
Discriminating active sites for the electrochemical synthesis of H2O2 by molecular functionalisation of carbon nanotubes†
Juyeon
Lee
a,
Yesol
Lee
a,
June Sung
Lim
b,
Sun Woo
Kim
a,
Hongje
Jang
 a,
Bora
Seo
cd,
Sang Hoon
Joo
b and
Young Jin
Sa
*a
a,
Bora
Seo
cd,
Sang Hoon
Joo
b and
Young Jin
Sa
*a
aDepartment of Chemistry, Kwangwoon University, Seoul 01897, Republic of Korea. E-mail: youngjinsa@kw.ac.kr
bDepartment of Chemistry, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
cHydrogen and Fuel Cell Research Center, Korea Institute of Science and Technology (KIST), Seoul 02792, Republic of Korea
dDivision of Energy & Environment Technology, KIST School, University of Science and Technology (UST), Seoul, Republic of Korea
First published on 17th November 2022
Abstract
The electrochemical production of H2O2via the two-electron oxygen reduction reaction (2e− ORR) has recently attracted attention as a promising alternative to the current anthraquinone process. Identification of active sites in O-doped carbon materials, which exhibit high activities and selectivities for the 2e− ORR, is important for understanding the selective electrocatalytic process and achieving the rational design of active electrocatalysts. However, this is impeded by the heterogeneous distribution of various active sites on these catalysts. In this study, we exploited the molecular functionalisation approach to implant anthraquinone, benzoic acid, and phenol groups on carbon nanotubes and systematically compared the electrocatalytic activities and selectivities of these functional groups. Among these oxygen functional groups, the anthraquinone group showed the highest surface-area-normalised and active-site-normalised activities.
 Young Jin Sa | Dr Young Jin Sa received his PhD degree in Chemistry from the Ulsan National Institute of Science and Technology (UNIST) in 2018. He participated in energy-related research about the development of active electrocatalytic nanomaterials for hydrogen fuel cells and electrolyzers. He then joined the clean energy research center at the Korea Institute of Science and Technology (KIST) as a postdoctoral fellow and conducted research on CO2 electroreduction. His current research interest is focused on designing nanomaterials and understanding the nature of active sites for selective electrocatalytic processes such as the electrochemical synthesis of H2O2via O2 reduction. |
Introduction
The electrosynthesis of H2O2 is an emerging technology for the clean and sustainable production and supply of H2O2, which is in high industrial demand.1,2 For the two-electron oxygen reduction reaction (2e− ORR), active and selective electrocatalysts are essential to convert O2 to H2O2 efficiently; otherwise, the competitive 4e− ORR occurs, which leads to the formation of H2O as the main product.3–6 So far, various design strategies have been developed to enhance the 2e− ORR activities of various classes of catalysts. For example, in the cases of noble metal and non-precious transition metal catalysts, it has been reported that alloys with secondary metals,7–10 compounds containing non-metal elements,11–13 and atomically dispersed active sites14–19 exhibit high 2e− ORR activities and selectivities owing to their distinct end-on O2 adsorption mode, while single metal-based surfaces prefer side-on adsorption leading to facile O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O breakage.
O breakage.
Metal-free carbon materials with oxygen functional groups (O-doped carbons) are highly promising 2e− ORR electrocatalysts that are particularly active under alkaline conditions.20–32 O-doped carbon materials are generally synthesized via a top-down approach, wherein oxygen functional groups are implanted in as-prepared carbon materials by oxidative treatments, such as HNO3, plasma, and heat treatments.20–28 In contrast, the bottom-up approach involves the transformation of O- and C-containing precursors via pyrolysis at high temperatures, which results in graphitised carbon materials with oxygen functionalities.29–32 Exploration and optimization of the parameters involved in both synthetic methods result in the discovery of highly active electrocatalysts. However, the fundamental questions regarding the nature of the active functional groups remain unsolved, because such synthetic methods usually lead to the formation of multiple types of oxygen species.
To date, only a few systematic investigations have been performed to identify the actual active functional groups in O-doped carbon materials. For example, McCloskey et al. demonstrated that epoxy and ring ether groups are active species in reduced graphene oxides.22 Baek et al. performed a systematic study of well-controlled edge-functionalised carbon nanoplatelets and found that quinone groups exhibit high selectivity and activity.24 In addition, Joo et al. prepared edge-rich graphitic nanoporous carbons with controlled oxygen functional groups and performed selective poisoning experiments, which revealed carboxyl and aromatic carbonyl groups as active species.31 The poisoning approach was also utilised by Liu et al. to identify CO groups as the active species in O-doped carbon nanosheets.32 However, despite these investigations, some inconsistencies related to active oxygen functional groups still remain unresolved.
Accurate identification of the active functional groups can be achieved by using structurally well-defined organic molecules. A conductive substrate is necessary for electrochemical applications. In this regard, direct loading of molecules on an electrode or physisorption of molecules on graphitic carbon supports via π–π interactions has been the main approach for electrocatalytic applications.24,33 Since the physical interaction between the molecules and the support/electrode is weaker than chemical bonding, the molecules are prone to be detached during electrocatalysis. Covalent molecular functionalisation of carbon materials offers chances to tailor the surface functionality with higher stability; however, this approach has been mostly applied to improve the physical, dispersion, and redox properties of carbon materials.34,35 In this study, we exploited the covalent molecular functionalisation of carbon nanotubes (CNTs) to provide target functionalities for identifying active oxygen functional groups for the 2e− ORR. More specifically, diazotisation and a subsequent radical coupling reaction are employed to fabricate CNT-based electrocatalysts bearing anthraquinone (AQ; CO), benzoic acid (COOH), and phenol (C–OH) groups. Among the prepared electrocatalysts, AQ-functionalised CNTs exhibited the highest intrinsic activity in terms of specific activity per surface area and O-atom-normalised activity. Meanwhile, the 2e− ORR selectivity was similarly high regardless of the functional groups, suggesting that the functional groups influence the rate of the rate-determining step (RDS) rather than the reaction pathway or mechanism.
