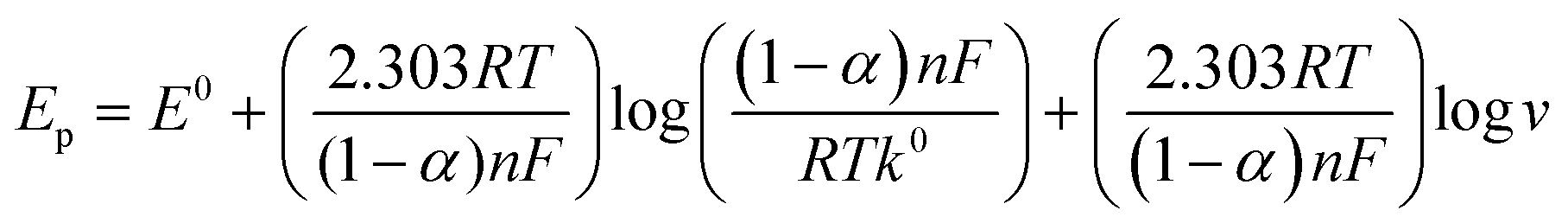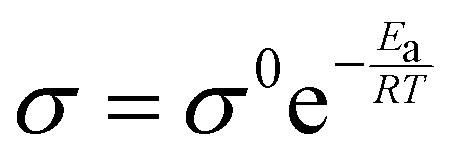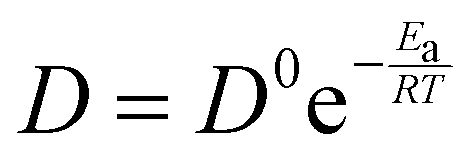DOI:
10.1039/D2NJ05060A
(Paper)
New J. Chem., 2023,
47, 315-323
Improved electrochemical detection of harmful 1-NAA molecules by a MWNTs/Ca–ZnO nanocomposite-modified paste electrode
Received
15th October 2022
, Accepted 21st November 2022
First published on 21st November 2022
Abstract
The electrochemical degradation of 1-naphthaleneacetic acid (1-NAA) was studied using multiwalled carbon nanotubes (MWNTs) and calcium-doped zinc oxide (Ca–ZnO) nanoparticles embedded in carbon paste electrodes (CPEs). Samples of water, fruits, soil, and vegetables were accurately analyzed using the square wave voltammetry (SWV) technique. Using cyclic voltammetry (CV) and SWV methods, we investigated the impact of various factors on 1-NAA. The sensor displayed exceptional catalytic properties and peak current in a pH 3.0 0.2 M phosphate buffer (PB) solution. The peak currents for the bare CPE (BCPE), MWNTs/CPE, Ca-doped ZnO/CPE, and MWNTs/Ca–ZnO/CPE were 2.53 μA, 4.42 μA, 5.96 μA, and 13.78 μA, respectively. Similarly, the peak potentials for BCPE, MWNTs/CPE, Ca-doped ZnO/CPE, and MWNTs/Ca–ZnO/CPE were measured to be 1.127 V, 1.131 V, 1.126 V, and 1.081 V, respectively. Electrokinetic parameters like temperature and the heterogeneous rate constant, as well as the scan rate, pH, accumulation time, the energy of activation, transfer coefficient, and electron involvement, were examined in the electrochemical oxidation of 1-NAA. So the detection and quantification limits for the 1-NAA molecule are 0.025 nM and 0.084 nM.
Introduction
A naphthalene derivative called 1-naphthaleneacetic acid (1-NAA), also known as 1-naphthylacetic acid, is a synthetic plant growth stimulant for many crops. These are frequently combined with synthetic compounds like naphthylacetamide (NAD) or naturally occurring auxins like indole-3-butyric acid (IBA) and indole-3-acetic acid (IAA).1 NAA is categorized as unlikely to be dangerous by the WHO and highly toxic by the U.S. EPA when it comes to toxicity. Based on the plant type, it is sprayed at a concentration of 20 to 100 mg L−1. Consequently, its residuals have been discovered in fruits such as apples, pears, and soil and water samples.1–3 For numerous fruits, vegetables, seeds, legumes, and others, the maximum residue limit (MRL) for the mixture of NAD and 1-NAA (expressed as a quantity of 1-NAA) within the range of 0.6 to 0.15 mg kg−1 has been established by the European Union. For nuts and citrus fruits, the MRL for 1-NAA is 0.05 mg kg−1.4
Numerous analytical methods were developed to determine the precise concentration of this phytohormone. A single analyte was first fixed on the gel matrix or employed in conjunction with the flow-injection approach, followed by direct fluorescence or phosphorescence detection of 1-NAA, commonly combined with NAD or another growth regulator, without any prior separation step.5–7 For the quantification of 1-NAA, only a few literature reports have been noted, like different chromatographic methods depending upon ultra- and high-performance liquid chromatography using tandem mass,1,8 UV,9 diode array,10 or fluorescence detection.11 But these techniques demand the use of high-priced equipment. In addition, the assays require highly trained professionals, lengthy, complex operations, and sophisticated processes. Consequently, laboratory-based approaches cannot meet the requirements of actual implementations in various domains in terms of low cost, high speed, and easy operation. The electrochemical method has proven to be an effective solution for overcoming these inevitable limitations.12–15 This method is sensitive, reliable, reproducible, easy to use, and economical. Advanced electrochemical techniques, particularly voltammetry, represent an intriguing alternative to the aforementioned analytical approaches due to the low costs of the equipment and analysis, broad application range, and analysis rate.16–18 According to our understanding, the electrochemical performance of NAA has only been studied on modified carbon matrix working electrodes, specifically a glassy carbon electrode (GCE) fabricated with a multi-walled carbon nanotube film or an acetylene black film as well as a CPE manufactured using the ionic liquid.
