–G6) with various π-conjugated moieties and charges were selected to study the binding abilities of 1. The six guests can be divided into two categories: pyridinium guests (G1–G3) and naphthalene guests (G4–G6), and each category contains singly charged small cations (G1, G4), singly charged large cations (G2, G5) and double charged cations (G3, G6). By carrying out a series of analytical methods, including 1H NMR, UV-vis spectroscopy, fluorescence titration, and X-ray crystallography, it could be realized how noncovalent interactions like π-stacking, charge attraction and C–H⋯O hydrogen bonding play their role in host–guest binding processes with high affinity. Moreover, a zwitterion bipyridinium guest G7 was also selected as a candidate because the complex of G3⊂1 could only exist at a very low concentration.
1H NMR spectroscopy
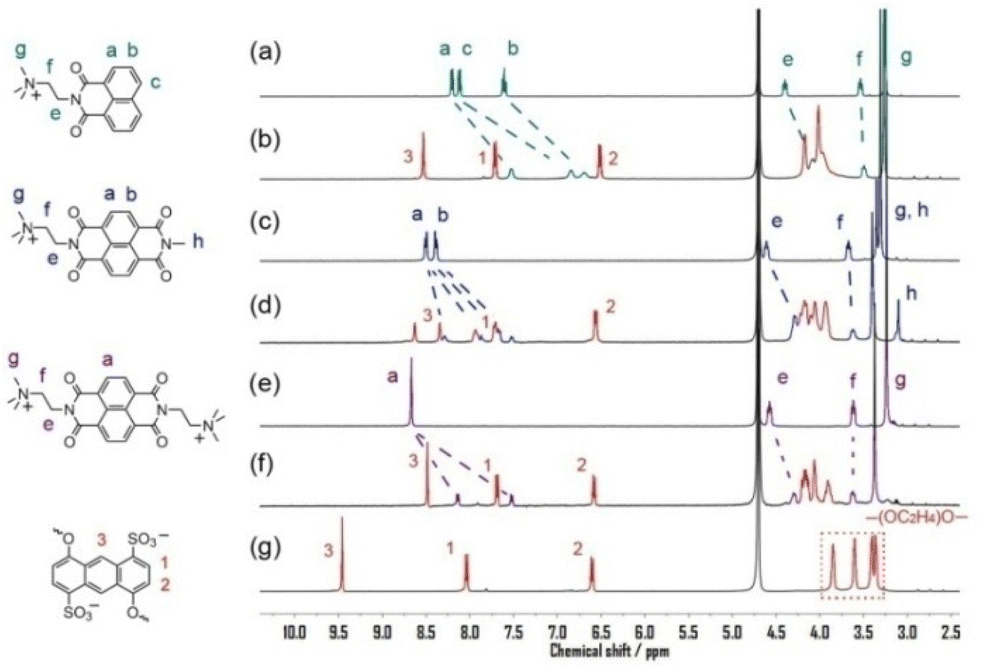
First, we examined the host–guest complexation of the crown ethers with the six guests (G1–G6) by means of 1H NMR spectroscopy. When 1 equiv. of G1 was added to the D2O solution of 1, a slight color change was observed. As can be seen in the spectra presented in Fig. 2, the exchange of G1⊂1 complexation between the free and bound states of the guest was fast relative to the NMR timescale because the ring of 1 is broad enough to allow the guests to enter and exit through the cavity freely. Complexation resulted in large upfield shifts of the proton signals for most of the aromatic protons of G1 (Ha and Hb, Fig. 2b) and 1 (H1–H3, Fig. 2b), the exception being H1 of G1⊂1, which even shifted slightly downfield (Δδ = 0.07). The upfield shifts clearly indicated that these aromatic protons experienced mutual diamagnetic shielding, which can be attributed to the sandwiched π-stacking structure in the host–guest complexes, similar to π-stacking structures for other macrocyclic hosts.20 The magnitudes of the upfield shifts for 1 decreased in the order of H3 > H2 > H1, implying that upon complexation, G1 interacted mostly with the central six-membered ring of the anthracene units of 1, whereas the outer rings of the anthracene units (those attached to the tetraethylene glycol chains) were located farther away from G1 and thus less shielded. On the other hand, the 1H NMR signals corresponding to tetraethylene glycol chains on crown ether 1 (3.0 < δ < 4.5) exhibited clearly downfield shift changes (Δδaverage = 0.38), which were mainly due to the decrease of the electron density on hydrogen atoms induced by C–H⋯O hydrogen bonds between G1 and 1. By carrying out 1H NMR titration experiments and analyzing sequential changes in the chemical shift of H3 (ΔδH3) (Fig. S18 in ESI†), we determined the Ka value for G1⊂1 to be (2.2 ± 0.1) × 103 M−1 in D2O. The 1H NMR spectral changes for the complexation of G2⊂1 closely resembled those observed for G1, except for the slightly small difference in chemical shift changes, as shown in Fig. 2d. The guest G3 was not investigated here because it formed a red precipitate with 1 at a concentration of 10−3 M. Here we investigated the binding behaviours of G7⊂1 instead. It could be found that the binding-induced upfield shift changes were similar to those of G1 and G2 except for the hydrogen neighbouring to the sulfonate group on G7. The small downfield shift change indicates that the sulfonate groups are located on the shielded area of the anthracene when bound by 1.
