
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoverage-modulated halogen bond geometry transformation in supramolecular assemblies†
Alejandro
Jiménez-Martín
 abc,
Aurelio
Gallardo
*cd and
Bruno
de la Torre
*ac
abc,
Aurelio
Gallardo
*cd and
Bruno
de la Torre
*ac
aRegional Centre of Advanced Technologies and Materials, Czech Advanced Technology and Research Institute (CATRIN), Palacký University Olomouc, 78371 Olomouc, Czech Republic. E-mail: bruno.de@upol.cz
bFaculty of Nuclear Sciences and Physical Engineering, Czech Technical University, 11519 Prague, Czech Republic
cInstitute of Physics, Czech Academy of Sciences, 16200 Prague, Czech Republic
dIMDEA Nanoscience, 28049 Madrid, Spain. E-mail: aurelio.gallardo@imdea.org
First published on 21st September 2023
Abstract
Halogen bonding (HB) has emerged as a promising route for designing supramolecular assemblies due to its directional nature and versatility in modifying interactions through the choice of halogens and molecular entities. Despite this, methods for tuning these interactions on surfaces, particularly in terms of directionality, are limited. In this study, we present a strategy for tuning the directionality of self-assembly processes in homomolecular organic compounds on inert metal surfaces. A variety of halogen–halogen geometries can promote highly-extended one-dimensional or two-dimensional self-assembly depending on the molecular coverage. Our results indicate that under lower molecular coverage conditions, robust one-dimensional (1D) structures promote the self-assembly of halogen-bonded molecules on Au(111). At certain coverage, a transformation from type-I to synthon halogen bonding is observed, leading to an extended hexagonal pattern of molecular assembly. The atomistic details of the structures are experimentally studied using high-resolution atomic force microscopy and supported by first-principle calculations. We employed DFT to evaluate the interplay between electrostatics and dispersion forces driving both type-I and synthon assemblies. The results reveal a halogen-bond geometry transformation induced by a subtle balance of molecule–molecule interaction. Finally, we investigate the capability of the halogen-bonded supramolecular assembly to periodically confine electronic quantum states and single atoms. Our findings demonstrate the versatility of sigma-bonding in regulating molecular assembly and provide new insights for tailoring functional molecular structures on an inert metal substrate.
Introduction
The engineering of supramolecular assemblies on solid supports heavily relies on non-covalent interactions. Halogen bonding is an excellent candidate for tailoring such structures due to its directional, hydrophobic and tuneable strength in combination with its length features.1,2 Fabrication of robust nanostructures of high complexity on surfaces can be achieved by appropriately designed building blocks and by tuning the hierarchical intermolecular interactions under ultrahigh vacuum,3–5 ambient conditions6 or at liquid–solid interfaces.7–9 Halogen-bonded supramolecular networks have been suggested for use in the construction of molecular nanopores for drug delivery applications owing to their structural rigidity. The entrapment of individual molecules on surfaces without the formation of strong covalent bonds is a prerequisite for molecular recognition and the utilization of molecular function.6,10–12 The self-assembly of nanometer-sized building blocks at surfaces and interfaces is of increasing interest in nanotechnology research due to its potential for applications in nanopatterning, surface templating, heterogeneous catalysis, sensing and quantum dots.6,11–14 Such two-dimensional porous networks provide the possibility of confinement of the Shockley surface state and immobilizing functional units as guest molecules in a repetitive and spatially defined manner. The ability to gain control over molecular ordering on surfaces has resulted in active research15 demonstrating the utility of halogen bonding for constructing supramolecular networks with well-defined structures and characteristics following bottom-up principles.16,17Although halogen bonding has been successfully utilized as a driving force for several monocomponent 2D supramolecular assemblies on surfaces,17–19 examples of geometrical transformations, polymorphism, or one-dimensional (1D) arrangements are rare. The development of 1D supramolecular structures through non-covalent interactions is particularly challenging, given the strong directionality involved, and demands attention in the area of molecular self-assembly. The formation of 1D organic architectures through non-covalent interactions has been proposed as potential prototypes for molecule-based logic gates that rely on controlled charge distribution or vibrational modes.20–22 The development of supramolecular structures on surfaces through weak non-covalent bonds relies on a delicate balance between molecule–molecule and molecule–substrate interactions, with the local polarizability of the supporting surface playing a crucial role in guiding the molecular arrangement.23 Therefore, achieving an optimal balance between intermolecular and substrate interactions is essential to the success of developing 1D non-covalent supramolecular structures using halogen bonding.24
Understanding the geometry-driving mechanism of halogen bonding has the potential to introduce new techniques for controlling these interactions and expanding their applicability in various fields.25–27 Recent advances in bond-resolved scanning probe microscopy28 and an expanded understanding of the supramolecular self-assembly mechanism15,29,30 offer valuable opportunities to explore the characteristics of non-covalent bonding31–34 and potential for leading exquisite control over surface-bound molecular structures.
We present a comprehensive study of a controlled geometrical transformation from 1D to 2D of a homomolecular network using halogen bonding. By exploiting Br⋯Br bonds between 2,2′,7,7′-tetrabromo-9,9′-biflurenylidene (TBF) molecules, we fabricated either a 1D robust molecular structure or a 2D atomic-scale porous network on an inert Au(111) surface. The detailed structures of these phases are addressed further in the discussion. Our investigation revealed that the interplay between intermolecular interactions plays a significant role in the formation of halogen-based supramolecular structures by modulating the halogen character of the bond. High-resolution non-contact AFM imaging confirmed that both networks rely on intermolecular halogen bonds. We quantified the interplay between electrostatic and dispersion forces in the network using density functional theory (DFT) calculations. Furthermore, we demonstrated that the 2D network can confine quantum electronic states and efficiently accommodate non-interacting Xe atoms. Our findings shed light on the physical chemistry nature of halogen bonds and offer innovative methods to engineer halogen-based supramolecular assemblies.
