
Efficient interlayer confined nitrate reduction reaction and oxygen generation enabled by interlayer expansion†
Ye
Zhang‡
a,
Mengqiu
Xu‡
a,
Xudong
Xu
a,
Xiaoyu
Li
a,
Genping
Zhu
*a,
Gan
Jia
 a,
Bingchuan
Yang
*bc,
Ruilian
Yin
d,
Peng
Gao
a and
Wei
Ye
*a
a,
Bingchuan
Yang
*bc,
Ruilian
Yin
d,
Peng
Gao
a and
Wei
Ye
*a
aCollege of Material, Chemistry and Chemical Engineering, Key Laboratory of Organosilicon Chemistry and Material Technology, Ministry of Education, Hangzhou Normal University, Hangzhou, Zhejiang 311121, China. E-mail: yewei@hznu.edu.cn; zhugenping@hznu.edu.cn
bSchool of Chemistry and Chemical Engineering, Liaocheng University, Liaocheng 252000, Shandong, China. E-mail: yangbingchuan@lcu.edu.cn
cSchool of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, China
dCollege of Chemical Engineering, Zhejiang University of Technology, Hangzhou, Zhejiang 310014, China
First published on 25th November 2022
Abstract
Electrochemically converting nitrate ions back to ammonia can not only eliminate water pollution but also obtain valuable ammonia without a serious carbon footprint, and is thus deemed as an efficient supplement to the traditional Haber–Bosch process. Currently reported catalysts can achieve a single electrode reaction in the electrochemical nitrate reduction reaction. However, the bifunctionality of a single catalyst for both cathodic and anodic reactions has not yet been reported. Herein, we report Fe-doped layered α-Ni(OH)2 with expanded interlayer spacing as an efficient bifunctional catalyst for the nitrate reduction reaction and oxygen evolution reaction. The expanded interlayer spacing facilitates in situ electrochemical potassium ion intercalation between layers. In situ Raman spectroscopy characterization confirms that both the nitrate reduction reaction and oxygen evolution reaction are confined between layers and are triggered by the accumulation of potassium ions. The obtained α-Ni0.881Fe0.119(OH)2 nanosheets deliver an ammonia yield rate of 8.1 mol gcat.−1 h−1 with a NO3−-to-NH3 faradaic efficiency of 97.5% at the cathode. The overpotential of oxygen generation at 10 mA cm−2 is reduced to 254 mV at the anode. As a bifunctional catalyst in overall electrolysis, the current density of α-Ni0.881Fe0.119(OH)2 reaches 24.8 mA cm−2 at a voltage of 2.0 V and performs continuously for 50 h with a current retention of 80.2%.
 Wei Ye | Ye Wei is an associate professor at the College of Material, Chemistry and Chemical Engineering, Hangzhou Normal University. He received his PhD from Shandong University in 2015. From 2015 to 2018, he worked as a Postdoctoral fellow at the University of Science and Technology of China. His research interest focuses on the synthesis of novel electrocatalysts for electrochemical energy conversion. |
Introduction
Industrial ammonia (NH3) synthesis plays a vital role in modern society as ammonia is a versatile and fundamental feedstock for the manufacture of chemical fertilizers, polymer materials, insecticides, medicine and so on.1–3 Ammonia is also deemed as a promising fuel and clean energy carrier, whose hydrogen content reaches 17.6 wt%.4 Currently, industrial ammonia synthesis mainly relies on the century-old Haber–Bosch process, in which N2 and steam-reformed H2 are converted to NH3 at elevated temperature (400–500 °C) and high pressure (20–30 MPa).5 At present, ammonia production reaches 170 million tons per year, consuming about 1%–2% of the global annual energy supply and releasing 1.44% of global CO2 emissions.6–8 Energy intensive ammonia synthesis aggravates the energy crisis and environmental problems. To reduce the carbon footprint, it is essential to seek energy efficient, eco-friendly ammonia synthesis methods to achieve carbon neutrality in terms of emissions.As a prevalent water pollutant, nitrate ions (NO3−) mainly originate from biological nitrogen fixation, agricultural fertilizers, industrial wastewater, and domestic sewage.9,10 The electrochemical nitrate reduction reaction (NTRR) back to valuable ammonia is a powerful supplement to the century-old Haber–Bosch process, as the energy intensive harsh conditions are eliminated in electrolysis. Moreover, the NTRR can be triggered with relatively low energy input because the bond energy of C–O in nitrate is only 204 kJ mol−1.11 In contrast to chemically inert N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N (bond energy: 941 kJ mol−1), the NTRR ammonia production rate exceeds 10 molNH3 gcat.−1 h−1 with ∼100% NO3−-to-NH3 faradaic efficiency (FE).12 It is about three orders of magnitude higher than that of the electrochemical nitrogen reduction reaction (NRR, NH3 production rate < 50 mgNH3 gcat.−1 h−1, FE < 20%).13–15 The electrochemical NTRR can not only eliminate the water pollutant NO3− but also obtain valuable ammonia with large production rate and negligible carbon footprint.
N (bond energy: 941 kJ mol−1), the NTRR ammonia production rate exceeds 10 molNH3 gcat.−1 h−1 with ∼100% NO3−-to-NH3 faradaic efficiency (FE).12 It is about three orders of magnitude higher than that of the electrochemical nitrogen reduction reaction (NRR, NH3 production rate < 50 mgNH3 gcat.−1 h−1, FE < 20%).13–15 The electrochemical NTRR can not only eliminate the water pollutant NO3− but also obtain valuable ammonia with large production rate and negligible carbon footprint.
To achieve an efficient electrochemical NTRR, catalysts play a central role, which determine both ammonia production rate and NO3−-to-NH3 FE at the cathode. However, the performance in real NTRR electrolysis is not only determined by NTRR activity, but also restricted by the oxygen evolution reaction (OER) at the anode. That is because the OER at the anode involves four electrons and multistep deprotonation coupled process, which suffers from sluggish kinetics and large overpotential.16 To improve energy utilization efficiency in overall electrolysis, a bifunctional catalyst is preferred, which should efficiently catalyze the cathodic NTRR and anodic OER, synchronously. Layered α-Ni(OH)2 is one kind of efficient OER catalyst, which can efficiently lower the overpotential of the OER.17 Fe doping in α-Ni(OH)2 is an ideal choice to boost the NTRR and OER performance, synchronously. That is because Fe is an efficient active element for the NTRR, especially the atomically dispersed Fe active centre. Furthermore, FeNi pairs show excellent ability to catalyze the OER.18–20
Inspired by the promoting role of Fe doping in the NTRR and OER, here we report Fe-doped α-Ni(OH)2 as a bifunctional catalyst to achieve an efficient NTRR and OER, synchronously. The results indicate that Fe doping expands the interlayer spacing of the layered structure, which facilitates in situ electrochemical potassium ion intercalation between layers both at the cathode and anode. The accumulation of potassium ions between layers compels the insertion of nitrate ions and hydroxyl ions, and the corresponding NTRR and OER are confined between layers. The interlayer confined NTRR and OER assisted by K+ greatly enhance ammonia production rate and lower the overpotential of the OER, concurrently. As a bifunctional catalyst, α-Ni0.881Fe0.119(OH)2 nanosheets stably operate for 50 h with a current density of about 25 mA cm−2 and 20.2% energy utilization efficiency in overall electrolysis.