Advanced electrochemical techniques, particularly voltammetry, represent an attractive alternative to the previously mentioned analytical techniques due to their low cost of analysis and instruments, analysis rate, and vast application range. Due to their superior qualities, such as high surface area, electrochemical properties, electrical conductivity, chemical stability, and electrical conductivity, carbon nanotubes and conducting polymers have been increasingly employed in sensor applications in recent years.19–21 As far as we are acquainted, the electrochemical performance of 1-NAA has only been discussed concerning carbon-based working electrodes that have been modified, such as a glassy carbon electrode (GCE) made from an acetylene black film22 or a multiwalled carbon nanotube film23 and a carbon paste electrode (CPE) prepared using the ionic liquid.24 To determine 1-NAA, we combined multi-walled carbon nanotubes (MWNTs) with a composite of calcium-doped zinc oxide nanoparticles (Ca–ZnO) on the carbon paste working electrode. Calcium (Ca) nanoparticles doped into zinc oxide (ZnO) nanoparticles change their physical properties without changing their crystalline structure. Electronic, magnetic, electrical, and optical elements are all improved by doping nanoparticles. It has varying band gap properties, piezoelectricity, magneto-optical properties, and ferromagnetism. Including the calcium dopant in zinc oxide nanoparticles increases the electrical conductivity because substitutional doping of Ca2+ at Zn2+ sites generates one additional carrier. The accumulation of calcium in zinc oxide NPs controls molecular adsorption through aqueous solutions.25,26
Graphene, carbon nanotubes, and fullerenes are a few examples of carbon nanomaterials used in various applications, such as electrochemical sensors, drug delivery, catalysis, and more.27–29 Since they can facilitate electron transport, encourage molecular adsorption, and increase the electroanalytical surface area, carbon nanostructures are advantageous for electrochemical sensor applications. Modifying and improving the physical, mechanical, and chemical, including optical, thermal, electrical, and adsorptive, properties of the overall produced nanohybrids for specific applications, metal oxides, and carbon nanomaterial hybridization is a plausible strategy.30,31
Electroanalysis also entails a simple sampling formula, inexpensive instruments, a quick reaction time, and high equipment precision. In the proposed research, we employed MWNTs/Ca–ZnO NPs mixed with carbon paste to construct a sensor to analyze 1-NAA. Regarding 1-NAA, the upgraded sensor MWNTs/Ca–ZnO/CPE exhibits the advantages of improved electrocatalytic properties, exceptional sensitivity, low detection limit, accuracy, easy procedure, fast response, and stability. There are currently no reports of 1-NAA being electrochemically oxidized at MWNTs/Ca–ZnO/CPE. In this case, the nanocomposite was successfully discovered, determined, and studied using 1-NAA electroanalysis. We observed remarkable recovery with lowered detection limits when compared to other approaches.
Experimental measurements
Standard chemicals and instrumentation
Multiwalled carbon nanotubes (MWNTs) and 1-NAA were acquired from Sigma-Aldrich. The stock solution of 1-NAA was prepared in ethanol. An endorsing buffer for the analysis was a phosphate buffer (PB) solution. Its pH ranges between 3.0 and 9.2, and we employed double distilled water for the experiment.
Electrochemical analyses were carried out using a CHI D630 electrochemical device in conjunction with a three-compartment electrode cell. Of the three electrodes, a platinum wire was used as the counter electrode, and (0.3 M KCl) AgCl was used as the reference electrode. The oxidation potential of the 1-NAA analyte was determined against the Ag|AgCl electrode.
Synthesis of 5% Ca–ZnO nanoparticles
To produce 5% Ca–ZnO NPs, the co-precipitation technique was used. To achieve total zinc nitrate disintegration, a solution containing 5% mole calcium nitrate, zinc nitrate (Zn(NO3)2·4H2O) (0.1 M), and 10 mg L−1 sodium dodecyl sulfate was rapidly mixed with a stirrer for an hour. We added 0.2 M NaOH (100 mL) dropwise to the previously collected solution and mixed vigorously for 45 minutes. The reaction was allowed to proceed for two hours after adding the NaOH solution. The solution was then left to rest overnight before being discretely extracted, and centrifugation was conducted for roughly 10 to 15 min. The generated ZnO NPs were washed multiple times with double distilled water and ethanol to eliminate any impurities that were adhered to the ZnO NPs. Later, they were air-dried at 60 °C for nearly 4 h. The nanoparticles were again heated inside a muffle oven at 500 °C for 3 h.26,32
Preparation of the bare CPE and MWNTs/Ca–ZnO/CPE
Graphite powder and paraffin oil (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) were manually combined in a mortar and pestle to obtain the bare CPE, which was then allowed to self-homogenize for about 24 h. The paste was then carefully packed into the PTFE cylindrical tube chamber of the electrode, and the electrode surface was polished on a filter paper sheet. The working electrode MWNTs/Ca–ZnO/CPE was constructed by adding 0.05 mg of MWNTs and Ca–ZnO NPs each in a mortar along with graphite powder and paraffin oil and blending uniformly. Afterward, inside the PTFE tube, this uniform mixture was loaded. MWNTs/Ca–ZnO/CPE was used as an efficacious working electrode, and the study was performed for analyzing various parameters utilizing a few voltammetric approaches with satisfactory results. The voltammograms were obtained using a cyclic voltammetry (CV) approach in the potential window of 0.5 V to 1.3 V in 0.2 M pH 3.0 PB at a sweep rate of 0.05 V s−1 (Scheme 1).
:3) were manually combined in a mortar and pestle to obtain the bare CPE, which was then allowed to self-homogenize for about 24 h. The paste was then carefully packed into the PTFE cylindrical tube chamber of the electrode, and the electrode surface was polished on a filter paper sheet. The working electrode MWNTs/Ca–ZnO/CPE was constructed by adding 0.05 mg of MWNTs and Ca–ZnO NPs each in a mortar along with graphite powder and paraffin oil and blending uniformly. Afterward, inside the PTFE tube, this uniform mixture was loaded. MWNTs/Ca–ZnO/CPE was used as an efficacious working electrode, and the study was performed for analyzing various parameters utilizing a few voltammetric approaches with satisfactory results. The voltammograms were obtained using a cyclic voltammetry (CV) approach in the potential window of 0.5 V to 1.3 V in 0.2 M pH 3.0 PB at a sweep rate of 0.05 V s−1 (Scheme 1).
 |
| | Scheme 1 The flowchart for preparing the bare and modified working electrodes. | |
The Randles–Sevcik equation was applied to determine the exact surface area of the sensor. Using the CV technique, the voltammograms were recorded for an irreversible response utilizing 1.0 mM [K3Fe(CN)6] at 298 K, along with a supporting electrolyte solution of 0.1 M KCl. The equation is shown below.
| | | Ip = (26900)n3/2AD01/2ν1/2C0 | (1) |
In eqn (1), “n” denotes the total number of electrons implied in the electrochemical reaction, “A” means the surface area of the sensor, “Ip” indicates the peak current, “ν” denotes the sweep rate, “D0” denotes the diffusion coefficient, and “C0” represents the K3[Fe(CN)6] concentration (1.0 mM). Accordingly, the bare CPE surface area was estimated to be 0.026 cm2, and the modified CPE surface area was 3 to 4 times greater than that of the bare CPE.
Soil sample arrangement
Different soils, such as black, brick, red, and agricultural soil, were collected from other sources. These soil types were sampled and divided into several constituents, such as sand particles and stones. Then, these samples were screened and homogenized. After adding the appropriate quantity of soil samples to the glass beaker containing 25 mL of ultrapure distilled water, followed by vigorous mixing, then allowing to stay for eight hours, the dirty layer settled over the top can be removed. The sample solutions were also screened before being placed into volumetric flasks. After adding the 1-NAA stock solution (0.1 mM), the solution samples were mixed with demineralized water. Square wave voltammetry (SWV) and the standard addition method were used to determine the amount of 1-NAA.