Results and discussion
For the functionalisation of CNTs, we adopted a diazotisation and subsequent radical coupling approach using sodium nitrite and aromatic amines containing the desired oxygen functional groups (see Fig. 1a and the Experimental section for details).36 In this study, CNTs were functionalised with AQ, benzoic acid (BA), and phenol (Ph) groups to identify the most active group. Initially, we first demonstrated the synthesis of functionalised CNTs with 1- and 2-aminoanthraquinones to investigate the effect of the substituent position. The CNTs functionalised with these molecules were denoted as AQ–CNT-1 and AQ–CNT-2, respectively. For comparison, CNTs containing physisorbed AQ molecules (AQ/CNT) were also prepared. The presence of AQ moieties in the catalysts was verified by an increase in the number of O atoms, evidenced by X-ray photoelectron spectroscopy (XPS) survey spectra and combustion elemental analysis (ESI Fig. S1a and Table S1†). The chemical states of the O and C atoms in the catalysts were investigated using detailed XPS scans. The O 1s XPS spectra were deconvoluted into two main peaks at 531.4–531.6 eV for CO and 533.0–533.2 eV for C–O (Fig. 1b).21,22 Compared with the bare CNT, all AQ–CNTs and AQ/CNT catalysts show higher CO peak intensities in their O 1s XPS spectra, which suggests the successful introduction of the AQ functional groups. The C 1s XPS analysis reveals a relatively higher proportion of CO in the AQ–CNTs and AQ/CNT catalysts than in the bare CNT, which is consistent with the O 1s XPS spectra (ESI Fig. S1b and Table S2†).37 The C K-edge near-edge X-ray absorption fine structure (NEXAFS) analysis of the AQ–CNTs and AQ/CNT catalysts exhibited more intense C–O and CO signals, indicating that the O content in these catalysts is higher than that in the bare CNT (ESI Fig. S2†). Moreover, the O K-edge NEXAFS spectra of the AQ–CNTs and AQ/CNT show sharp π* peaks at 531.5 eV, which correspond to the quinone CO moieties (Fig. 1c).38
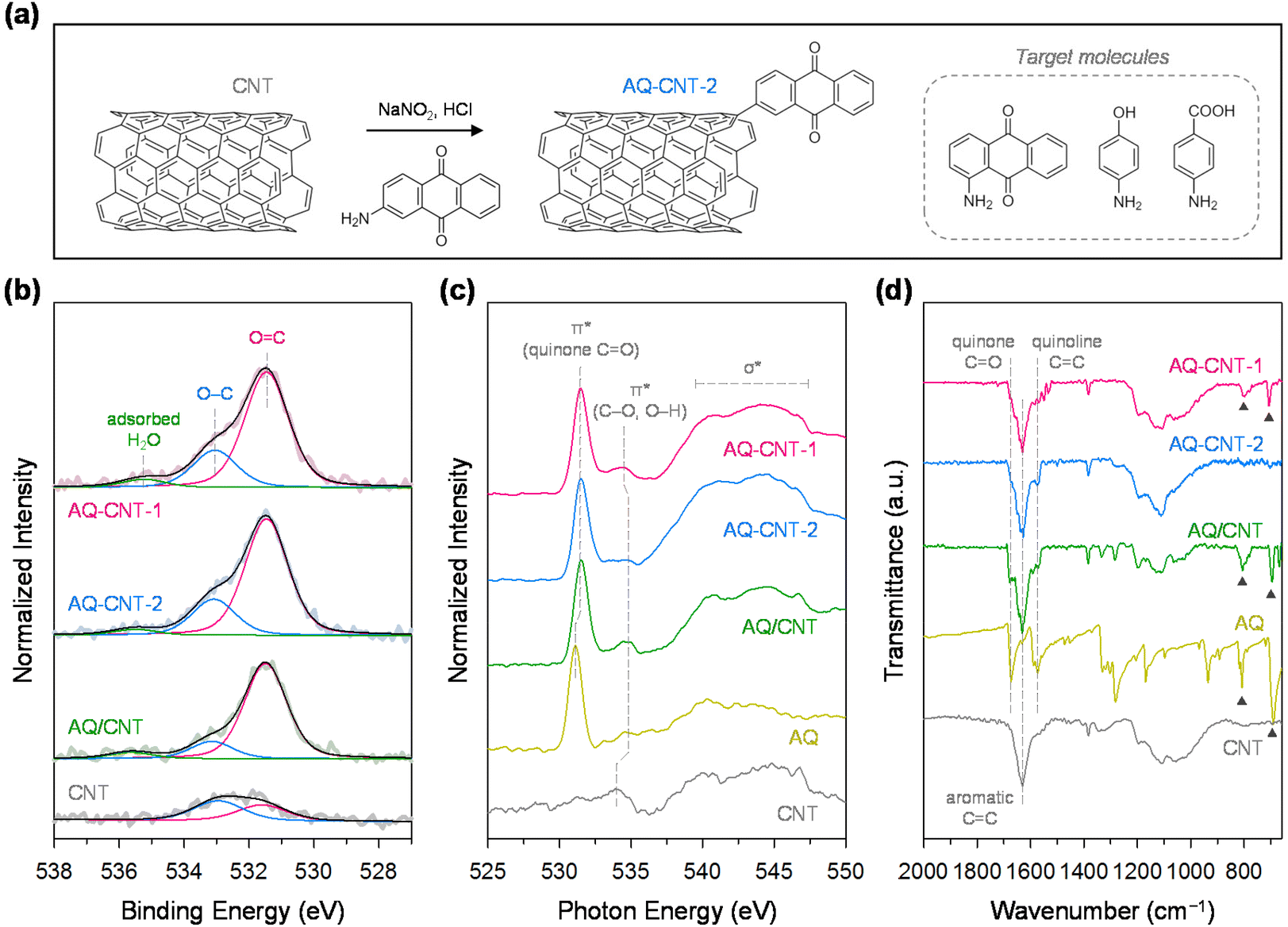 | ||
| Fig. 1 (a) Schematic illustration of the synthesis of functionalised CNTs. (b) Deconvoluted O 1s XPS spectra of AQ–CNT-1, AQ–CNT-2, the AQ/CNT, and the CNT. (c) O K-edge NEXAFS spectra and (d) FT-IR spectra of AQ–CNT-1, AQ–CNT-2, the AQ/CNT, AQ, and the CNT. | ||
The presence of AQ species in the AQ–CNTs and AQ/CNT was further confirmed by Fourier transform infrared (FT-IR) spectroscopy. Compared with the bare CNT, the FT-IR spectra of the AQ–CNTs and AQ/CNT show stronger signals for the quinone CO and quinoid CC moieties.39 In AQ–CNT-1, there are characteristic IR peaks at 700 and 800 cm−1, which are also observed in the FT-IR spectra of the AQ molecules and AQ/CNT (marked by black triangles, Fig. 1d). This result implies that some AQ molecules are present in the physisorbed state in AQ–CNT-1, whereas AQ–CNT-2 is essentially free from adsorbed AQ molecules. The X-ray diffraction (XRD) patterns of the AQ–CNTs reveal the absence of crystalline phases different from that of the CNT; the XRD pattern of the AQ/CNT indicates the presence of crystalline AQ molecules that are not fully dispersed during synthesis (ESI Fig. S3†). The Raman spectra of AQ–CNT-1 and AQ–CNT-2 exhibit G band peaks (1566–1571 cm−1), and the D band (1338–1341 cm−1) (ESI Fig. S4†).40 The relative G/D peak intensity of AQ–CNT-1 and AQ–CNT-2 is higher than those of the AQ/CNT and bare CNT, which may be indicative of the formation of covalent bonds between the CNT and AQ in AQ–CNT-1 and AQ–CNT-2. Thermogravimetric analysis coupled with differential thermal analysis (TGA/DTA) was performed to confirm the thermal resistance of the catalysts (ESI Fig. S5†). The bare CNT exhibits an exothermic peak at ∼700 °C, whereas AQ shows a strong endothermic peak at ∼230 °C along with a drastically decreased mass; these features originate from the thermal decomposition of the CNT and AQ, respectively (ESI Fig. S5d and e†). In the case of the AQ/CNT, the mass decrease starts from ∼200 to 600 °C because of the decomposition of the AQ and CNT, respectively (ESI Fig. S5c†). In contrast, AQ–CNT-1 and AQ–CNT-2 exhibit new exothermic DTA peaks at ∼800 °C, which presumably indicates the breakage of the CNT–AQ covalent bond (ESI Fig. S5a and b†).