Results and discussion
Catalyst synthesis and characterization
Fe-doped layered α-Ni(OH)2 nanosheets are prepared by in situ Fe doping in the formation process of α-Ni(OH)2. The desired Fe doping level is controlled by regulating the ratio of Ni/Fe in the precursors. The Fe doping level in the final samples is measured by inductively coupled plasma atomic emission spectroscopy (ICP-AES), and the final Fe-doped α-Ni(OH)2 with different Fe doping amounts are denoted as α-Ni1−xFex(OH)2. As summarized in Table S1,† the ratio of Ni/Fe changes from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.1 to 1:0.5, and the corresponding x is 0.053, 0.098, 0.119, 0.150 and 0.171, respectively, suggesting the successful Fe doping in α-Ni(OH)2. Fig. 1a shows the powder X-ray diffraction (XRD) patterns of α-Ni1−xFex(OH)2 samples. The diffraction peaks of the sample without Fe doping coincide with that of layered α-Ni(OH)2 (JCPDS No. 38-0715). For α-Ni1−xFex(OH)2 (x = 0.053, 0.098, 0.119, 0.150, and 0.171) samples, the diffraction peaks of the (003) plane are all shifted from 11.8° towards a smaller angle 10.5°, indicating the expanded interlayer spacing after Fe doping. That is because Fe3+ doping (see the XPS results below) destroys the charge balance of Ni(OH)2, and electrostatic repulsion between layers expands the interlayer spacing. Interlayer expansion for layered α-Ni(OH)2 facilitates the insertion of reaction species, which may boost electrochemical performance.21 The thermogravimetric (TG) curve of α-Ni0.881Fe0.119(OH)2 in Fig. 1b exhibits three weight loss regions. Weight loss (8.8%) below 150 °C can be attributed to the loss of absorbed water molecules and/or triethylene glycol molecules. The following region (17.0%) between 150 and 300 °C represents weight loss of endothermic removal of interlayer crystallization water. The last region (10.7%) between 300 and 600 °C is assigned to the conversion of Ni(OH)2 to NiO.22
:0.1 to 1:0.5, and the corresponding x is 0.053, 0.098, 0.119, 0.150 and 0.171, respectively, suggesting the successful Fe doping in α-Ni(OH)2. Fig. 1a shows the powder X-ray diffraction (XRD) patterns of α-Ni1−xFex(OH)2 samples. The diffraction peaks of the sample without Fe doping coincide with that of layered α-Ni(OH)2 (JCPDS No. 38-0715). For α-Ni1−xFex(OH)2 (x = 0.053, 0.098, 0.119, 0.150, and 0.171) samples, the diffraction peaks of the (003) plane are all shifted from 11.8° towards a smaller angle 10.5°, indicating the expanded interlayer spacing after Fe doping. That is because Fe3+ doping (see the XPS results below) destroys the charge balance of Ni(OH)2, and electrostatic repulsion between layers expands the interlayer spacing. Interlayer expansion for layered α-Ni(OH)2 facilitates the insertion of reaction species, which may boost electrochemical performance.21 The thermogravimetric (TG) curve of α-Ni0.881Fe0.119(OH)2 in Fig. 1b exhibits three weight loss regions. Weight loss (8.8%) below 150 °C can be attributed to the loss of absorbed water molecules and/or triethylene glycol molecules. The following region (17.0%) between 150 and 300 °C represents weight loss of endothermic removal of interlayer crystallization water. The last region (10.7%) between 300 and 600 °C is assigned to the conversion of Ni(OH)2 to NiO.22
 | ||
| Fig. 1 (a) XRD patterns of layered α-Ni1−xFex(OH)2 samples with different Fe doping levels. (b) TG curve, (c and d) TEM images, (e) HAADF-STEM image, (f) HRTEM image and (g) EDS elemental mapping profiles of α-Ni0.881Fe0.119(OH)2 nanosheets. (h) Fe 2p and (i) Ni 2p spectra of α-Ni0.881Fe0.119(OH)2 nanosheets in contrast with Fe(OH)x and Ni(OH)2. | ||
Then, transmission electron microscopy (TEM) characterization is carried out to reveal the morphology of the sample. As an example, TEM images in Fig. 1c and d reveal the ultrathin nanosheet structure for α-Ni0.881Fe0.119(OH)2 with a thickness of about 3.0 nm, consistent with the atomic force microscopy result (2.2 nm, Fig. S1†). The scanning electron microscopy (SEM, Fig. S2†) image shows that α-Ni0.881Fe0.119(OH)2 is a nanoflower structure composed of ultrathin nanosheets. The high-angle annular dark-field scanning TEM (HAADF-STEM, Fig. 1e) image further confirms the conclusion. Notably, the size of the nanosheet for α-Ni1−xFex(OH)2 (x = 0.053, 0.098, 0.119, 0.150, and 0.171) decreases from about 3 μm to 200 nm after Fe doping, due to the destroyed charge balance (Fig. S3†). The decreased nanosheet size further facilitates the insertion of ion species between layers in catalysis. The number of layers for the α-Ni0.881Fe0.119(OH)2 nanosheet is estimated to be 4, considering that the thickness of a single α-Ni(OH)2 layer is about 0.8 nm.23 The N2 adsorption–desorption measurement indicates that the α-Ni0.881Fe0.119(OH)2 sample possesses a surface area of 267.5 m2 g−1, larger than that of pristine α-Ni(OH)2 (93.2 m2 g−1, Fig. S4†), consistent with TEM results. The high-resolution TEM (HRTEM) image in Fig. 1f shows poor crystallinity of the α-Ni0.881Fe0.119(OH)2 sample, and the lattice distance of 0.25 nm is assigned to the (012) plane of α-Ni0.881Fe0.119(OH)2, suggesting that Fe doping will not change the layered structure of α-Ni(OH)2.