Results and discussion
Coverage-modulated molecular assembly
During gradual increasing of TBF coverage on Au(111) we have identified two self-assembled molecular phases comprising molecules with distinct geometries of halogen bonding: 1D molecular chains (due to intermolecular type I halogen-bonding) and 2D hexagonal pattern (due to intermolecular synthon halogen-bonding). After the initial deposition at room temperature, the source of molecules is turned off and the sample is moved to a different chamber under UHV conditions. Molecular decorated samples are characterized at 4.8 K in order to achieve bond resolution. As a reference, the chemical structure of the TBF molecule is displayed in Fig. 1a. On surface, the molecule exhibits a non-planar conformation due to the strong steric repulsion between adjacent hydrogen atoms. As the molecule contains a double bond connecting both fluorene moieties, it relaxes by symmetrically bending its structure perpendicular to the molecular plane (Fig. 1b).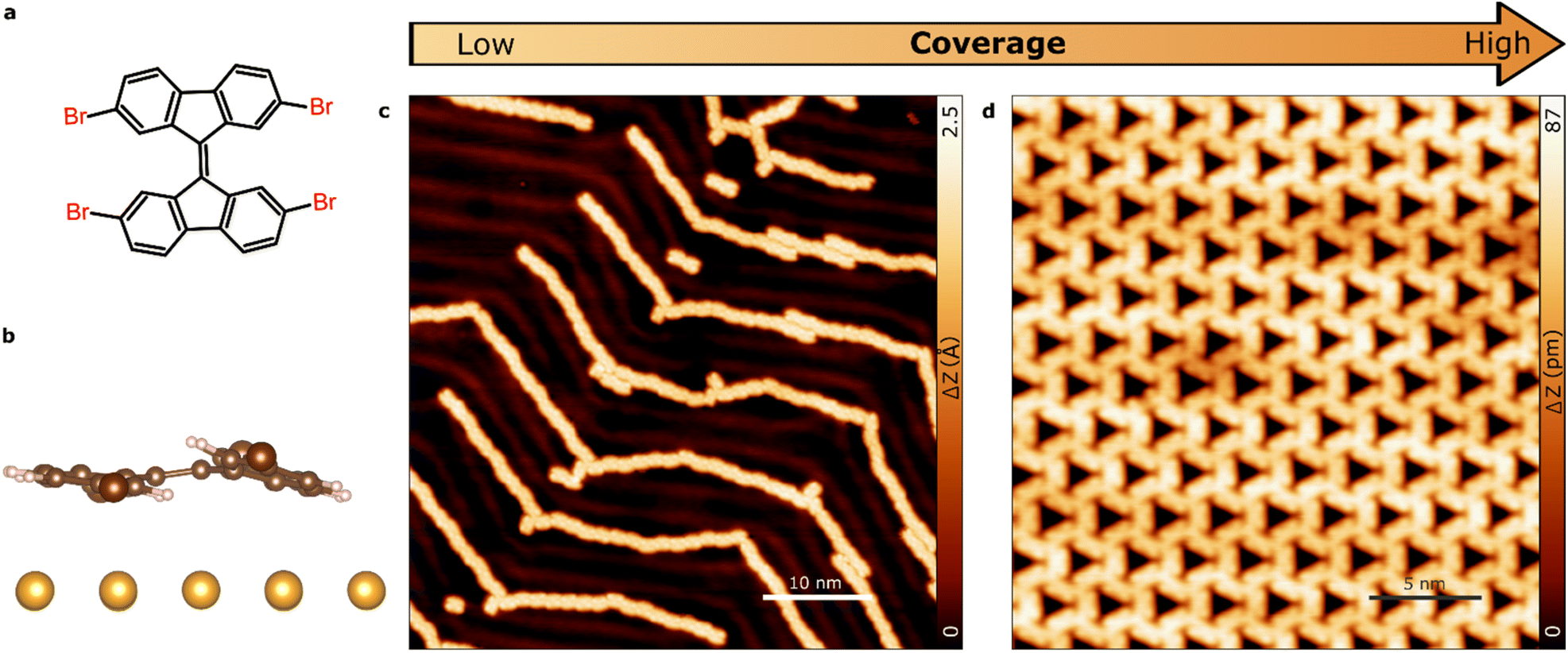 | ||
| Fig. 1 Supramolecular assembly of TBF molecule on Au(111). (a) Molecular model of the TBF. (b) Side view calculation of the relaxed TBF on the Au(111) surface. (c and d) STM topography overview after (c) low-, (d) high-coverage deposition at RT on Au(111). (c) (0.1 V; 10 pA) (d) (1.2 V; 0.1 nA). | ||
Fig. 1 provides a comprehensive summary of the sample morphology observed upon increasing the molecular coverage. We observed a clear dependence of the intermolecular interactions on the coverage, as evidenced by the emergence of distinct morphological features. In particular, we observed the formation of ordered molecular chains at lower coverage, while at higher coverage, we observed the coalescence of these to form continuous films. These observations suggest that the intermolecular interactions are significantly influenced by the coverage and play a crucial role in determining the morphology of the self-assembled monolayers. Considering that the TBF molecule contains four bromine and eight outer hydrogen atoms, it is reasonable to expect the formation of halogen bonds between neighbouring molecules on the surface. Indeed, the calculated spatial distribution of the electrostatic potential for an isolated TBF molecule on the Au surface (see Fig. S1†) shows significant anisotropy of the charge distribution on Br atoms. That is, a negative belt surrounding a positive area with cylindrical symmetry about the axis of the C–Br covalent bond. This unique potential distribution is the result of interplay between the Schrodinger equation and the Poisson equation, and it is the physical origin of the halogen bonds.2,35
Our observations reveal that the molecule undergoes diffusion to form extended one-dimensional structures on the surface at low coverage regime (<0.4 ML), as confirmed by scanning tunneling microscopy (STM) images (Fig. 1c). The well-defined supramolecular chains, consisting of up to 50 molecules, follow the herringbone corrugation of Au(111). This might indicate a non-negligible influence of the surface in the formation of the molecular chains. Upon increasing the molecular coverage to the range of 0.4–0.8 ML, we observed the emergence of two-dimensional molecular arrangements at the middle of the substrate terraces, which coexist with the 1D structures (see Fig. S2†). As the coverage is further increased, a fully covered substrate is obtained with closed-packed self-assembled monolayers, where the molecules arrange themselves in a highly-ordered hexagonal superstructure (Fig. 1d). Quantitatively, the ratio of molecules on 1D and 2D phases corresponds to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 for 0.1 ML, 0.45:0.55 for 0.4 ML, 0.15:0.85 for 0.6 ML and 0:1 for 1 ML, respectively. It is interesting to remark that the system prefers the 2D phase with respect to the 1D because the former structure only dominates at low coverages.