Water sample arrangement
Reverse osmosis (RO), tap, and lake water samples were collected. The water samples were filtered before the examination to eliminate any suspended pollutants. In an electrochemical cell, each water sample was combined with PB (5.0 mL) of pH 3.0, and the blank performance was determined using SWV. The stock solution of 1-NAA (0.1 mM) was spiked into 50 mL water to prepare the samples for analysis, and consecutive voltammograms were recorded.
Vegetable and fruit sample arrangement
Vegetables and fruits like carrots, cucumbers, potatoes, radishes, tomatoes, and grapes were obtained from the local market. The determination of 1-NAA was conducted in these vegetable and fruit samples. 5.0 g of vegetable and fruit samples each was put in a mortar containing 5.0 mL of pH 3.0 PB and carefully crushed with a pestle until a fine liquid sample was obtained. Using the standard addition method, 1-NAA was then determined using the extract solutions of the vegetables and fruits.
Results and discussion
Characterization of 5% Ca–ZnO NPs
XRD, EDX, SEM, and TEM were used to study the structure and morphology of the synthesized Ca–ZnO nanoparticles. The band gap transitions, blue shift, as well as other optical characteristics of ZnO are influenced by Ca doping. To stabilize lattice systems and enhance the chemical bond ionicity in ZnO systems, ZnO doping with Ca2+ species in particular is more successful since the Ca2+ element has a larger ionic size than the host Zn2+ cation. Fig. 1(A) demonstrates the XRD spectra with a prominent peak for zinc oxide, and the lower 2-theta degrees correspond to calcium content peaks. The lattice planes and the diffraction peaks all seem to be compatible. Lattice planes show that the synthesized nanoparticles have a hexagonal wurtzite structure. This pattern, caused by the difference between Ca (1.94 Å) and Zn (1.42 Å) ion sizes, shows a local strain in the hexagonal lattice caused by some residual focus in the nanoparticles. The average crystal size, d, was calculated using Scherrer's formula from the XRD spectra.| | 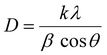 | (2) |
Here k is a constant, θ denotes the Bragg angle, and λ represents the wavelength. The atomic composition determined by TEM energy-dispersive X-ray (EDX) spectroscopy examination of the specific calcium-doped zinc oxide nanoparticles is Ca:Zn:O = 5.37:58.74:35.88. The sample consists of Ca, Zn, and O, and the ratio of Zn/O equals 1.6 (Fig. 1(B)). The surface structure of 5% Ca-doped zinc oxide NPs were examined using a scanning electron microscope (SEM). From the SEM analysis, they were found to have varying sizes and shapes, indicating that the surface area is relatively vast (Fig. 1(C)). Calcium doping using the precursor solution, particularly facilitating the nucleation phase, makes the doped layers dense. The size of the particles and also the shape of 5% Ca–ZnO nanoparticles were examined through TEM analysis. The grains in the nanoparticle sample have an unusually dispersed particle size, as shown in Fig. 1(D). The dark specks in the TEM image are thought to be dopants of Ca that are attached to ZnO nanoparticles that are between 30 and 75 nm in length and width.
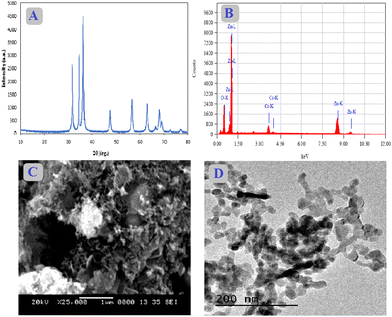 |
| | Fig. 1 (A) XRD image, (B) EDX image, (C) SEM image, and (D) TEM image of Ca–ZnO NPs. | |
Electrochemical performance of 1-NAA
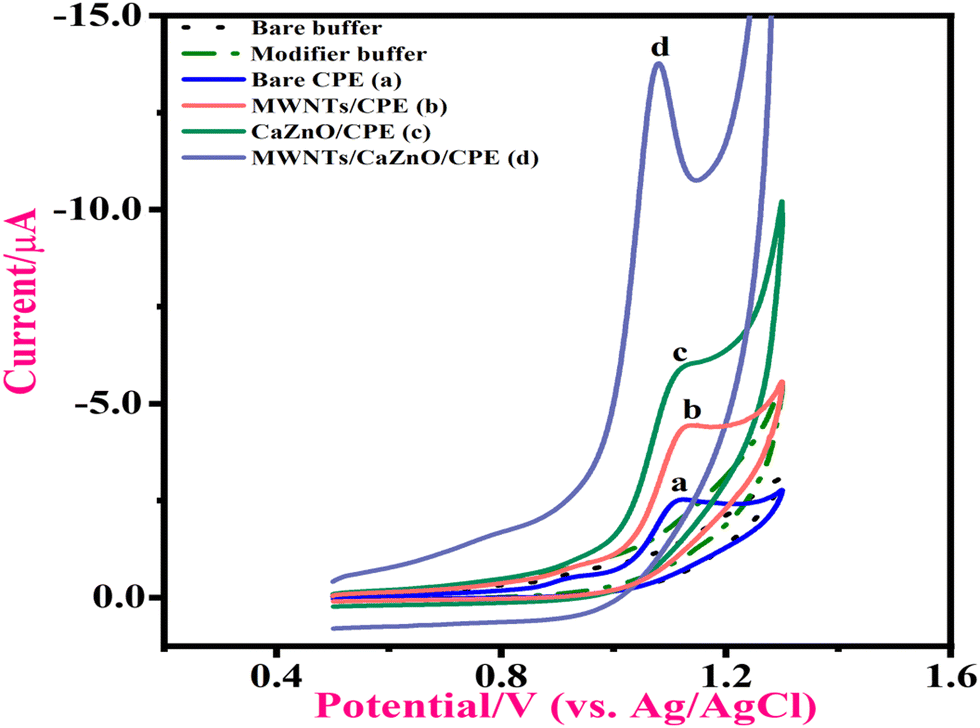
The CV scans showed no peaks at the bare CPE as well as MWNTs/Ca–ZnO/CPE in the presence of PB and in the absence of 1-NAA. But in the presence of 1-NAA, its electrochemical performance was studied at MWNTs/Ca–ZnO/CPE at a 50 mV s−1 scan rate. However, with the inclusion of 1-NAA in pH 3.0 PB, the analyte's peak current was significantly higher than that of the bare CPE at MWNTs/Ca–ZnO/CPE. Fig. 2 shows that the increased peak current (Ipa) of 1-NAA at the modified sensor was 13.78 μA at Epa 1.081 V, and at the bare CPE, it was found to be 2.52 μA at 1.127 V. The peak current of the upgraded electrode appeared to be 5 or 6 times greater than that of the bare CPE. There were no peaks present in the reverse scan. Thus, the reaction is irreversible. Because of the prominent area and high electrical conductivity of the MWNTs/Ca–ZnO NPs, the electrochemical oxidation peak current of 1-NAA at MWNTs/Ca–ZnO/CPE was enhanced.