Elemental mapping images (Fig. 1g) show that Ni, Fe and O are uniformly distributed across nanosheets, indicating uniform Fe doping in α-Ni(OH)2. Then, X-ray photoelectron spectroscopy (XPS) examination is performed. The survey spectrum indicates no impurities of the sample (Fig. S5†). The Fe 2p spectrum of the α-Ni0.881Fe0.119(OH)2 sample in Fig. 1h shows a pair of peaks, which are located at 712.5 and 725.6 eV, higher than the binding energies (711.2 eV for 2p3/2 and 724.9 eV for 2p1/2 of Fe3+) of Fe3+(OH)x.24 Previous reports have indicated that the higher binding energies of Fe 2p in Fe-doped Ni(OH)2 may be due to the strong Fe–Ni interaction, and/or the changes of the coordination environment enabled by the amorphous structure of Ni(OH)2 induced by Fe doping.25 The Ni 2p spectrum (Fig. 1i) shows one pair of peaks with binding energies located at 855.5 and 873.0 eV, which can be attributed to 2p3/2 and 2p1/2 of Ni2+, respectively. The binding energies located at 861.2 and 879.2 eV can be assigned to the satellite peaks of 2p3/2 and 2p1/2 of Ni2+, respectively.26 The binding energies of Ni 2p spectra are lower than those of Ni(OH)2, which reveals that Fe donates electrons to the adjacent Ni atoms, confirming Fe–Ni interaction. Charge polarization between Fe and Ni atoms may facilitate the NTRR.27–31
Electrochemical NTRR performance evaluation
Then, electrochemical NTRR performance of α-Ni1−xFex(OH)2 samples is assessed using a three-electrode system, in which cathode and anode chambers are divided by the Nafion 117 membrane. Firstly, we screen the relationship of NTRR performance with Fe doping level using potentiostatic electrolysis at −0.6 V versus reversible hydrogen electrode (RHE). The produced amount of NH3 in the cathode chamber is quantified by spectrophotometry using the Nessler's reagent method and indophenol method (Fig. S6 and S7†). As shown in Fig. 2a, NH3 yield rates and NO3−-to-NH3 FEs all increase as x increases from 0 to 0.119, and then decline with x further increasing to 0.150 and 0.171. Linear sweep voltammetry (LSV) curves show that the current density is greatly enhanced after Fe doping in α-Ni(OH)2, suggesting that the Fe dopant acts as the active centre for the NTRR (Fig. S8†). Upon confirming the optimal Fe doping level in α-Ni(OH)2, we further assess the potential-dependent NTRR performance of α-Ni0.881Fe0.119(OH)2 nanosheets. It is found that NH3 yield rates quickly decline from 8.1, 6.0, 4.9, 2.6, 0.5 to 0.2 mol gcat.−1 h−1 as the applied potential positively shifts from −0.6 to −0.1 V (Fig. 2b). Correspondingly, NO3−-to-NH3 FEs show a similar decreasing trend in the potential range between −0.6 and −0.1 V. Surprisingly, NO3−-to-NH3 FE reaches 97.5% for α-Ni0.881Fe0.119(OH)2 nanosheets at −0.6 V, which implies that almost all electrons are used for the NTRR, and the competing hydrogen evolution reaction (HER) is completely inhibited. Notably, the catalyst is unstable by further decreasing the applied potential to −0.7 V (Fig. S9†). As a comparison, NTRR performance of the pristine α-Ni(OH)2 nanosheets is also obtained (Fig. S10†). NH3 yield rate and NO3−-to-NH3 FE are 2.3 mol gcat.−1 h−1 and 43.8%, and the performance of the Fe-doped sample is about 3.5 and 2.2 times larger than that of the pristine α-Ni(OH)2 nanosheets, respectively. This result indicates that Fe doping in the α-Ni(OH)2 lattice effectively enhances NTRR performance.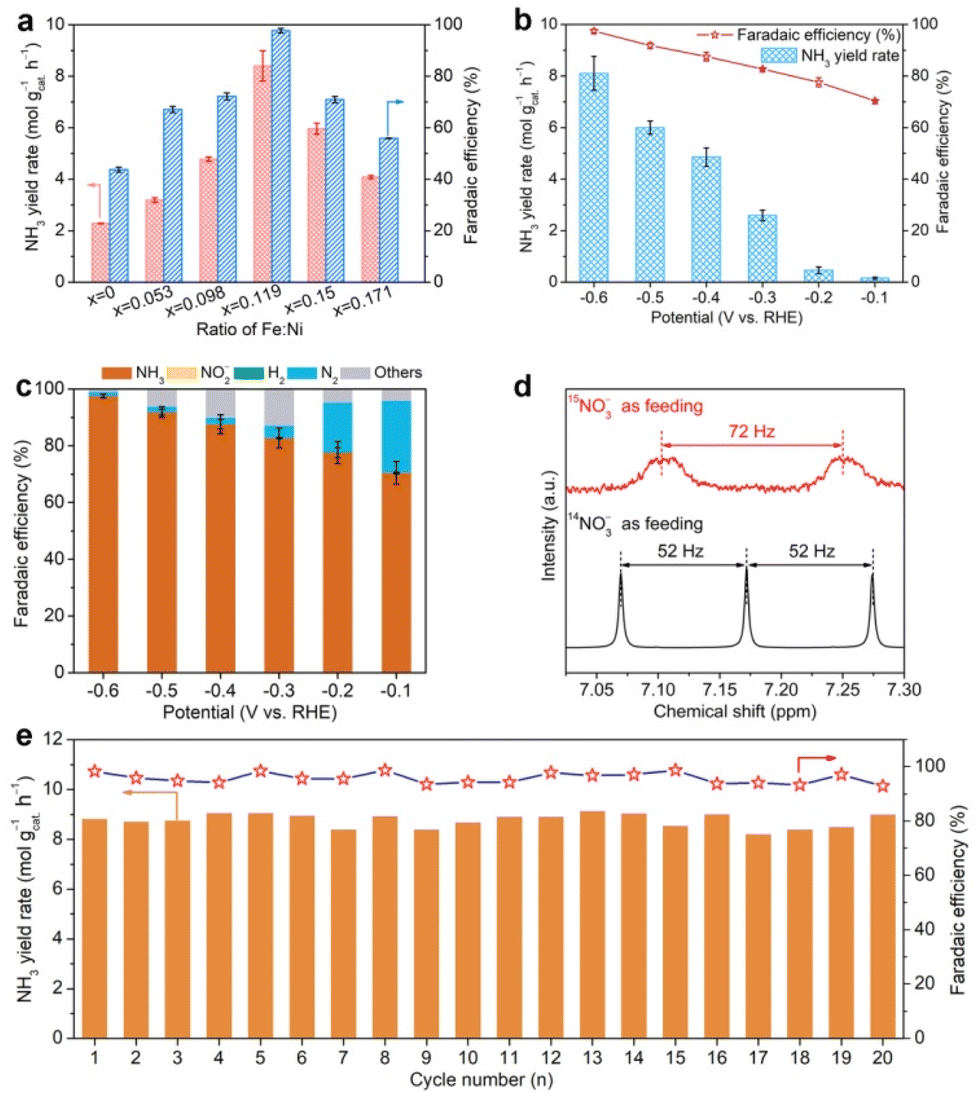 | ||
| Fig. 2 (a) Screening NH3 yield rates and NO3−-to-NH3 FEs of α-Ni1−xFex(OH)2 nanosheets at −0.6 V in a mixture of KOH (1.0 M) and KNO3 (1.0 M), (b) potential-dependent NH3 yield rates and FEs of α-Ni0.881Fe0.119(OH)2 nanosheets. (c) FEs of the possible products, i.e., NH3, NO2−, N2 and H2 for α-Ni0.881Fe0.119(OH)2 nanosheets in the NTRR. (d) 1H-NMR spectra recorded in the electrolytes using 14NO3− or 15NO3− as feeding. (e) Cycling stability. | ||
Apart from the main product (NH3), some by-products, e.g., liquid product NO2− and gas products N2 and H2, also exist, which are also quantified by spectrophotometry using the Griess's reagent method (Fig. S11†) and gas chromatography (GC, Fig. S12†), respectively. As shown in Fig. 2c, NO3−-to-N2 FEs quickly decline from 25.3% to 17.4%, 4.3%, 2.3%, 1.9% and 1.4% as the potential shifts from −0.1 to −0.6 V. Previous reports have pointed out that the formation of N2 involves the N–N coupling process in the NTRR, and the coupling process more likely occurs on the adjacent catalytic sites.12,32,33 In our case, Fe doping in the α-Ni(OH)2 lattice ensures atomic dispersion of Fe sites, which effectively inhibits the N–N coupling process to N2.34 In addition, NO2− and the competing HER are negligible between −0.1 and −0.6 V.
To further confirm that the produced NH3 comes from the NTRR, 15N isotope labeling experiments are carried out. As shown in Fig. 2d, the 1H-NMR spectrum of the electrolyte shows a nearly 1:1:1 triplet peak with a coupling constant of 52 Hz using 14NO3− as feeding, verifying that the obtained product is NH3.35 When the 14NO3− is replaced by 15NO3− solution (100 mM) as feeding, a 1:1 double pattern with a coupling constant of 72 Hz is observed, consistent with the reported result of 15NH4+.36 This result confirms that the produced NH3 really originates from the NTRR, not from the contamination of nitrogenous species. Beyond that, cycling stability is another parameter in real application. α-Ni0.881Fe0.119(OH)2 nanosheets can sustain continuous twenty cycles without obvious activity and FE decay (Fig. 2e). After twenty cycles of continuous electrolysis, the catalyst still retains the nanosheet structure and same phase structure (Fig. S13†). To further show the broad application prospect, the NTRR is further carried out in different concentrations of NO3− and pH. It is found that NH3 yield rates and FEs are positively correlated with concentrations and pH (Fig. S14†).