:0 for 0.1 ML, 0.45:0.55 for 0.4 ML, 0.15:0.85 for 0.6 ML and 0:1 for 1 ML, respectively. It is interesting to remark that the system prefers the 2D phase with respect to the 1D because the former structure only dominates at low coverages.
Since no other structures of assemblies have been observed on the surface, our interest was immediately focused on understanding the intermolecular interaction driving both different arrangements. To this end, we perform a comprehensive structural analysis using high-resolution noncontact atomic force microscopy (nc-AFM) images acquired with a functionalized CO-tip. This enabled us to unambiguously determine the atomic structure of the molecules and to investigate their orientation and spatial arrangements on the surface. The intriguing geometry of the subsequent phases is described separately in the following.
Phase 1D
Fig. 2a and b presents high-resolution nc-AFM and STM constant height images of the linear conformation of TFB molecules. The molecules are arranged in a head-to-head fashion, forming chains with an intermolecular distance of 13.0 ± 0.3 Å. Interestingly, consecutive molecules were not perfectly aligned but instead showed a ladder chain arrangement. Furthermore, our nc-AFM images allowed us to resolve intramolecular features that provided insights into the molecular geometry. Specifically, one of the fluorene motifs appeared brighter than the other in each individual molecule. This was a consequence of the nc-AFM imaging mechanism with CO-tip, where the contrast is generated by the subtle interplay between attractive and repulsive tip–sample forces and it is thus extremely sensitive to tip–sample distance variations.36 DFT calculations shows that one of the fluorene motifs in TBF molecule, is tilted on the Au(111) surface (see Fig. S3†) resembling the relaxed adsorption of a single TBF molecule (as depicted in Fig. 1b). This observation is further confirmed by the perfect match between experimental and simulated nc-AFM images (Fig. 2b and c). A noteworthy observation is that adjacent molecules may possess identical fluorene motifs (same tilt: AA, see Fig. 2c), resulting in a similar appearance, or distinct tilt configurations (alternate tilt: AB, see Fig. 2b) leading to dissimilar motifs. Analysis of a dataset comprising over 1300 molecules indicates that the majority of sequential molecules are characterized by opposing tilt orientations.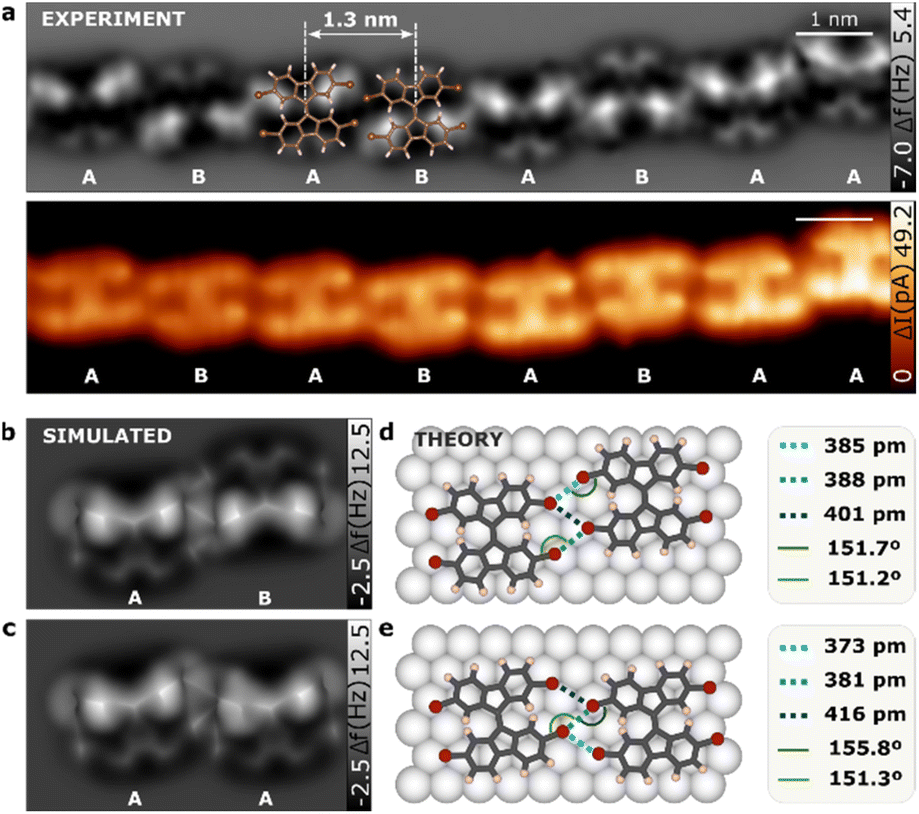 | ||
| Fig. 2 TBF molecules in one-dimensional conformation. (a) nc-AFM and STM constant-height images of a linear structure of TBF with a superimposed ball-stick. The letters A and B refer to the relative tilt that the TBF molecule adopts on the surface. (b and c) Simulated nc-AFM images of a dimer when consecutive TBF molecules have (b) the opposite tilt, (c) the same tilt. (d and e) DFT model of relaxed TBF dimers on Au(111) surface when the molecules have (d) opposite tilt (AB) and (e) same tilt (AA). | ||
The imaging of TBF on a Au(111) surface using nc-AFM presents certain challenges in determining the intermolecular bond length and angle due to the molecule's tilted orientation upon adsorption. Although the CO-tip tilt has shown sensitivity in detecting bond order37 and extended electronic cloud,38 the precise positions of atomic cores remain indeterminate through direct measurement of the nc-AFM image. To overcome these limitations, we have employed computational modelling techniques to extract quantitative bond distances and angles. Through a comparison of the characteristic features of our models with the results of calculations, we have arrived at a set of values that are in good agreement with the observed data. The resulting quantitative measurements have been presented in Fig. 2d and e; which provides an in-depth view of the atomic structure and intermolecular interactions of TBF molecules. We carried out a separate analysis of consecutive molecules with same and alternate tilts. In the case of molecules exhibiting the alternate tilt, our analysis revealed the presence of three Br⋯Br bonds in the dimer, with respective bond distances of 385 ± 1 pm, 388 ± 1 pm and 402 ± 1 pm. Meanwhile, for molecules exhibiting the same tilt, our analysis indicated that three Br⋯Br bonds could be connected with respective bond distances of 373 ± 1 pm, 381 ± 1 pm and 416 ± 1 pm. The reported values for Br⋯Br bonds suggest that it is highly unlikely for Br⋯Br bonds with a length exceeding 400 pm to be classified as halogen bonds.39 As such, in the 1D superstructure each TBF molecule is stabilized by four Br⋯Br halogen bonds, with two such bonds formed with each of the adjacent molecules in the chain. By comparing both AA and AB stacking, we show that in AA, 3 Br atoms per molecule participate in the formation of 2 halogen bonds, while for AB stacking are 4 Br atoms per molecule. Consequently, we can infer that AB stacking reduces the bond-energy per atom, being more favorable, as observed experimentally.