 |
| | Fig. 2 Cyclic voltammetry performance of 1-NAA in pH 3.0 PB; ν = 50 mV s−1, tacc = 25 s. The dotted lines are for the buffer solution. (a) Bare CPE, (b) MWNTs/CPE, (c) Ca–ZnO/CPE, and (d) MWNTs/Ca–ZnO/CPE. | |
The importance of accumulation time
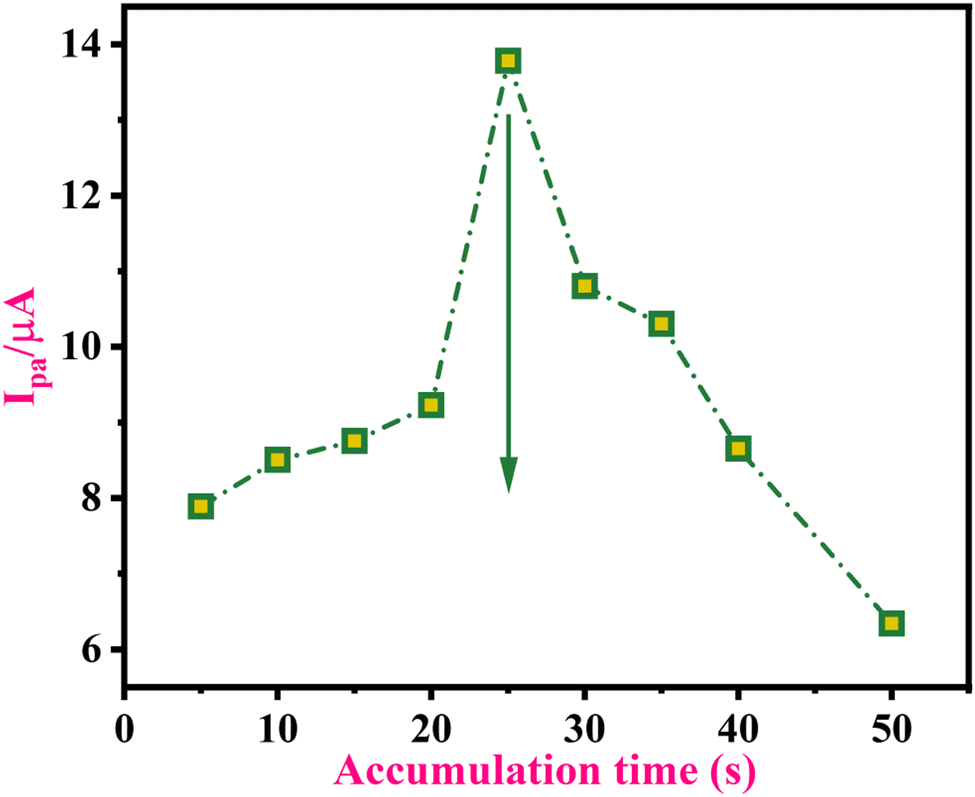
The electrochemical behaviour of the analyte mainly depends on its presence on the sensor surface. Diffusion results from a change in the concentration between two sites within the electrochemical cell; this concentration gradient causes the analyte to move from high to low concentration areas. In an electrolysis study, diffusion is vital because the conversion reaction solely occurs at the electrode surface. When voltage is not applied, the diffusion gradient causes the analyte to move from the bulk solution to the surface of the electrode. An electrode-solution interface is significant in electro-analysis because the concentration of the analyte near the electrode surface might impact the electrochemical parameters. Therefore, in the present experiment, voltammograms were obtained at various time intervals to examine the effect of the vicinity concentration. The impact of accumulation time was examined for 5–50 s at the MWNTs/Ca–ZnO/CPE surface in PB at pH 3.0 to measure efficiency. The best performance of the analyte was observed at 25 s, as shown in Fig. 3. Thus, 25 s was chosen as the optimal time for each parameter throughout the research.
 |
| | Fig. 3 The effect of accumulation time of 0.1 mM 1-NAA at MWNTs/Ca–ZnO/CPE at a 50 mV s−1 sweep rate in pH 3.0 PB at different time points: 5 s, 10 s, 15 s, 20 s, 25 s, 30 s, 35 s, 40 s, and 50 s. | |
The significance of pH variation
The pH of the electrolyte may influence the electrode surface response.24,25 The modified sensor showed cyclic voltammograms in the 0.5–1.3 V potential window. The CV approach was used to investigate the significance of the pH variation of 1-NAA at MWNTs/Ca–ZnO/CPE from pH 3.0 to 9.2 of PB with a 0.05 V s−1 scan rate. Fig. 4 shows that as the pH increased from 3.0 to 4.2, there was a sudden decrease in the peak potential, and then the peak potential was almost constant until pH 9.2. The linear regression equation was found for pH 3.0 and 4.2 as Epa (V) = 1.23–0.052 (pH); R2 = 1. The slope (−0.052 V per pH) was remarkably close to the theoretical slope value (−0.055 V per pH) of the Nernst equation, showing that the same number of protons and electrons are engaged in this electrode response mechanism. Simultaneously, the peak current vs. pH curve and the peak potential against pH curve had nearly the same tendency, i.e., there was a sudden decrease in the peak current from pH 3.0 to 4.2, and then nearly constant peak currents were observed from pH 4.2 to 9.2. Thus, the electrochemical research findings conclude that the 1-NAA oxidation is an irreversible reaction.