OER performance evaluation
Upon assessing NTRR performance at the cathode, the performance of the OER at the anode is also evaluated in 1.0 M KOH. Fig. 3a shows the polarization curves of α-Ni0.881Fe0.119(OH)2 nanosheets and α-Ni(OH)2 nanosheets in reference with commercial RuO2. It is found that the onset potential of α-Ni0.881Fe0.119(OH)2 nanosheets is 1.40 V, which is lower than that of pristine α-Ni(OH)2 nanosheets (1.52 V) and commercial RuO2 (1.44 V). At a current density of 10 mA cm−2, the overpotential of α-Ni0.881Fe0.119(OH)2 nanosheets (Fig. 3b, 254 mV) is lower than that of α-Ni(OH)2 nanosheets (381 mV) and commercial RuO2 (289 mV), suggesting that Fe doping in α-Ni(OH)2 really boosts the OER performance. The Fe-doped α-Ni(OH)2 sample also surpasses commercial RuO2. To elucidate the reaction kinetics of the above catalysts, Tafel slopes of the samples are derived from polarization curves (Fig. 3c). α-Ni0.881Fe0.119(OH)2 nanosheets exhibit a Tafel slope of 69.1 mV dec−1, lower than that of RuO2 (88.0 mV dec−1) and α-Ni(OH)2 nanosheets (101.5 mV dec−1), indicating the fastest oxygen evolution kinetics among the samples. Moreover, α-Ni0.881Fe0.119(OH)2 nanosheets also show long-term cycling stability without obvious current density decay for 50 h (Fig. 3d). | ||
| Fig. 3 (a) Polarization curves of α-Ni0.881Fe0.119(OH)2 NSs, α-Ni(OH)2 NSs and RuO2 in O2-saturated 1.0 M KOH solution. (b) Overpotentials of α-Ni0.881Fe0.119(OH)2 NSs, α-Ni(OH)2 NSs and RuO2 in the OER at a current density of 10 mA cm−2. (c) Tafel plots of α-Ni0.881Fe0.119(OH)2 NSs, α-Ni(OH)2 NSs and RuO2. (d) Cycling stability. | ||
Integration as a bifunctional catalyst for the NTRR and OER
Considering the greatly enhanced NTRR and OER performance after Fe doping both at the cathode and anode, α-Ni0.881Fe0.119(OH)2 nanosheets are further used as a bifunctional catalyst in NTRR overall electrolysis. In order to meet the requirement of real application, α-Ni0.881Fe0.119(OH)2 are firstly grown on nickel foam to form nanosheet arrays (Fig. 4a and Fig. S15†). Electrochemical NTRR performance is assessed in a three-electrode system, in which α-Ni0.881Fe0.119(OH)2 nanosheet powder is used as the cathode catalyst and nanosheet arrays are used as the anode catalyst (denoted as α-Ni0.881Fe0.119(OH)2 NSs(±)), respectively. Before the bifunctional test, the single electrochemical NTRR or OER performance of α-Ni0.881Fe0.119(OH)2 nanosheet arrays on nickel foam is firstly assessed (Fig. S16†). As shown in Fig. 4b, NH3 production rates are 11.1, 9.2, 7.7, 4.6, 3.1 and 2.4 mol gcat.−1 h−1 at −0.6, −0.5, −0.4, −0.3, −0.2 and −0.1 V, respectively. The corresponding FEs are 97.7%, 95.0%, 92.2%, 93.4%, 90.7% and 87.5% in the potential range of −0.6 and −0.1 V. Notably, NH3 yield rates and FEs are all higher than those of α-Ni0.881Fe0.119(OH)2 NSs(−)/Pt mesh(+), especially at more positive applied potentials, i.e., −0.1 V and −0.2 V. The enhanced NTRR performance is attributed to the enhanced oxygen evolution activity at the anode and the reduced overpotential in the OER. The faster oxygen evolution kinetics at the anode provides more electrons for the NTRR at the cathode; therefore, the NTRR at the cathode is boosted (Fig. S17†).37,38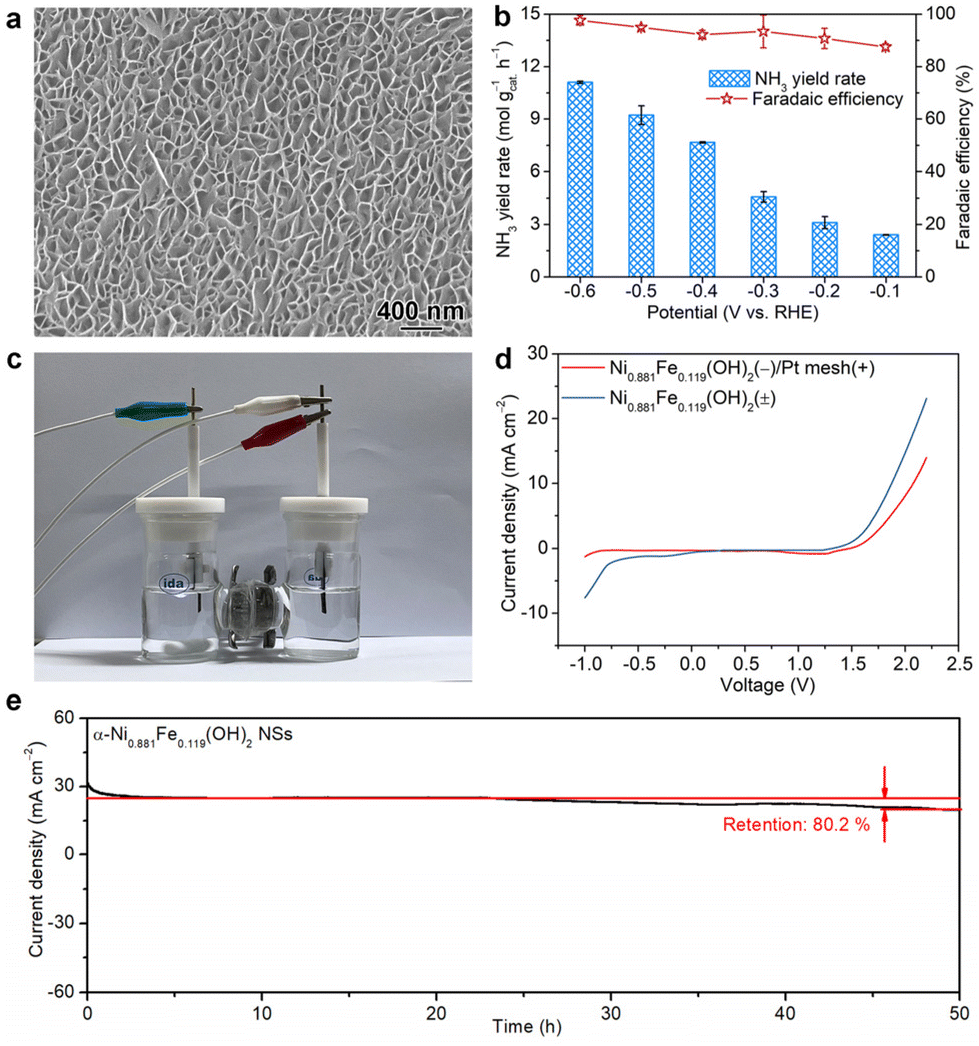 | ||
| Fig. 4 (a) SEM image of α-Ni0.881Fe0.119(OH)2 nanosheet arrays on nickel foam, (b) ammonia production rates and FEs for α-Ni0.881Fe0.119(OH)2 NSs(±) analyzed in a three-electrode system. (c) The electrochemical setup in the two-electrode system. (d) LSV curves recorded in the two-electrode system using α-Ni0.881Fe0.119(OH)2 nanosheets as bifunctional catalysts for the NTRR at the cathode and OER at the anode, respectively. (e) Cycling stability. | ||
Then, the bifunctional electrochemical performance is further assessed using a two-electrode system, as illustrated in Fig. 4c. LSV curves in Fig. 4d indicate that the NTRR and OER are triggered at a voltage of −0.20 and 1.40 V for the α-Ni0.881Fe0.119(OH)2 NS(±) system, respectively. In comparison, when α-Ni0.881Fe0.119(OH)2 NSs and Pt mesh are used as the cathode and anode, respectively, the onset voltage is shifted to −0.84 and 1.47 V, respectively. This result indicates that with the replacement of common Pt mesh with α-Ni0.881Fe0.119(OH)2 nanosheet arrays as the anode, the overall electrolysis is boosted. To further show the advantage of the bifunctional catalyst for the NTRR, energy utilization efficiency of NO3−-to-NH3 is further obtained. Energy utilization efficiency reaches 20.2% for the α-Ni0.881Fe0.119(OH)2 NS(±) system at a voltage of 1.6 V, higher than that of the α-Ni0.881Fe0.119(OH)2 NS(−)/Pt mesh(+) system (13.8%, Fig. S18†). In addition, cycling stability for the α-Ni0.881Fe0.119(OH)2 NS(±) system in the two-electrode setup is further assessed. After continuous 50 h of electrolysis, the current density slightly declines from 24.8 to 19.9 mA cm−2, with a current density retention of 80.2% (Fig. 4e).