Subsequently, we examined the electrochemical redox properties of the AQ–CNTs and AQ/CNT catalysts. Cyclic voltammetry (CV) was carried out (Fig. 2a), wherein the CV curve of AQ–CNT-1 shows main reversible redox peaks (A1) at ∼0.13 V (vs. reversible hydrogen electrode; RHE) and relatively less reversible redox peaks (A2), centred at ∼0.53 V (vs. RHE). In contrast, the CV curve of AQ–CNT-2 exhibits only A1 peaks with small shoulder redox peaks at ∼0.45 V (vs. RHE). Similarly, the AQ/CNT catalyst also displayed only A1 peaks, although the peak current was smaller than that of the AQ–CNTs. The redox peak intensities of the AQ/CNT catalyst drastically decrease during successive potential cycling (ESI Fig. S6†), indicating an irreversible transformation of the AQ molecules into other species. On the other hand, the CV responses of the other covalently functionalized samples were stable (ESI Fig. S6†). Bélanger et al. showed that AQ-physisorbed carbon black exhibited constantly decreasing peak current during CV in 1 M KOH.41 The authors attributed this result to the loss of the AQ molecules from the carbon black because of repulsion between cathodically generated AQ dianions. Thus, the CV instability of the AQ/CNT appears to result from the detachment of the AQ molecules from the CNT. This was verified by UV-vis analysis of the used electrolyte after 100 potential cycles. It was revealed that the used electrolyte contained the AQ molecules, evidenced by the absorption peak at 218 nm (ESI Fig. S7†).41 Therefore, the detachment of the AQ molecules can partly contribute to the decrease in the CV peaks of the AQ/CNT. The remaining adsorbed AQ might participate in electrocatalysis as shown below.
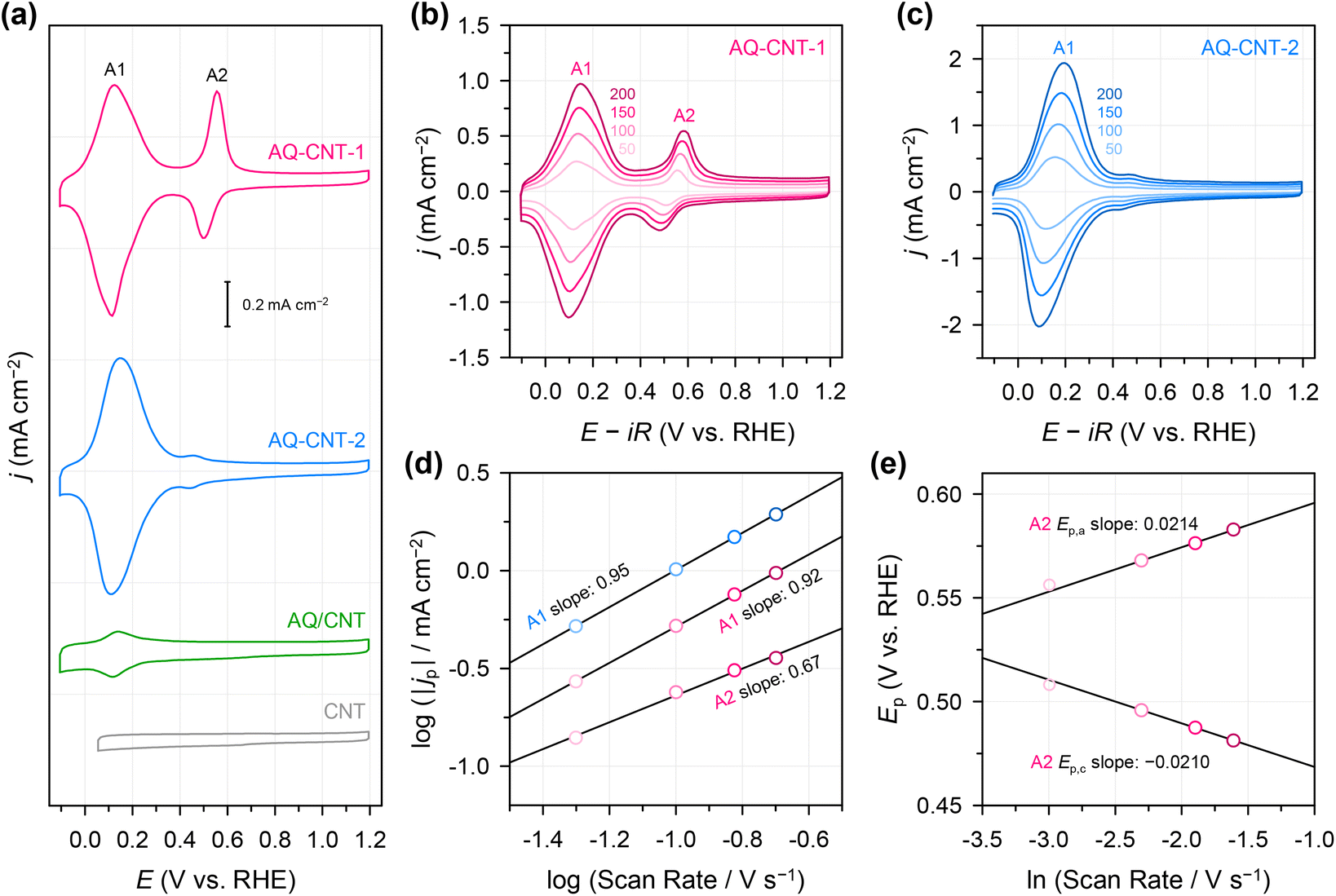 | ||
| Fig. 2 (a) CV curves of AQ–CNTs, the AQ/CNT, and the CNT at a potential scan rate of 50 mV s−1. CV curves of (b) AQ–CNT-1 and (c) AQ–CNT-2 recorded at different scan rates (in mV s−1) as indicated by the numbers in the plot. (d) A1 peak currents of AQ–CNT-1 and AQ–CNT-2 and (e) A2 peak positions of AQ–CNT-1 at different scan rates. The measurements were performed in N2-saturated 0.1 M KOH. | ||
To examine the nature of the A1 and A2 redox processes, scan rate-dependent CV was performed (Fig. 2b and c). The plot of the scan rate versus the peak current on the logarithmic scale shows straight lines for both the A1 and A2 redox peaks (Fig. 2d). For AQ–CNT-1 and AQ–CNT-2, the slopes of the regression lines for the A1 peaks were determined to be 0.92 and 0.95, respectively. These close-to-unity slopes indicate that the redox reaction occurs on surface-adsorbed species, i.e., covalently bonded AQ molecules.42 However, the line for the A2 redox peaks (only shown for AQ–CNT-1) exhibits a slope of 0.62. In addition, A2 peak separation becomes larger at higher scan rates (Fig. 2e). These observations imply the quasi-reversibility of the A2 redox process.42 The plot of the peak potential versus the logarithmic scan rate also shows straight lines, from which the electron transfer number (n) and the transfer coefficient (α) can be estimated using Laviron's equation (see the Experimental section for details).43 In our case, n = 2.4 and α = 0.5 are calculated, which confirm that AQ typically undergoes a redox transformation via a two-electron transfer process.41
