
Supramolecular structures from structurally persistent and surface active carbon dots in water†
Aoxue
Xu
,
Ning
Feng
,
Keyang
Yin
,
Hongguang
Li
 * and
Jingcheng
Hao
* and
Jingcheng
Hao
Key Laboratory of Colloid and Interface Chemistry, Ministry of education, School of Chemistry and Chemical Engineering, Shandong University, Jinan, 250100, China. E-mail: hgli@sdu.edu.cn
First published on 28th November 2022
Abstract
Carbon dots (CDs) have developed into an important class of nanomaterials that have attracted increasing attention during the past decades. Despite numerous types of CDs reported to date, research on their self-assembly is still limited. Herein, we report for the first time the self-assembly of CDs in water, which show concentration-dependent aggregation behavior. The CDs used have a structural motif of a fully carbonized core surrounded by a highly condensed, polymeric network, to which triethylene glycol monomethyl ether (TGME) chains are grafted. When dissolved in water, they show a low critical aggregation concentration (cac) of 0.07 mg mL−1 with the lowest surface tension of ∼37 mN m−1. Above this cac, nanoclusters and vesicles are observed at relatively low and high concentrations, respectively. At an intermediate concentration, polymorphism is noticed where nanotubes coexist with nanorods. At an elevated temperature, the CDs become more hydrophobic due to the dehydration of peripheral TGME, which decreases the cac and triggers phase transfer from water to toluene. These surface active CDs were used to disperse and stabilize multi-walled carbon nanotubes in water, which showed much better performance than that of both traditional ionic and nonionic surfactants. Our work indicates that with a careful structural design, CDs can be developed into a new type of amphiphiles with properties superior to those of traditional surfactants in specific aspects.
1 Introduction
Nearly two decades have passed since carbon dots (CDs) were accidentally discovered from the purification of oxidized carbon nanotubes.1 Later, similar synthetic procedures were applied on a variety of carbon-rich materials,2,3 yielding water-soluble CDs rich in hydroxyl and carboxyl groups on the surface. These CDs are characterized by their high solubility in water, which normally tend to stay individually. An alternative way to produce CDs relies on the pyrolysis of organic molecules, during which CDs with carbonized cores and various surface functional groups are produced. The precursors are diverse and different combinations could be used besides the single components. This feature makes pyrolysis highly flexible to produce CDs with varying surface chemistries and intriguing photoluminescent (PL) properties.4–7To date, research on CDs is largely restricted to their PL properties and related applications. Being a new type of nanomaterial, they are also attractive building blocks for supramolecular chemistry, which has, however, received trace attention. We have recently showed that the organization of alkylated CDs in a solvent-free state had an obvious influence on their nonlinear optical properties.8 In bulk solutions, the assembly of CDs largely depend on the assistance of additional components such as lipid gelator in ethanol9 and oppositely charged surfactants in water.10–12 To achieve the self-assembly of CDs in water without any auxiliary component, a careful structural design is needed to impart surface activity, which is a big challenge. CDs lack atomic precision, which makes it difficult to achieve accurate chemical modification as performed on fullerene C60.13 Furthermore, the rather small sizes of CDs hinder asymmetric functionalization like the Janus particles.14–16 It should be noted that there are some studies reported in the literature discussing the amphiphilicity of CDs.17–21 However, the discussion is limited to the solubilities of CDs in different solvents, which is far from the connotation of “amphiphilicity” with regard to colloid and interface science.
It is known that attaching polymer chains to nanoparticles would produce hairy nanoparticles, which could exhibit amphiphilicity in bulk solutions.22 Using them as building blocks, nanoparticle vesicles were produced.23–30 Adopting similar structural motifs, Liu et al. prepared CDs co-functionalized with an inner layer of quaternary ammonium salts covered by an outer alkyl chain layer, which showed surface activity at the air/water interface.31 However, no aggregation behaviour was addressed, presumably due to the limited solubility of CDs in water caused by the intrinsic hydrophobicity of the peripheral alkyl chains. In another work, Das et al. attached cholesterol moieties to CDs via amide bonds by post-functionalization.32 Unfortunately, CDs showed ever poorer solubility in water, and investigations were performed in dimethyl sulfoxide (DMSO) or DMSO/water binary mixtures.
Oligoxyethylene groups act as hydrophilic parts in non-ionic surfactants. We envision that capping CDs with oligoxyethylene groups may lead to the formation of a new class of surface-active CDs, which will have much higher solubility in water than that of their counterparts bearing alkyl chains or cholesterol moieties. Bearing this in mind, in this work, we used citric acid (CA) as the carbon source and triethylene glycol monomethyl ether (TGME) as the modifying group to prepare CDs, which show considerable surface activity in water with the lowest surface tension of ∼37 mN m−1. The high solubility of CDs in water (>30 mg mL−1) enables us to investigate their concentration-dependent self-assembly for the first time, where a variety of aggregates including nanoclusters, nanorods, nanotubes and vesicles were found. As a typical example of their applications, the CDs were also used to disperse and stabilize multi-walled carbon nanotubes (MWCNTs) in water, which showed much better performance than that of both traditional ionic surfactants, such as sodium dodecyl sulphate (SDS), and non-ionic surfactants, such as Pluronic F127.
2. Experimental section
2.1 Materials
TGME (99%) and CA (95%) were purchased from Sigma. MWCNTs with diameters of 5–15 nm and lengths of 10–30 μm were bought from Jiangsu XFNANO Materials Tech Co., Ltd (Nanjing, China). SDS (CP) and NaOH (99%) were purchased from Sinopharm Chemical Reagent Co., Ltd. Pluronic F127 was bought from Sigma. The chemicals were used without further purification unless other stated. Ultrapure water with a resistivity of 18.25 MΩ cm−1 used in the experiments was obtained using a UPH-IV ultrapure water purifier (China).2.2 Characterization
Fluorescence spectra were recorded using a spectrofluorometer (FluoroMax-4, Horiba). Fourier transform infrared (FTIR) spectra were recorded using a spectrometer (Tensor II) with KBr pellets (Germany Bruker). UV-vis measurements were carried out using a UV-vis-NIR spectrometer (Cary 5000, Agilent). Thermogravimetric analysis (TGA) was carried out using a TGA 5500 (TA). The absolute fluorescence quantum yield (Φ) and fluorescence lifetime (τ) were measured using a spectrofluorometer (FLSP920, Edinburgh Instruments Ltd). X-Ray photoelectron spectroscopy (XPS) spectra were recorded using an X-ray photoelectron spectrometer (ESCALAB 250) equipped with a monochromatic Al Kα X-ray source (1486.71 eV). Raman spectra were recorded using a confocal microscopic Raman spectrometer (Lab Ram HR Evolution). X-ray diffraction (XRD) patterns were acquired using a D8 ADVANCE (Germany Bruker) diffractometer equipped with a Cu Kα radiation source and a graphite monochromator. Nuclear magnetic resonance (NMR) spectra were recorded using a 400 MHz nuclear magnetic resonance instrument (Switzerland Bruker). Surface tension measurements were performed by a plate method using a Surface tension meter (Germany Biolin Scientific). Differential scanning calorimetry (DSC) measurements were carried out using a DSC250 (TA Instruments). Particle size and zeta potential (ζ) analysis were carried out using a NanoZS (Malvern). For the measurement of the dispersed MWCNTs, the dispersion obtained three days after preparation was centrifuged for 1 min at 5000 rpm. Measurements were performed on the supernatant after removing the precipitated tubes at the bottom.High-resolution transmission electron microscopic (HRTEM) images were recorded using a JEM2100 instrument operating at 200 kV (JEOL). For sample preparation, a drop of sample solution (∼20 μL) was placed on a holey-carbon-coated copper grid (300 meshes) and dried using an infrared lamp for 30 min. For cryo-TEM observations, the sample (∼4 μL) was dropped onto a micro grid under high humidity (>80%). The excess sample was removed with two pieces of filter paper, leaving a thin film sprawling on the micro grid, which was plunged into liquid ethane pre-liquefied with liquid nitrogen. The vitrified sample was transferred into a sample holder (Gatan 626) and observed using a JEOL JEM-1400 TEM (120 kV) at ∼−174 °C. The images were recorded using a Gatan multiscan CCD. In freeze-fracture TEM (FF-TEM) observations, a small amount of sample (∼4 μL) was dropped onto the specimen carrier. The sample was frozen by plunging into liquid propane cooled by liquid nitrogen. Fracturing and replication were carried out using a freeze-fracture apparatus (EM BAF 060, Leica, Germany) at a temperature of −160 °C. Pt/C was deposited at an angle of 45° to shadow the fracture surface and C was deposited at an angle of 90° to consolidate the fracture surface. The replicas were transferred onto a copper grid and then checked using a JEOL JEM-1400 TEM (Japan) at an accelerating voltage of 120 kV.