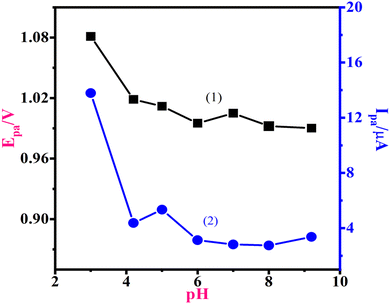 |
| | Fig. 4 Significance of pH on the (1) Epa and (2) Ipa of 1-NAA. | |
The impact of the scan rate
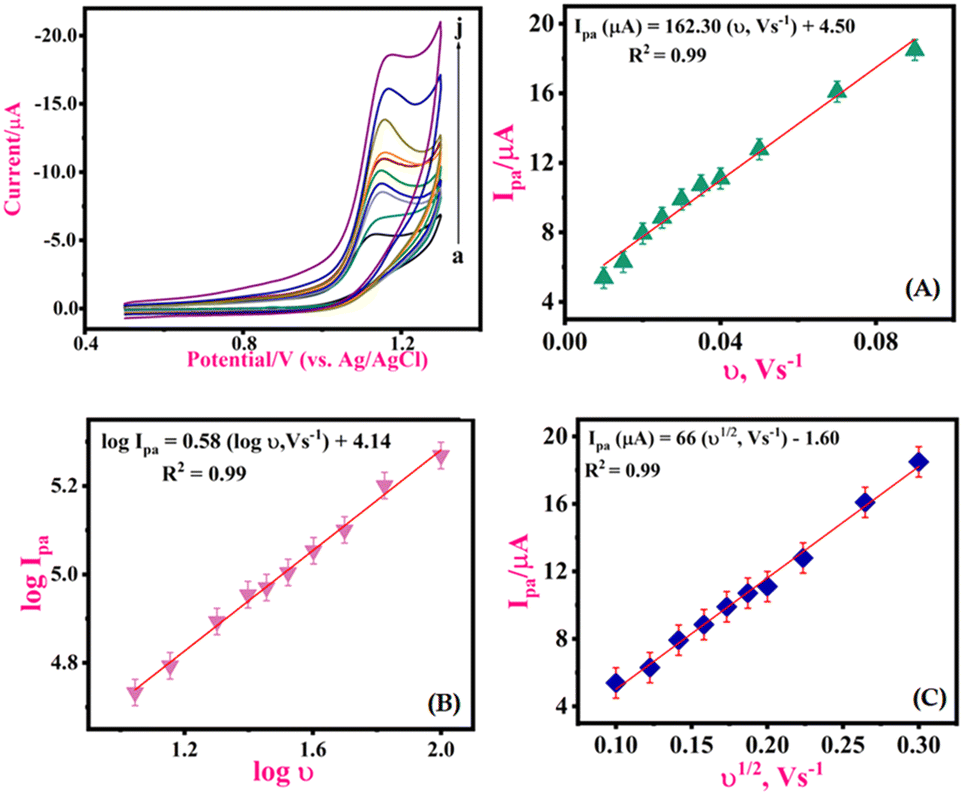
By analyzing the variation of the scan rate, the electrochemical process of the 1-NAA analyte at the MWNTs/Ca–ZnO/CPE was explained. An elevation in anodic peak current was observed when the sweep rate was varied between 0.01 and 0.09 V s−1 (Fig. 5). To study the variance of the sweep rate, the CV approach was used for 0.1 mM 1-NAA in pH 3.0 PB at MWNTs/Ca–ZnO/CPE. So, as the scan rate was increased, there was a linear elevation in the anodic peak current, as in the equation Ipa (μA) = 162.30 (ν, V s−1) + 4.50; R2 = 0.99, which can be seen from Fig. 5(A). The regression representation of the plot of logIpavs. logν is reported to have a linear relationship (Fig. 5(B)): logIpa (μA) = 0.58 (logν, V s−1) + 4.14; R2 = 0.99. and Ipavs. ν1/2 (Fig. 5(C)); Ipa (μA) = 66 (ν1/2, V s−1) – 1.60; R2 = 0.99. The slope of the logIpavs. logν plot shows that the mechanism is a diffusion-controlled process. Peak potential (Epa) was shown to be linearly correlated with logν: Epa (V) = 1.19–0.02 logν; R2 = 0.99. For an irreversible reaction, the anodic peak potential (Epa) and scan rate are correlated, as shown by the Laviron equation.33–35| |  | (3) |
| | 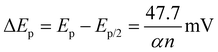 | (4) |
The formal redox potential is denoted as “E0”, the contributing number of electrons in the electrode process is marked as “n = 2”, the charge transfer coefficient is designated as “α”, the scan rate is denoted as “ν” and the standard heterogeneous rate constant is marked as “k0”, which is determined to be 0.055 s−1 in the Laviron equation (eqn (3)). Ep/2 signifies the half of peak potential, and the current equals the half of the peak value; however, in the Bard and Faulkner formula (eqn (4)),36 the “α” value is 0.55.
 |
| | Fig. 5 CVs for 1-NAA (0.1 mM) at MWNTs/Ca–ZnO/CPE for multiple scan rates (in V s−1): (a–j) 0.01, 0.015, 0.02, 0.025, 0.03, 0.035, 0.04, 0.05, 0.07, and 0.09; pH = 3.0, tacc = 25 s. Plots of (A) Ipavs. logν, (B) logIpavs. logν and (C) Ipa μA−1vs. ν1/2. | |
The conversion of alcohol 2 to aldehyde 3 results in the oxidation peak of 1-NAA. At the measured potential, the oxidation of both 1-NAA to alcohol 2 and alcohol 2 to aldehyde 3 takes place. This involves decarboxylating 1-NAA on the electrode surface, producing an alkyl radical, which then transforms into alcohol 2. The possible mechanism of 1-NAA oxidation is shown in Scheme 2. It demonstrates that the side chain of 1-NAA undergoes oxidation.37
 |
| | Scheme 2 The possible electro-oxidation mechanism of 1-NAA. | |
Analytical significance
The effect of concentration variance
The SWV approach was used to modify the 1-NAA concentration between 0.1 × 10−7 M and 1.6 × 10−7 M to investigate the sensitivity of the MWNTs/Ca–ZnO/CPE sensor at pH 3.0. The equation Ipa (μA) = 0.83 (C0, 10−7 M L−1) + 1.22; R2 = 0.99 was observed to possess a linear relationship with the plot of peak current vs. variance of 1-NAA concentration (Fig. 6). The formulae LOD = 3 S/M and LOQ = 10 S/M, respectively, were used to compute the limits of detection (LOD) and quantification (LOQ), where S stands for the blank standard deviation and M for the slope.38,39 According to calculations, the LOD and LOQ are 0.025 nM and 0.084 nM. The LOD and LOQ are low, indicating that the developed sensor possesses excellent sensitivity. So the present investigation is competent in detecting and validating the 1-NAA analyte. Table 1 summarizes the results of the reported methods with the current research work.
 |
| | Fig. 6 SWV curves were measured in the potential window of +0.8 V to +1.3 V for multiple concentrations of 1-NAA at MWNTs/Ca–ZnO/CPE: (a–k) from 0.1 × 10−7 M to 1.6 × 10−7 M. (A) Plot of Ipa/A vs. [1-NAA]/10−7 M. | |
Table 1 Comparison of LODs for 1-NAA detection
| Electrode material or substance |
Linear range |
LOD |
Ref. |
| DHP – dihexadecyl hydrogen phosphate, BPPF6 – N-butyl-pyridinium hexafluorophosphate, AB – acetylene black, CA – citric acid. |
| A non-ionic solid polymeric resin, Amberlite XAD-7 |
0–500 ng mL−1 |
1.2 ng mL−1 |
11
|
| Air-stable lipid films containing the auxin-binding protein 1 receptor |
1–10 μM |
0.1 μM |
40
|
| MBT-PVP Cu NPs |
0.5–50 μM |
9.6 nM |
41
|
| MWCNT/GCE |
1.0 × 10−8 to 2.0 × 10−6 mol L−1 |
2.5 × 10−9 mol L−1 |
42
|
| AB/DHP/GCE |
4.0 × 10−8–5.0 × 10−6 mol L−1 |
1.0 × 10−8 mol L−1 |
43
|
| BPPF6/CPE |
2.0 × 10−5–4.0 × 10−4 mol L−1 |
1.7 × 10−7 mol L−1 |
44
|
| NAA immunosensor |
2.7 × 10−7–1.1 × 10−5 mol L−1 |
1.9 × 10−7 mol L−1 |
45
|
| MWCNTs/Ca-doped ZnO/CPE |
0.1–1.6 × 10−7 M |
0.025 nM |
This work |
Analysis of soil samples
The agricultural and ecological applications were determined by examining the soil samples. As revealed by the satisfactory recovery results shown in Table 2, the proposed sensor is adequate to analyze 1-NAA. Between 84.83 and 99.92% of the 1-NAA analyte was recovered. Thus, based on our observations, the concentrations ranged from 0.47 μM to 1.49 μM.