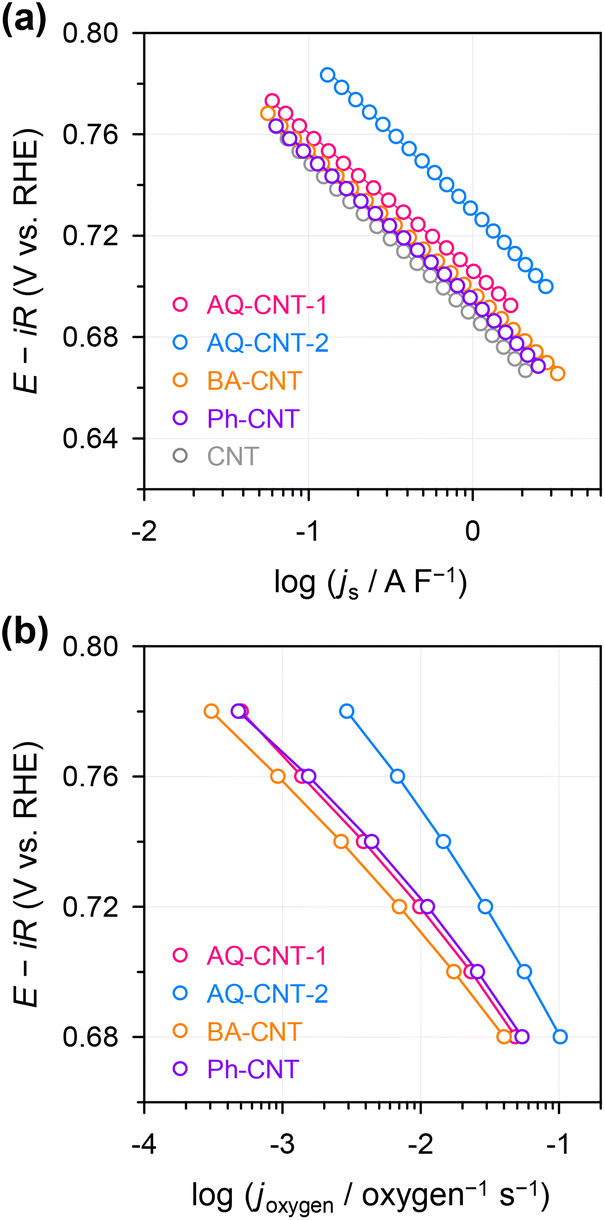
The ORR activities and selectivities of the prepared catalysts were then tested by the rotating ring disk electrode (RRDE) method in 0.1 M KOH (Fig. 3). The ORR activities of all the AQ–CNTs and AQ/CNT catalysts are higher than that of the bare CNT, as evidenced by the more positive onset potentials of AQ-containing catalysts compared to that of the bare CNT. This result demonstrates the catalytic ability of the AQ moieties in the catalysts (Fig. 3a). Among them, AQ–CNT-2 exhibits the highest activity with an onset potential of 0.78 V (vs. RHE). H2O2 selectivity was assessed using the current ratio obtained from the disk (id) and ring (ir) electrodes during linear sweep voltammetry. All the catalysts possess a high H2O2 molar selectivity of ∼80%, indicating that AQ sites have little influence on the 2e−/4e− reaction pathway (Fig. 3b). Instead, AQ-based active sites are considered to facilitate the RDS, and this is confirmed by similar Tafel slopes of the catalysts (55–62 mV dec−1). The Tafel slope close to 59 mV dec−1 suggests that the RDS is the first electron transfer to an O2 molecule.44
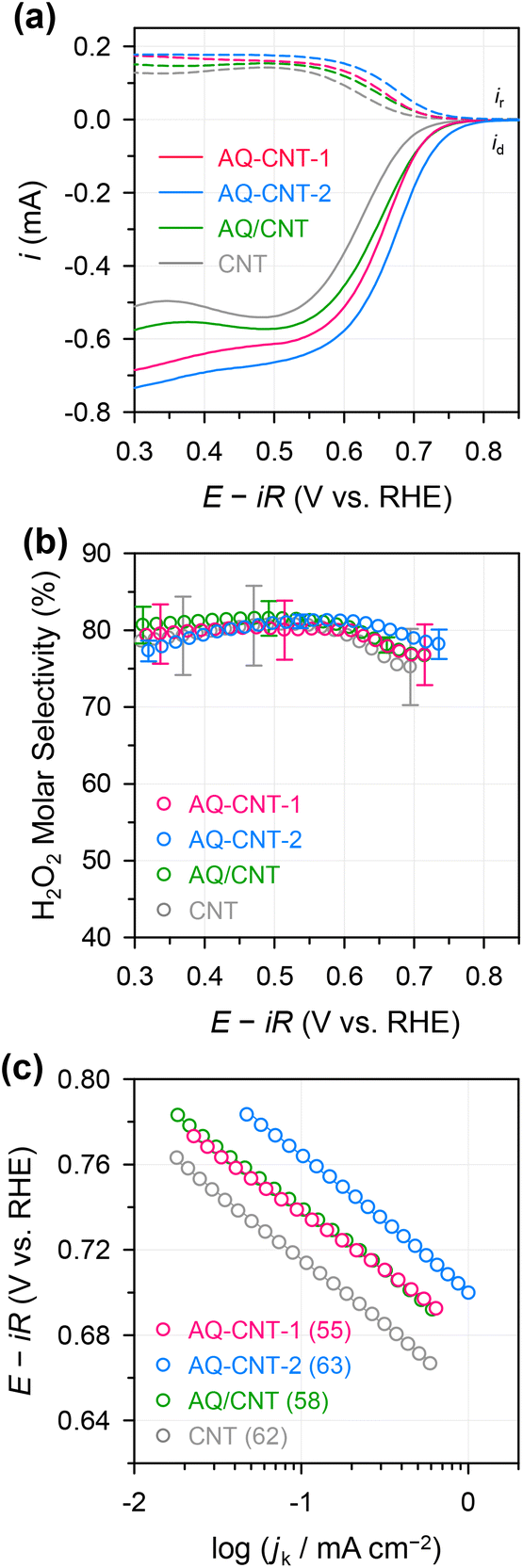 | ||
| Fig. 3 (a) Linear sweep voltammetry (LSV) curves of the AQ–CNTs, AQ/CNT, and CNT measured in N2-saturated 0.1 M KOH using the RRDE method at an electrode rotation speed of 1600 rpm. (b) H2O2 molar selectivity as a function of the applied potential calculated from the RRDE responses. (c) Tafel plot; the numbers in the parenthesis indicate the Tafel slope (in mV dec−1). | ||
We then explored the possibility that changes in the surface charge could be induced by the implanted AQ molecules since this could potentially affect the rate of electron transfer (RDS for this reaction).45 Thus, from differential capacitance measurements, the potential of zero charge (PZC) was evaluated in a 10 mM NaF electrolyte. The AQ–CNTs show slightly higher PZC values than the AQ/CNT and CNT (ESI Fig. S8 and Table S3†). The higher ORR activity of AQ–CNT-2 possibly results from its higher PZC value, because a more negative surface charge is advantageous for a higher cation population at the catalyst interface during the ORR, and this higher cation population results in higher activity.28 Notably, the 2e− ORR is unlikely to be mediated by the redox reaction of the AQ moieties because: (i) there are significant differences between the ORR onset potentials and the redox potentials of the catalysts and (ii) the AQ/CNT showed decreased redox signals, yet exhibited good 2e− ORR activity (ESI Fig. S6† and Fig. 2).