2.3 Synthesis of CDs
The CDs were synthesized by pyrolyzing TGME and CA in a reaction kettle with a volume of 20 mL. In a typical experiment, TGME and CA were introduced into the reaction kettle. TGME is in large excess, with a molar ratio to CA (nTGME/nCA) of 21/1. The total volume of the mixture was controlled to be about two-thirds of the volume of the reaction kettle. At room temperature, TGME is a liquid, while CA is a solid. To fully mix these two components, the reaction kettle was put into a water bath, and the mixture was stirred at 60 °C until CA was dissolved totally. The reaction kettle was then closed and heated in an oven to 240 °C. After 1 h, pyrolysis was stopped and the reaction kettle was cooled down to room temperature. The crude product was diluted with an equal volume of water, and the pH was adjusted to ∼7 using an aqueous solution of NaOH (100 mg mL−1). The as-obtained solution was then subjected to dialysis using a dialysis bag with a molecular weight cut-off of 100 Da. The dialysis was continued with water exchange every day. The final conductivity of water outside of the dialysis bag is below 3.0 μS cm−1. The aqueous solution of the purified CDs was then lyophilized for further use. In control experiments, pyrolysis of the TGME/CA mixtures with different mixing ratios as well as TGME alone was also carried out.2.4 Dispersion of MWCNTs
In the dispersion of MWCNTs, 0.5 mg of MWCNTs and 3 mL of aqueous solution of CDs were used in each sample. The concentration of CDs (cCDs) was varied between 0.001 and 0.08 mg mL−1. The samples were sonicated at 30 kHz for 2 h (keeping the temperature no more than 25 °C by changing water outside whenever necessary). After this treatment, the samples were transferred into a thermostat and kept at 25 °C to check the change of the appearance of the samples with time. In control experiments, MWCNTs were also treated under the same condition using aqueous solutions of SDS and F127 as well as pure water.3. Results and discussion
3.1 Synthesis and structural characterization of CDs
The CDs were facilely synthesized by pyrolyzing TGME and CA with a nTGME-to-nCA proportion of 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in a reaction kettle under 240 °C for 1 h, followed by neutralization with NaOH and dialysis against water (for details, see Experimental section). After these procedures, an aqueous solution of CDs was obtained, which showed faint yellow colour at room temperature (Fig. S1†) and blue luminescence under 365 nm UV irradiation (Scheme 1). Two possible paths exist for the formation of CDs. Generally, for pyrolysis of two precursor molecules, condensation between them via active functional groups is expected before carbonization.33 A similar process is expected for TGME and CA here via esterification (Path I), yielding various intermediates. The structures of two representative intermediates are given in Scheme 1, and more possibilities can be found in Scheme S1.† However, in the current work, stepwise heating was not adopted as we did previously for the synthesis of imidazolium-modified CDs.10,34 Instead, the temperature was directly increased to 240 °C in an oven, which has well exceeded the temperature needed for the carbonization of CA (∼180 °C). Thus, another path is also possible (Path II) where CA-derived CDs form first, to which TGME is grafted via esterification.
:1 in a reaction kettle under 240 °C for 1 h, followed by neutralization with NaOH and dialysis against water (for details, see Experimental section). After these procedures, an aqueous solution of CDs was obtained, which showed faint yellow colour at room temperature (Fig. S1†) and blue luminescence under 365 nm UV irradiation (Scheme 1). Two possible paths exist for the formation of CDs. Generally, for pyrolysis of two precursor molecules, condensation between them via active functional groups is expected before carbonization.33 A similar process is expected for TGME and CA here via esterification (Path I), yielding various intermediates. The structures of two representative intermediates are given in Scheme 1, and more possibilities can be found in Scheme S1.† However, in the current work, stepwise heating was not adopted as we did previously for the synthesis of imidazolium-modified CDs.10,34 Instead, the temperature was directly increased to 240 °C in an oven, which has well exceeded the temperature needed for the carbonization of CA (∼180 °C). Thus, another path is also possible (Path II) where CA-derived CDs form first, to which TGME is grafted via esterification.
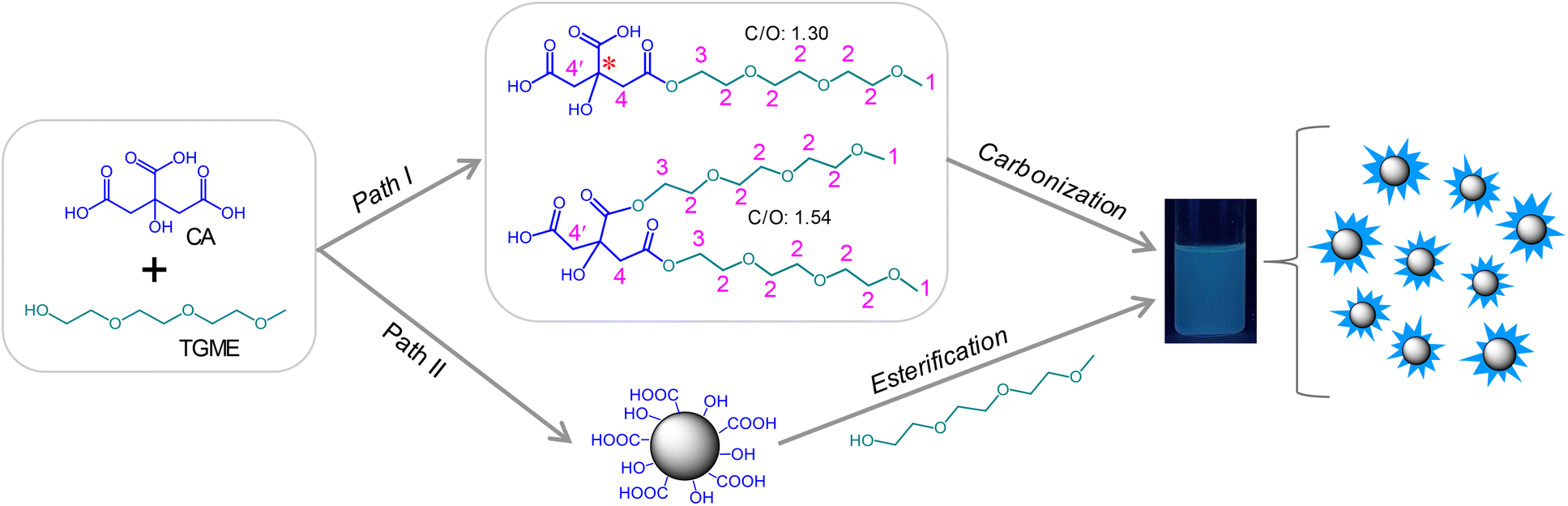 | ||
| Scheme 1 Two paths possibly occurring during the pyrolysis of CA and TGME. | ||
The structures of the CDs were fully investigated by a variety of techniques. HRTEM observation on a 5 mg mL−1 CD dispersion in ethanol shows that the CDs are quasi-spherical (Fig. 1a). The majority of the CDs have sizes ranging from 1.5 to 4 nm, with a few of larger ones up to 6 nm (Fig. 1b). The magnification of a representative CD reveals its well-ordered structure in the core with a lattice spacing of 0.27 nm (Fig. 1c), which corresponds to the (020) diffraction plane of graphitic carbon.35 From the 1H NMR spectrum (Fig. 1d), all the protons on the TGME chain can be found with perfect expected ratios, which unambiguously confirmed that TGME serves mainly as a surface-modifying agent and remains intact after pyrolysis. In the control experiment where pyrolysis was performed solely on TGME, the colour of the mixture is almost colourless (Fig. S1†), affirming negligible carbonization of TGME. As a main component for carbonization, the NMR spectra related to CA were analysed carefully. It was found that signals from the protons of positions 4 and 4′ are still seen in the 1H NMR spectrum, and the quaternary carbon atom can be still found in the 13C NMR spectrum (Fig. 1e, marked by the star). These observations indicate that in between the fully carbonized cores and the peripheral TGME chains, there is a zone of transition where CA is only partially carbonized and acts as a glue to unite the inner and outer parts of CDs. This partial carbonization causes splitting of signals for both the two carbon atoms (4 and 4′) and the protons on them, as seen from both 1H NMR and 13C NMR spectra. It is generally accepted that carbonization of organic precursors is a continuous process, and a prolonged time of pyrolysis will produce CDs with heavier carbonization and larger sizes. The incomplete carbonization of CA in the current study is thus understandable considering that the time of pyrolysis is limited to 1 h. The XRD profile of CDs gives a bulge at 21.5° together with a smaller one at 41.2° (Fig. S2†), indicating the amorphous structure of the CDs. This observation indicates that although ordered structures are presented in the carbonized center of the dot as proven by HRTEM, the content should be small and the structural feature of the dot is dominated by the polymeric network as well as the peripheral oligoxyethylene chain.