Table 2 Detection of 1-NAA in spiked soil samples
| Sample |
Spiked (10−6 M) |
Detected (10−6 M) |
Recovery (%) |
RSD (%) |
| Brick soil |
0.5 |
0.49 |
99.18 |
2.33 |
| 1.0 |
0.99 |
99.25 |
2.33 |
| 1.5 |
1.42 |
95.20 |
2.43 |
| Farm soil |
0.5 |
0.48 |
96.28 |
0.48 |
| 1.0 |
0.96 |
96.50 |
0.48 |
| 1.5 |
1.45 |
97.17 |
0.47 |
| Black soil |
0.5 |
0.47 |
94.53 |
5.13 |
| 1.0 |
0.84 |
84.83 |
5.72 |
| 1.5 |
1.35 |
90.26 |
5.38 |
| Red soil |
0.5 |
0.49 |
99.92 |
1.50 |
| 1.0 |
0.97 |
97.10 |
1.54 |
| 1.5 |
1.49 |
99.40 |
1.50 |
Analysis of water samples
The SWV approach has been used to analyze the 1-NAA molecule in water samples. Standard solutions of 1-NAA in known amounts were added to pretreated water samples to assess the analyte recovery. Table 3 summarizes the findings of the water sample analysis. The percentage of recovery varied from 97.52 to 99.70%.
Table 3 Detection of 1-NAA in water samples
| Sample |
Spiked (10−6 M) |
Detected (10−6 M) |
Recovery (%) |
RSD (%) |
| RO water |
0.5 |
0.49 |
99.46 |
0.34 |
| 1.0 |
0.99 |
98.43 |
0.34 |
| 1.5 |
1.48 |
98.79 |
0.34 |
| Tap water |
0.5 |
0.49 |
98.85 |
1.09 |
| 1.0 |
0.99 |
99.7 |
1.08 |
| 1.5 |
1.46 |
97.55 |
1.10 |
| Lake water |
0.5 |
0.49 |
97.52 |
0.17 |
| 1.0 |
0.99 |
99.7 |
0.17 |
| 1.5 |
1.49 |
99.52 |
0.17 |
Analysis of vegetable and fruit samples
Due to its improved sensitivity and stability, the modified MWNTs/Ca–ZnO/CPE sensor may be used in actual analytical applications. The recovery data were acquired using the SWV technique, which required adding multiple amounts of fruit and vegetable samples to a specific amount of the 1-NAA sample in pH 3.0 PB. The percentage of recovery varied from 97.20 to 99.68% (Table 4). Depending on the excellent outcomes, the proposed approach is effective, precise, economical, and favorable for 1-NAA analysis in real samples.
Table 4 Detection of 1-NAA in vegetable and fruit samples
| Sample |
Spiked (10−6 M) |
Detected (10−6 M) |
Recovery (%) |
RSD (%) |
| Carrot |
0.5 |
0.49 |
98.05 |
0.64 |
| 1.0 |
0.98 |
98.44 |
0.64 |
| 1.5 |
1.48 |
99.29 |
0.64 |
| Cucumber |
0.5 |
0.48 |
97.96 |
0.30 |
| 1.0 |
0.98 |
98.09 |
0.30 |
| 1.5 |
1.47 |
98.53 |
0.30 |
| Grapes |
0.5 |
0.49 |
98.84 |
0.16 |
| 1.0 |
0.99 |
99.06 |
0.16 |
| 1.5 |
1.48 |
99.15 |
0.16 |
| Potato |
0.5 |
0.49 |
99.68 |
1.13 |
| 1.0 |
0.97 |
97.50 |
1.15 |
| 1.5 |
1.47 |
98.08 |
1.15 |
| Radish |
0.5 |
0.48 |
97.47 |
0.14 |
| 1.0 |
0.97 |
97.34 |
0.14 |
| 1.5 |
1.45 |
97.20 |
0.14 |
| Tomato |
0.5 |
0.49 |
98.80 |
0.38 |
| 1.0 |
0.98 |
98.05 |
0.38 |
| 1.5 |
1.47 |
98.53 |
0.38 |
The effect of temperature variation
To investigate the effect of temperature on 1-NAA degradation and electrochemical oxidation, CV was performed in PB with a 0.05 V s−1 scan rate at MWNTs/Ca–ZnO/CPE and an accumulation time of 25 s. The peak current of 1-NAA increased linearly as its temperature increased from 288 K to 308 K (Fig. 7). With thermodynamic equations (eqn (5) and (6)), the graph of log (Ipa) of the analyte versus 1/temperature (T) was analyzed to evaluate its linearity. Deviations in the diffusibility of the 1-NAA molecule may have affected the increase in oxidation current. When the temperature increases, the activation energy (Ea) of the harmful chemical 1-NAA is influenced significantly by the conductivity properties. Furthermore, as per the Arrhenius equation,46–48 the activation energy of the 1-NAA peak current is 28.31 kJ mol−1.| |  | (5) |
| |  | (6) |
Here, σ/D represents the conductivity/diffusivity, σ0/D represents the normal conductivity/initial diffusivity, Ea refers to the activation energy, R indicates the universal gas constant (8.314 J mol k−1), and T stands for the temperature (kelvin). The Eyring equation, along with the thermodynamic equations (eqn (7) and (8)), was used to determine the enthalpy shift (H*) and entropy change (S*) of 1-NAA at the MWNTs/Ca–ZnO/CPE.| | 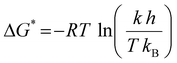 | (7) |
Here, H* is required to check if the electrode response is spontaneous, and even when S* is positive and H* is negative, then G* is negative as per the Gibbs free energy. The Boltzmann constant, kB (1.381 × 10−23 m2 kg s−2 k−1), and the Planck constant, h (6.66 × 10−34 J s), are also included in the equations mentioned above. The electrochemical reaction of 1-NAA was inquired at different temperatures to see if the electrode reactions were spontaneous throughout the electrode's response. The working sensor acted as an electrocatalyst to drive the processes as MWNTs/Ca–ZnO NPs were used, elevating the system's peak current. Table 5 lists the thermodynamic variables and the Ea* values.