The molecular functionalisation strategy was then extended to BA (carboxyl group) and Ph (hydroxyl group), and this was achieved using 4-aminobenzoic acid and 4-aminophenol precursors to give BA–CNT and Ph–CNT, respectively. Elemental analysis showed that compared with the bare CNT, BA–CNT and Ph–CNT contain a larger amount of O, implying that oxygen functional groups were introduced on these catalysts. The deconvoluted O 1s XPS spectrum of the BA–CNT shows an increased amount of C–O and CO with a relatively higher content of CO. By contrast, the O 1s XPS spectrum of the Ph–CNT shows a higher C–O signal than CO signals, which confirms the introduction of the desired functionalities (Fig. 4a). Furthermore, the O K-edge NEXAFS spectrum of the BA–CNT (Fig. 4b) contains σ* peaks for C–O or O–H at 537.2 eV and for CO at 545.7 eV, whereas that of the Ph–CNT contains a π* peak for C–O or O–H at 534.0 eV, consistent with XPS analysis.46,47 Introduction of oxygen functional groups in the BA–CNT and Ph–CNT were also verified in C 1s XPS spectra (ESI Fig. S9 and Table S4†).37 The FT-IR and Raman spectra of the BA–CNT and Ph–CNT indicate the formation of covalent bonds and the presence of the corresponding oxygen functional groups (ESI Fig. S10 and S11†).48,49 The PZC measurement results reveal that the PZC value of the BA–CNT is higher than that of the bare CNT, and this higher PZC value of the BA–CNT originates from the negatively charged carboxyl functional groups on it (ESI Fig. S12 and Table S5†).
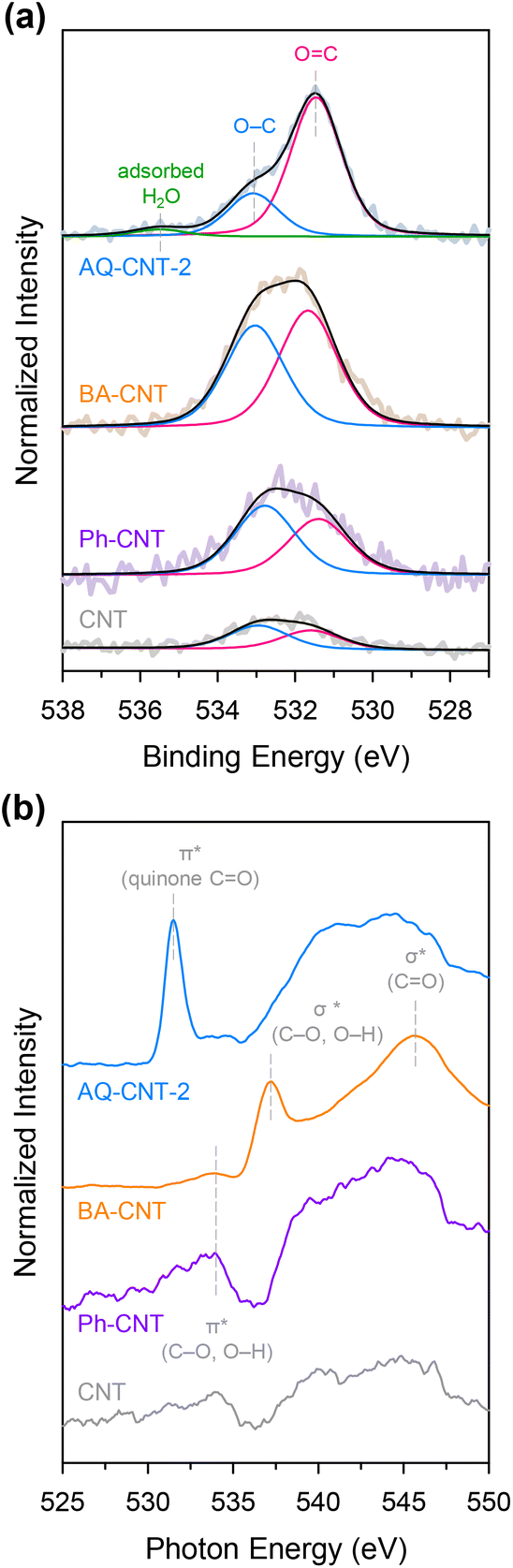 | ||
| Fig. 4 (a) Deconvoluted O 1s XPS spectra and (b) O K-edge NEXAFS spectra of the AQ–CNT-2, BA–CNT, Ph–CNT, and CNT. | ||
Next, we examined the electrochemical and electrocatalytic properties of the BA–CNT and Ph–CNT. The CV curves of the BA–CNT and Ph–CNT do not show any redox peaks. However, a slight increase in the capacitive current is seen, indicating a higher electrochemically active surface area (ECSA), which results from the incorporation of the functional groups (ESI Fig. S13a†). The ORR activities and selectivities of the BA–CNT and Ph–CNT were tested by the RRDE method. Compared to the bare CNT, the BA–CNT and Ph–CNT exhibit higher onset potentials because of their respective functional groups (ESI Fig. S13b†). However, the carboxyl and phenol functional groups seem not to affect the reaction pathway, as revealed by their similar H2O2 selectivities and Tafel slopes (ESI Fig. S13c and d†).
Since the functionalised CNTs possess different ECSAs and different numbers of oxygen functional groups, the apparent activity should be normalised to extract their intrinsic activities. Accordingly, the ECSAs were estimated from double-layer capacitance measurements by potential cycling in non-faradaic potential regions (ESI Fig. S14†). Compared to the bare CNT, all functionalised CNTs have 28–43% higher ECSA values, which result from the additional functional groups. The ECSA-normalised current plot shows that the specific activity increases in the following order: AQ–CNT-2 > AQ–CNT-1 ≥ BA–CNT ≥ Ph–CNT ≥ bare CNT (Fig. 5a). These activity trends are consistent with those observed in previous studies;24,31,32 however, the difference between the species is modest. Thus, the intrinsic activity per oxygen site was evaluated by normalising the activity with the amount of oxygen functional groups. This calculation is based on the assumption that all the oxygen sites are electrochemically accessible and equally active. As a result, the following activity trend was obtained: AQ–CNT-2 > Ph–CNT ≈ AQ–CNT-1 > BA–CNT (Fig. 5b). This result is contradictory to the previous results, which suggests that the hydroxyl groups are the least active. We assume that this discrepancy from the previous works is observed because the oxygen functional groups in typical carbon materials are located at the carbon sites with an extended π-network. In contrast, in the BA–CNT and Ph–CNT, each carboxyl or hydroxyl functional group is attached to a single aromatic ring. It has been suggested that the π-network size is important for O-doped carbon materials in several electrocatalytic reactions.30,50 Such differences in the local carbon structures can have significant influences on the electrical conductivity and electronic structures of the active sites. Systematic investigations to ascertain the importance of the π-network size are ongoing to further investigate this discrepancy.