Interlayer confined NTRR
After confirming the enhanced NTRR and OER performance both at the cathode and anode, a question naturally arises: what is the role of the Fe dopant in the α-Ni(OH)2 lattice in the enhanced NTRR and OER performance? At the cathode, the Fe dopant in α-Ni(OH)2 is used as the active site for the NTRR, which is confirmed by LSV curves. Beyond that, electrochemical active surface areas (ECSA) are also obtained derived from cyclic voltammetry curves recorded at different sweep rates to investigate the impact of Fe doping (Fig. S19†). Cdl for α-Ni0.881Fe0.119(OH)2 nanosheets (0.66 mF cm−2) is lower than that for α-Ni(OH)2 nanosheets (1.24 mF cm−2), suggesting the decreased ECSA after Fe doping. The decreased ECSA indicates that the enhanced electrochemical performance of the α-Ni0.881Fe0.119(OH)2 sample is due to the higher intrinsic activity of the Fe dopant, not ECSA. To further examine the impact of Fe doping on conductivity for α-Ni1−xFex(OH)2 samples, EIS Nyquist tests are performed (Fig. S20†). It is found that charge transfer resistances for α-Ni1−xFex(OH)2 follow the tendency of α-Ni(OH)2 > α-Ni0.947Fe0.053(OH)2 > α-Ni0.902Fe0.098(OH)2 > α-Ni0.850Fe0.150(OH)2 > α-Ni0.829Fe0.171(OH)2 > α-Ni0.881Fe0.119(OH)2. The result indicates that Fe doping improves the electron transportation ability, and x = 0.119 is the best doping level.39To further confirm that the expanded interlayer spacing triggers the interlayer confined NTRR, we cover the outmost layers of α-Ni0.881Fe0.119(OH)2 nanosheets with a relatively inert TiO2 layer using atomic layer deposition (ALD) technology.40 The advantage of ALD technology is that only the outmost layers of α-Ni0.881Fe0.119(OH)2 are blocked by TiO2, while the interior layers are still exposed (Fig. S21†). In theory, about 25%–33% of active sites located on the outmost layers are blocked after the ALD process for 3–4 layers of α-Ni0.881Fe0.119(OH)2 nanosheets (Fig. S21†). After 50 cycles of deposition of TiO2 on the sample (about 2.25 nm), NH3 yield rate declines from 8.1 to 5.7 mol gcat.−1 h−1 (29.6% decline), while NO3−-to-NH3 FE slightly declines from 97.5 to 92.2% (Fig. 5a). The result indicates that the NTRR mainly occurs confined between layers, instead of the traditional viewpoint that catalytic reactions only occur on the outside of the nanosheets. To further confirm the interlayer confined NTRR, the premise is to prove that nitrate ions are intercalated into the layers during the electrochemical process. Then, in situ Raman spectroscopy characterization is carried out. Fig. 5b shows the time-dependent Raman spectra of the α-Ni0.881Fe0.119(OH)2 sample at −0.6 V, in contrast with that of pristine α-Ni(OH)2. Raman shifts located at 1047 and 1380 cm−1 are attributed to ν1 and ν3 modes of free NO3−.41,42 Obviously, the intensity of the two vibration modes for the α-Ni0.881Fe0.119(OH)2 sample increases gradually with the electrolysis time, verifying the continuous insertion of nitrate ions into layers. The continuous intercalation of nitrate ions accumulates and concentrates NO3− in the interior of nanosheets, which boosts the NTRR performance. The novel interlayer confined NTRR also indicates that the layered structure does not need to be stripped to few layers or a single layer to maximally expose the active sites. The expanded interlayer spacing caused by Fe doping accelerates the intercalation of nitrate ions. This conclusion can be further confirmed by the higher intensity of ν1 and ν3 modes for α-Ni0.881Fe0.119(OH)2 than that for α-Ni(OH)2 nanosheets.
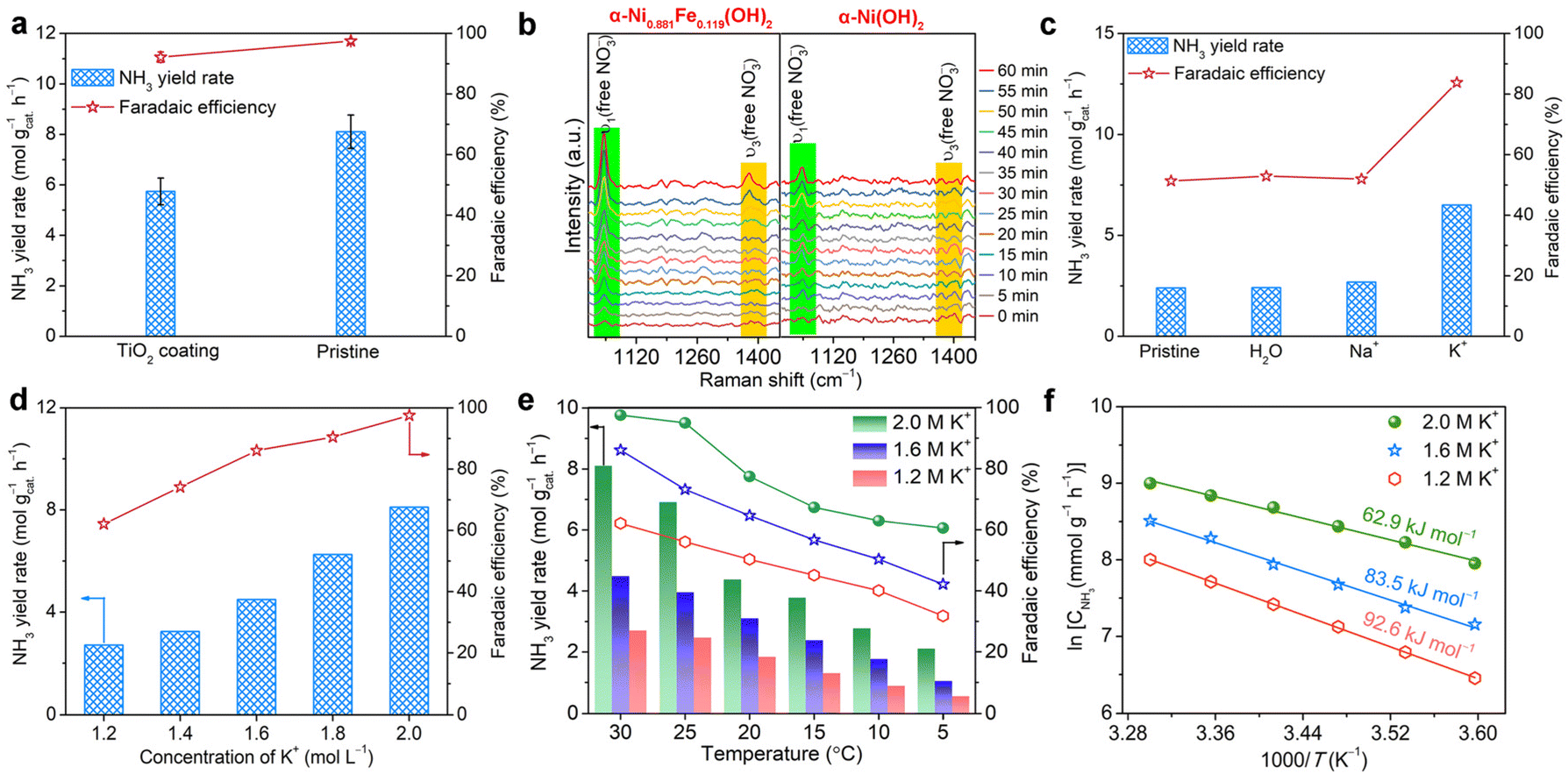 | ||
| Fig. 5 (a) Comparison of NH3 yield rates and FEs for α-Ni0.881Fe0.119(OH)2 nanosheets with or without TiO2 coating. (b) Time-dependent Raman spectra recorded in a mixture of 1.0 M KOH and 1.0 KNO3 at −0.6 V. (c) NH3 yield rates and FEs for α-Ni0.881Fe0.119(OH)2 nanosheets pre-inserted with different species. (d) NH3 yield rates and FEs of α-Ni0.881Fe0.119(OH)2 nanosheets recorded at different concentrations of potassium ions. (e) NH3 yield rates and FEs acquired at different reaction temperatures. (f) Apparent activation energy (Ea) for the NTRR. | ||
Upon confirming the interlayer confined NTRR, we wondered whether there is possibility of insertion of potassium ions into layers. It is generally reported that potassium ions greatly affect the local microenvironment of the active sites, water structure and local electric field, which determine the water splitting and the related proton-coupled electron transfer reactions.43–47 Therefore, X-ray photoelectron spectroscopy (XPS) measurement is employed to determine the trace of the inserted potassium ions. The K 2p spectrum (Fig. S22†) of the α-Ni0.881Fe0.119(OH)2 sample after the NTRR test confirms the existence of K+.48 It should be noted that the trace of K+ does not come from surface adsorption, as the sample is rinsed thoroughly, indicating that potassium ions are really inserted into layers during the electrochemical reaction. To further reveal the contribution of the intercalated potassium ions to the NTRR, K+ is firstly pre-inserted into layers through electrolysis in KOH solution for 10 h. Nanosheets pre-inserted with Na+ or H2O species are used as control samples. Electrochemical NTRR performance for α-Ni0.881Fe0.119(OH)2 samples pre-inserted with different species, i.e., K+, Na+ or H2O, is assessed in the mixture of NaOH (1 M) and NaNO3 (1 M) to exclude the impact of potassium ions from the electrolyte. As shown in Fig. 5c, NH3 yield rate for the sample pre-inserted with K+ reaches 6.5 mol gcat.−1 h−1, higher than that of Na+ (2.7 mol gcat.−1 h−1), H2O (2.4 mol gcat.−1 h−1) and pristine α-Ni(OH)2 nanosheets (2.4 mol gcat.−1 h−1). In addition, NH3 FE for the sample pre-inserted with K+ is obviously higher than that for Na+, H2O and pristine samples. This result confirms that the inserted K+ between layers enhances the NTRR performance by inhibiting the side reactions, i.e., HER and NO3− → NO2−. It also highlights that the impact of sodium ions on the NTRR is much lower than that of potassium ion.