 |
| | Fig. 7 The temperature effect on the 1-NAA (0.1 mM) peak current at MWNTs/Ca–ZnO/CPE (288–308 K). (A) logIpavs. 1/T K−1. | |
Table 5 Thermodynamic parameters for 1-NAA at MWNTs/Ca–ZnO/CPE
| Thermodynamic parameters |
Obtained values |
|
E
a* (kJ mol−1) |
28.31 |
| ΔH* (kJ mol−1) |
25.84 |
| ΔS* (kJ mol−1) |
−136 |
| ΔG* (kJ mol−1) |
66.35 |
| logA |
6.11 |
Repeatability, reproducibility, and long-term stability of the proposed sensor
The constructed electrode was stored in a sealed jar for around 21 days to ensure its protracted reliability and repeatability. With this stored sensor, the voltammograms of 0.1 mM 1-NAA were recorded. The electrode exhibited a 97–98% peak value, showing a steady MWNTs/Ca–ZnO/CPE sensor. An intra-day research was performed with five different electrodes. Inter-day research for five days with five new electrodes was conducted at ambient temperature to ensure reproducibility, short-term stability, and long-term stability. The peak potentials remained almost unchanged, and the current signals retained 99.85% (Fig. 8(A)) and 99.06% (Fig. 8(B)) of their first early current response, which shows the long-term stability of the sensor. The 1-NAA concentration was maintained steady. With a relative standard deviation (RSD) of 2.07%, the developed MWNTs/Ca–ZnO/CPE displayed exceptional reproducibility and long-term stability (Fig. 8 and 9).
 |
| | Fig. 8 Stability study to ensure (A) short-term stability and (B) long-term stability. | |
 |
| | Fig. 9 (A) Repeatability and (B) reproducibility of the MWNTs/Ca–ZnO/CPE sensor. | |
Interference
The ability of a sensor to differentiate between the particular analyte molecule and interfering molecules in the sample is a crucial factor. To assess the selectivity of the devised sensor, the metal ions, excipients, and stimulator were individually spiked in pH 3.0 PB (0.2 M) along with 0.1 mM of 1-NAA. SWV was adopted to examine the results. Fig. 10 demonstrates that MnSO4, KNO3, FeSO4, CuSO4, citric acid, glycine, ascorbic acid, starch, and indole-2-acetic acid are 100 times more concentrated than the harmful molecule, and the 1-NAA signal maintains its integrity.
 |
| | Fig. 10 Selectivity of the sensor in the presence of various interferents. | |
Conclusions
Toxic chemicals cause harm to biological systems by disrupting biochemical processes and causing deleterious, even fatal, consequences. To avoid the effect of toxic molecules and safeguard human health and the environment, advanced harmful molecule detection methods are crucial. MWNTs/Ca–ZnO/CPE was developed to investigate the electrochemical behaviour of 1-NAA, a potentially hazardous compound. XRD, EDX, SEM, and TEM methods enabled the characterization of nanoparticles. The low limit of detection, good sensitivity, reproducibility, rapid response, electrocatalytic effectiveness, and repeatability of the proposed sensor were all established using SWV and CV approaches. The 1-NAA molecule was detected in real samples such as vegetables, soil, fruits, and water. The constructed MWNTs/Ca–ZnO/CPE sensor had a low detection limit of 0.025 nM for the 1-NAA analyte. The modified sensor is economical, simple, ecological, effective, productive, and captivating from a reasonable standpoint, and therefore it fits the ecological requirements for sensors.
Author contributions
Keerthi Prabhu: conceptualization; data curation; formal analysis; investigation; methodology; writing – original draft. Shweta J. Malode: resources; methodology; supervision; validation; visualization; writing – original draft; writing – review & editing. Raviraj M. Kulkarni: resources; methodology; investigation. Nagaraj P. Shetti: resources; methodology; supervision; validation; visualization; writing – review & editing.
Conflicts of interest
There are no conflicts of interest to declare.
References
- M. Zhang, Y. Yang, Y. Wang, B. Zhang, H. Wang, G. Fang and S. Wang, Food Control, 2022, 132, 108532, DOI:10.1016/j.foodcont.2021.108532.
- J. Blaškovičová and J. Labuda, Sensors, 2022, 22(12), 4332, DOI:10.3390/s22124332.
- H. Li, Z. Deng, Q. Tian, L. Lun, P. Zhao, X. Yang, J. Shen, B. Jiang, Y. Zhou and T. Zhou, Int. J. Environ. Anal. Chem., 2022, 102(4), 987–1000, DOI:10.1080/03067319.2020.1730338.
- L. Janíková, J. Chýlková, R. Šelešovská, M. Sedlák, J. Váňa, L. Dušek and J. Bartáček, J. Electroanal. Chem., 2020, 859, 113855, DOI:10.1016/j.jelechem.2020.113855.
- Z. Wang, H. Wu, Y. He, Y. Yan, W. Zhou, G. Zhang, D. Liu, Z. Ye and F. Qiu, ChemistrySelect, 2022, 7(22), e202201141, DOI:10.1002/slct.202201141.
- H. Zhou and Q. Lu, Microchem. J., 2022, 175, 107144, DOI:10.1016/j.microc.2021.107144.
- X. Huang, A. Jiang, X. Jin, H. Yu, Y. Zhang, S. Xia, Z. Liu, G. Zhang and D. D. Dionysiou, J. Environ. Chem. Eng., 2021, 9(5), 106022, DOI:10.1016/j.jece.2021.106022.
- W. Guan, P. Xu, K. Wang, Y. Song and H. Zhang, Food Chem. Toxicol., 2011, 49(11), 2869–2874, DOI:10.1016/j.fct.2011.08.009.
- Z.-H. Wang, J.-F. Xia, Q. Han, H.-N. Shi, X.-M. Guo, H. Wang and M.-Y. Ding, Chin. Chem. Lett., 2013, 24(7), 588–592, DOI:10.1016/j.cclet.2013.04.015.
- W. K. Li, J. Chen, H. X. Zhang and Y. P. Shi, Talanta, 2017, 168, 136–145, DOI:10.1016/j.talanta.2017.03.034.
- G. Li, S. Liu, Z. Sun, L. Xia, G. Chen and J. You, Food Chem., 2015, 170, 123–130, DOI:10.1016/j.foodchem.2014.07.146.
- G. Manasa, A. K. Bhakta, J. Bafna, R. J. Mascarenhas, S. J. Malode and N. P. Shetti, Environ. Res., 2022, 212, 113541, DOI:10.1016/j.envres.2022.113541.
- V. Erady, R. J. Mascarenhas, A. K. Satpati, A. K. Bhakta, Z. Mekhalif and J. Delhalle, Surf. Interfaces, 2020, 19, 100517, DOI:10.1016/j.surfin.2020.100517.
- G. Manasa, R. J. Mascarenhas, N. P. Shetti, S. J. Malode, A. Mishra, S. Basu and T. M. Aminabhavi, ACS Appl. Bio Mater., 2022, 5(3), 945–970, DOI:10.1021/acsabm.1c01289.