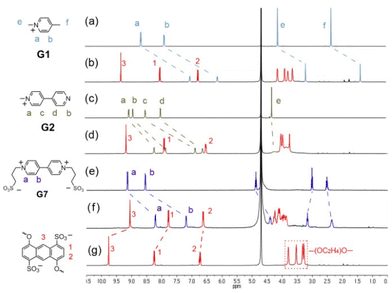 |
| | Fig. 2 Partial 1H NMR spectra (400 MHz, D2O, 25 °C) of (a) G1; (b) G1⊂1 complex; (c) G2; (d) G2⊂1 complex; (e) G7; (f) G7⊂1; (g) host 1 ([G1] = [G2] = [G7] = [1] = 3 mM). | |
The complexation between naphthalene imide guests (G4, G5, and G6) and tetrasulfonated crown ether 1 was also investigated by 1H NMR spectroscopy (Fig. 3). For the most part, the chemical shift changes induced by the host–guest interactions were similar to those described for the pyridinium guests. That is, for the three guests, all the aromatic protons on both the host and guests exhibited a large upfield shift except for the H1 protons with slightly small chemical shift changes, while the proton resonances of tetraethylene glycol of 1 showed considerable downfield shift changes. This observation indicated that although the three naphthalene imide guests had more extensive π-conjugation than the corresponding pyridinium guests, they exhibited similar binding features like π-stacking, C–H⋯O hydrogen bonding and inner binding in the cavity nearby the central six-membered rings of anthracenes. On the other hand, appreciable disparities were still observed for the associated 1H NMR spectra of these naphthalene imide guests. After being combined with 1, the peaks of aromatic protons on the naphthalene imide moiety of G4 broadened obviously (Fig. 3b). More distinct resonances could even be observed for the complexes of G5⊂1 and G6⊂1. Otherwise, when the guests G5 and G6 were bound in the cavity of 1, the proton resonances of naphthalene diimide splitted into two sets of upfield shifted signals. The two sets of signals did not correspond to any signals of free G5 or G6, indicating that these splitting signals were not due to the kinetically slow exchange between free and bound guests on the NMR time scale. Considering these broadening or splitting was not found upon the complexation of pyridinium guests G1 or G2 with smaller molecular sizes (ESI, Fig. S1†), these 1H NMR spectra indicated that when the guests G4–G6 were bound in the cavity of 1, their large aromatic moieties of naphthalene imide caused the binding to be of low degrees of conformational freedom, and constrained tightly in the cavity.
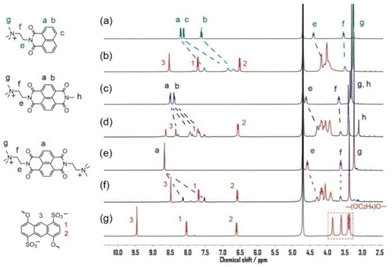 |
| | Fig. 3 Partial 1H NMR spectra (400 MHz, D2O, 25 °C) of (a) G4; (b) G4⊂1 complex; (c) G5; (d) G5⊂1 complex; (e) G6; (f) G6⊂1 complex; ((g) host 1 ([G4] = [G5] = [G6] = [1] = 3 mM). | |
UV-vis spectroscopy
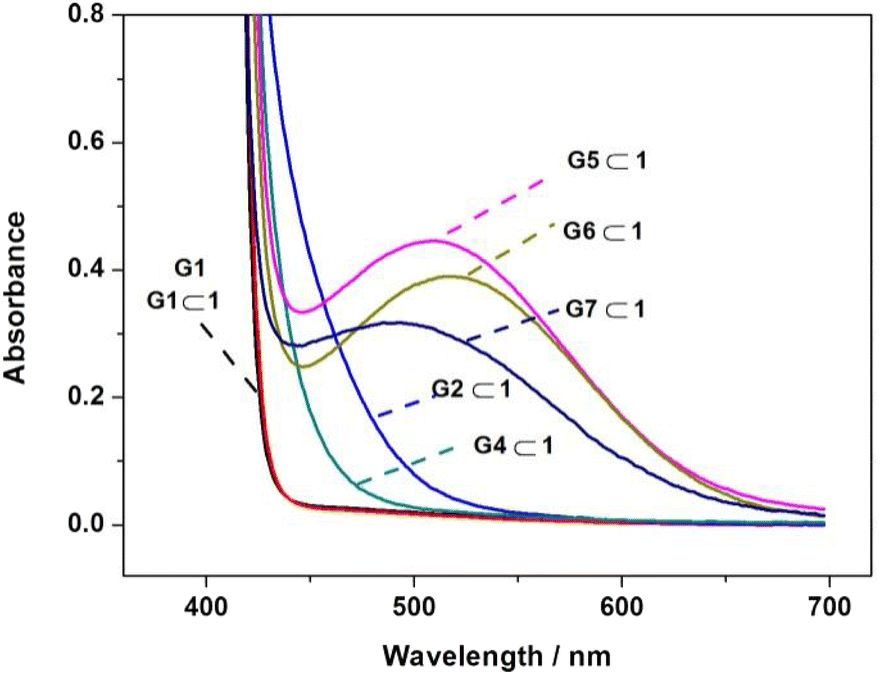