Interestingly, the angles between these C–Br⋯Br bonds are 151.7 ± 0.1 deg and 151.2 ± 0.1 deg in the AB conformation and 155.8 ± 0.1 deg and 151.3 ± 0.1 deg in the AA form, indicating that is equal at both sides of the bond which agrees with the definition of type I for the halogen bond.40 This strongly directional C–Br⋯Br conformation leads to the formation of 1D supramolecular structures. This discovery is particularly noteworthy as the observed head-to-head configuration is typically only found in densely packed crystals, and not as a result of net attractive electrostatic interactions.41
Phase 2D
Upon increasing molecular coverage, a different scenario is observed where the molecules arrange in a hexagonal pattern (see Fig. 1d). nc-AFM and STM imaging provide a detailed look at the molecular assembly as depicted in Fig. 3a. The self-assembled molecular network corresponds to a honeycomb lattice with a cell size of 2.4 × 2.4 ± 0.1 nm. Close-up high-resolution nc-AFM image reveals that TBF molecules in the 2D phase are adsorbed again tilted with respect to the surface (see Fig. 3b). Although the TBF molecule retains the adsorption geometry from 1D phase, the tilt of the molecule has significantly changed from the 1D phase (see calculated structures in Fig. S3†), leading to a difference adsorption height of 110 pm between fluorene motifs in the same molecule (comparatively, in the 1D phase this difference is 70 pm). As we will see, this change results crucial to stabilize the 2D phase on the surface since it enables the emergence of new intermolecular Br⋯H bonds. It is worthy to note that images acquired at intermediate coverage show the emergence of the 2D phase from the confluence of three 1D chains with distinct angles (see Fig. S4†).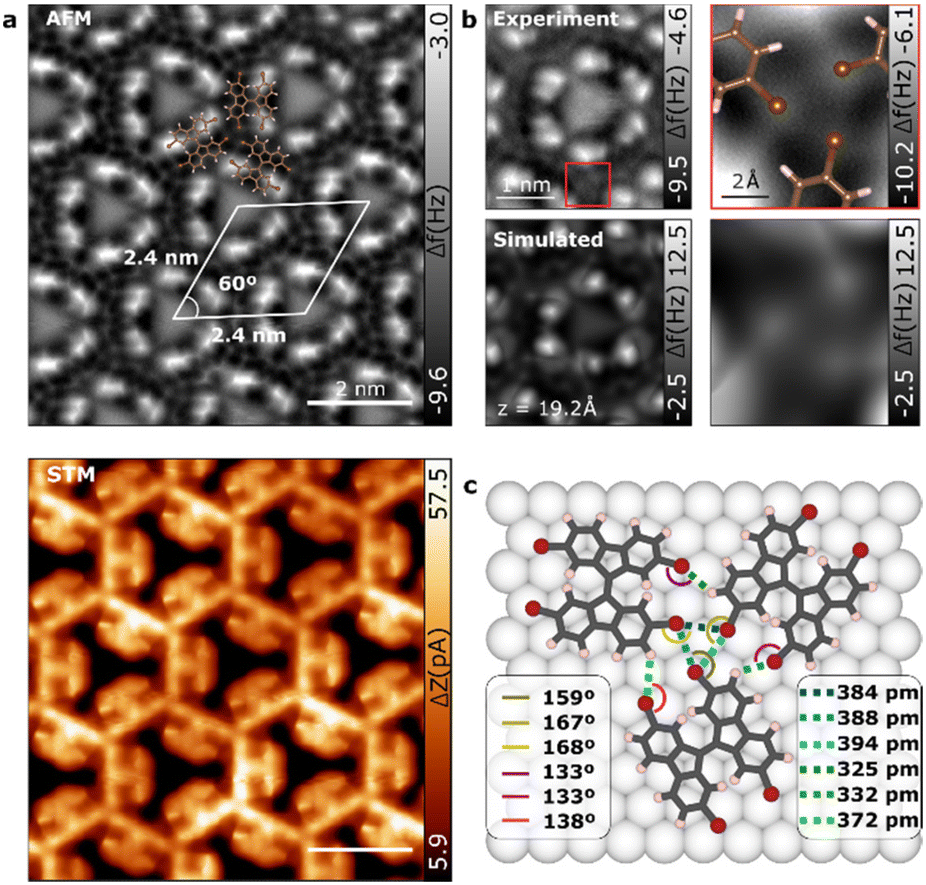 | ||
| Fig. 3 Characterization of the two-dimensional phase of TBF molecules on Au(111). (a) nc-AFM and STM constant height images of the hexagonal structure with a lattice of 2.4 nm × 2.4 nm with a superimposed ball-stick model of three TBF molecules. (b) Experimental (top) and simulated (down) nc-AFM images of a close-up view of the center of the pore (left) and the Br⋯Br bond (right) of the TBF molecules. (c) DFT model of the relaxed TBF molecules on Au(111) surface. | ||
In 2D network structures with halogen bonds, three Br of adjacent molecules combine to form the triplet nodes of the windmill shapes giving rise to the triangular motifs. nc-AFM close-up images depicted in Fig. 3b (right panels) confirmed the windmill conformation of the Br atoms in the 2D phase of TBF. The hexagonal shape of the molecular arrangement and the pinwheel conformation of the Br atoms suggest a chiral type II synthon halogen bond between the Br atoms of three adjacent molecules featuring both chiral forms. The lateral proximity to adjacent molecules may allow Br⋯H bonds that further stabilize the system. To disentangle the effects associated with both contributions to the net molecule–molecule interaction and to shed light on the details of the supramolecular network, the 2D phase has been studied by means of density functional theory (DFT) calculations. The fully relaxed molecular structure on the Au(111) substrate is shown in Fig. 3c. Interestingly, in this case, TBF molecules relax in a flatter conformation (see Fig. S3†) probably due to larger intermolecular interactions. Indeed, in the assembled structure we found two possible intermolecular bonds: C–Br⋯Br and C–Br⋯H, as depicted with dotted green and yellow lines in Fig. 3c, respectively. The direction of the Br⋯Br (∼165 deg) and Br⋯H bonds (∼135 deg) is determined by the negative potential region of the Br atom (i.e. the negative belt around the sigma-hole). Br⋯Br and Br⋯H bonds form a triangular structure, making an acute angle which is very similar in the three Br atom as expected for a halogen-bonded synthon according with its definition.40 Such acute-triangular structures were found in various supramolecular structures such as molecular chains, ladders and layered structures.42,43 Quantitatively, we found Br⋯Br bonds with a length of 384 ± 1 pm, 388 ± 1 pm and 394 ± 1 pm while H⋯Br bonds extend to 325 ± 1 pm, 331 ± 1 pm and 372 ± 1 pm in good agreement with other C–H⋯Br computed distances in crystal structures.44
The relationship between concentration dependence and polymorph stability, as well as molecular density, is widely recognized in the scientific community. High-density structures are generally favoured from a free-energy perspective. However, halogen-bonding interactions are typically weaker than hydrogen-bonding interactions,41 and the former may be compromised in order to maximize packing density.