- G. Manasa, R. J. Mascarenhas, A. K. Bhakta and Z. Mekhalif, MWCNT, Electroanalysis, 2020, 32(5), 939–948, DOI:10.1002/elan.201900573.
- A. K. Bhakta, S. Kumari, S. Hussain, P. Martis, R. J. Mascarenhas, J. Delhalle and Z. Mekhalif, J. Mater. Sci., 2019, 54(1), 200–216, DOI:10.1007/s10853-018-2818-y.
- G. Manasa, R. J. Mascarenhas, N. P. Shetti, S. J. Malode and T. M. Aminabhavi, ACS Biomater. Sci. Eng., 2022, 8(7), 2726–2746, DOI:10.1021/acsbiomaterials.2c00390.
-
S. J. Malode, P. Sharma, M. R. Hasan, N. P. Shetti and R. J. Mascarenhas, Carbon and Carbon Paste Electrodes, Electrochemical Sensors, Woodhead Publishing, 2022, pp. 79–114. DOI:10.1016/B978-0-12-823148-7.00004-0.
- Y. Wang and W. Liang, Curr. Pollut. Rep., 2021, 7(1), 31–39, DOI:10.1007/s40726-021-00173-9.
- O. I. Dar, R. Aslam, D. Pan, S. Sharma, M. Andotra, A. Kaur, A.-Q. Jia and C. Faggio, Environ. Technol. Innovation, 2022, 25, 102122, DOI:10.1016/j.eti.2021.102122.
- H. V. Pereira-Maróstica, L. Bracht, J. F. Comar, R. M. Peralta, A. Bracht and A. B. Sá-Nakanishi, Toxicol. Appl. Pharmacol., 2022, 442, 115987, DOI:10.1016/j.taap.2022.115987.
- W. Huang, W. Qu and D. Zhu, Bull. Korean Chem. Soc., 2008, 29(7), 1323–1325, DOI:10.5012/bkcs.2008.29.7.1323.
- S. Lü, Anal. Lett., 2003, 36(8), 1523–1534, DOI:10.1081/AL-120021534.
- C.-Q. Duan, Y.-M. Zhang and Z.-N. Gao, Croat. Chem. Acta, 2012, 85, 27–32, DOI:10.5562/cca1901.
- G. Manasa, R. J. Mascarenhas, S. J. Malode and N. P. Shetti, Biosens. Bioelectron., 2022, 11, 100189, DOI:10.1016/j.biosx.2022.100189.
- S. J. Malode, P. K. Keerthi, N. P. Shetti and R. M. Kulkarni, Electroanalysis, 2020, 32(7), 1590–1599, DOI:10.1002/elan.201900776.
- N. Sebastian, W. C. Yu and D. Balram, Anal. Chim. Acta, 2020, 1095, 71–81, DOI:10.1016/j.aca.2019.10.026.
- N. P. Shetti, D. S. Nayak, S. J. Malode, R. R. Kakarla, S. S. Shukla and T. M. Aminabhavi, Anal. Chim. Acta, 2019, 1051, 58–72, DOI:10.1016/j.aca.2018.11.041.
- E. Rozhina, S. Batasheva, R. Miftakhova, X. Yan, A. Vikulina, D. Volodkin and R. Fakhrullin, Appl. Clay Sci., 2021, 205, 106041, DOI:10.1016/j.clay.2021.106041.
- N. P. Shetti, S. J. Malode, D. S. Nayak, C. Venkata Reddy and K. R. Raghava Reddy, Mater. Res. Express, 2019, 6(11), 116308, DOI:10.1088/2053-1591/ab4471.
- N. P. Shetti, S. J. Malode, D. S. Nayak and K. R. Reddy, Mater. Res. Express, 2019, 6(11), 115085, DOI:10.1088/2053-1591/ab4b92.
- D. R. Kulkarni, S. J. Malode, K. Keerthi Prabhu, N. H. Ayachit, R. M. Kulkarni and N. P. Shetti, Mater. Chem. Phys., 2020, 246, 122791, DOI:10.1016/j.matchemphys.2020.122791.
- E. Laviron, J. Electroanal. Chem. Interfacial Electrochem., 1979, 101(1), 19–28, DOI:10.1016/S0022-0728(79)80075-3.
- S. J. Malode, K. Keerthi Prabhu and N. P. Shetti, New J. Chem., 2020, 44(44), 19376–19384, 10.1039/D0NJ04644B.
- S. J. Malode, K. Keerthi Prabhu, N. P. Shetti and K. R. Reddy, Environ. Technol. Innovation, 2021, 21, 101222, DOI:10.1016/j.eti.2020.101222.
-
A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, 2nd edn, Wiley, New York, 2001 Search PubMed.
- L. Janikova, J. Chylkova, R. Selesovska, M. Sedlak and J. Vana, J. Electroanal. Chem., 2020, 859, 113855, DOI:10.1016/j.jelechem.2020.113855.
- K. Prabhu, S. J. Malode and N. P. Shetti, Environ. Technol. Innovation, 2021, 23, 101687, DOI:10.1016/j.eti.2021.101687.
- N. P. Shetti, D. Ilager, S. J. Malode, D. Monga, S. Basu and K. R. Reddy, Mater. Sci. Semicond. Process., 2020, 120, 105261, DOI:10.1016/j.mssp.2020.105261.
- L. An, J. Ma, D. Qin, H. Wang, Y. Yuan, H. Li, R. Na and X. Wu, J. Agric. Food Chem., 2019, 67(4), 1292–1301, DOI:10.1021/acs.jafc.8b05233.
- L. Li, J. Chen, Y. Li, N. Song, L. Zhu and Z. Li, Spectrochim. Acta, Part A, 2020, 224, 117433, DOI:10.1016/j.saa.2019.117433.
- S. Lü, Anal. Lett., 2003, 36, 1523–1534 CrossRef.
- W. Huang, W. Qu and D. Zhu, Bull. Korean Chem. Soc., 2008, 29, 1323–1326 CrossRef CAS.
- C.-Q. Duan, Y.-M. Zhang and Z.-N. Gao, Croat. Chem. Acta, 2012, 85, 27–32 CrossRef CAS.
- S. Z. Liang, J. R. Pan, X. M. Yin and K. Sui, Adv. J. Food Sci. Technol., 2015, 9, 439–443 CrossRef CAS.
- S. J. Malode, N. P. Shetti and S. T. Nandibewoor, J. Solution Chem., 2010, 39(3), 417–430, DOI:10.1007/s10953-010-9501-5.
- K. Prabhu, S. J. Malode, N. P. Shetti and R. M. Kulkarni, Chemosphere, 2022, 287, 132086, DOI:10.1016/j.chemosphere.2021.132086.
- S. J. Malode, N. P. Shetti and S. T. Nandibewoor, Catal. Lett., 2011, 141, 1526–1540, DOI:10.1007/s10562-011-0623-1.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Shweta J.
Malode
a,
Shweta J.
Malode