Then, we further carry out the electrochemical NTRR test in the electrolytes (1−x M NaOH + x M KOH + 1 M KNO3, x = 0.2, 0.4, 0.6, 0.8, and 1.0, Fig. 5d) with different K+ concentrations. The pH of the electrolytes and concentrations of NO3− are maintained. It is found that NH3 yield rates and FEs all show an increasing trend with K+ concentrations. This result indicates that K+ really enhances NH3 production rate by inhibiting the competing HER. High concentration of potassium ions means that more K+ are intercalated into the layers to boost the interlayer confined NTRR. The promotion effect of potassium ions on the interlayer confined NTRR can be summarized as follows: (1) the accumulation of potassium ions between layers elevates the electrochemical potential at the electrode surface, which promotes the intercalation of nitrate ions through electrostatic interaction (K+ ↔ NO3−, Fig. S23†).49 (2) The intercalated potassium ions between layers changes the water structure to accelerate the cleavage of water; therefore, more protons are produced and the proton-coupled NTRR is also enhanced.46
NTRR performance is further assessed at different temperatures and concentrations of potassium ions to estimate the apparent activation energy (Ea), and to evaluate the effect of potassium ions. As shown in Fig. 5e, NH3 yield rates and FEs all show an increasing trend with temperatures and the concentrations of potassium ions. In particular, in the electrolyte of 2.0 M K+, NH3 yield rates and FEs are increased from 2.1 mol gcat.−1 h−1 and 60.6% at 5 °C to 8.1 mol gcat.−1 h−1 and 97.5% at 30 °C. The estimated apparent activation energy Ea is 92.6, 83.5 and 62.9 kJ mol−1 at potassium ion concentrations of 1.2, 1.6 and 2.0 M, respectively (Fig. 5f). The result indicates that high concentration of K+ can decrease the apparent activation energy Ea of the NTRR.
Upon clarifying the impact of K+ on the cathodic NTRR, we wondered whether potassium ions affect the OER at the anode. The OER in alkaline media is started by formation of M–OH according to the equation M + OH− → M–OH + e− (M = Ni and Fe).50,51 Therefore, the accumulation of K+ between layers also has a tendency to force OH− to insert into layers to participate in the OER. Then, the OER is assessed in the electrolytes (1−x M NaOH + x M KOH, x = 0.2, 0.6 and 1.0) with different potassium ion concentrations. It is found that the onset potential is decreased and polarization current density is enhanced as the concentration of K+ is increased from 0.2 to 0.6 and 1.0 M (Fig. 6a). Accordingly, the Tafel slopes slowly decline from 78.9 mV dec−1 at 0.2 M K+ to 75.3 mV dec−1 (0.6 M) and 69.1 mV dec−1 (1.0 M, Fig. 6b). This result suggests that high concentration of potassium ions enhances the OER performance by increasing OER kinetics. To further confirm the accumulation of OH− between layers, which contributes to the enhanced OER performance, in situ Raman spectroscopy characterization is carried out. Fig. 6c shows the time-dependent Raman spectra of α-Ni0.881Fe0.119(OH)2 and α-Ni (OH)2 samples in KOH solution. The low frequency band located at 288 cm−1 can be attributed to the K+ ↔ OH− pair in the concentrated KOH aqueous solution.52 It is found that the intensity for α-Ni0.881Fe0.119(OH)2 nanosheets is obviously stronger than that for the α-Ni(OH)2 counterpart, suggesting that more OH− and K+ are inserted into layers. It should be noted that the vibration mode of the K+ ↔ OH− pair only appears in concentrated KOH solution; therefore, the concentration of K+/OH− between layers is higher than that in the electrolyte. Notably, we cannot exclude the possibility that OH− is inserted into the layers of the α-Ni(OH)2 sample.
 | ||
| Fig. 6 (a) LSV curves recorded at different concentrations of potassium ions. (b) Potassium ion-dependent Tafel plots. (c) Time-dependent in situ Raman spectra of α-Ni0.881Fe0.119(OH)2 and α-Ni(OH)2 samples recorded in 1.0 M KOH at 1.25 V. | ||
Conclusions
In summary, the interlayer confined NTRR to ammonia and OER are confirmed in layered Fe-doped α-Ni(OH)2. Fe doping in α-Ni(OH)2 expands the interlayer spacing, which facilitates the intercalation of potassium ions during the electrochemical process. The accumulation of potassium ions between layers provides an extra driving force to compel the intercalation of nitrate ions at the cathode and hydroxide ions at the anode into the interlayer through electrostatic interaction, respectively. Benefitting from the enrichment of the reactants, the obtained α-Ni0.881Fe0.119(OH)2 nanosheets show excellent bifunctional performance for the NTRR to ammonia at the cathode and OER at the anode. As a cathode catalyst, NH3 yield rate and FE reach 8.1 mol gcat.−1 h−1 and 97.5% at −0.6 V, as well as remarkable cycling stability. As an anode catalyst, the overpotential declines from 381 to 254 mV after Fe doping. The energy utilization efficiency of NO3−-to-NH3 is increased to 20.2% as a bifunctional catalyst in the overall NTRR. The findings provide new insights into the rational design of highly active, selective and durable bifunctional electrocatalysts for the NTRR from the angle of the interlayer confinement effect.Experimental
Materials
Nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O, A.R.), iron(III) trichloride hexahydrate (FeCl3·6H2O, A.R.), urea (CH4N2O, A.R.), triethylene glycol (C6H14O4, A.R.), ammonium chloride (NH4Cl, A.R.), sodium nitrite (NaNO2, A.R.), ethanol (EtOH, A.R.), hydrochloric acid (36 wt% in H2O), acetone (C3H6O, A.R.), 15N-labeled potassium nitrate (K15NO3, ≥99.5%), potassium nitrate (KNO3, A.R.), potassium hydroxide (KOH, A.R.), ultra-high purity Ar (99.999%), ruthenium(IV) oxide (RuO2, ≥99.5%), Nessler reagent and Griess reagent were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). All chemicals were used without further purification. All aqueous solutions were prepared using deionized (DI) water with a resistivity of 18.25 MΩ cm−1.Synthesis of α-Ni1−xFex(OH)2 nanosheets
In a typical synthesis of α-Ni0.881Fe0.119(OH)2 nanosheets, Ni(NO3)2·6H2O (1.45 g) was firstly dissolved in 10 mL deionized water. After that, 40 mL triethylene glycol was added into the above solution. The mixture was sonicated for 30 minutes, during which urea (0.6 g) and FeCl3·6H2O (405.5 mg, mole ratio: Ni:Fe = 1:0.3) were added into the above mixture in turn to form a homogeneous solution. Finally, the solution was transferred and sealed in an autoclave with a Teflon liner, and was heated at 120 °C for 24 h. After it was cooled to room temperature, the products were collected by centrifugation, rinsed with DI water and absolute ethanol several times, and dried at 60 °C in an oven. The protocol for the synthesis of α-Ni1−xFex(OH)2 (x = 0, 0.098, 0.150, and 0.171) nanosheets was similar to that of α-Ni0.881Fe0.119(OH)2 nanosheets, except with different ratios of Ni:Fe (1:0, 1:0.1, 1:0.2, 1:0.4, and 1:0.5).