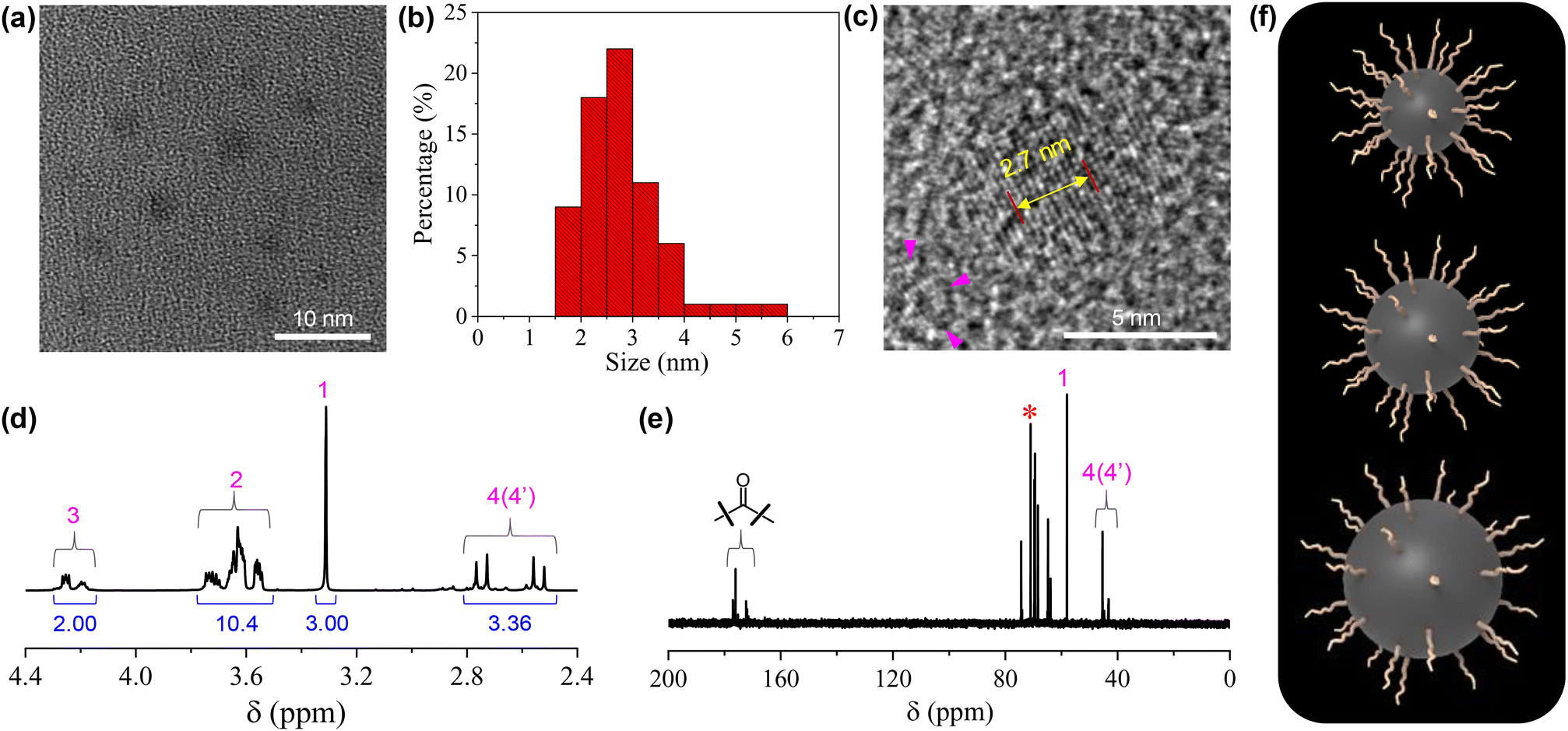 | ||
| Fig. 1 (a) Typical HRTEM image of the CDs (5 mg mL−1 in ethanol). (b) Size distribution of the CDs based on the statistics of 70 single particles. (c) Magnified image that highlights the internal structure of the carbon dots. The distance between 10 layers of the graphite-like lattice is also given. (d and e) 1H NMR and 13C NMR spectra of the CDs. For the assignment of the peaks, see Scheme 1. (f) Cartoons of three CDs with the same numbers of TGME on the surfaces but different sizes of the cores. | ||
Our conclusion on the structure of the CDs gains further support from FTIR measurements. As shown in Fig. 2a, the characteristic absorptions of TGME are largely retained. Specifically, the peaks at 2904 cm−1 and 1108 cm−1 confirm the survival of the –CH3 and C–O–C groups, respectively.36 In comparison, the spectrum of CA, in particular the peaks over 3000 cm−1 that are assigned to the stretching vibrations of –OH from the hydroxyl and carboxyl groups,37 changed a lot due to its condensation/carbonization during the pyrolysis. The sharp peak at 1730 cm−1 of the CDs proves the presence of the carbonyl group from the ester group, affirming the proposed paths for the formation of the CDs shown in Scheme 1. The XPS spectra were recorded to further probe the components and bonding details of the CDs. Signals from C 1s and O 1s were found in the survey at 285.6 eV and 533.3 eV, respectively (Fig. S3†). The high-resolution spectrum of C 1s can be divided into three peaks at 284.7 eV, 286.3 eV, and 288.1 eV, respectively (Fig. 2b), which are assigned to the presence of C–C, C–O, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds.19 The high-resolution spectrum of O 1s can be divided into two peaks at 532.3 eV and 530.7 eV (Fig. 2c), proving the presence of C–O and CO bonds. Besides C 1s and O 1s, the presence of Na 1s was also confirmed by the peak at 1068.8 eV together with an Auger peak at 498 eV (Fig. S3†). The atomic ratio of Na/C/O was determined to be approximately 1/4.9/8.3. This trace amount of Na should be introduced by the neutralization process (for details, see the Experimental section) via the reaction of NaOH with the remaining acidic groups on the surfaces of the CDs. Indeed, the zeta potential measurement shows that the CDs are weakly charged, with a ζ value of −5.6 mV. Considering that the –OH group in CA is less chemically active than –COOH, it is possible that not all of them are under condensation during pyrolysis and partially left on the surfaces. Based on all of the characterizations, a profile map of the CD is proposed and given in Scheme 2, which includes the whole subunits of the CD from the inner carbonized core to the outer TGME chains across the intermediate polymeric layer. According to the classifications summarized by Otyepka et al. in a recent review,38 our CDs belong to the subclass of carbonized polymer dots (CPDs).
O bonds.19 The high-resolution spectrum of O 1s can be divided into two peaks at 532.3 eV and 530.7 eV (Fig. 2c), proving the presence of C–O and CO bonds. Besides C 1s and O 1s, the presence of Na 1s was also confirmed by the peak at 1068.8 eV together with an Auger peak at 498 eV (Fig. S3†). The atomic ratio of Na/C/O was determined to be approximately 1/4.9/8.3. This trace amount of Na should be introduced by the neutralization process (for details, see the Experimental section) via the reaction of NaOH with the remaining acidic groups on the surfaces of the CDs. Indeed, the zeta potential measurement shows that the CDs are weakly charged, with a ζ value of −5.6 mV. Considering that the –OH group in CA is less chemically active than –COOH, it is possible that not all of them are under condensation during pyrolysis and partially left on the surfaces. Based on all of the characterizations, a profile map of the CD is proposed and given in Scheme 2, which includes the whole subunits of the CD from the inner carbonized core to the outer TGME chains across the intermediate polymeric layer. According to the classifications summarized by Otyepka et al. in a recent review,38 our CDs belong to the subclass of carbonized polymer dots (CPDs).