 | ||
| Fig. 5 (a) ECSA-normalised activity and (b) O-atom-normalised activity of the functionalised CNTs and bare CNT. | ||
Conclusions
In conclusion, covalent molecular functionalisation on CNTs was utilised to identify the nature of active oxygen functional groups in the selective 2e− ORR. It was found that the intrinsic activity of the AQ groups is higher than that of the carboxyl and phenol groups in terms of ECSA-normalised and O-atom-normalised currents, whereas high H2O2 selectivity was obtained for all functional groups. We expect that molecular functionalisation will be an effective strategy for revealing the electrochemical and electrocatalytic properties of various functional groups, not limited to oxygen-containing groups, as well as the effect of the local structures of adjacent carbon atoms.Experimental
Chemicals
KOH (99.99%), NaF (99.99%), Nafion solution (5 wt%), K3[Fe(CN)6] (99%), K4[Fe(CN)6]·3H2O (98.5–102.0%), 1-aminoanthraquinone (97%), 4-aminobenzoic acid (99%), 4-aminophenol (98%), and NaNO2 (99%) were purchased from Sigma Aldrich. 2-Aminoanthraquionone (80%) was purchased from TCI Chemicals. HCl (35–37 wt%), acetonitrile (99.8%), and anhydrous ethanol (EtOH) were purchased from Samchun Chemicals. Acetone (99.8%) was purchased from Daejung Chemicals. The Nafion solution (5 wt%) was diluted with deionised (DI) water to a concentration of 0.5 wt%.Synthesis of functionalised CNTs
To remove any metallic impurities, CNTs were first pre-treated with 6 M HCl, washed with a large amount of DI water, and lyophilised. For functionalisation, the purified CNTs (50 mg) were sonicated in DI water (50 mL). A diazonium ion solution was prepared by mixing NaNO2 (4.8 mmol), the target molecule (AQ, BA, or Ph; 0.48 mmol), and HCl (10 mL) for 30 min in an ice bath with vigorous stirring. The diazonium ion solution was then poured into the CNT dispersion and stirred at room temperature for 1 h to form covalent bonds between the CNTs and the target molecules. For the AQ–CNT, the mixture was vacuum filtered and washed with DI water and acetonitrile prior to freeze-drying. During acetonitrile washing, the unbonded physisorbed molecules were washed off from the product as evidenced by the yellow filtrate. The resulting functionalized CNTs were freeze-dried. In the cases of BA–CNT and Ph–CNT, washing was done with DI water and ethanol. To prepare the AQ/CNT, the purified CNT (90 mg) and acetone (10 mL) were mixed in an agate mortar with a pestle.Characterisation methods
XPS spectra were recorded using a K-alpha X-ray photoelectron spectrometer (Thermo Fisher Scientific) with monochromated Al Kα radiation (1486.6 eV) and deconvoluted using XPSPeak41 software. Shirley-type background removal and mixed Gaussian–Lorentzian functions were used for deconvolution. Raman spectra were recorded using an XperRam200 Raman spectrometer (Nanobase, Republic of Korea). An Olympus microscope equipped with a 40× objective lens was used for focused measurements. A 532 nm laser with a spot size of ∼1 μm was irradiated on the samples with an integration time of 5 s (600 lines per mm grating). At least 10 Raman spectra were recorded between 0 and 3000 cm−1 using a charge-coupled device detector and averaged for display. FT-IR spectra were acquired in transmittance mode using an FT-IR spectrometer (Thermo Fisher Scientific). For FT-IR measurements, a catalyst powder was mixed with KBr to form a pellet using a force of 5 tons for 30 s (PIKE Technologies). TGA/DTA analysis was performed using a thermal analysis system (DTG-60H, Shimadzu) at a temperature ramping rate of 10 °C min−1 under Ar flow. NEXAFS spectra were recorded at the 10D beamline of the Pohang accelerator laboratory at a current of 300 mA and an acceleration voltage of 3 GeV. UV-vis spectra were recorded using a UV-vis spectrophotometer (Lambda 465, PerkinElmer).Electrochemical characterisation
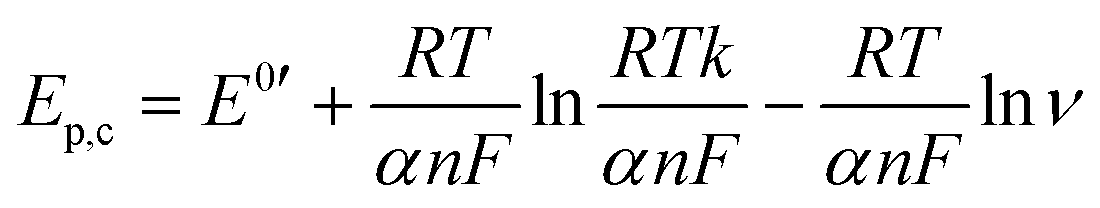
where Ep,c, Ep,a, E0′, k, and ν represent the cathodic peak current, anodic peak current, formal potential, heterogeneous rate constant for the redox process, and scan rate, respectively. The n and α values could be calculated using the slopes of ln
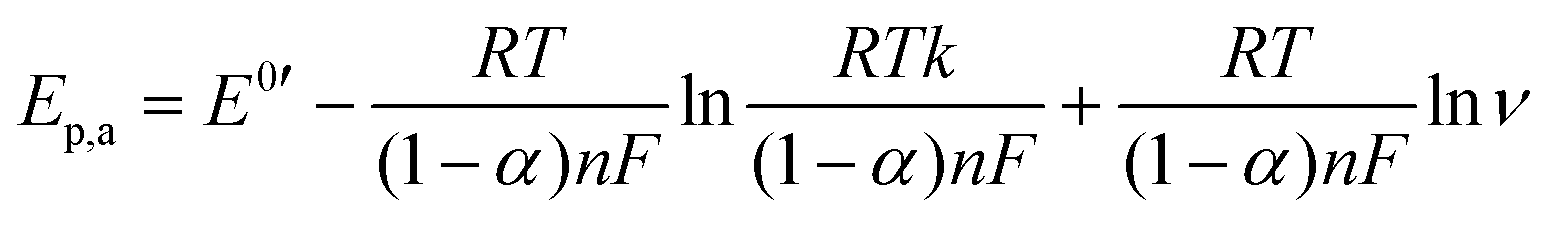
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ν versus Ep curves (Fig. 2e).
ν versus Ep curves (Fig. 2e).
The Pt ring was then cleaned electrochemically five times by CV in the range of 0.05–1.20 V (vs. RHE) at a scan rate of 500 mV s−1. In RRDE measurements, Pt ring cleaning is important for accurately estimating the H2O2 selectivity.6 After O2 bubbling for 10 min, electrochemical impedance spectroscopy (EIS) was performed from 100000 to 1 Hz at 0.68 V (vs. RHE) with a potential amplitude of 10 mV. The Nyquist plot of the EIS spectra was analysed, and series resistance (Rs) was obtained from the real part of the impedance in the high-frequency region. The Rs value was used to correct the ohmic potential drop; 90% of the value was applied according to the following equation:
| EiR-corrected = EHg/HgO + 0.89 − 0.9iRs |
where N, id, and ir denote the collection efficiency of the RRDE, disk current, and ring current, respectively. The collection efficiency was experimentally determined using a three-electrode setup, composed of a polished RRDE, Pt coil, and Ag/AgCl (RE-5B, BASi, filled with 3 M KCl) as the working, counter, and reference electrodes, respectively, in a 2 mM K3[Fe(CN)6] + 0.1 M KOH electrolyte. After purging the electrolyte with N2 gas, chronoamperometry was performed at −0.3 V (vs. Ag/AgCl) with a constant ring potential of 0.5 V (vs. Ag/AgCl) and an electrode rotation speed of 1600 rpm. To exclude the background ring current, another chronoamperometry measurement was carried out at 0.5 V (vs. Ag/AgCl) with a constant ring potential of 0.5 V (vs. Ag/AgCl). The collection efficiency was calculated by averaging the current ratio collected at the ring and disk in the final 5 s, according to the following equation:
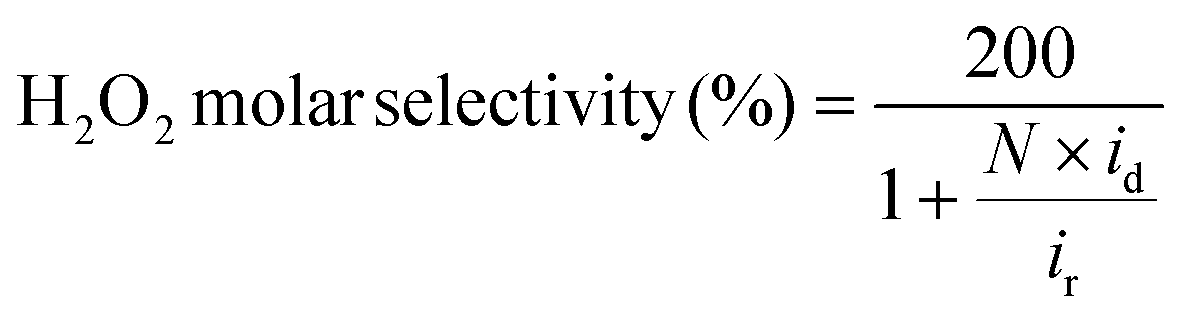
where ir,bg indicates the background ring current. The resulting value (38.9%) was similar to that provided by the manufacturer (ESI Fig. S15†).