Synthesis of α-Ni0.881Fe0.119(OH)2 nanosheet arrays on nickel foam
Before the synthesis of nanosheet arrays, a piece of nickel foam with an area of 1 × 3 cm2 was rinsed thoroughly with HCl solution (3 M), acetone, ethanol and deionized water to remove nickel oxide and organic residues. After that, the nickel foam was dried in a vacuum oven at 60 °C. In a typical synthesis of α-Ni0.881Fe0.119(OH)2 nanosheet arrays, Ni(NO3)2·6H2O (1.45 g) was firstly dissolved in 10 mL deionized water. Then, 40 mL triethylene glycol was added into the above solution. The mixture was sonicated for 30 minutes, during which urea (0.6 g) and FeCl3·6H2O (405.5 mg, mole ratio: Ni:Fe = 1:0.3) were added into the above mixture in turn to form a homogeneous solution. Finally, the solution was transferred, and a piece of clean nickel foam was added into the mixture and sealed in an autoclave with a Teflon liner, and was heated at 120 °C for 24 h. After it was cooled to room temperature, the nickel foam was washed with deionized water and ethanol three times, respectively. The nanosheet arrays were dried at 60 °C in an oven.
Sample characterization
Prior to electron microscopy characterization, a drop of the suspension of nanostructures in ethanol was placed on a piece of a carbon-coated copper grid, and dried under ambient conditions. Transmission electron microscopy (TEM), high-resolution TEM (HRTEM) images, high-angle annular dark-field scanning TEM (HAADF-STEM) images and the corresponding energy-dispersive X-ray spectroscopy (EDS) mapping profiles were taken on a JEOL JEM-2100F field-emission high-resolution transmission electron microscope operated at 200 kV. Scanning electron microscopy (SEM) images were collected on an FEI XL-30 ESEM scanning electron microscope operated at 5 kV. The specific surface area and the porosity distributions were calculated using Brunauer–Emmett–Teller (BET) nitrogen adsorption–desorption isotherms and Barrett–Joyner–Halenda (BJH) method, respectively, by using a Micromeritics 3 Flex™ surface characterization analyzer at 77 K. Structural and chemical analyses of the as-prepared samples were performed by powder X-ray diffraction (XRD). X-ray photoelectron spectra (XPS) were collected on an ESCALab 250 X-ray photoelectron spectrometer with nonmonochromatized Al-Kα X-ray as the excitation source. The in situ Raman measurement was conducted in a home-made electrolytic tank on an integrated Raman spectroelectrochemical instrument.Electrochemical NTRR test
2 mg α-Ni(1−x)Fex(OH)2 samples were dispersed in the mixture of deionized water (0.7 mL), isopropanol (0.25 mL) and Nafion (50 μL, 5 wt%) to form the catalyst ink. Then, 50 μL catalyst ink was coated on carbon paper with an area of 1 × 1 cm2 and catalyst loading of 0.1 mg cm−2, which was used as the working electrode. All electrochemical NTRR tests were conducted in an H-type cell using a three-electrode system. The anode and cathode chambers were separated by the Nafion 117 membrane. Ag/AgCl (saturated KCl solution) and platinum mesh were used as the reference electrode and counter electrode, respectively. The potentials in the article were calibrated relative to the reversible hydrogen electrode (RHE) using the following equation:| ERHE = EAg/AgCl + 0.198 V + 0.059 × pH | (1) |
The electrolytes in the anode and cathode chambers are both the mixture of 40 mL KOH (1 M) and KNO3 (1 M). Before the electrochemical NTRR test, the electrolyte was pumped with ultra-pure Ar flow for 30 min to remove the dissolved O2 and N2. The electrochemical NTRR was triggered by applying a fixed potential for 1 h under continuous stirring. The possible liquid products, i.e., NH3, and NO2−, and gas products, i.e., N2 and H2, in the cathode chamber were quantified by the spectrophotometry method and gas chromatography (GC), respectively.
Determination of produced NH3 and NO2−
Owing to the large concentration of NH3, the electrolyte was diluted two or one hundred times. The amount of NH3 in the electrolyte was determined by spectrophotometry using the Nessler's reagent method. Specifically, 5 mL of the diluted electrolyte and 0.1 mL of potassium sodium tartrate solution (0.2 M) were mixed, and then 0.15 mL Nessler reagent was added into the above solution. After standing for 10 min at room temperature, the UV-visible absorption spectrum was recorded. The concentration of produced NH3 was determined using the absorbance at a wavelength of 420 nm according to the working curve of NH3.The concentration of NO2− in the electrolyte was determined using the Griess's regent method. Typically, the Griess reagent (1.0 mL) was mixed with the nitrite-containing solution (5.0 mL), and allowed to react at room temperature for 15 min to obtain a magenta solution. Finally, the concentration of NO2− was determined using the absorbance at a wavelength of 540 nm according to the working curve of NO2−.
15NO3− isotopic labeling tracer experiment
The produced NH3 was qualitatively confirmed by an isotope-labeled tracer experiment by using a 100 mM solution of 15NO3− (≥99.5%) as the N source. After 1 h of electrocatalysis, 15NH4+ in the reaction solution was tested using 1H-NMR (500 MHz) spectroscopy.Calculation of NO3−-to-NH3 FE
Faradaic efficiency (FE) was calculated by dividing the charge used for the given product by the total charge consumption (Q):For NO3−-to-NH3:
| FE = (8 × F × C(NH3) × V)/Q | (2) |
For NO3−-to-NO2−:
| FE = (2 × F × C(NO2−) × V)/Q | (3) |
For H2O-to-H2:
| FE = (2 × F × V(H2)/Vm)/Q | (4) |
485 C mol−1), C is the measured concentration (mol L−1), V is the volume of the electrolyte (L), V(H2) is the volume of produced H2 (L), and Vm is molar volume of gas in the standard state (22.4 L mol−1).
Electrochemical OER test
All OER tests were carried out in a three-electrode system. The α-Ni0.881Fe0.119(OH)2 catalyst was coated on carbon paper, and used as the working electrode, and Ag/AgCl (saturated KCl solution) and platinum mesh were used as the reference electrode and counter electrode, respectively. To prepare the catalyst ink, 5 mg α-Ni0.881Fe0.119(OH)2 nanosheets were dispersed in the mixture of deionized water (0.5 mL), isopropanol (0.48 mL) and Nafion (20 μL, 5 wt%). Then, 60 μL catalyst ink was coated on carbon paper with an area of 1 × 1 cm2, and the catalyst loading was 0.3 mg cm−2.Before the OER test, cyclic voltammetry curves were recorded in 1.0 M KOH from 0.8 to 1.5 V at a sweep rate of 50 mV s−1 until two adjacent curves exactly coincided. Polarization curves were recorded in a fresh oxygen saturated 1.0 M KOH solution with iR compensation in the potential range of 1.0 to 2.0 V and a sweep rate of 5 mV s−1. The overpotential (η) is calculated according to the following equation:
| η = ERHE − 1.23 V | (5) |
| η = blogj + a | (6) |
Author contributions
W. Ye, Y. Zhang and M. Q. Xu conceived the idea, and designed and carried out the experiments, as well as data analysis. X. D. Xu, X. Y. Li and R. L. Yin contributed to the TEM and SEM measurements. W. Ye wrote the manuscript. G. Jia, G. P. Zhu, B. C. Yang, R. L. Yin and P. Gao performed analysis, supervised, and supported the work.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was financially supported in part by the Natural Science Foundation of China (No. 51902077), Innovation and Entrepreneurship Project of High-Level Overseas Students in Hangzhou in 2021, University Leading Talents Program of Zhejiang province, Medical Health Science and Technology Project of Zhejiang Provincial Health Commission (No. 2023KY1009) and Research Start-Up Fund of Hangzhou Normal University (No. 4095C5021920466).References
- Y. Yao, J. Wang, U. B. Shahid, M. Gu, H. Wang, H. Li and M. Shao, Electrochem. Energy Rev., 2020, 3, 239–270 CrossRef CAS.
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C. Chen, M. R. Smith and T. W. Hamann, Chem. Rev., 2020, 120, 5437–5516 CrossRef CAS PubMed.
- R. Verma, Y. Jing, H. Liu, V. Aggarwal, H. K. Goswami, E. Bala, Z. Ke and P. K. Verma, Eur. J. Org. Chem., 2022, e202200298 CAS.
- F. Jiao and B. J. Xu, Adv. Mater., 2019, 31, 1805173 CrossRef.
- T. Kandemir, M. E. Schuster, A. Senyshyn, M. Behrens and R. Schlçgl, Angew. Chem., Int. Ed., 2013, 52, 12723–12726 CrossRef CAS PubMed.
- F. Chen, Z. Wu, S. Gupta, D. J. Rivera, S. V. Lambeets, S. Pecaut, J. Kim, P. Zhu, Y. Z. Finfrock, D. M. Meira, G. King, G. Gao, W. Xu, D. A. Cullen, H. Zhou, Y. Han, D. E. Perea, C. L. Muhich and H. Wang, Nat. Nanotechnol., 2022, 17, 759–767 CrossRef CAS PubMed.