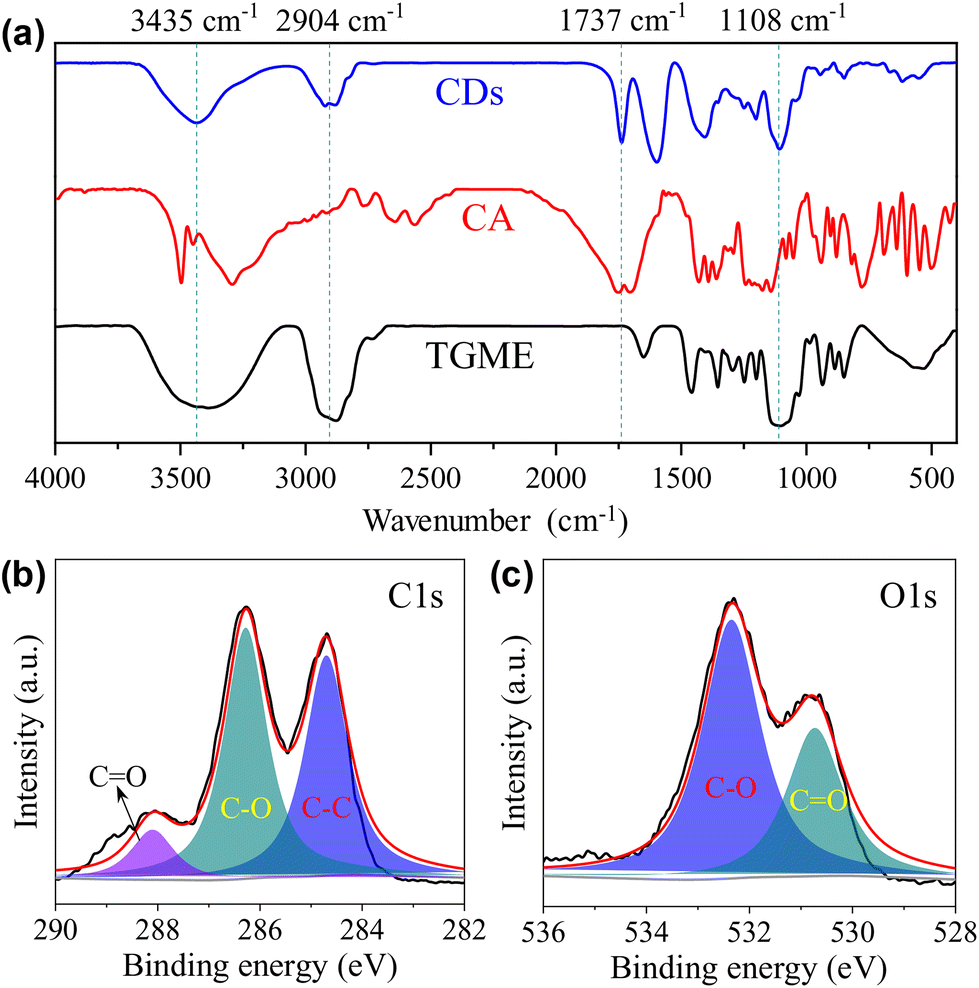 | ||
| Fig. 2 (a) FTIR spectrum of the CDs. Data from CA and TGME are also given for comparison. High-resolution XPS spectra of C 1s (b) and O 1s (c). | ||
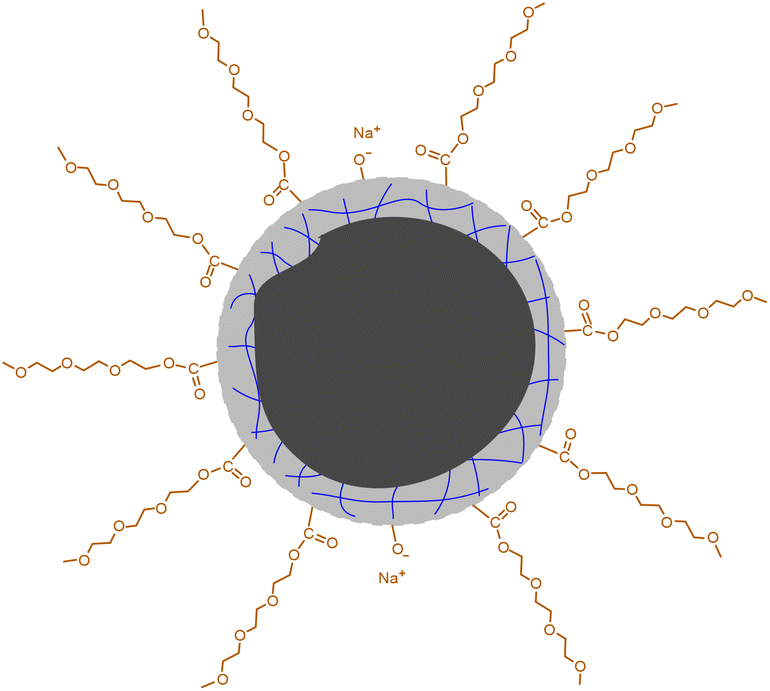 | ||
| Scheme 2 Proposed profile map of the CD. The black part in the center represents the carbonized core. The gray part with blue lines represents the polymeric network formed by partially carbonized CA. | ||
3.2 Optical properties of CDs
The absorption of the aqueous solution of CDs was recorded within the wavelength range from 200 to 800 nm, which shows that the absorption mainly occurs in the UV region (Fig. 3a). The sharp peak at 210 nm could be ascribed to π–π* transition of the conjugated structures,39 including the CO group and possibly also the sp2 carbons inside the cores of the CDs. A weak, shoulder peak was also observed at 260 nm, which could be partially assigned to the n–π* transition of the CO group.37 Of course, contributions from the carbonized core and the condensed polymeric layer cannot be excluded, considering the rather complicated structures of the CDs.38 The emission of CDs in water with a concentration of 1 mg mL−1 is given in Fig. 3b, which exhibits excitation-dependent behaviour. The maximum emission occurs at 442 nm under an excitation wavelength of 365 nm, and the absolute fluorescence quantum yield (Φ) was determined to be 2.17%. Maser et al. pointed out that for many CDs, the supramolecular-enhanced charge transfer within entangled polyamide chains is the origin of the universal blue fluorescence.40 However, the fast growth of the knowledge on cluster-triggered emission (CTE)41 also indicated that the clustering of heteroatoms including O and N could also lead to the enhancement of the photoluminescence. The blue-emission character with relatively low Φ indicates that the CDs presented in the current work probably follow a similar mechanism, i.e., the emission should mainly originate from the closely arranged ester groups. Concentration-dependent PL measurements indicate that with the increase in cCDs, the position of the emission under excitation at 365 nm remains unchanged, all of which locate in the blue light region (insets of Fig. 3b and Fig. S4†). Excitation-dependent photoluminescence was observed as well in a solvent-free state (Fig. 3c). Compared to the aqueous solution, the maximum emission red-shifts to 542 nm and Φ decreases to 1.66%. The PL decays of the CDs in water and the solvent-free state were also tested. In both cases, the decay could be fit by a biexponential model with the average lifetime (〈τ〉) to be 4.22 ns and 4.48 ns, respectively (Fig. 3d and Table S1†).
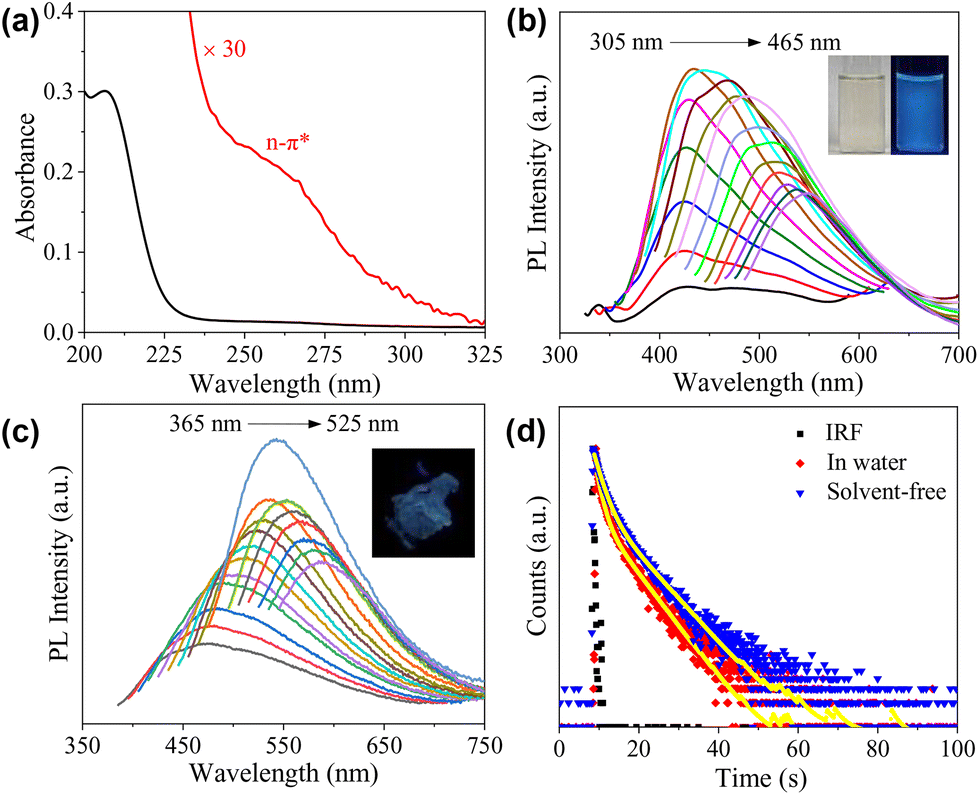 | ||
| Fig. 3 (a) UV-vis absorption spectra of the aqueous solution of CDs (0.01 mg mL−1). (b) Emission spectra recorded at different excitation wavelengths ranging from 305 nm to 465 nm with 10 nm increment of the aqueous solution of CDs (1 mg mL−1). Insets are photos of a 5 mg mL−1 sample under room light (left) and 365 nm UV irradiation (right). (c) Emission spectra recorded at different excitation wavelengths ranging from 365 nm to 525 nm with 10 nm increment of the CDs in solvent-free state. Inset is the photo of the solvent-free sample under 365 nm UV irradiation. (d) PL decay profiles of the CDs recorded in an aqueous solution and a solvent-free state. | ||
3.3 Surface activity and self-assembly behaviour
The surface activity of the CDs was evaluated by the standard surface tension measurements. As shown in Fig. 4a, the semi-log plot of the surface tension (γ) vs. concentration shows an obvious inflection point at 0.07 mg mL−1, which corresponds to the critical aggregation concentration (cac) of the CDs. The lowest value of γ within the investigated concentration range reaches ∼37 mN m−1, which is comparable to that obtained by traditional ionic surfactants such as sodium dodecyl sulphate (SDS),42 but is higher than that achieved by non-ionic surfactants such as tetraethylene glycol monododecyl ether (C12EO4).43,44 As the CDs are also negatively charged, conductivity measurements were performed, which gives a well-defined inflection point designated as the cac at 0.09 mg mL−1 (Fig. 4b). The slightly varied cac values derived from surface tension and conductivity measurements might be caused by the differences in these two techniques. For the CDs presented in the current study, the carbonized cores act as the hydrophobic part, and the peripheral TGME chains as well as the remaining hydrophilic groups on the cores behave as the hydrophilic moieties. The considerably high surface activity of the CDs indicates that the hydrophilic moieties must not fully cover the hydrophobic cores. At the air/water interface, the flexible TGME chains should try to stretch into the water phase, exposing the area of the hydrophobic core to the air as much as possible (see the illustration shown in Scheme 3). It should be emphasized that nTGME/nCA in the current study, which is 21:1, is an optimized value obtained by evaluating the γ value at a fixed concentration of 0.1 mg mL−1, which is ∼37 mN m−1 at nTGME/nCA = 21:1. Increasing nTGME/nCA to 24:1 or decreasing it to 18:1 will both lead to larger γ values, which are ∼42 and ∼39 mN m−1, respectively. The surface activity of two other CDs bearing carboxyl or imidazolium groups, developed in our group previously, was also tested. Both of them totally lack surface activity (Fig. S5†), highlighting the importance of our structural design in the current study to produce surface active CDs.
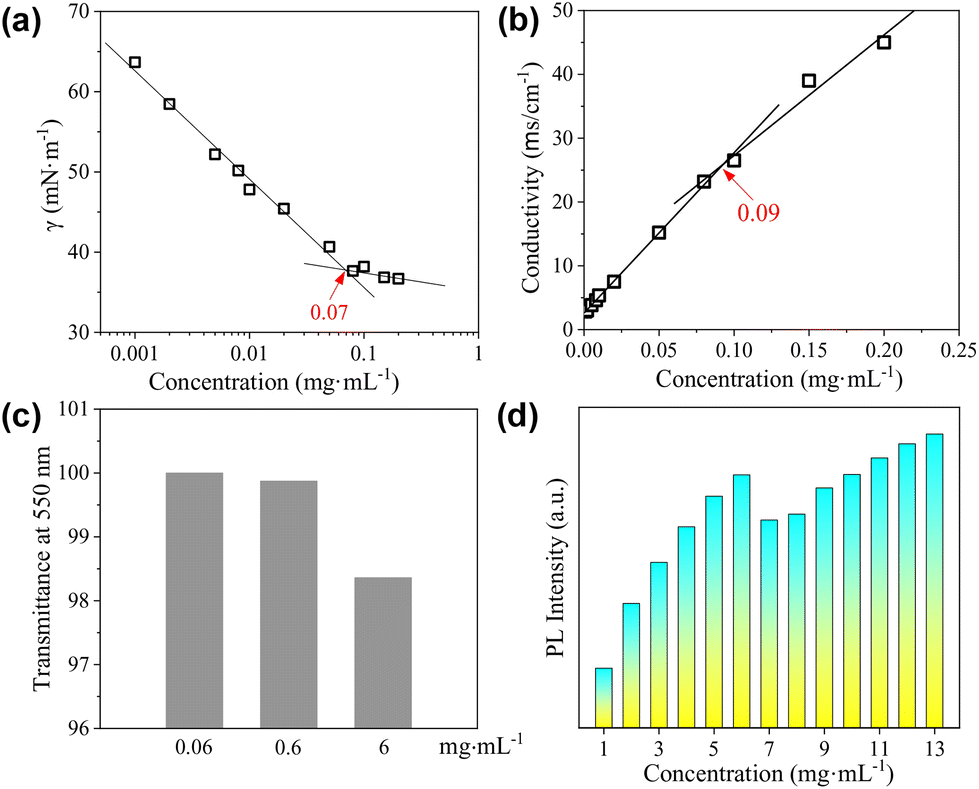 | ||
| Fig. 4 (a and b) Surface tension and conductivity as a function of cCDs in water. The straight lines are guides for the eyes. (c) Transmittance at 550 nm of the aqueous solutions of the CDs at three representative concentrations as indicated. (d) Statistics of the PL intensity of the aqueous solution of the CDs with the increase in concentration. | ||
 | ||
| Scheme 3 Illustration of the concentration-dependent self-assembly of the CDs in water. For better clarity, the CDs involved in different supramolecular structures are not drawn in scale. | ||
The CDs have high solubility in water (>30 mg mL−1), which enables a detailed study on their self-assembly behaviour in water that is hard or impossible for the CDs bearing peripheral hydrophobic groups.31,32 The sample with a concentration of 0.06 mg mL−1, which is below the cac, is transparent with transmittance at 550 nm nearly 100%. Increasing cCDs to 0.6 and 6 mg mL−1, respectively, causes a continuous decrease in the transmittance (Fig. 4c). The PL intensity basically increases with the increase in cCDs within the investigated concentration range. However, a sudden drop was observed at cCDs of 6 mg mL−1 (Fig. 4d). These observations are indicative of the concentration-dependent self-assembly behaviour of the CDs in water.
The morphologies of the aggregates formed by the CDs in water were investigated by imaging studies. For this purpose, three cCDs above cac were selected, which are 1, 6 and 20 mg mL−1. Typical results are summarized in Fig. 5. At cCDs = 1 mg mL−1, nanoclusters with irregular shapes and an average size of ∼25 nm were found (Fig. 5a). A magnified image showing a typical nanocluster which consists of about six individual CDs is given in Fig. 5b. At cCDs = 6 mg mL−1, polymorphism was noticed seen from the typical image shown in Fig. 5c, where nanorods coexist with nanotubes. In addition, black dots were noticed whose sizes are larger than those of individual CDs observed in ethanol (see Fig. 1a), indicating that they are probably oligomers of CDs. Fig. 5d–f give the magnified image focusing respectively on the nanorods, oligomers and nanotubes. The nanorods have lengths ranging from 20 to 140 nm and diameters of 3–10 nm. The length of the nanotube can approach several hundreds of nanometres with a diameter of ∼20 nm. The polymorphism, which has also been observed for amphiphilic fullerene C60 derivatives,45 should be ascribed to the rather complicated structure of the CDs41 which leads to a high conformational freedom. At cCDs = 20 mg mL−1, vesicles were detected exclusively, which have polydisperse sizes ranging from 22 to 108 nm (Fig. 5g). Analysis of three typical vesicles with different sizes (Fig. 5h–j) shows that the walls of the vesicles all contain a dark interior and a lighter margin, which fits perfectly the model of the organization of CDs with the hydrophobic cores sitting inside and the hydrophilic TGME chains outside. Considering that the thickness of the dark region of the wall is less than 3 nm, a monolayer arrangement of the CDs is preferred. The formation of the vesicles could also be confirmed by DLS measurement, which gives consistent size distribution with that obtained from imaging study (Fig. 5k). An illustration of the self-assembly of CDs in water as a function of the concentration is given in Scheme 3 with proposed structures of the observed aggregates. A cartoon of the CDs at cac is also included, where a monolayer of CDs is shown at the air/water interface that is balanced with individual CDs and the oligomers of CDs in the bulk aqueous phase.
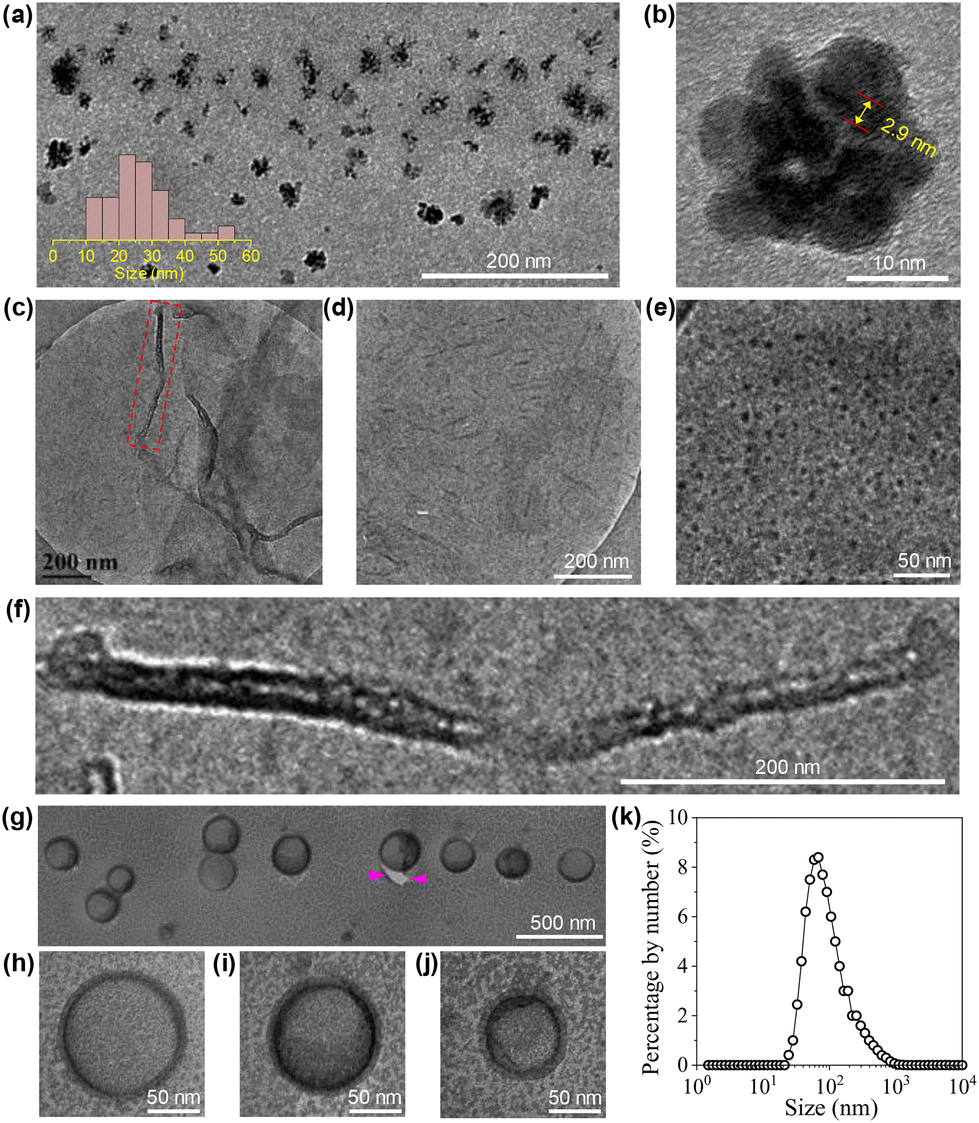 | ||
| Fig. 5 HRTEM (a and b), cryo-TEM (c–f), FFTEM (g–j) images and DLS result (k) of the CDs in water with concentrations of 1 (a and b), 6 (c–f) and 20 (g–j) mg mL−1. In image (b), the distance between 10 layers of the lattice inside the core of a CD is given. Image (f) corresponds to the square region in image (c). Between the arrow heads in image (g) is the crack of the film. | ||
3.4 Temperature effect
The thermal decomposition of the CDs was investigated by TGA. As shown in Fig. 6a, the curve can be roughly divided into four parts. The first small weight loss before 160 °C (∼3 wt%) can be attributed to the presence of trace water. The second weight loss occurs between 160 °C and 246 °C and occupies ∼30 wt% of the total weight. As this temperature range overlaps with the decomposition of CA, we tentatively ascribed it to the continued condensation of the incompletely carbonized polymer networks locating in between the totally carbonized cores and the peripheral TGME chains (the grey part with blue lines in Scheme 2). The third weight loss appears within a rather narrow temperature range of 246 °C to 270 °C and accounts for ∼29 wt% of the total weight, which is ascribed to the cleavage of the ester links and subsequent volatilization of the disconnected TGME chains. The remaining weight loss up to 800 °C is less obvious and accounts for ∼17 wt% of the total weight, which could be ascribed to the heavier carbonization at elevated temperatures. No phase transition could be detected from DSC measurements up to 110 °C (Fig. S6†), indicating that the CDs remain in the amorphous state with disordered TGME chains.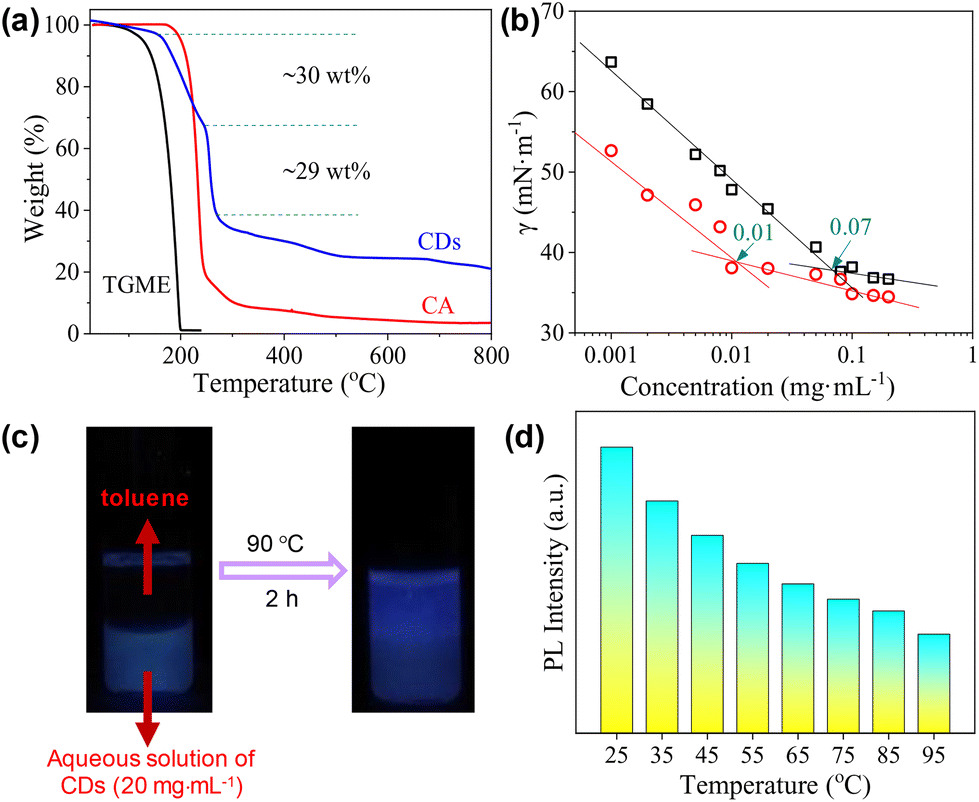 | ||
| Fig. 6 (a) TGA curve of the CDs. Data from CA and TGME are also given for comparison. Note that the sharp weight loss of TGME over 122 °C (the boiling point of TGME) is caused by its evaporation rather than decomposition. (b) Surface tension as a function of cCDs in water measured at 70 °C (circles). For comparison, data obtained at 25 °C (squares) are also shown. (c) Photos of a typical sample denoting the partial phase transfer of the CDs triggered by temperature. (d) Statistics of the PL intensity of the CDs in the solvent-free state with the increase in temperature, excited at 465 nm. | ||
It is known that for ionic surfactants, increasing the temperature will increase its hydrophilicity due to the enhanced dissociation of the ionic moiety. For non-ionic surfactants containing methylene oxide (EO) groups, increasing the temperature will increase its hydrophobicity due to the dehydration of the EO groups. The CDs presented in the current work bear both ionic and non-ionic parts. Therefore, their response towards temperature is interesting. The surface activity of the CDs in water at 70 °C was evaluated. As shown in Fig. 6b, the γ values are lower than those obtained at 25 °C within the whole concentration range, and the cac is only one-seventh that at 25 °C. This observation indicates that the CDs become more hydrophobic at elevated temperatures, i.e., their response towards temperature is dominated by the non-ionic moieties instead of ionic ones. The increase in the hydrophobicity of the CDs at elevated temperatures can be also reflected by phase transfer between water and toluene. As displayed in Fig. 6c, the CDs stayed in the water phase at room temperature, which partially transferred to the top toluene phase when the sample was heated to 90 °C. The incomplete phase transfer can be understood considering the polydispersity of the CDs, both in sizes and amphiphilicity. Finally, the emission of the CDs at different temperatures was tested where the PL intensity decreases continuously (Fig. 6d), a phenomenon commonly found for most of the chromophores.