It is well documented that the π-stacking interaction between electron-rich and electron-poor aromatic rings could generate a charge transfer (CT) absorbance band in the visible light region.21 To further understand the π-stacking interaction in the binding process, we investigated the UV-vis absorption of the complexes with host 1. As shown in Fig. 4, the absorption differs considerably between the complexes containing different pyridinium guests (G1–G7, except G3) in the visible light range, which mainly follow the order of G1 < (G4, G2) < (G7, G6, G5). Since G1 has only a simply small pyridinium moiety, the absorption of G1⊂1 is almost the same as the free host 1 (ESI, Fig. S16†), indicating that no charge transfer interaction occurs during the binding of G1. In contrast, complexes G2⊂1 and G4⊂1 showed a wide and weak CT absorption band at 430 nm, while complexesG5⊂1, G6⊂1 and G7⊂1 showed clearly strong CT absorption with peaks at 510, 517 and 484 nm, respectively. In general, the CT absorption wavelengths and intensities were associated directly with the electron-deficiency of the guests. G2 and G4 had larger π-conjugation than G1, while G5, G6 and G7 had a naphthalene diimide or bipyridinium moiety to support the stronger CT effect.
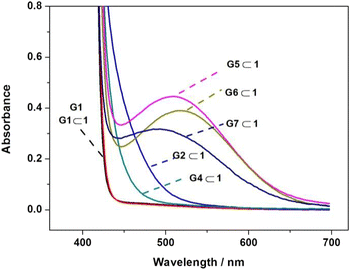 |
| | Fig. 4 Charge-transfer absorption spectra of G1–G7 (1 mM) upon complexation with equimolar host 1 (except the complex G3⊂1 with solubility far less than 1 mM). | |
Crystal structures
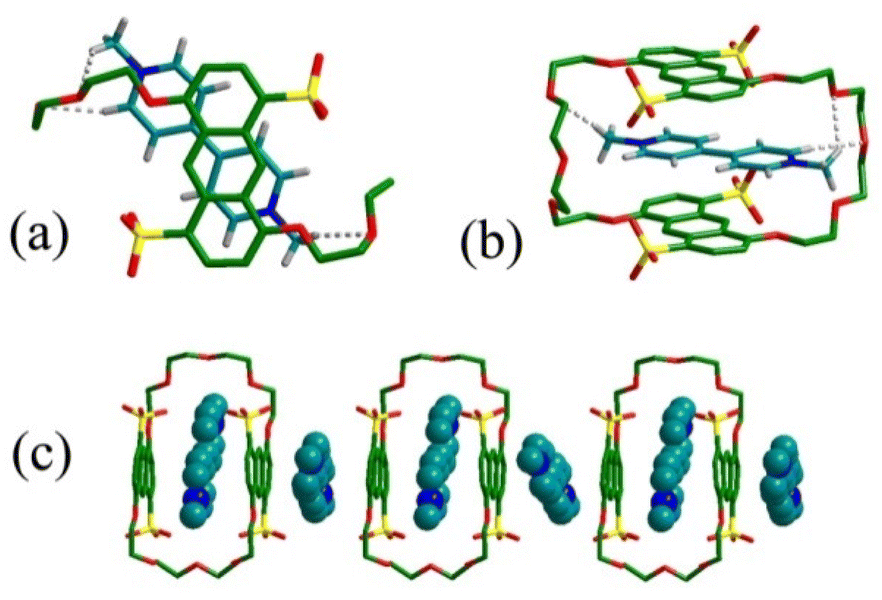
To better understand the binding geometry of tetrasulfonated crown ether 1, we obtained the single crystal of G3⊂1 using the hydrothermal method. As shown in Fig. 5, the crystal structure of G3⊂1 adopts a threading conformation to form a [2]pseudorotaxane structure. Two pyridinium groups of G3 adopt almost a fully planar conformation to form face-to-face π-stacking with the two anthracene units of host 1 with interplanar distances of 3.41 Å. In addition, the guest G3 does not occupy the symmetric center of the cavity of host 1. One N-methyl group of G3 comes close to the neighbouring tetraethylene glycol chain to form two C–H⋯O hydrogen bonds, another tetraethylene glycol curls up to get close to its neighbouring N-methyl group and only one C–H⋯O hydrogen bond is formed. This unsymmetrical binding conformation is mainly due to the fact that the cavity of host 1 is too large for G3, so one tetraethylene glycol chain on crown ether 1 needs to be curled up for more noncovalent interaction with G3. It is interesting to note that the positive charge center of the nitrogen atom on pyridinium is very far away from the negative charge center of the sulfonate group on 1, and the N–S distances are 5.64, 5.80, 6.81, and 7.11 Å, respectively, which is farther than our previously reported tetrasulfonated crown ethers.4 This means that electrostatic interaction might have less influence on the binding of G3⊂1 as compared to other effects like π-stacking interaction. This result should be attributed to the fact that anthracene is a large π-group which can form strong π-stacking interaction with π-electron-deficient G3 and this effect is very strong in an aqueous environment.21 The guest G3 needs to be located in the cavity of 1 for stacking with the anthracene plane as efficiently as possible, which causes the N-atom and sulfonate group move far away from each other.
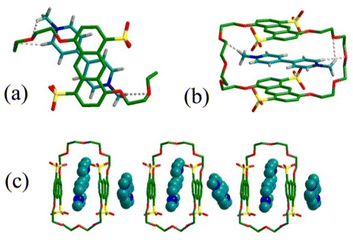 |
| | Fig. 5 Crystal structure of G3⊂1: (a) top view; (b) side view; (c) packing structure. Some hydrogen atoms are omitted for clarity. | |
Dyes binding and association constants
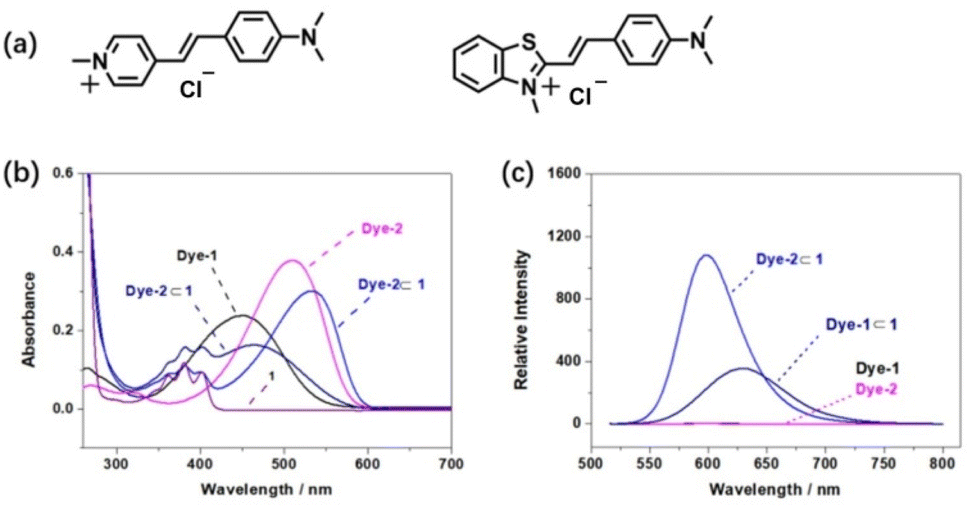
From the preliminary 1H NMR, UV-vis spectrum and single crystal structure, a general description of the association of tetrasulfonated crown ether 1 could be obtained. It is important to further measure their association constants to confirm the relationship between structures and association constants in the host–guest complexation process. Due the relatively large Ka values, the binding strength for the complexation between the host 1 and guests G2–G7 was difficult to be measured by 1H NMR titration experiments; thus the fluorescence titration method was employed to evaluate the molecular recognition process.22 Two singly charged dyes (dye-1 and dye-2, Fig. 6a) that have high affinity with tetrasulfonated crown ethers were introduced to act as competitive guests and fluorescent indicators in experiments of competitive fluorescence titration. Our previous data have shown that the fluorescence of dye-1 and dye-2 can be highly enhanced upon binding with TS32C8.5d,15e On the other hand, dye-1 and dye-2 are classical dipolar push–pull dyes that exhibit strong intramolecular charge–transfer interactions owing to the presence of electron-donor and electron-acceptor groups positioned at the opposite end of linear π-conjugated moieties. This effect generates a significant absorption peak at wavelengths more than 500 nm, and perfectly avoids the absorption of the host 1 making them suitable for fluorescent titration experiments. Binding of such highly dipolar dyes can be expected to markedly change the π-electron-withdrawing ability of the positively charged end of the molecules and thus result in appreciable shifts in the UV–vis spectra of the dyes. In fact, the absorption peak for Dye-1, which was centered at 451 nm, shifted to 464 nm in the presence of equimolar 1 (Fig. 6b), accompanied by a modest decrease in the absorbance. Similar changes in the UV–vis spectra were observed upon the binding of Dye-2 (Fig. 6b). These bathochromic shifts reflect electronic perturbations of intermolecular charge-transfer effects, arising from the restriction of solvent–solute dipole interactions by the reduction of exposure of dissolved Dye-1 or Dye-2 to water when bound in the cavity of 1.
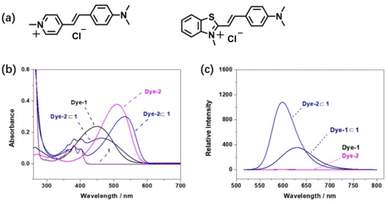 |
| | Fig. 6 Molecular structures of two dyes and their complexation induced a change in the spectra, λex = 500 nm: (a) molecular structures; (b) UV-vis spectra of two dyes in the presence or absence of equimolar amount of 1 in water, [Dye-1] = [Dye-2] = 0.01 mM; (c) fluorescence spectra of two dyes in the presence or absence of equimolar amount of 1 in water, [Dye-1] = [Dye-2] = 0.005 mM. | |
Binding in the cavity of crown ether 1 not only induced a change in the UV-vis absorbance of Dye-1 and Dye-2, but also significant enhancement of the fluorescent emission was observed. As shown in Fig. 6c, free Dye-1 and Dye-2 showed weak emission bands at a low concentration (0.005 mM). When an equimolar amount of 1 was added to a solution of Dye-1 or Dye-2, a moderate enhancement was observed in the fluorescence of Dye-1⊂1 (at 628 nm). The fluorescence intensity of Dye-2⊂1 (at 596 nm) was approximately twice that of Dye-1⊂1, indicating that the large positive group on dye-2 interacted with host 1 more strongly than that of the N-methyl pyridinium group of dye-1.
The inclusion complexes G3⊂1, G5⊂1 and G6⊂1 precipitated gradually at relatively higher concentrations. Therefore, isothermal titration calorimetry was not applicable for the determination of association constants. Thus we further investigated the Ka values of G2–G7 through fluorescence titration experiments (except G1 with small association constants). Job's plots confirmed the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding stoichiometry of the host–guest complexation. The Ka values of Dye-1 and Dye-2 with host 1 were determined to be 2.2 × 106 and 5.9 × 108 M−1, respectively. Using Dye-1 and Dye-2 as binding competitors and fluorescent indicators, the Ka values of G2–G7 were determined by fluorescence titration experiments (Fig. S19–S26 in the ESI†), and the values are listed in Table 1.
:1 binding stoichiometry of the host–guest complexation. The Ka values of Dye-1 and Dye-2 with host 1 were determined to be 2.2 × 106 and 5.9 × 108 M−1, respectively. Using Dye-1 and Dye-2 as binding competitors and fluorescent indicators, the Ka values of G2–G7 were determined by fluorescence titration experiments (Fig. S19–S26 in the ESI†), and the values are listed in Table 1.
Table 1 Association constants (Ka in M−1) and changes of Gibbs free energy (ΔG in kJ mol−1) for inclusion complexes between host 1 and guest molecules in water at 25 °C
| Guest |
Charge number |
K
a
|
ΔG |
|
K
a was determined by 1H NMR titration due to the small value. The others were determined by the fluorescence titration method.
|
|
G1
|
+1 |
(2.2 ± 0.1) × 103,a |
13.8 |
|
G2
|
+1 |
(2.8 ± 0.3) × 106 |
24.8 |
|
G3
|
+2 |
(2.0 ± 0.4) × 107 |
31.7 |
|
G4
|
+1 |
(4.3 ± 0.4) × 105 |
26.8 |
|
G5
|
+1 |
(5.5 ± 0.3) × 107 |
35.6 |
|
G6
|
+2 |
(7.5 ± 0.5) × 108 |
36.6 |
|
G7
|
0 |
(2.9 ± 0.4) × 105 |
31.2 |
|
Dye-1
|
+1 |
(2.2 ± 0.2) × 106 |
27.7 |
|
Dye-2
|
+1 |
(5.9 ± 0.3) × 108 |
38.1 |
The Ka values for all the complexes varied from a low value of 2.2 × 103 M−1 (G1⊂1) to a high value of 7.2 × 108 M−1 (G6⊂1). The values for the nine guests decreased in the following order (the charges of the guests are indicated in parentheses): G6(2+), Dye-2(1+) > G3(2+), G5(1+)>G2(1+), Dye-1(1+) > G4(1+), G7(0+) > G1(1+), showing significant binding selectivity. That is, for all the guests, the Ka values were mainly dependent on the size of guests, but not very related to the charge numbers. For example, the value of Ka,G1⊂1 was only 2.2 × 103 M−1, due to the simple small π-group of G1 with only one pyridinium. The G2 guest has a structure containing one more pyridyl group extended from the 4-position of G1, its Ka,G2⊂1 value was 3 orders of magnitude higher than that of Ka,G1⊂1, despite both the guests having the same positive charge. In contrast, the G3 guest has the same aromatic skeleton as G2 but one more charge; the Ka,G3⊂1 increases by only 1 order of magnitude compared to Ka,G2⊂1, well indicating that the extended pyridyl group plays a more important role in the complexation of G2⊂1 and G3⊂1 due to the stronger π-stacking effect. Moreover, when the electrically neutral guest G7 associates with 1, its Ka,G7⊂1 is one order of magnitude smaller than that of G2 with single charge, and two orders of magnitude smaller than that of G3⊂1. The Ka values of G2, G3 and G7 indicated that each charge contributed about one order of magnitude to the association constants for the guests G1–G3. Very similar results were also observed toward the naphthalene imide guests G4, G5 and G6, which also revealed that one charge contributed about one order of magnitude, and exerted less impact as compared to the π-stacking interaction between these three guests and 1. These results showed that host 1 had good guest binding selectivity toward the guests with a large π-moiety; the strongest binding always took place between host 1 and the guests with a large π-moiety but not with the guests with more charge.
In summary, we have demonstrated that anthracene-based sulfonated crown ether 1 is a powerful π-electron-accepting receptor that can form inclusion complexes with π-electron-deficient guests, as compared to previously reported sulfonated naphtho crown ethers (Fig. 7). Interestingly, TS32C8 bound small guests such as G1 (1.13 × 105 M−1) more strongly than 1 did,4b whereas 1 tended to bind complex guests with relatively large aromatic skeletons, such as G6, Dye-1 and Dye-2, more strongly than TS32C8 did (9.81 × 105 M−1, 8.1 × 105 M−1 and 4.94 × 106 M−1, respectively).5d These differences indicated that host 1 preferred to form strong complexes with large π-guests, mainly because 1 had larger anthracene units which could contribute more to π-stacking. Our previous work can explain this: the naphtho host TS32C8 had a small cavity and thus exhibited a better size-match effect to form a more stable complex with the small-sized guest G1, while 1 had a large cavity with low size matching to the small-sized guests.
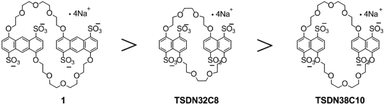 |
| | Fig. 7 Tetrasulfonated anthracenyl and naphtho crown ethers in the order of decreasing average guest-binding ability. | |
Conclusions
In conclusion, we have successfully synthesized an anthracene-based tetrasulfonated crown ether 1, which has more π-extended aromatic rings than the known tetrasulfonated crown ethers. The molecular binding ability of this new type of water-soluble macrocyclic host 1 is greatly enhanced toward seven typical cationic guests, especially the symmetrical ones possessing two cations, mainly due to the extensive π-stacking interaction with anthracene units. These findings are beneficial for the synthesis of more π-conjugated sulfonated crown ethers with a larger π-aromatic group such as pyrene and perylene, and are expected to develop crown ether-based supramolecular devices and sensors that can well function in aqueous media and biological fluids.
Author contributions
J.-L. Zhou, Y.-H. Li, L. Chen and Y. Liu conceived and directed this study. J.-L. Zhou and Y.-H. Li conducted the synthesis, characterization, and analysis of all materials described with assistance from Y.-M. Zhang. L. Chen and Y.-M. Zhang wrote and edited this manuscript. All authors discussed and reviewed the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grants 21861021, 22061022, and 22131008), Haihe Laboratory of Sustainable Chemical Transformation, and the Fundamental Research Funds for the Central Universities, Nankai University.
References
-
(a) M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398–7501 Search PubMed;
(b) Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC;
(c) X.-N. Han, Y. Han and C.-F. Chen, Nat. Commun., 2021, 12, 6378 CrossRef CAS PubMed;
(d) W.-L. Zhou, W. Lin, Y. Chen and Y. Liu, Chem. Sci., 2022, 13, 7976–7989 RSC.
-
(a) V. Balzani, A. Credi and M. Venturi, Chem. Soc. Rev., 2009, 38, 1542–1550 Search PubMed;
(b) J. Zhang, Q. Zou and H. Tian, Adv. Mater., 2013, 25, 378–399 CrossRef CAS PubMed;
(c) J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 Search PubMed.