Analysis of directional halogen bond (interaction energy analysis)
Now, we aimed to gain a comprehensive understanding of the directional transformation of Br⋯Br bonds. To achieve this goal, we examined the cohesive energies of TBF molecules in both 1D and 2D configurations by total energy DFT calculations, analysing the electrostatic and van der Waals (vdW) contributions separately. Our analysis of the interaction energy per molecule revealed that the 2D arrangement exhibited greater stability than the linear configuration, with a total energy per molecule of −203 meV compared to −138 meV for the latter. As demonstrated by the results presented in Table 1, the electrostatic contribution to the total interaction energy decreased as the molecules organized into clusters. This was due to the fact that while each molecule shared six Br⋯Br halogen bonds with its neighbours in the linear configuration, in the clustered configuration, they only shared four such bonds with surrounding molecules. Simultaneously, vdW interactions increased in the cluster configuration likely due to the appearance of the halogen–hydrogen interactions and the more closed packed arrangement of the molecules.| Energy (meV) | 1D E/mol | 1D E/Br⋯Br | 2D E/mol | 2D E/Br⋯Br |
|---|---|---|---|---|
| Total | −138 | −45.9 | −203 | −68.8 |
| Electrostatic | −710 | −236.8 | −617 | −372.1 |
| van der Waals | −81 | −26.9 | −153 | −41.6 |
To gain further insight into the energetic evolution of the halogen bonds as the system transitioned from type-I halogen bonds in chains to synthon bonds present in the cluster configuration, we compared the energy per halogen bond in a dimer with that present in a synthon in a cluster (see models in Fig. 4). To isolate the synthon bond from the rest of the hydrogen and halogen–hydrogen bonds present in the cluster configuration, we created a fictitious system as shown in Fig. 4b. In this system, we isolated, in a cluster of three molecules, the portion of these molecules that created the synthon bond and passivated other bonds with H atoms. Our findings indicated that the interaction energy per bond increased by approximately 50% during the transition, as did the electrostatic and vdW energies, indicating an increase in the halogen character of the bond45 (see Table 1).
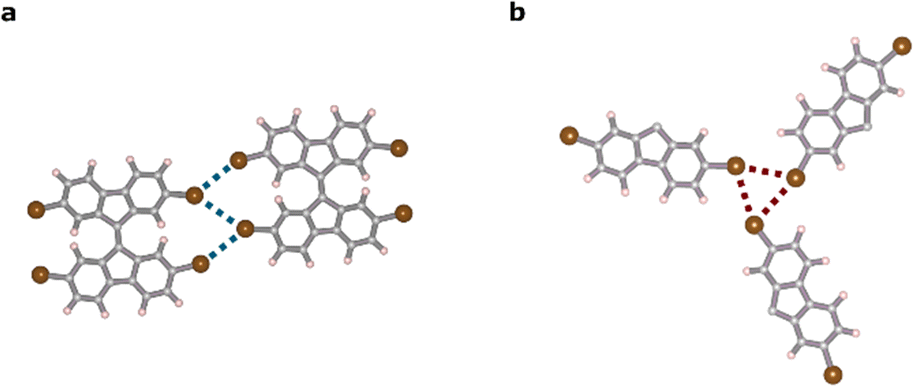 | ||
| Fig. 4 DFT models of the relaxed TBF molecules for (a) 1D phase and (b) 2D phase. | ||
Confinement properties of 2D structure
Finally, we explore the confinement properties of the 2D supramolecular structure. The 2D self-assembly of TBF on Au(111) exhibits a sub-micrometer size of nano-porous assembly with a low degree of defects, only grain boundaries corresponding to the concurrence of patches with different angles (see Fig. S5†).First, we aimed to investigate the potential of confining periodic electronic states in two dimensions by utilizing the regularly nanopatterned structure of TBF on Au(111). Our experimental procedure consisted of acquiring scanning tunneling spectra at different points of the supramolecular structure (see inset Fig. 5c). These spectra displayed a new unoccupied electronic state at 1.4 eV at the center of the pore (in addition to the surface state), whereas spectra obtained on the TBF molecule or on the clean Au(111) surface exhibited the characteristic steplike onset of the Shockley surface state at approximately −0.5 V (Fig. 5c). Interestingly, while previous research has reported several quantum states on Ag and Cu surfaces using molecular networks, these states are usually localized above the surface state by only tens or hundreds of meV.46–48 Furthermore, conducting dI/dV maps acquired close to the energy of the newly discovered electronic state in the same area as the topographic image depicted in Fig. 5a revealed that this state is localized within the pores of the 2D network (Fig. 5b). Therefore, it can be concluded that each pore represents a single quantum dot within a regular “quantum dot array,” which confines the surface state electrons (see Fig. S6† for a complete set of dI/dV images at different bias).
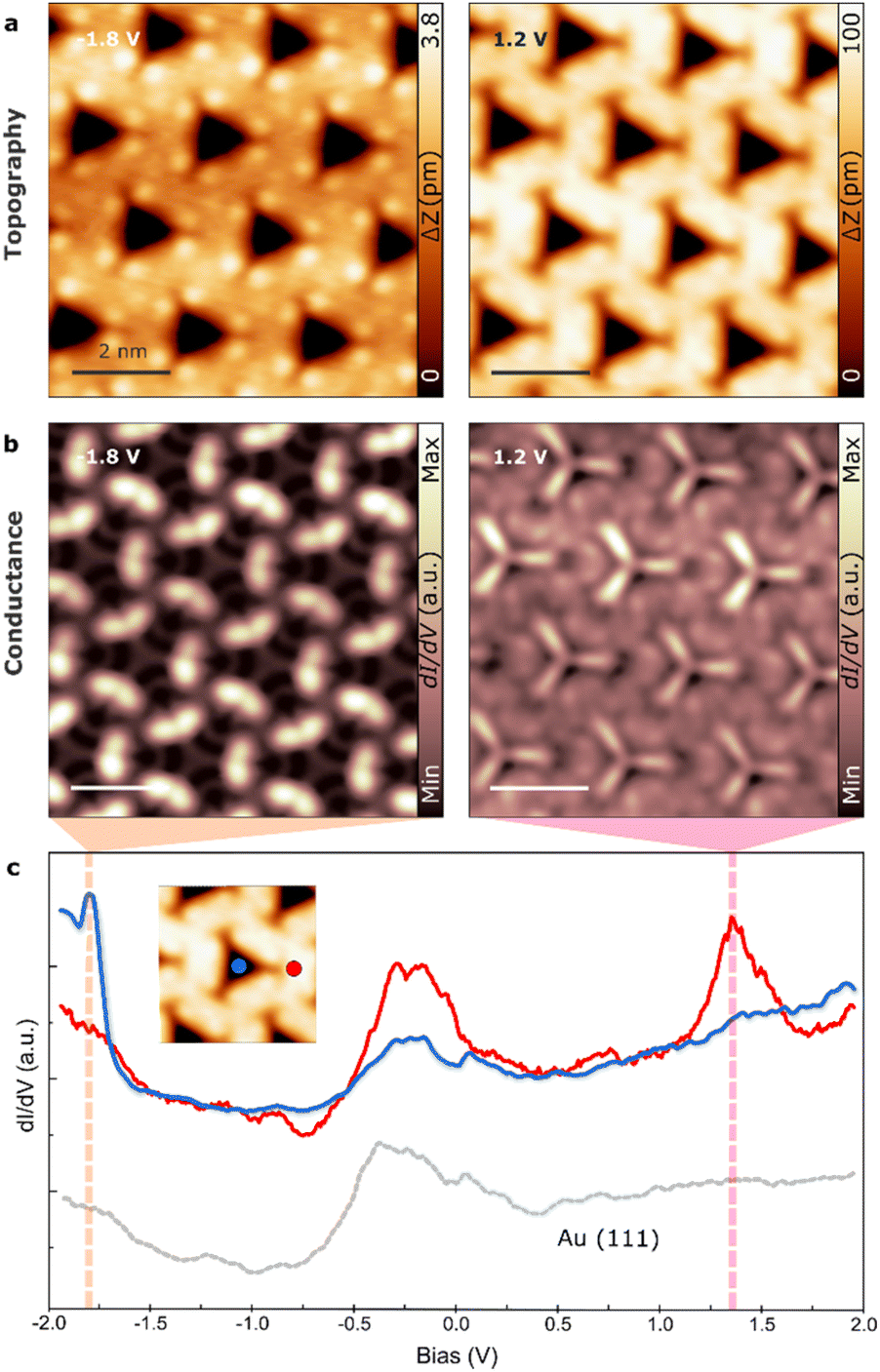 | ||
| Fig. 5 Electronic characterization of the 2D supramolecular structure. (a) STM topography overview images acquired at −1.8 V (left) and 1.2 V (right). (b) Differential conductance images of the 2D structure at the indicated voltages. (c) dI/dV spectroscopies at the positions marked with points in the inset image. | ||
Lastly, we investigated the confinement of adsorbates in the molecular porous network. The small size of the TBF pores (∼1 nm2) implies their functionality as hosts for single atoms. A means of evaluating the potential of the engineered 2D network for organizing single-atom functional materials and host–guest chemistry is to investigate its structural stability and steering effects in the coadsorption of atomic-scale entities. As a test species, Xe atoms were selected, as they can be readily deposited onto the surface at low temperatures and they exhibit a closed-shell structure and thus, can only undergo weak van der Waals interactions. The control of Xe-surface interactions and positioning is of both scientific and technological interest. As an example, investigating the mechanism of particle clustering can be better understood through the examination of noble gas condensates of varying sizes, as they interact with each other through isotropic van der Waals forces.49,50
The atomically-defined cavities of the 2D TBF network are capable of accommodating small, compact clusters of Xe atoms, as demonstrated by the STM and high-resolution nc-AFM images presented in Fig. 6. The blue arrows in the image indicate the location of Xe atoms within the pore. The histogram of the occupancies, which occur spontaneously (Fig. S7†), indicates that the presence of certain numbers of Xe atoms in the pores is preferred. The highest probability was observed for 4 atoms, followed by 8, 5, and 3 atoms. We found that for such a small number of atoms, their preferential placement at the center of the pore can be explained by weak electrostatic attraction between Xe and the ground state of the confined surface state. This contrasts with the observations made for Xe adsorbed in the pores of supramolecular networks grown on Cu(111), which experienced a repulsive interaction with the quantum well state of the pore.51 Notably, certain pores contain a substantial number of Xe atoms, and even more may accommodate Xe atoms atop those already situated within the pore forming a second Xe layer (as indicated by green arrows in Fig. 6a). These 3D clusters are observed in pores that are not necessarily fully filled, indicating stronger Xe–Xe interactions attributable to large dispersion forces. Interestingly, repositioning experiments in which individual Xe atoms were manipulated with the STM tip only resulted in the movement of Xe atoms in the second layer from one pore to another thus indicating the high stability of the Xe clusters within the pore (see Fig. S7†).
 | ||
| Fig. 6 Confinement of Xe atoms in the 2D phase of the TBF molecules. (a) STM topography overview of the Xe coverage within the hexagonal structure on the Au(111) surface. The blue(green) arrows indicate the position hole(top) of the Xenon in the 2D pores of the TBF structure. (0.5 V; 5 pA) (b) STM images showing the number of Xe atoms (3 and 7) confined by the TBF molecules and an AFM image of the N 3 Xe atoms within the hole of the pore. (0.01 V; 10 pA). (c) Profile of the Xe layers in the pore. | ||
Conclusions
In conclusion, we demonstrate the versatility of halogen bonding in regulating molecular assembly on an inert metal substrate by tuning the directionality of supramolecular assemblies of tetra-bromo fluorene molecules on Au(111) using a combination of STM/nc-AFM experiments supported by DFT calculations. A transition between two different halogen-bonded molecular structures—linear and hexagonal—were observed upon increasing the molecular coverage. This discovery is particularly noteworthy since the observed “head-on” configuration with an effective bonding angle is typically only observed in densely packed crystals, and not as a result of attractive electrostatic interactions. Based on high-resolution AFM images, we proposed molecular models that are in good agreement with DFT studies and can be explained by a geometric transition of the intermolecular motif consisting of Br⋯Br and Br⋯H bonds from trans- to Synthon. The measured bonding distances and strengths are consistent with existing bulk data. These results demonstrate the potential of halogen-bonding as a promising tool for the fabrication of complex organic architectures with tuneable properties.Finally, we demonstrated the ability of the hexagonal supramolecular to act as electronic confiner and single atom porous by exposing the system to Xe. The results of the study contribute to the understanding of how directionality can be controlled in halogen-bonded supramolecular structures, which can inform the design of future materials for various applications. We envision that the dimension of these porous structures maybe useful to motivate further studies to stabilize individual atoms on surface for single-atom catalysis studies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support of Czech Science Foundation 23-06781M and CzechNanoLab Research Infrastructure supported by MEYS CR (LM2023051). We acknowledge the Research Infrastructure NanoEnviCz, supported by the Ministry of Education, Youth and Sports of the Czech Republic under Project No. LM2023066.References
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46(11), 2686–2695 CrossRef CAS PubMed.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed.
- J. V. Barth, Annu. Rev. Phys. Chem., 2007, 58, 375–407 CrossRef CAS PubMed.
- G. Pawin, K. L. Wong, K.-Y. Kwon and L. Bartels, Science, 2006, 313, 961–962 CrossRef CAS PubMed.
- S. Stepanow, N. Lin, D. Payer, U. Schlickum, F. Klappenberger, G. Zoppellaro, M. Ruben, H. Brune, J. V. Barth and K. Kern, Angew. Chem., 2007, 119, 724–727 CrossRef.
- J. V. Barth, G. Costantini and K. Kern, Nature, 2005, 437, 671–679 CrossRef CAS PubMed.
- H. Zhou, H. Dang, J.-H. Yi, A. Nanci, A. Rochefort and J. D. Wuest, J. Am. Chem. Soc., 2007, 129, 13774–13775 CrossRef CAS PubMed.
- D. Bléger, D. Kreher, F. Mathevet, A.-J. Attias, G. Schull, A. Huard, L. Douillard, C. Fiorini-Debuischert and F. Charra, Angew. Chem., Int. Ed., 2007, 46, 7404–7407 CrossRef PubMed.
- Y. Wei, W. Tong and M. B. Zimmt, J. Am. Chem. Soc., 2008, 130, 3399–3405 CrossRef CAS PubMed.
- N. Katsonis, T. Kudernac, M. Walko, S. J. van der Molen, B. J. van Wees and B. L. Feringa, Adv. Mater., 2006, 18, 1397–1400 CrossRef CAS.
- A. Llanes-Pallas, C.-A. Palma, L. Piot, A. Belbakra, A. Listorti, M. Prato, P. Samorì, N. Armaroli and D. Bonifazi, J. Am. Chem. Soc., 2009, 131, 509–520 CrossRef CAS PubMed.
- S. De Feyter and F. De Schryver, Two-Dimensional Dye Assemblies on Surfaces Studied by Scanning Tunneling Microscopy, Springer, Berlin, 2005 Search PubMed.
- B. Hulsken, R. Van Hameren, J. W. Gerritsen, T. Khoury, P. Thordarson, M. J. Crossley, A. E. Rowan, R. J. M. Nolte, J. A. A. W. Elemans and S. Speller, Nat. Nanotechnol., 2007, 2, 285–289 CrossRef CAS PubMed.
- I. Piquero-Zulaica, J. Lobo-Checa, A. Sadeghi, Z. M. A. El-Fattah, C. Mitsui, T. Okamoto, R. Pawlak, T. Meier, A. Arnau, J. E. Ortega, J. Takeya, S. Goedecker, E. Meyer and S. Kawai, Nat. Commun., 2017, 8, 787 CrossRef PubMed.
- D. P. Goronzy, M. Ebrahimi, F. Rosei, A. Arramel, Y. Fang, S. De Feyter, S. L. Tait, C. Wang, P. H. Beton, A. T. S. Wee, P. S. Weiss and D. F. Perepichka, ACS Nano, 2018, 12, 7445–7481 CrossRef CAS PubMed.
- J.-M. Lehn, Science, 1993, 260(5115), 1762–1763 CrossRef CAS PubMed.
- R. Gutzler, C. Fu, A. Dadvand, Y. Hua, J. M. MacLeod, F. Rosei and D. F. Perepichka, Nanoscale, 2012, 4, 5965–5971 RSC.
- R. Gutzler, O. Ivasenko, C. Fu, J. L. Brusso, F. Rosei and D. F. Perepichka, Chem. Commun., 2011, 47, 9453–9455 RSC.
- F. Silly, J. Phys. Chem. C, 2013, 117, 20244–20249 CrossRef CAS.
- M. Leisegang, A. Christ, S. Haldar, S. Heinze and M. Bode, Nano Lett., 2021, 21, 550–555 CrossRef CAS PubMed.
- H.-Z. Tsai, J. Lischner, A. A. Omrani, F. Liou, A. S. Aikawa, C. Karrasch, S. Wickenburg, A. Riss, K. C. Natividad, J. Chen, W.-W. Choi, K. Watanabe, T. Taniguchi, C. Su, S. G. Louie, A. Zettl, J. Lu and M. F. Crommie, Nat. Electron., 2020, 3, 598–603 CrossRef CAS.
- C. Li, Z. Wang, Y. Lu, X. Liu and L. Wang, Nat. Nanotechnol., 2017, 12, 1071–1076 CrossRef CAS PubMed.
- A. Kumar, K. Banerjee and P. Liljeroth, Nanotechnology, 2017, 28, 082001 CrossRef PubMed.
- F. Zordan, L. Brammer and P. Sherwood, J. Am. Chem. Soc., 2005, 127, 5979–5989 CrossRef CAS PubMed.
- L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2013, 5, 42–47 CrossRef CAS PubMed.
- A. Priimagi, M. Saccone, G. Cavallo, A. Shishido, T. Pilati, P. Metrangolo and G. Resnati, Adv. Mater., 2012, 24(44), OP345–OP352 CrossRef CAS PubMed.
- O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed.
- L. Gross, F. Mohn, N. Moll, P. Liljeroth and G. Meyer, Science, 2009, 325, 1110–1114 CrossRef CAS PubMed.
- P. Procházka, M. A. Gosalvez, L. Kormoš, B. de la Torre, A. Gallardo, J. Alberdi-Rodriguez, T. Chutora, A. O. Makoveev, A. Shahsavar, A. Arnau, P. Jelínek and J. Čechal, ACS Nano, 2020, 14, 7269–7279 CrossRef PubMed.
- J. A. A. W. Elemans, S. Lei and S. De Feyter, Angew. Chem., Int. Ed., 2009, 48, 7298–7332 CrossRef CAS PubMed.
- S. Kawai, A. Sadeghi, F. Xu, L. Peng, A. Orita, J. Otera, S. Goedecker and E. Meyer, ACS Nano, 2015, 9, 2574–2583 CrossRef CAS PubMed.
- Z. Han, G. Czap, C. Chiang, C. Xu, P. J. Wagner, X. Wei, Y. Zhang, R. Wu and W. Ho, Science, 2017, 358, 206–210 CrossRef CAS PubMed.
- A. Gallardo, J. Fanfrlík, P. Hobza and P. Jelínek, J. Phys. Chem. C, 2019, 123, 8379–8386 CrossRef CAS.
- B. Mallada, A. Gallardo, M. Lamanec, B. de la Torre, V. Špirko, P. Hobza and P. Jelinek, Science, 2021, 374, 863–867 CrossRef CAS PubMed.
- O. Hassel, Science, 1970, 170, 497–502 CrossRef CAS PubMed.
- B. Schuler, W. Liu, A. Tkatchenko, N. Moll, G. Meyer, A. Mistry, D. Fox and L. Gross, Phys. Rev. Lett., 2013, 111, 106103 CrossRef PubMed.
- L. Gross, F. Mohn, N. Moll, B. Schuler, A. Criado, E. Guitián, D. Peña, A. Gourdon and G. Meyer, Science, 2012, 337(6100), 1326–1329 CrossRef CAS PubMed.
- B. de la Torre, M. Švec, P. Hapala, J. Redondo, O. Krejčí, R. Lo, D. Manna, A. Sarmah, D. Nachtigallová, J. Tuček, P. Błoński, M. Otyepka, R. Zbořil, P. Hobza and P. Jelínek, Nat. Commun., 2018, 9, 2831 CrossRef PubMed.
- R. S. Rowland and R. Taylor, J. Phys. Chem., 1996, 100, 7384–7391 CrossRef CAS.
- B. Li, S.-Q. Zang, L.-Y. Wang and T. C. W. Mak, Coord. Chem. Rev., 2016, 308, 1–21 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116(4), 2478–2601 CrossRef CAS PubMed.
- O. Navon, J. Bernstein and V. Khodorkovsky, Angew. Chem., Int. Ed. Engl., 1997, 36, 601–603 CrossRef CAS.
- H. F. Lieberman, R. J. Davey and D. M. T. Newsham, Chem. Mater., 2000, 12, 490–494 CrossRef CAS.
- V. Rajnikant, D. Jasrotia and B. Chand, J. Chem. Crystallogr., 2008, 38, 211–230 CrossRef CAS.
- M. H. Kolář and P. Hobza, Chem. Rev., 2016, 116, 5155–5187 CrossRef PubMed.
- J. Lobo-Checa, M. Matena, K. Müller, J. H. Dil, F. Meier, L. H. Gade, T. A. Jung and M. Stöhr, Science, 2009, 325, 300–303 CrossRef CAS PubMed.
- F. Klappenberger, D. Kühne, W. Krenner, I. Silanes, A. Arnau, F. J. García de Abajo, S. Klyatskaya, M. Ruben and J. V. Barth, Nano Lett., 2009, 9, 3509–3514 CrossRef CAS PubMed.
- A. Shchyrba, S. C. Martens, C. Wäckerlin, M. Matena, T. Ivas, H. Wadepohl, M. Stöhr, T. A. Jung and L. H. Gade, Chem. Commun., 2014, 50, 7628–7631 RSC.
- O. Echt, K. Sattler and E. Recknagel, Phys. Rev. Lett., 1981, 47, 1121–1124 CrossRef CAS.
- K. D. Sattler, Handbook of nanophysics: clusters and fullerenes, CRC press, Florida, 2010 Search PubMed.
- S. Nowakowska, A. Wäckerlin, S. Kawai, T. Ivas, J. Nowakowski, S. Fatayer, C. Wäckerlin, T. Nijs, E. Meyer, J. Björk, M. Stöhr, L. H. Gade and T. A. Jung, Nat. Commun., 2015, 6, 6071 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr03899h |
| This journal is © The Royal Society of Chemistry 2023 |