3.5 Applications in the dispersion of MWCNTs
Carbon nanotubes (CNTs) exhibit inherently high mechanical strength46 and electrical/thermal conductivity47 that originated from their unique one-dimensional structure. Therefore, they have a wide range of applications in fields such as lithium-ion battery fabrication48 and biosensing.49 However, the strong van der Waals forces and π–π stacking among individual tubes make them easily clog, aggregate, and entangle into bundles, leading to a poor wettability in water.47,50–52 Therefore, to disperse CNTs in water is a prerequisite for their applications.51 Non-covalent wrapping of the tubes using dispersants is an effective way to the dispersion and stabilization of CNTs,46,51,52 where surfactants are among the most popular dispersants.53To further demonstrate the amphiphilicity of the CDs as well as to present an example of their practical applications, the capability of our CDs in dispersing CNTs was then tested with close comparison to the typical ionic surfactant SDS and the typical non-ionic, polymeric surfactant Pluronic F127. The CNTs used are MWCNTs with diameters of 5–15 nm and lengths of 10–30 μm. In the presence of the CDs, MWCNTs can be well dispersed into water at very low cCDs of 0.006–0.02 mg mL−1 (Fig. 7a). In comparison, the lowest concentrations needed for SDS and Pluronic F127 to effectively disperse MECNTs are much higher under the same experimental condition (for details, see the Experimental section), which are 0.1 mg mL−1 (Fig. 7b) and 0.5 mg mL−1 (Fig. 7c), respectively. The CD-stabilized sample with the lowest amount of CDs, i.e., 0.006 mg mL−1, was selected for further study. TEM (Fig. 7d) and SEM (Fig. 7e) observations on the supernatant three days after preparation show well-dispersed tubes, which are in sharp contrast to the heavily entangled tubes when disperse in the absence of CDs (Fig. S7 and S8†). The impressively high capability of the CDs to disperse MWCNTs is first attributed to their three-dimensional structure with huge size, which can create a very effective steric hindrance. The advantage of such structure in stabilizing CNTs has also been observed previously in starlike block polymers.52 Second, the CDs bear a small amount of negative charges, which will provide electrostatic repulsion between the dispersed MWCNTs. Although ζ of individual CD is only −5.6 mV, measurement on the supernatant of the representative dispersion gives a relatively high value of −26.8 mV due to the multiple adsorption of CDs. Finally, as the hydrophobic parts of CDs are carbon-rich polymeric networks and carbonized cores, we suppose that they have higher affinity with surfaces of MWCNTs compared to the alkyl chains of SDS and the propylene oxide groups of Pluronic F127.
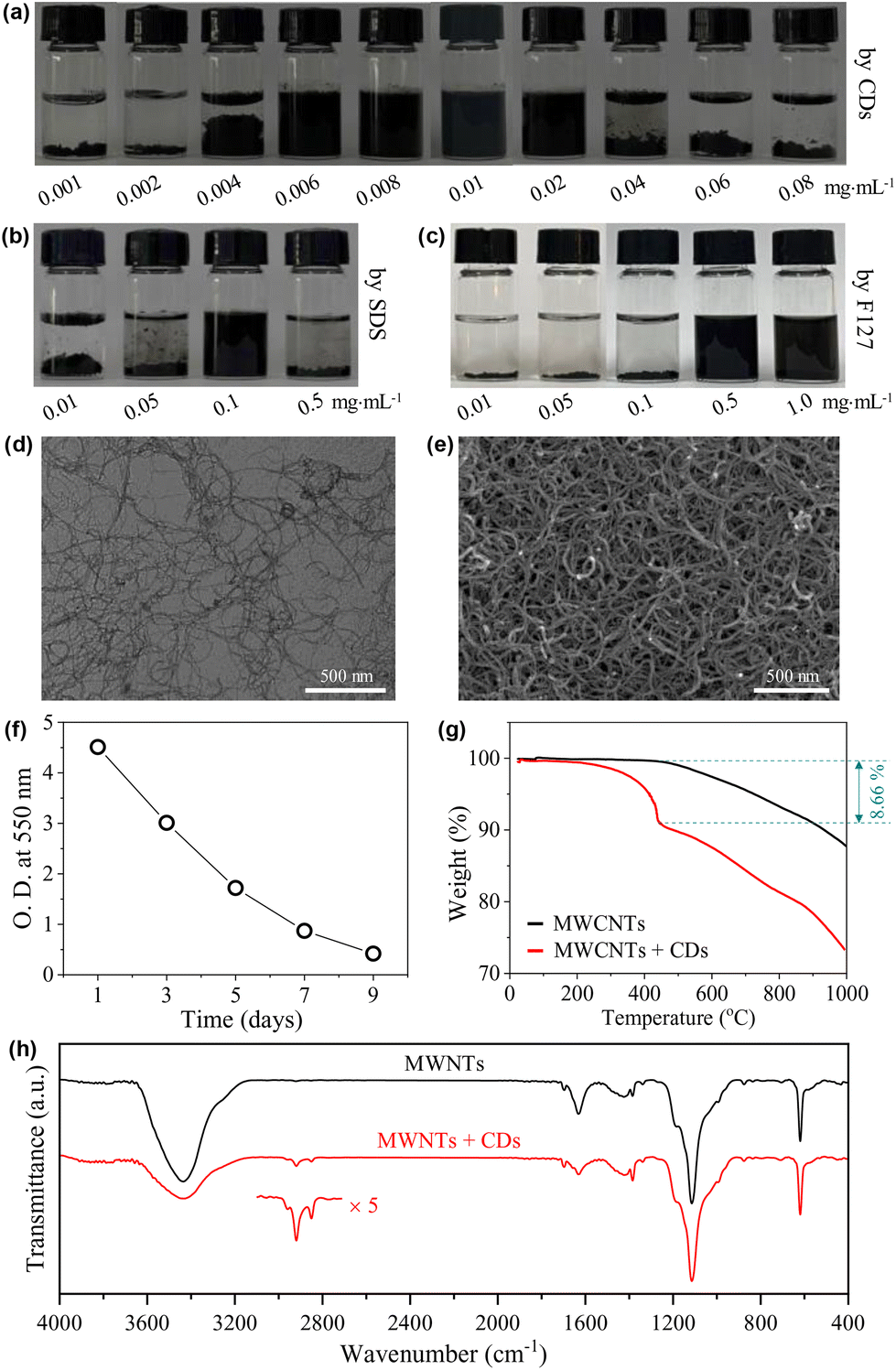 | ||
| Fig. 7 (a–c) Photos of aqueous dispersions of MWCNTs with different concentrations of CDs, SDS and F127, respectively, taken seven days after sample preparation. (d and e) Typical TEM and SEM images of the supernatant of the MWCNT dispersion with 0.006 mg mL−1 CDs. Observations were carried out on the 3rd day after the preparation of the dispersion. (f) Variation in the optical density (O.D.) at 550 nm of the supernatant of a typical sample of MWCNTs dispersed with 0.006 mg mL−1 CDs. (g and h) TGA curves and FTIR spectra of the dried supernatant of the MWCNT dispersion without and with 0.006 mg mL−1 CDs, lyophilized three days after the preparation of the dispersions. | ||
Note that the dispersions of MWCNTs are not thermodynamically stable systems, and they are only kinetically stable. If standing for a sufficiently long time, the dispersed tubes will finally deposit at the bottom. The time-dependent deposition of the tubes was checked. As shown in Fig. 7f, the dispersion is highly opaque one day after preparation with an optical density (O.D.) at 550 nm approaching 5. The O.D. decreased gradually with time, but is still >0.4 even on the 9th day. This time scale of stability is enough for most of the practical applications.
To get further information on the MWCNT-CD hybrid, the supernatant of the dispersion obtained three days after preparation was lyophilized. From TGA (Fig. 7g), the hybrid shows a heavier weight loss compared to pure MWCNTs due to the adsorption of CDs. The difference, calculated at 443 °C, occupies ∼8.66% of the total weight. After correction with the TGA curve of CDs (∼28% weight remaining at 443 °C from Fig. 6a), the weight ratio of the CDs to MWCNTs can be calculated to be ∼1/7.3. This value is much higher than that obtained from the initially added CDs (0.006 × 3 mg) and MWCNTs (5 mg), which is ∼1:277.8, indicating that the tubes with trace amounts of CDs adsorbed have deposited during the storage. The presence of the CDs in the hybrid was also detected by FTIR measurement. Two weak peaks at 2921 and 2849 cm−1 were noticed (Fig. 7h), which are assigned to the asymmetric and symmetric methylene stretching bands from TGME.
4 Conclusions
In summary, we showed that linking TGME to carbonized CA is an effective strategy to produce structurally persistent CDs that show high surface activity in water. Due to the presence of hydrated, peripheral TGME chains as well as negatively charged surface groups, these CDs exhibit the properties of both non-ionic and ionic amphiphiles. They spontaneously form aggregates in water with different morphologies including nanoclusters, nanorods, nanotubes and vesicles, which has not been achieved for other CDs reported to date. The CDs are temperature sensitive, providing a means to further tune their properties. When being applied to disperse and stabilize MWCNTs in water, the effective steric hindrance originating from the bulky structure as well as the electrostatic repulsion caused by the remaining surface charges leads to a much better performance of the CDs than that of traditional surfactants. We hope this work serves as a modest spur to trigger the enthusiasm in the near future on the research of surface active CDs, which can fill up the gap between luminescent carbon nanomaterials and surfactant science.Author contributions
Aoxue Xu: investigation, writing – original draft. Ning Feng: investigation, data curation. Keyang Yin: data curation. Hongguang Li: conceptualization, writing – review & editing. Jingcheng Hao: resources, supervision.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors are grateful for the financial supports from the National Natural Science Foundation of China (No. 21875129, 22032003). The authors acknowledge the assistance of Center for Structural and Property Analysis, Core Facilities Sharing Platform, Shandong University.References
- X. Xu, R. Ray, Y. Gu, H. Ploehn, L. Gearheart, K. Raker and W. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736 CrossRef CAS PubMed.
- N. Feng, H. Li and J. Hao, Acta Phys.-Chim. Sin., 2021, 37, 2005004 Search PubMed.