-
(a) Z. Qi and C. A. Schalley, Acc. Chem. Res., 2014, 47, 2222–2233 Search PubMed;
(b) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
-
(a) G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366–2393 CrossRef CAS PubMed;
(b) L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed;
(c) Y. Wu, C. Zhang, S. Fang, D. Zhu, Y. Chen, C. Ge, H. Tang and H. Li, Angew. Chem., Int. Ed., 2022, 61, e202209078 Search PubMed.
-
(a) G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723–2750 Search PubMed;
(b) N. Sakai and S. Matile, Langmuir, 2013, 29, 9031–9040 Search PubMed;
(c) H.-G. Fu, Y. Chen and Y. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 16117–16122 CrossRef CAS PubMed;
(d) H.-G. Li and G.-W. Wang, J. Org. Chem., 2017, 82, 6341–6348 CrossRef CAS PubMed.
-
(a) L. Stricker, E.-C. Fritz, M. Peterlechner, N. L. Doltsinis and B. J. Ravoo, J. Am. Chem. Soc., 2016, 138, 4547–4554 CrossRef CAS PubMed;
(b) D. Li, F. Lu, J. Wang, W. Hu, X.-M. Cao, X. Ma and H. Tian, J. Am. Chem. Soc., 2018, 140, 1916–1923 CrossRef CAS PubMed;
(c) Y.-M. Zhang, Y.-H. Liu and Y. Liu, Adv. Mater., 2019, 31, 806158 Search PubMed;
(d) M. Osaki, T. Sekine, H. Yamaguchi, Y. Takashima and A. Harada, ACS Appl. Polym. Mater., 2021, 3, 2189–2196 Search PubMed.
-
(a) A. Barba-Bon, Y.-C. Pan, F. Biedermann, D.-S. Guo, W. M. Nau and A. Hennig, J. Am. Chem. Soc., 2019, 141, 20137–20145 CrossRef CAS PubMed;
(b) S. Li, R. Ma, X.-Y. Hu, H.-B. Li, W.-C. Geng, X. Kong, C. Zhang and D.-S. Guo, Adv. Mater., 2022, 34, 2203765 CrossRef CAS PubMed.
-
(a) Z.-Y. Zhang, Y. Chen and Y. Liu, Angew. Chem., Int. Ed., 2019, 58, 6028–6032 Search PubMed;
(b) A. C. Garcia, L. N. Zakharov and M. D. Pluth, J. Am. Chem. Soc., 2022, 144(33), 15324–15332 CrossRef CAS PubMed;
(c) Z.-T. Li, S.-B. Yu, Y. Liu, J. Tian and D.-W. Zhang, Acc. Chem. Res., 2022, 55, 2316–2325 CrossRef CAS PubMed.
-
(a) H.-B. Cheng, Y.-M. Zhang, Y. Liu and J. Yoon, Chem, 2019, 5, 553–574 CrossRef CAS;
(b) P. Peluso and B. Chankvetadze, Chem. Rev., 2022, 122, 13235–13400 CrossRef CAS PubMed.
-
(a) X. Ji, J. Li, J. Chen, X. Chi, K. Zhu, X. Yan, M. Zhang and F. Huang, Macromolecules, 2012, 45, 6457–6463 CrossRef CAS;
(b) D.-Y. Zhang, Z. Zheng, H. Zhao, H.-Y. Wang, F. Ding, H.-B. Li, Y.-C. Pan and D.-S. Guo, Chem. Commun., 2021, 57, 12627–12630 RSC.
-
(a) X. Xu, E. A. Appel, X. Liu, R. M. Parker, O. A. Scherman and C. Abell, Biomacromolecules, 2015, 16, 2743–2749 Search PubMed;
(b) T. Kakuta, Y. Takashima, M. Nakahata, M. Otsubo, H. Yamaguchi and A. Harada, Adv. Mater., 2013, 25, 2849–2853 CrossRef CAS PubMed;
(c) Q. Yang, P. Wang, C. Zhao, W. Wang, J. Yang and Q. Liu, Macromol. Rapid Commun., 2017, 38, 1600741 Search PubMed;
(d) A. Roy, K. Manna, P. G. Ray, S. Dhara and S. Pal, ACS Appl. Mater. Interfaces, 2022, 14, 17065–17080 CrossRef CAS PubMed.
-
(a) R. M. Izatt, K. Pawlak, J. S. Bradshaw and R. L. Bruening, Chem. Rev., 1995, 95, 2529–2586 Search PubMed;
(b)
J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley, Chichester, 2nd edn, 2009 Search PubMed.
-
(a) D. J. Hoffart;, J. Tiburcio, A. de la Torre, L. K. Knight and S. J. Loeb, Angew. Chem., Int. Ed., 2008, 47, 97–101 Search PubMed;
(b) E. Lestini, K. Nikitin, H. Müller-Bunz and D. Fitzmaurice, Chem. – Eur. J., 2008, 14, 1095–1106 Search PubMed.