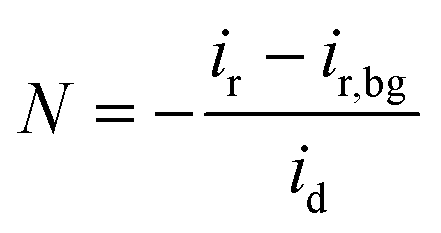
For the Tafel plot, the kinetic current density (jk) was extracted from the measured LSV curve by correcting the diffusion-limited current density (jl) according to the Koutecky–Levich equation:
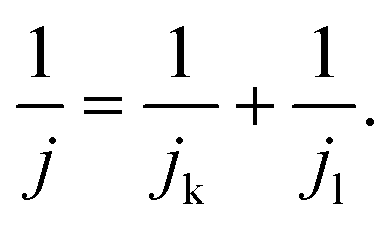
The plot of logarithmic jkversus the applied potential generates a Tafel plot, which can be described by the following Tafel equation:
| E = blogjk + C |
To assess ECSA-normalised specific activity, the ECSA was estimated by scan-rate-dependent CV measurements in capacitive potential regions in N2-saturated 0.1 M KOH (ESI Fig. S14†). The plot of scan rate versus half of the CV height (this is capacitive current, ic) gave a straight line whose slope is double layer capacitance (Cdl).51
| ic = Cdl × ν |
| ik = ik,CNT + ik,funct |
where nO,catal and nO,CNT indicate the number of O atoms in the functionalised catalyst and bare CNT, respectively, which were determined by combustion elemental analysis (ESI Table S1†).
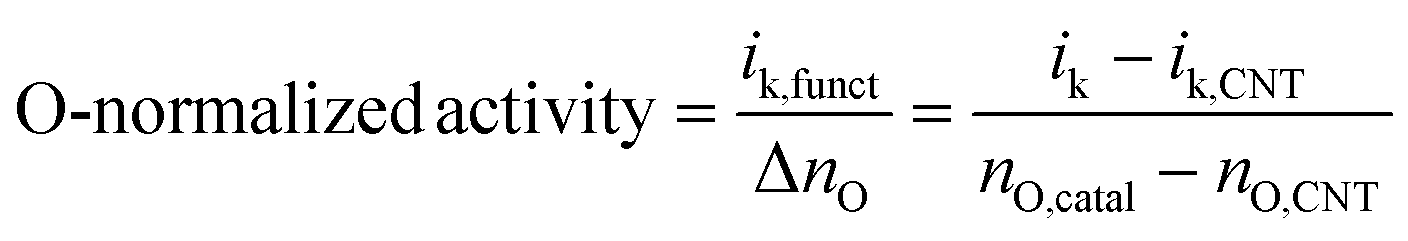
where C, ω, and Zim represent the differential capacitance, frequency, and imaginary part of the impedance, respectively. Finally, the potential was converted into the normal hydrogen electrode potential by adding 0.21 V to the Ag/AgCl potential scale.
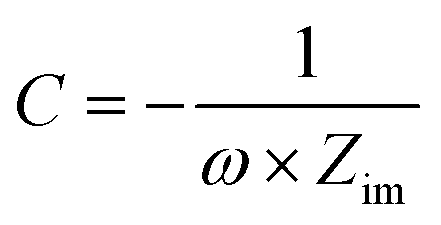
Author contributions
J. Lee: data curation, formal analysis, investigation, and writing – original draft. Y. Lee: data curation, formal analysis, and writing – original draft. J. S. Lim: data curation and formal analysis. S. W. Kim: data curation and formal analysis. H. Jang: writing – review & editing. B. Seo: data curation and formal analysis. S. H. Joo: writing – review & editing. Y. J. Sa: conceptualization, funding acquisition, investigation, project administration, supervision, writing – original draft, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. J. S. was supported by the National Research Foundation (NRF) of Korea funded by the Ministry of Science and ICT (NRF-2020R1C1C1006766). The present research has been conducted by the Research Grant of Kwangwoon University in 2019.References
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS
.
- S. Fukuzumi, Y. Yamada and K. D. Karlin, Electrochim. Acta, 2012, 82, 493–511 CrossRef CAS PubMed
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Čolić, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, ACS Catal., 2018, 8, 4064–4081 CrossRef CAS
- X. Hu, X. Zeng, Y. Liu, J. Lu and X. Zhang, Nanoscale, 2020, 12, 16008–16027 RSC
- Y. Zhou, G. Chen and J. Zhang, J. Mater. Chem. A, 2020, 8, 20849–20869 RSC
- J. S. Lim, Y. J. Sa and S. H. Joo, Cell Rep. Phys. Sci., 2022, 3, 100987 CrossRef CAS
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. L. Stephens and J. Rossmeisl, Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS PubMed
- A. Verdaguer-Casadevall, D. Deiana, M. Karamad, S. Siahrostami, P. Malacrida, T. W. Hansen, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, Nano Lett., 2014, 14, 1603–1608 CrossRef CAS
- Z. Zheng, Y. H. Ng, D. W. Wang and R. Amal, Adv. Mater., 2016, 28, 9949–9955 CrossRef CAS PubMed
- E. Pizzutilo, S. J. Freakley, S. Cherevko, S. Venkatesan, G. J. Hutchings, C. H. Liebscher, G. Dehm and K. J. J. Mayrhofer, ACS Catal., 2017, 7, 5699–5705 CrossRef CAS
- H. Li, P. Wen, D. S. Itanze, Z. D. Hood, S. Adhikari, C. Lu, X. Ma, C. Dun, L. Jiang, D. L. Carroll, Y. Qiu and S. M. Geyer, Nat. Commun., 2020, 11, 3928 CrossRef CAS PubMed
- J. Lee, S. W. Choi, S. Back, H. Jang and Y. J. Sa, Appl. Catal., B, 2022, 309, 121265 CrossRef CAS
- H. Sheng, A. N. Janes, R. D. Ross, D. Kaiman, J. Huang, B. Song, J. R. Schmidt and S. Jin, Energy Environ. Sci., 2020, 13, 4189–4203 RSC
- S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Angew. Chem., Int. Ed., 2016, 55, 2058–2062 CrossRef CAS
- C. H. Choi, M. Kim, H. C. Kwon, S. J. Cho, S. Yun, H.-T. Kim, K. J. J. Mayrhofer, H. Kim and M. Choi, Nat. Commun., 2016, 7, 10922 CrossRef CAS
- J. H. Kim, D. Shin, J. Lee, D. S. Baek, T. J. Shin, Y.-T. Kim, H. Y. Jeong, J. H. Kwak, H. Kim and S. H. Joo, ACS Nano, 2020, 14, 1990–2001 CrossRef CAS PubMed
- K. Jiang, S. Back, A. J. Akey, C. Xia, Y. Hu, W. Liang, D. Schaak, E. Stavitski, J. K. Nørskov, S. Siahrostami and H. Wang, Nat. Commun., 2019, 10, 3997 CrossRef
- C. Tang, L. Chen, H. Li, L. Li, Y. Jiao, Y. Zheng, H. Xu, K. Davey and S.-Z. Qiao, J. Am. Chem. Soc., 2021, 143, 7819–7827 CrossRef CAS