- A. Doring, E. Ushakova and A. Rogach, Light: Sci. Appl., 2022, 11, 75 CrossRef CAS PubMed.
- B. Wang, Z. Wei, L. Sui, J. Yu, B. Zhang, X. Wang, S. Feng, H. Song, X. Yong, Y. Tian, B. Yang and S. Lu, Light: Sci. Appl., 2022, 11, 172 CrossRef CAS PubMed.
- X. Yang, L. Ai, J. Yu, G. Waterhouse, L. Sui, J. Ding, B. Zhang, X. Yong and S. Lu, Sci. Bull., 2022, 67, 1450 CrossRef CAS.
- A. Pundi and C. Chang, Polymers, 2022, 14, 2153 CrossRef CAS PubMed.
- X. Yang, X. Li, B. Wang, L. Ai, G. Li, B. Yang and S. Lu, Chin. Chem. Lett., 2022, 33, 613 CrossRef CAS.
- K. Yin, D. Lu, W. Tian, R. Zhang, H. Yu, E. Gorecka, D. Pociecha, N. Godbert, J. Hao and H. Li, J. Mater. Chem. C, 2020, 8, 8980 RSC.
- Y. Ru, L. Sui, H. Song, X. Liu, Z. Tang, S. Zang, B. Yang and S. Lu, Angew. Chem., Int. Ed., 2021, 60, 14091 CrossRef CAS PubMed.
- X. Sun, Q. Zhang, K. Yin, S. Zhou and H. Li, Chem. Commun., 2016, 52, 12024 RSC.
- X. Sun, M. Chen, Y. Zhang, Y. Yin, L. Zhang, H. Li and J. Hao, J. Mater. Chem. B, 2018, 6, 7021 RSC.
- X. Sun, X. Dai, S. Zhou, H. Wang, A. Xu, L. Yan, H. Li and J. Hao, J. Phys. Chem. C, 2021, 125, 17291 CrossRef CAS.
- J. Li, M. Chen, S. Zhou, H. Li and J. Hao, Chem. Soc. Rev., 2020, 51, 3226 RSC.
- Y. Park, J. Yoo, B. Lim, W. Kwon and S. Rhee, J. Mater. Chem. A, 2016, 4, 11582 RSC.
- P. Namdari, B. Negahdari and A. Eatemadi, Biomed. Pharmacother., 2017, 87, 209 CrossRef CAS PubMed.
- B. Chen, M. Liu, C. Li and C. Huang, Adv. Colloid Interface Sci., 2019, 270, 165 CrossRef CAS PubMed.
- J. Wang, C. Wang and S. Chen, Angew. Chem., Int. Ed., 2012, 51, 9297 CrossRef CAS PubMed.
- M. Wang, B. Zheng, F. Yang, J. Du, Y. Guo, J. Dai, L. Yan and D. Xiao, Analyst, 2016, 141, 2508 RSC.
- Z. Zhu, P. Yang, M. Chen, T. Zhang, Y. Cao, W. Wang, X. Zhou and W. Chen, J. Lumin., 2019, 213, 474 CrossRef CAS.
- S. Pandit, S. Mondal and M. De, J. Mater. Chem. B, 2021, 9, 1432 RSC.
- O. Kozak, K. Datta, M. Greplova, V. Ranc, J. Kaslik and R. Zboril, J. Phys. Chem. C, 2013, 117, 24991 CrossRef CAS.
- C. Yi, S. Zhang, T. Webb and Z. Nie, Acc. Chem. Res., 2017, 50, 12 CrossRef CAS PubMed.
- J. Jang, S. Jeon, Y. Yoon, J. Bang, Y. Han and T. Kim, Polym. Chem., 2019, 10, 6269 RSC.
- C. Huo, M. Li, X. Huang, H. Yang and S. Mann, Langmuir, 2014, 30, 15047 CrossRef CAS PubMed.
- K. Niikura, N. Lyo, Y. Matsuo, H. Mitomo and K. Ljiro, ACS Appl. Mater. Interfaces, 2013, 5, 3900 CrossRef CAS PubMed.
- K. Niikura, N. Lyo, Y. Matsuo, H. Mitomo and K. Ljiro, J. Am. Chem. Soc., 2012, 134, 7632 CrossRef CAS PubMed.
- J. Song, X. Yang, O. Jacobson, P. Huang, X. Sun, L. Lin, X. Yan, G. Niu, Q. Ma and X. Chen, Adv. Mater., 2015, 27, 4910 CrossRef CAS PubMed.
- J. Hu, T. Wu, G. Zhang and S. Liu, J. Am. Chem. Soc., 2012, 134, 7624 CrossRef CAS PubMed.
- J. Song, B. Wu, Z. Zhou, G. Zhou, Y. Liu, Z. Yang, L. Lin, G. Yu, F. Zhang and G. Zhang, Angew. Chem., Int. Ed., 2017, 56, 8114 Search PubMed.
- M. Pang, A. Cairns, Y. Liu, Y. Belmabkhout, H. Zeng and M. Eddaoudi, J. Am. Chem. Soc., 2013, 135, 10234 CrossRef CAS PubMed.
- P. Cui, Y. Kuai, Q. Wu, Y. Zheng and X. Liu, Mater. Lett., 2019, 235, 161 CrossRef CAS.
- S. Sarkar, S. Dinda, P. Choudhury and P. Das, Soft Matter, 2019, 15, 2863 RSC.
- M. Krysmann, A. Kelarakis, P. Dallas and E. Giannelis, J. Am. Chem. Soc., 2012, 134, 747 CrossRef CAS PubMed.
- B. Wang, A. Song, L. Feng, H. Ruan, H. Li, S. Dong and J. Hao, ACS Appl. Mater. Interfaces, 2015, 7, 6919 CrossRef CAS PubMed.
- L. Wang, T. Lu, F. Ruan, D. Deng and S. Xu, Chin. J. Lumin., 2014, 35, 706 CrossRef CAS.
- B. Zheng, T. Liu, M. Paau, M. Wang, Y. Liu, L. Liu, C. Wu, J. Du, D. Xiao and M. Choi, RSC Adv., 2015, 5, 11667 RSC.
- Y. Zhou, P. Liyanage, D. Devadoss, L. Guevara, L. Cheng, R. Graham, H. Chand, A. Al-Youbi, A. Bashammakh and M. El-Shahawi, Nanoscale, 2019, 11, 22387 RSC.
- M. Langer, M. Paloncýová, M. Medveď, M. Pykal, D. Nachtigallová, B. Shi, A. J. A. Aquino, H. Lischka and M. Otyepka, Appl. Mater. Today, 2021, 100924 CrossRef.
- B. Moon, Y. Oh, D. Shin, S. Kim, S. Lee, T. Kim, M. Park and S. Bae, Chem. Mater., 2016, 28, 1481 CrossRef CAS.
- L. Vallan, E. Urriolabeitia, F. Ruiperez, J. Matxain, R. Canton, N. Tagmatarchis, A. Benito and W. Maser, J. Am. Chem. Soc., 2018, 140, 12862 CrossRef CAS PubMed.
- S. Tang, T. Yang, Z. Zhao, T. Zhu, Q. Zhang, W. Hou and W. Z. Yuan, Chem. Soc. Rev., 2021, 50, 12616 RSC.
- K. Mysels, Langmuir, 1986, 2, 423 CrossRef CAS.
- R. Dong, Z. Zhong and J. Hao, Soft Matter, 2012, 8, 7812 RSC.
- R. Dong, J. Wu, S. Dong, S. Song, F. Tian and J. Hao, Chem. – Asian J., 2013, 8, 863 Search PubMed.
- M. Chen, S. Zhou, L. Guo, L. Wang, F. Yao, Y. Hu, H. Li and J. Hao, Langmuir, 2019, 35, 6939 CrossRef CAS PubMed.
- A. Borode, N. Ahmed and P. Olubambi, Phys. Fluids, 2019, 31, 071301 CrossRef.
- H. Li, C. Wei, D. Zhang and B. Pan, Sci. Total Environ., 2019, 655, 807 CrossRef CAS PubMed.
- A. Hirano, W. Gao, X. He and J. Kono, Nanoscale Res. Lett., 2017, 12, 81 CrossRef PubMed.
- P. Lin, Y. Cong and B. Zhang, Polym. Chem., 2015, 6, 2909 RSC.
- L. Jiang, L. Gao and J. Sun, J. Colloid Interface Sci., 2003, 260, 89 CrossRef CAS PubMed.
- A. Breitwieser, U. Sleytr and D. Pum, Nanomaterials, 2021, 11, 1346 CrossRef CAS PubMed.
- X. Xin, G. Xu, T. Zhao, Y. Zhu, X. Shi, H. Gong and Z. Zhang, J. Phys. Chem. C, 2008, 112, 16377 CrossRef CAS.
- V. Moore, M. Strano, E. Haroz, R. Hauge, R. Smalley, J. Schmidt and Y. Talmon, Nano Lett., 2003, 3, 1379 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr05008k |
| This journal is © The Royal Society of Chemistry 2023 |