-
(a) L. Chen, Y.-M. Zhang and Y. Liu, J. Phys. Chem. B, 2012, 116, 9500–9506 Search PubMed;
(b) L. Chen, H.-Y. Zhang and Y. Liu, J. Org. Chem., 2012, 77, 9766–9773 Search PubMed.
-
(a) L. Chen, Y.-M. Zhang, L.-H. Wang and Y. Liu, J. Org. Chem., 2013, 78, 5357–5363 CrossRef CAS PubMed;
(b) R. Cervantes, R. I. Sánchez and J. Tiburcio, Chem. – Eur. J., 2013, 19, 4051–4057 Search PubMed;
(c) Y.-M. Zhang, Z. Wang, L. Chen, H.-B. Song and Y. Liu, J. Phys. Chem. B, 2014, 118, 2433–2441 Search PubMed;
(d) Y.-M. Zhang, X.-J. Zhang, X. Xu, X.-N. Fu, H.-B. Hou and Y. Liu, J. Phys. Chem. B, 2016, 120, 3932–3940 Search PubMed;
(e) X. Dong, C. Zhang, X. Dai, Q. Wang, Y.-M. Zhang, X. Xu and Y. Liu, Chem. – Eur. J., 2022, 28, e202200005 CAS.
-
(a) U. Choudhary and B. H. Northrop, Chem. – Eur. J., 2014, 20, 999–1009 Search PubMed;
(b) Z. Meng, Y. Han, L.-N. Wang, J.-F. Xiang, S.-G. He and C.-F. Chen, J. Am. Chem. Soc., 2015, 137, 9739–9745 Search PubMed;
(c) M. Gauthier and F. Coutrot, Eur. J. Org. Chem., 2022, e202101201 Search PubMed;
(d) S. Zhu, S. Zhu and F. Xing, Chem. – Asian J., 2022, 17, e202200564 CAS.
-
(a) L. K. Knight, V. N. Vukotic, E. Viljoen, C. B. Caputo and S. J. Loeb, Chem. Commun., 2009, 5585–5587 RSC;
(b) V. N. Vukotic and S. J. Loeb, Chem. – Eur. J., 2010, 16, 13630–13637 CrossRef CAS PubMed;
(c) J. Wang, H.-Y. Zhang, X.-J. Zhang, Z.-H. Song, X.-J. Zhao and Y. Liu, Chem. Commun., 2015, 51, 7329–7332 Search PubMed;
(d) A. Carrasco-Ruiz and J. Tiburcio, Org. Lett., 2015, 17, 1858–1861 Search PubMed;
(e) W. Zhang, Y.-M. Zhang, S.-H. Li, Y.-L. Cui, J. Yu and Y. Liu, Angew. Chem., Int. Ed., 2016, 55, 11452–11456 Search PubMed.
-
(a) M. V. Rekharsky, T. Mori, C. Yang, Y. H. Ko, N. Selvapalam, H. Kim, D. Sobransingh, A. E. Kaifer, S. Liu, L. Isaacs, W. Chen, S. Moghaddam, M. K. Gilson, K. Kim and Y. Inoue, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20737–20742 CrossRef CAS PubMed;
(b) F. Jia, H. Hupatz, L. P. Yang, H. V. Schröder, D.-H. Li, S. Xin, D. Lentz, F. Witte, X. Xie, B. Paulus, C. A. Schalley and W. Jiang, J. Am. Chem. Soc., 2019, 141, 4468–4473 Search PubMed;
(c) Y. Liu, W. Zhao, C. H. Chen and A. H. Flood, Science, 2019, 365, 159–161 CAS.
-
(a) B. Tang, H.-M. Yang, W.-J. Hu, M.-L. Ma, Y. A. Liu, J.-S. Li, B. Jiang and K. Wen, Eur. J. Org. Chem., 2014, 6925–6934 Search PubMed;
(b) X.-N. Han, Y. Han and C.-F. Chen, J. Am. Chem. Soc., 2020, 142, 8262–8269 Search PubMed;
(c) X.-N. Han, Q.-S. Zong, Y. Han and C.-F. Chen, CCS Chem., 2022, 4, 318–330 Search PubMed.
-
(a) W. Jiang, M. Han, H.-Yi. Zhang, Z.-J. Zhang and Y. Liu, Chem. – Eur. J., 2009, 15, 9938–9945 Search PubMed;
(b) Y.-S. Su and C.-F. Chen, Org. Lett., 2010, 12, 1888–1891 CrossRef CAS PubMed.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- Y.-C. Liu, W. M. Nau and A. Hennig, Chem. Commun., 2019, 55, 14123–14126 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization. CCDC 2216935. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ob02010f |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *b
*b