- C. Liu, H. Li, F. Liu, J. Chen, Z. Yu, Z. Yuan, C. Wang, H. Zheng, G. Henkelman, L. Wei and Y. Chen, J. Am. Chem. Soc., 2020, 142, 21861–21871 CrossRef CAS PubMed
- M. Melchionna, P. Fornasiero and M. Prato, Adv. Mater., 2019, 31, 1802920 CrossRef PubMed
- Z. Lu, G. Chen, S. Siahrostami, Z. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. Lin, Y. Liu, T. F. Jaramillo, J. K. Nørskov and Y. Cui, Nat. Catal., 2018, 1, 156–162 CrossRef CAS
- H. W. Kim, M. B. Ross, N. Kornienko, L. Zhang, J. Guo, P. Yang and B. D. McCloskey, Nat. Catal., 2018, 1, 282–290 CrossRef
- Y.-L. Wang, S.-S. Li, X.-H. Yang, G.-Y. Xu, Z.-C. Zhu, P. Chen and S.-Q. Li, J. Mater. Chem. A, 2019, 7, 21329–21337 RSC
- G.-F. Han, F. Li, W. Zou, M. Karamad, J.-P. Jeon, S.-W. Kim, S.-J. Kim, Y. Bu, Z. Fu, Y. Lu, S. Siahrostami and J.-B. Baek, Nat. Commun., 2020, 11, 2209 CrossRef CAS
- Y. Pang, K. Wang, H. Xie, Y. Sun, M.-M. Titirici and G.-L. Chai, ACS Catal., 2020, 10, 7434–7442 CrossRef CAS
- Z. Wang, Q.-K. Li, C. Zhang, Z. Cheng, W. Chen, E. A. McHugh, R. A. Carter, B. I. Yakobson and J. M. Tour, ACS Catal., 2021, 11, 2454–2459 CrossRef CAS
- A. Yu, G. Ma, L. Zhu, Y. Hu, R. Zhang, H.-Y. Hsu, P. Peng and F.-F. Li, Nanoscale, 2021, 13, 15973–15980 RSC
- J. Lee, J. S. Lim, G. Yim, H. Jang, S. H. Joo and Y. J. Sa, ACS Appl. Mater. Interfaces, 2021, 13, 59904–59914 CrossRef CAS PubMed
- Y. J. Sa, J. H. Kim and S. H. Joo, Angew. Chem., Int. Ed., 2019, 58, 1100–1105 CrossRef CAS PubMed
- H. Liu, S. Zhu, Z. Cui, Z. Li, S. Wu and Y. Liang, J. Catal., 2020, 392, 1–7 CrossRef CAS
- J. S. Lim, J. H. Kim, J. Woo, D. S. Baek, K. Ihm, T. J. Shin, Y. J. Sa and S. H. Joo, Chem, 2021, 7, 3114–3130 CAS
- S. Chen, T. Luo, K. Chen, Y. Lin, J. Fu, K. Liu, C. Cai, Q. Wang, H. Li, X. Li, J. Hu, H. Li, M. Zhu and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 16607–16614 CrossRef CAS
- B. Reuillard, S. Gentil, M. Carrière, A. L. Goff and S. Cosnier, Chem. Sci., 2015, 6, 5139–5143 RSC
- S. T. R. Naqvi, T. Rasheed, D. Hussain, M. N. ul Haq, S. Majeed, S. Shafi, N. Shmed and R. Nawaz, J. Mol. Liq., 2020, 297, 111919 CrossRef CAS
- N. M. Nurazzi, M. R. M. Asyraf, A. Khalina, N. Abdullah, F. A. Sabaruddin, S. H. Kamarudin, S. Ahmad, A. M. Mahat, C. L. Lee, H. A. Aisyah, M. N. F. Norrrahim, R. A. Ilyas, M. M. Harussani, M. R. Ishak and S. M. Sapuan, Polymers, 2021, 13, 1047 CrossRef CAS
- T. Jurzinsky, E. D. Gomez-Villa, M. Kübler, M. Bruns, P. Elsässer, J. Melke, F. Scheiba and C. Cremers, Electrochim. Acta, 2019, 298, 884–892 CrossRef CAS
- D. J. Morgan, C, 2021, 7, 51 CAS
- B. J. Schultz, R. V. Dennis, J. P. Aldinger, C. Jaye, X. Wang, D. A. Fischer, A. N. Cartwright and S. Banerjee, RSC Adv., 2014, 4, 634–644 RSC
- L. Sieuw, A. Jouhara, É. Quarez, C. Auger, J.-F. Gohy, P. Poizot and A. Vlad, Chem. Sci., 2019, 10, 418–426 RSC
- M. S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus and R. Saito, Nano Lett., 2010, 10, 751–758 CrossRef CAS PubMed
- A. L. Comte, T. Brousse and D. Bélanger, Electrochim. Acta, 2014, 137, 447–453 CrossRef CAS
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS
- E. Laviron, J. Electroanal. Chem., 1979, 101, 19–28 CrossRef CAS
- N. Ramaswamy and S. Mukerjee, J. Phys. Chem. C, 2011, 115, 18015–18026 CrossRef CAS
- T. Inagaki, N. Takendaka and M. Nagaoka, Phys. Chem. Chem. Phys., 2018, 20, 29362–29373 RSC
- H.-C. Youn, S.-M. Bak, M.-S. Kim, C. Jaye, D. A. Fischer, C.-W. Lee, X.-Q. Yang, K. C. Roh and K.-B. Kim, ChemSusChem, 2015, 8, 1875–1884 CrossRef CAS PubMed
- H. Ju, X. Feng, Y. Ye, L. Zhang, H. Pan, C. T. Campbell and J. Zhu, J. Phys. Chem. C, 2012, 116, 20465–20471 CrossRef CAS
- K. Bustos-Ramírez, A. L. Martínez-Hernández, G. Martínez-Barrera, M. de Icaza, V. M. Castaño and C. Velasco-Santos, Materials, 2013, 6, 911–926 CrossRef
- C. Galande, A. D. Mohite, A. V. Naumov, W. Gao, L. Ci, A. Ajayan, H. Gao, A. Srivastava, R. B. Weisman and P. M. Ajayan, Sci. Rep., 2011, 1, 85 CrossRef PubMed
- Y. Lin, K.-H. Wu, Q. Lu, Q. Gu, L. Zhang, B. Zhang, D. Su, M. Plodinec, R. Schlögl and S. Heumann, J. Am. Chem. Soc., 2018, 140, 14717–14724 CrossRef CAS PubMed
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed
- L.-H. Shao, J. Biener, D. Kramer, R. N. Viswanath, T. F. Baumann, A. V. Hamza and J. Weissmüller, Phys. Chem. Chem. Phys., 2010, 12, 7580–7587 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr04652k |
| This journal is © The Royal Society of Chemistry 2023 |