- V. Kyriakou, I. Garagounis, A. Vourros, E. Vasileiou and M. Stoukides, Joule, 2020, 4, 142–158 CrossRef CAS.
- G. Soloveichik, Nat. Catal., 2019, 2, 377–380 CrossRef CAS.
- P. Brimblecombe and D. H. Stedman, Nature, 1982, 298, 460–462 CrossRef CAS.
- M. Alahi and S. C. Mukhopadhyay, Sens. Actuators, A, 2018, 280, 210–221 CrossRef CAS.
- J. Li, G. Zhan, J. Yang, F. Quan, C. Mao, Y. Liu, B. Wang, F. Lei, L. Li, A. W. M. Chan, L. Xu, Y. Shi, Y. Du, W. Hao, P. K. Wong, J. Wang, S. Dou, L. Zhang and J. C. Yu, J. Am. Chem. Soc., 2020, 142, 7036–7046 CrossRef CAS PubMed.
- M. Xu, Q. Xie, D. Duan, Y. Zhang, Y. Zhou, H. Zhou, X. Li, Y. Wang, P. Gao and W. Ye, ChemSusChem, 2022, e202200231 CAS.
- X. Liu, P. V. Kumar, Q. Chen, L. Zhao, F. Ye, X. Ma, D. Liu, X. Chen, L. Dai and C. Hu, Appl. Catal., B, 2022, 316, 121618 CrossRef CAS.
- W. Guo, K. Zhang, Z. Liang, R. Zou and Q. Xu, Chem. Soc. Rev., 2019, 48, 5658–5716 RSC.
- J. Wang, S. Chen, Z. Li, G. Li and X. Liu, ChemElectroChem, 2020, 7, 1067–1079 CrossRef CAS.
- N. Suen, S. Hung, Q. Quan, N. Zhang, Y. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed.
- J. Hu, Y. Q. Liang, S. L. Wu, Z. Y. Li, C. S. Shi, S. Y. Luo, H. J. Sun, S. L. Zhu and Z. D. Cui, Mater. Today Nano, 2022, 17, 100150 CrossRef CAS.
- C. Huang, H. Wang and P. Chiu, Water Res., 1998, 32, 2257–2264 CrossRef CAS.
- P. Li, Z. Jin, Z. Fang and G. Yu, Energy Environ. Sci., 2021, 14, 3522–3531 RSC.
- D. Jia, H. Gao, W. Dong, S. Fan, R. Dang and G. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 20476–20483 CrossRef CAS.
- Y. Cai, J. Ma and T. Wang, J. Alloys Compd., 2014, 582, 328–333 CrossRef CAS.
- J. Kang, Y. Xue, J. Yang, Q. Hu, Q. Zhang, L. Gu, A. Selloni, L. Liu and L. Guo, J. Am. Chem. Soc., 2022, 144, 8969–8976 CrossRef CAS PubMed.
- B. J. Tan, K. J. Klabunde and P. A. Sherwood, Chem. Mater., 1990, 2, 186–191 CrossRef CAS.
- Y. Song, M. Song, P. Liu, W. Liu, L. Yuan, X. Hao, L. Pei, B. Xu, J. Guo and Z. Sun, J. Mater. Chem. A, 2021, 9, 14372–14380 RSC.
- A. N. Mansour and C. A. Melendres, Surf. Sci. Spectra, 1994, 3, 255–262 CrossRef CAS.
- L. Li, D. Yu, P. Li, H. Huang, D. Xie, C. Lin, F. Hu, H. Chen and S. Peng, Energy Environ. Sci., 2021, 14, 6419–6427 RSC.
- L. Wang, Y. Hao, L. Deng, F. Hu, S. Zhao, L. Li and S. Peng, Nat. Commun., 2022, 13, 5785 CrossRef CAS.
- L. Deng, F. Hu, M. Ma, S. Huang, Y. Xiong, H. Chen, L. Li and S. Peng, Angew. Chem., Int. Ed., 2021, 60, 22276 CrossRef CAS.
- H. Huang, D. Yu, F. Hu, S. Huang, J. Song, H. Chen, L. Li and S. Peng, Angew. Chem., 2022, 134, e202116068 Search PubMed.
- F. Hu, D. Yu, M. Ye, H. Wang, Y. Hao, L. Wang, L. Li, X. Han and S. Peng, Adv. Energy Mater., 2022, 12, 2200067 CrossRef CAS.
- J. Liu, D. Richards, N. Singh and B. R. Goldsmith, ACS Catal., 2019, 9, 7052–7064 CrossRef CAS.
- J. Wang, W. Teng, L. Ling, J. Fan, W. Zhang and Z. Deng, Environ. Sci.: Nano, 2020, 7, 1496–1506 RSC.
- Z. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F. Chen, J. Y. Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Nat. Commun., 2021, 12, 2870 CrossRef CAS PubMed.
- R. Y. Hodgetts, A. S. Kiryutin, P. Nichols, H. Du, J. M. Bakker, D. R. Macfarlane and A. N. Simonov, ACS Energy Lett., 2020, 5, 736–741 CrossRef CAS.
- F. Xu, F. Wu, K. Zhu, Z. Fang, D. Jia, Y. Wang, G. Jia, J. Low, W. Ye, Z. Sun, P. Gao and Y. Xiong, Appl. Catal., B, 2021, 284, 119689 CrossRef CAS.
- H. Zhang, A. Maijenburg, X. Li, S. L. Schweizer and R. B. Wehrspohn, Adv. Funct. Mater., 2020, 30, 2003261 CrossRef CAS.
- S. Ghosh and R. N. Basu, Nanoscale, 2018, 10, 11241 RSC.
- Y. Wu, S. Ji, H. Wang, B. G. Pollet, X. Wang and R. Wang, Appl. Surf. Sci., 2020, 528, 146972 CrossRef CAS.
- M. Xu, F. Xu, K. Zhu, X. Xu, P. Deng, W. Wu, W. Ye, Z. Sun and P. Gao, Comput. Commun., 2022, 29, 101037 Search PubMed.
- N. Sergent, M. Epifani and T. Pagnier, J. Raman Spectrosc., 2006, 37, 1272–1277 CrossRef CAS.
- K. Hadjiivanov, Catal. Rev.: Sci. Eng., 2000, 42, 71–144 CrossRef CAS.
- L. D. Chen, M. Urushihara, K. Chan and J. K. Nørskov, ACS Catal., 2016, 6, 7133–7139 CrossRef CAS.
- N. C. Kani, A. Prajapati, B. A. Collins, J. D. Goodpaster and M. R. Singh, ACS Catal., 2020, 10, 14592–14603 CrossRef CAS.
- K. Zhu, J. Ma, L. Chen, F. Wu, X. Xu, M. Xu, W. Ye, Y. Wang, P. Gao and Y. Xiong, ACS Catal., 2022, 12, 4840–4847 CrossRef CAS.
- Y. Wang, S. Zheng, W. Yang, R. Zhou, Q. He, P. Radjenovic, J. Dong, S. Li, J. Zheng, Z. Yang, G. Attard, F. Pan, Z. Tian and J. Li, Nature, 2021, 600, 81–85 CrossRef CAS.
- Y. Hao, Y. Guo, L. Chen, M. Shu, X. Wang, T. Bu, W. Gao, N. Zhang, X. Su, X. Feng, J. Zhou, B. Wang, C. Hu, A. Yin, R. Si, Y. Zhang and C. Yan, Nat. Catal., 2019, 2, 448–456 CrossRef CAS.
- O. Rosseler, M. Sleiman, V. N. Montesinos, A. Shavorskiy, V. Keller, N. Keller, M. I. Litter, H. Bluhm, M. Salmeron and H. Destaillats, J. Phys. Chem. Lett., 2013, 4, 536–541 CrossRef CAS.
- J. Yu, J. Yin, R. Li, Y. Ma and Z. Fan, Chem. Catal., 2022, 2, 1–24 CrossRef.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS.
- J. Zhao, J. Zhang, Z. Li and X. Bu, Small, 2020, 16, 2003916 CrossRef CAS.
- S. K. Sharma and S. C. Kashyap, Nucl. Chem., 1972, 34, 3623 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr05001c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |