
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From benzofuro-, benzothieno- and 10-methylindolo-[2,3-b]-fused benzothiopyrano[4,3,2-de]quinolines to the corresponding benzothiopyrano[4,3,2-de]1,8-naphthyridines: synthesis and properties of these hexacyclic heteroaromatic compounds†
Nicolas
Mast
 a,
William
Erb
a,
Lionel
Nauton
b,
Pascale
Moreau
*b,
Olivier
Mongin
a,
Thierry
Roisnel
a,
Margaux
Macaigne
c,
Thomas
Robert
cd,
Stéphane
Bach
cde,
Laurent
Picot
f,
Valérie
Thiéry
f,
Jean-Pierre
Hurvois
a and
Florence
Mongin
*a
a,
William
Erb
a,
Lionel
Nauton
b,
Pascale
Moreau
*b,
Olivier
Mongin
a,
Thierry
Roisnel
a,
Margaux
Macaigne
c,
Thomas
Robert
cd,
Stéphane
Bach
cde,
Laurent
Picot
f,
Valérie
Thiéry
f,
Jean-Pierre
Hurvois
a and
Florence
Mongin
*a
aUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, F-35000 Rennes, France. E-mail: florence.mongin@univ-rennes1.fr
bUniversité Clermont Auvergne, CNRS, Clermont Auvergne INP, ICCF, F-63000 Clermont-Ferrand, France. E-mail: pascale.moreau@uca.fr
cSorbonne Université, CNRS, FR2424, Plateforme de criblage KISSf (Kinase Inhibitor Specialized Screening facility), Station Biologique de Roscoff, Place Georges Tessier, 29680 Roscoff, France
dSorbonne Université, CNRS, UMR8227, Integrative Biology of Marine Models Laboratory (LBI2M), Station Biologique de Roscoff, 29680 Roscoff, France
eCentre of Excellence for Pharmaceutical Sciences, North-West University, Private Bag X6001, Potchefstroom 2520, South Africa
fLa Rochelle Université, Laboratoire Littoral Environnement et Sociétés, UMRi CNRS 7266, Université de La Rochelle, 17042 La Rochelle, France
First published on 28th November 2022
Abstract
Polycyclic heterocycles such as 1- and 2-aminothioxanthones are important precursors to molecules endowed with different properties and original syntheses are always required. Here, 1-aminothioxanthone was obtained either in four steps from commercially available thioxanthone, or in five steps from 3-fluoroaniline. As for 2-aminothioxanthone, direct N-arylation using commercial 2-chlorothioxanthone gave its Boc-protected derivative. From 1-aminothioxanthone, original 10-methylbenzothiopyrano[4,3,2-de]indolo[2,3-b]quinoline was synthesized by tandem N-arylation-cyclization using 2-iodo-N-methylindole in the presence of copper. However, this approach being less efficient with 2-iodobenzothiophene, we also studied another strategy. Our efforts were next concentrated on the synthesis of benzofuro-, benzothieno- and indolo[2,3-b]benzothiopyrano[4,3,2-de]1,8-naphthyridines which are original hexacycles of helical nature. Different approaches were designed and we achieved our goal from 2-chloro-4-fluoropyridine, in five steps. The original polycyclic compounds obtained were evaluated as protein kinase inhibitors, and the results were rationalized by molecular modeling experiments. Finally, their photophysical properties were measured for the purpose of structure-property relationships.
Introduction
Polyaromatic hydrocarbons (PAHs) are organic scaffolds that exhibit significant biological or/and physicochemical properties, especially when they incorporate heteroatoms such as nitrogen, oxygen and sulfur.1,2 Moreover, compared to planar ones, heterocyclic PAHs that contain helical structures are more soluble, and therefore more suitable for applications.3Thioxanthones are heterocycles that can be found in bioactive compounds such as lucanthone, hycanthone, and related compounds SR233377 and SR271425, which interact with DNA.4,5 Additionally, as thioxanthone derivatives are P-glycoprotein modulators, they may impact drug bioavailability.6 Due to their specific photophysical properties, thioxanthones have also caught the attention of chemists for applications in the field of photocatalysis (e.g. as photoinitiators in polymerization reactions and chemical transformations).7 However, although synthetic routes exist,8,9 they are limited to specific patterns, and original approaches are still needed to access and functionalize thioxanthones.
During the development of lithium-zinc bases10 for the functionalization of aromatic ketones,11 we reported a one-pot synthesis of 1-iodothioxanthone (1) and its subsequent functionalization by copper-catalyzed C–N bond formation,12 possibly followed by a cyclization.13,14 The hexacycles MN34-20 and MN42-62, obtained in this way, showed high fluorescence and were further established as promising inhibitors of the disease-related Provirus Integration site for Moloney leukemia virus (PIM) serine/threonine protein kinases (Fig. 1).14
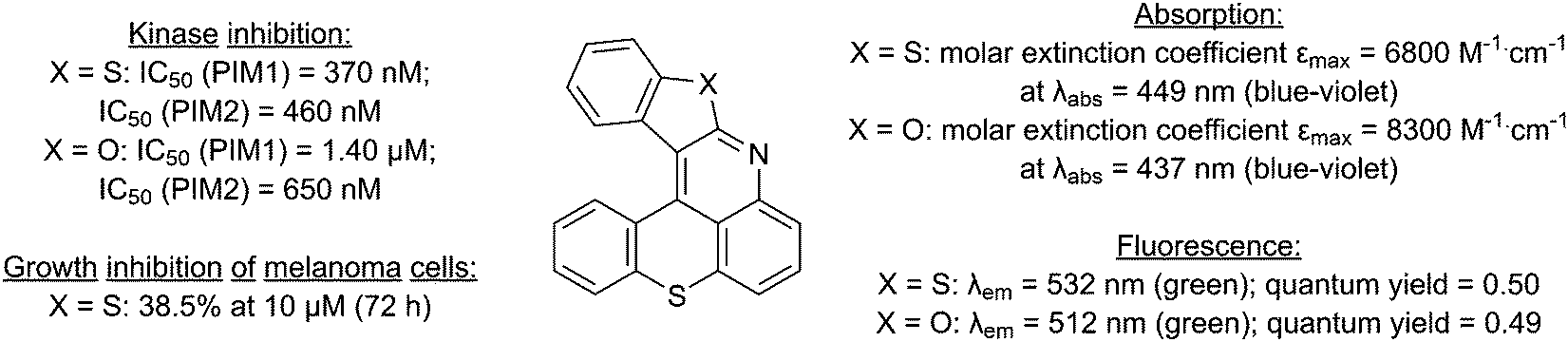 | ||
| Fig. 1 Properties of benzothieno[2,3-b]benzothiopyrano[4,3,2-de]quinoline (MN34-20; X = S) and benzofuro[2,3-b]benzothiopyrano[4,3,2-de]quinoline (MN42-62; X = O). | ||
These preliminary results convinced us to explore different accesses to 1-aminothioxanthone (2), the precursor of these hexacycles, and to consider the synthesis of analogs of MN34-20 and MN42-62 for property evaluation.
Results and discussion
Exploration of alternative routes to 1-aminothioxanthone
In our previous study,14 1-aminothioxanthone (2) was obtained in 50% yield from the corresponding iodide 1 by slightly modifying a literature procedure.15 Thus, 1 was treated with excess aqueous ammonia, in the presence of catalytic copper(I) iodide and N,N′-dimethylethylenediamine, in dimethylsulfoxide (DMSO) at 130 °C. However, the use of a sealed tube limited the scaling up, leading us to investigate alternative approaches.Inspired by a patent,16 we first attempted to prepare 2 by stirring a tetrahydrofuran (THF) solution of thiosalicylic acid and aniline (1 equiv.) containing H2SO4 (0.08 equiv.) overnight at 65 °C, and then for 20 min at 90 °C in the presence of H2O. However, we did not observe the formation of the expected product (not shown).
We therefore reverted to using 1-iodothioxanthone (1) as starting material, and chose to couple it with phthalimide in the presence of copper(I) oxide in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetonitrile–pyridine at 90 °C, by adapting a procedure reported previously.17 The phthalimide 3, obtained in 54% yield, was identified unambiguously by X-ray diffraction (Scheme 1, top left and bottom left). It was conventionally18 converted into 2 in 97% yield by treating it with hydrazine hydrate in 1:1 ethanol–dichloromethane (not shown). However, we faced reproducibility issues when scaling up (especially when we attempted to purify 2 by column chromatography over silica gel), and a two-step protocol was therefore preferred19 (Scheme 1, top right). As the intermediate hydrazinamide 4 is insoluble in chloroform, it proved easy to isolate it and then heat it to 70 °C in 1:1 ethanol–dichloromethane. Thus, after filtration of phthalhydrazide, the pure 1-aminothioxanthone (2) was obtained with an overall yield of 74%.
:1 acetonitrile–pyridine at 90 °C, by adapting a procedure reported previously.17 The phthalimide 3, obtained in 54% yield, was identified unambiguously by X-ray diffraction (Scheme 1, top left and bottom left). It was conventionally18 converted into 2 in 97% yield by treating it with hydrazine hydrate in 1:1 ethanol–dichloromethane (not shown). However, we faced reproducibility issues when scaling up (especially when we attempted to purify 2 by column chromatography over silica gel), and a two-step protocol was therefore preferred19 (Scheme 1, top right). As the intermediate hydrazinamide 4 is insoluble in chloroform, it proved easy to isolate it and then heat it to 70 °C in 1:1 ethanol–dichloromethane. Thus, after filtration of phthalhydrazide, the pure 1-aminothioxanthone (2) was obtained with an overall yield of 74%.
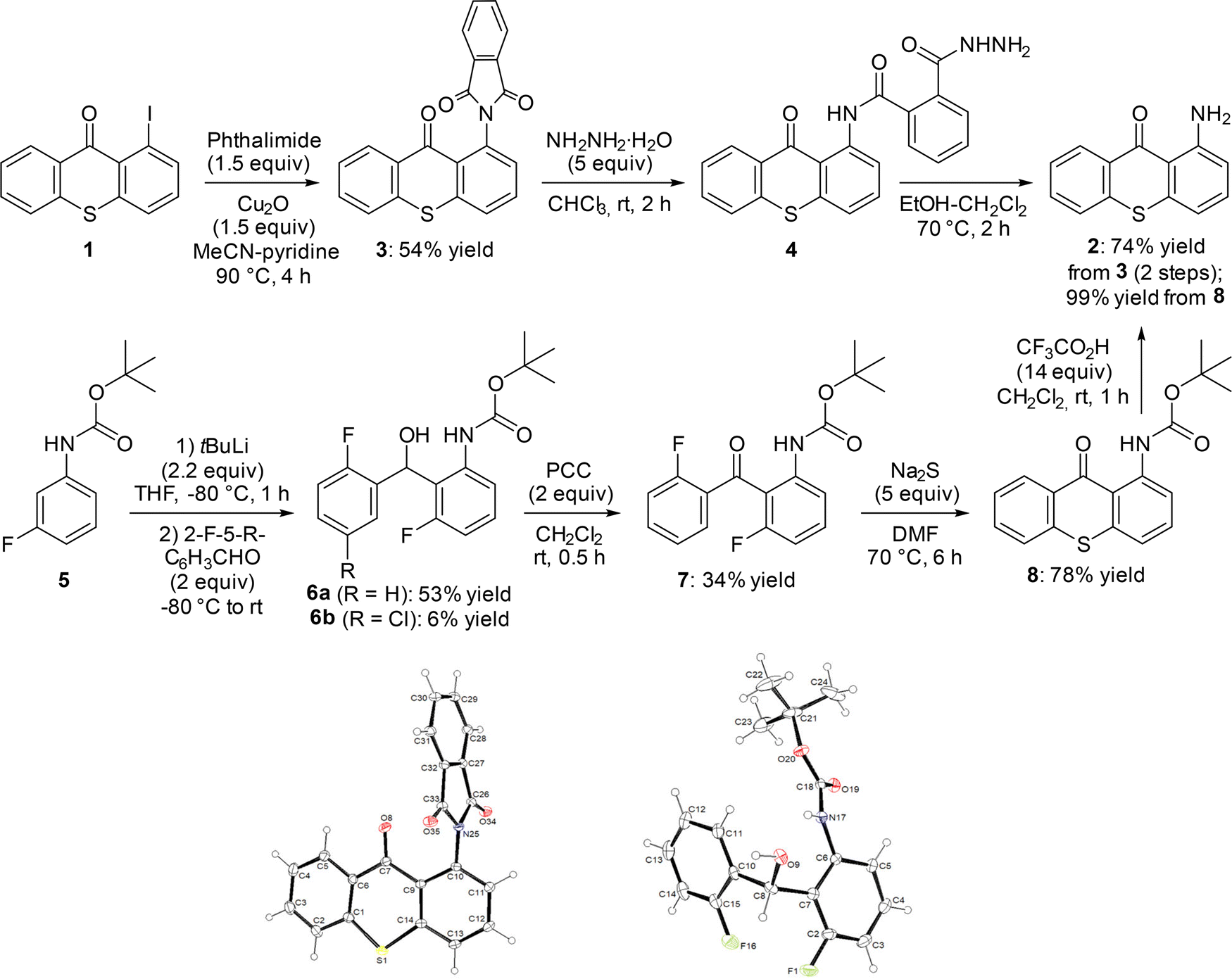 | ||
| Scheme 1 Alternative routes to 1-aminothioxanthone (2) and ORTEP diagrams (30% probability) of the compounds 3 and 6a. | ||
An alternative way to access 2 involves building the thioxanthone ring from an appropriately substituted diarylketone. To explore this more general approach, we started with tert-butyl N-(3-fluorophenyl)carbamate (5), prepared quantitatively from 3-fluoroaniline by adapting a reported procedure.20 Deprotolithiation of 5 was carried out by using tert-butyllithium in THF under conditions described by Warawa and co-workers,21 and subsequent electrophilic trapping with 2-fluorobenzaldehyde afforded the alcohol 6a in 53% yield (Scheme 1, middle left and bottom right). Replacing 2-fluorobenzaldehyde with 5-chloro-2-fluorobenzaldehyde (more acidic), to then access a substituted 1-aminothioxanthone, also led to 6b, but in a low 6% yield. In order to progress to 2, the alcohol 6a was oxidized in the presence of pyridinium chlorochromate22 (PCC; reaction not optimized) and the resulting ketone 7 was cyclized by using excess sodium sulfide under conditions documented by Peterson and co-workers.23 The target compound 2 was finally obtained after quantitative aniline deprotection24 (Scheme 1, right).
In order to directly obtain the ketone 7 from 5, we also attempted to use 2-fluorobenzoyl chloride in place of 2-fluorobenzaldehyde after the deprotolithiation step. However, only traces of the expected ketone were detected and vinyl 2-chloro-6-fluorobenzoate (9) was the only isolatable product (Scheme 2, left). The latter probably results from the reaction of lithium enolate (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHOLi), coming from the degradation of THF in the presence of strong bases,25 with the acyl chloride used, as already reported during the butyllithium-mediated deprotolithiation of N-phenylpivalamide.26
CHOLi), coming from the degradation of THF in the presence of strong bases,25 with the acyl chloride used, as already reported during the butyllithium-mediated deprotolithiation of N-phenylpivalamide.26
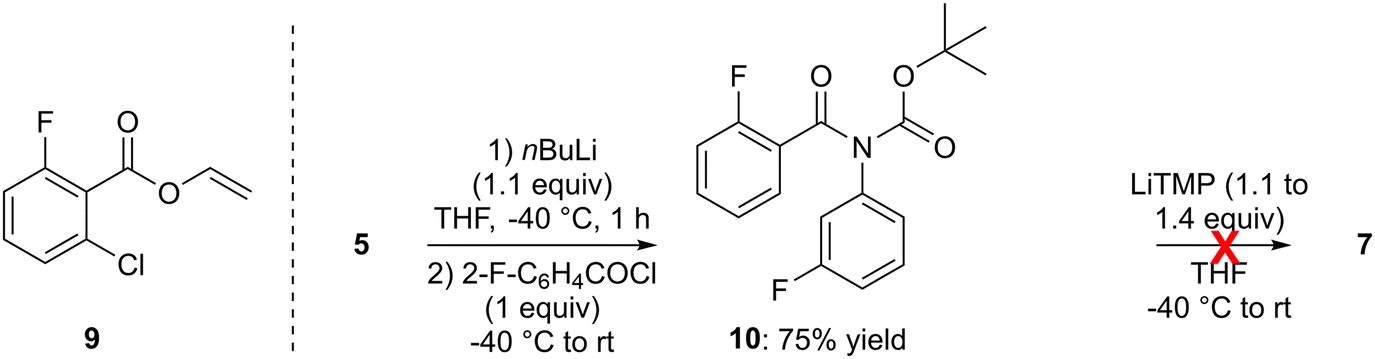 | ||
| Scheme 2 Left: Example of a vinyl ester obtained as a side product using an acyl chloride to intercept an aryllithium prepared in THF. Right: Attempted rearrangement of tert-butyl N-(2-fluorobenzoyl)-N-(3-fluorophenyl)carbamate (10) and ORTEP diagram (30% probability) of 10. | ||
Inspired by related work of Horne and Rodrigo,27 we also prepared tert-butyl N-(2-fluorobenzoyl)-N-(3-fluorophenyl)carbamate (10) from 5, and attempted a rearrangement to 7 by treatment with lithium 2,2,6,6-tetramethylpiperidide (LiTMP; 1.1 to 1.4 equiv.) at −40 °C before slowly warming (overnight) to room temperature. However, the expected reaction did not occur under these conditions (Scheme 2, right).
As some sulfur-containing rings can be formed by intramolecular electrophilic aromatic substitution with a remote methylsulfinyl group,28–30 we successfully prepared 5-chloro-2-(methylthio)benzaldehyde (11) from 5-chloro-2-fluorobenzaldehyde by substitution of the fluorine using sodium sulfide,31 then carried out its S-methylation. However, an attempt coupling of 11 (2 equiv.) with acetanilide in the presence of excess tert-butyl hydroperoxide (TBHP) and catalytic palladium(II) trifluoroacetate, in toluene at 90 °C,32 did not provide the expected N-(2-(5-chloro-2-methylthio)benzoyl)phenylacetamide (Scheme 3, top). Additionally, our attempts to intercept the lithiated derivative of tert-butyl N-phenylcarbamate (prepared by using tert-butyllithium in THF between −70 and −40 °C)33 with 11 yielded only small amounts of the expected alcohol (Scheme 3, bottom).
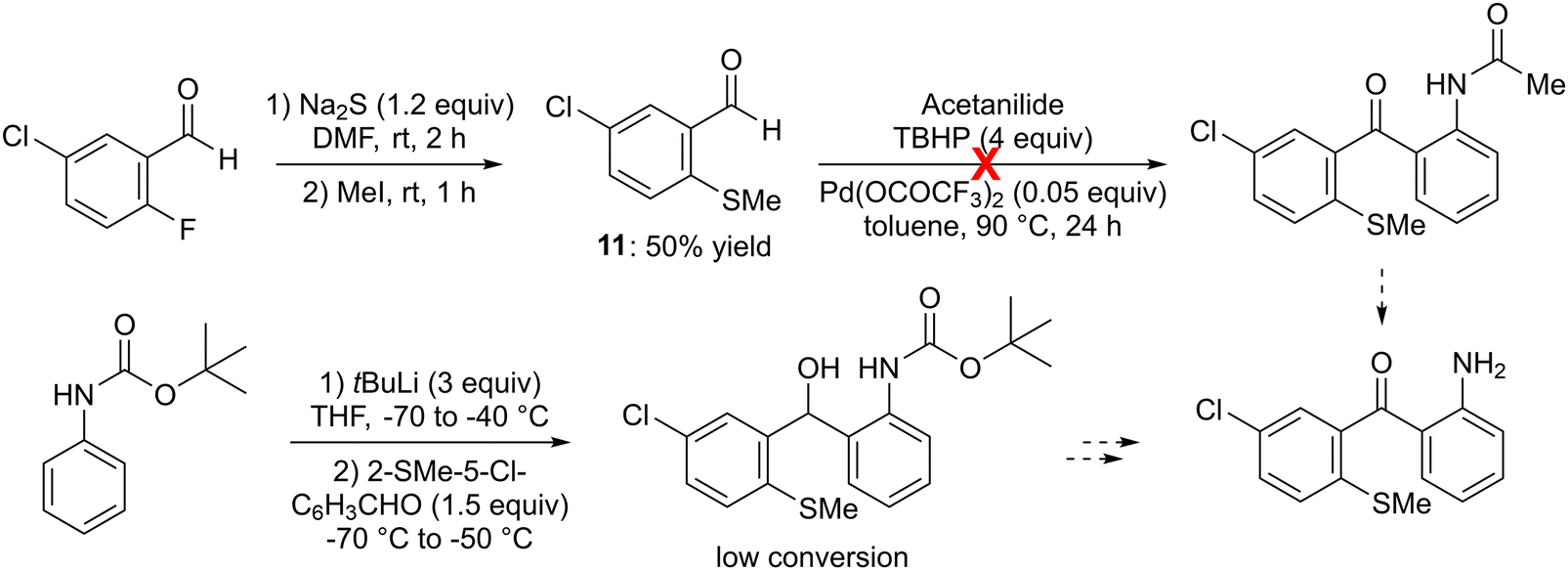 | ||
| Scheme 3 Unsuccessful routes to 2-(5-chloro-2-methylthiobenzoyl)aniline, a potential precursor of 7-substituted 1-aminothioxanthone. | ||
Exploration of alternative routes to 2-aminothioxanthone
Despite the interest of 2-aminothioxanthone (12) for further functionalization,34 very few well-described preparations are reported.35,36 Therefore, we considered straightforward accesses from the available 2-chlorothioxanthone. First, it was treated with aqueous ammonia (10 equiv.) and copper(I) oxide (0.05 equiv.) in N-methyl-2-pyrrolidinone (NMP) at 80 °C overnight.37 However, these conditions did not lead to the expected amine (not shown). The starting material was also recovered by treating 2-chlorothioxanthone with copper(II) sulfate or copper(I) oxide or copper(I) iodide (1.2 equiv.), in the presence of potassium carbonate (2.5 equiv.) in formamide at 160 °C for 2.5 h38 (not shown). Similarly, the use of copper(I) iodide (0.20 equiv.), N,N′-dimethylethylenediamine (0.30 equiv.), potassium hydroxide (1.5 equiv.) and cesium carbonate (0.50 equiv.) in acetonitrile–water at 100 °C for 15 h39 did not yield the acetamide (not shown). To our satisfaction, the reaction with tert-butyl carbamate (1.2 equiv.) in the presence of cesium carbonate (1.4 equiv.), palladium(II) acetate (0.03 equiv.) and 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (XPhos; 0.09 equiv.), in dioxane at 100 °C for 48 h,40 in turn, gave the expected protected amine 12-Boc in 72% yield (Scheme 4).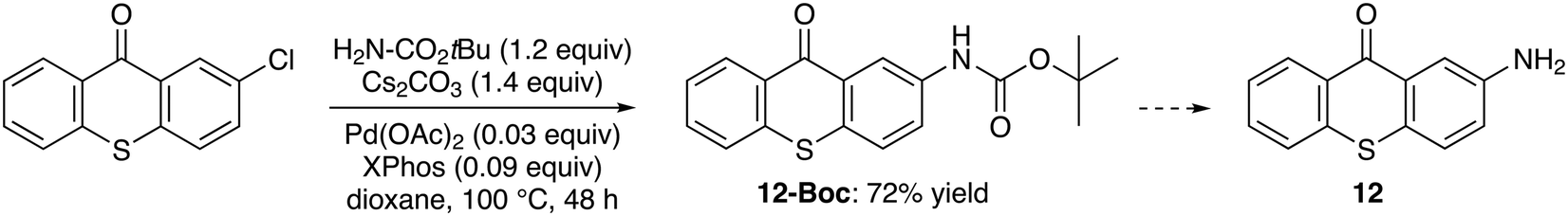 | ||
| Scheme 4 Alternative route to (Boc-protected) 2-aminothioxanthone (12). | ||
On the conversion of 1-aminothioxanthone to 10-methylbenzothiopyrano[4,3,2-de]indolo[2,3-b]quinoline, benzothieno[2,3-b]benzothiopyrano[4,3,2-de]quinoline and benzofuro[2,3-b]benzothiopyrano[4,3,2-de]quinoline
In 2020, benzothieno[2,3-b]benzothiopyrano[4,3,2-de]quinoline and benzofuro[2,3-b]benzothiopyrano[4,3,2-de]quinoline (MN34-20 and MN42-62) were obtained as side products upon N-arylation of 2 with 2-iodothiophene and 2-iodofuran, respectively.14 These coupling reactions were carried out in the presence of potassium carbonate (2 equiv.) and activated copper41 (Cu*; 0.2 equiv.) in dibutyl ether at 140 °C. Here we selected 2-iodo-N-methylindole42 to optimize the reaction using three different solvents (dibutyl ether, dimethylformamide (DMF) and DMSO) and two bases (potassium carbonate and tripotassium phosphate) (Table 1). While tripotassium phosphate mainly led to deiodination, replacing dibutyl ether with DMSO resulted in marked improvement.|
|
|||
|---|---|---|---|
| Entry | Solvent, T | Base | Yield of 13b |
| a All the reactions (0.5 mmol) were run in a sealed tube in 2 mL of solvent. b Yield after purification (see experimental part). c In the presence of 4 Å molecular sieves (0.25 g). d Not purified since deiodination of 2-iodo-N-methylindole was observed by NMR. | |||
| 1 | Bu2O, 160 °C | K2CO3 | 42%, 73%c |
| 2 | Bu2O, 160 °C | K3PO4 | —d |
| 3 | DMF, 170 °C | K2CO3 | 35% |
| 4 | DMSO, 170 °C | K2CO3 | 94% |
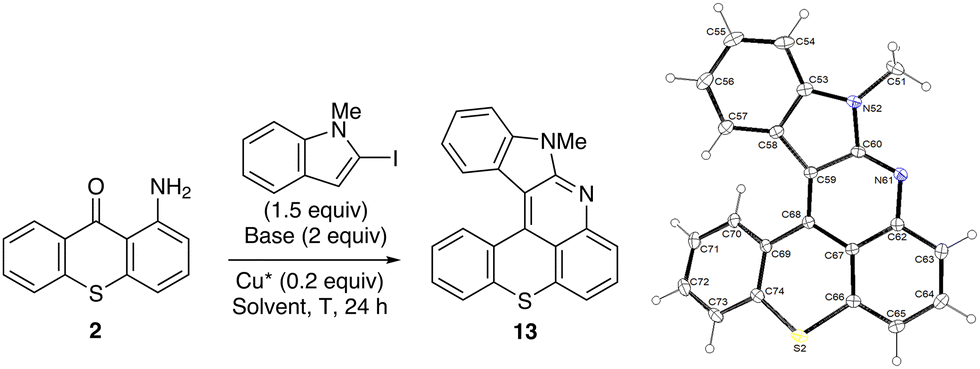
However, these conditions did not allow benzothieno[2,3-b]benzothiopyrano[4,3,2-de]quinoline (MN34-20) to be obtained efficiently (<20% yield). Without the sulfur bridge, 11-phenylbenzothieno[2,3-b]quinoline (NM-049) formed similarly in a modest 30% yield, suggesting that 2-iodobenzothiophene43,44 is not a good coupling partner in these tandem N-arylation-cyclizations. Based on findings by Hartwig and co-workers of related palladium-catalyzed couplings in these series,45,46 copper-catalyzed N-arylation reactions might be favored in the case of less electron-rich aryl groups as follows:47 2-benzofuryl (C2 Mulliken charge48 = +0.07) > 2-indolyl (+0.02) > 2-benzothienyl (−0.38). On the contrary, the ability to cyclize must be favored in the case of more electron-rich aryl groups as follows: 3-indolyl (C3 Mulliken charge48 = −0.24) ≥ 3-benzofuryl (−0.235) > 3-benzothienyl (−0.14). This might help explain why 2-iodo-N-methylindole is a more suitable substrate than 2-iodobenzothiophene for these N-arylation-cyclization reactions.
The fact that not all iodides react in the same way in these N-arylation-cyclizations prompted us to develop an alternative approach to MN34-20 from tert-butyl N-(3-fluoro-2-(2-fluorobenzoyl)phenyl)carbamate (7) with the formation of the sulfur bridge as the final step. Once deprotected,24 the aniline 14 (prone to cyclization to 1-fluoroacridone) was directly subjected to the tandem coupling-cyclization with 2-iodobenzothiophene. Despite the use of a stoichiometric amount of copper(I) iodide in order to obtain more reproducible results,47 the benzothienoquinoline 15a was isolated in a low 17% yield. This confirms the low reactivity of this particular halide since the use of 2-iodo-N-(methoxymethyl)indole49 under the same conditions afforded the expected product 15b in 85% yield (Scheme 5).
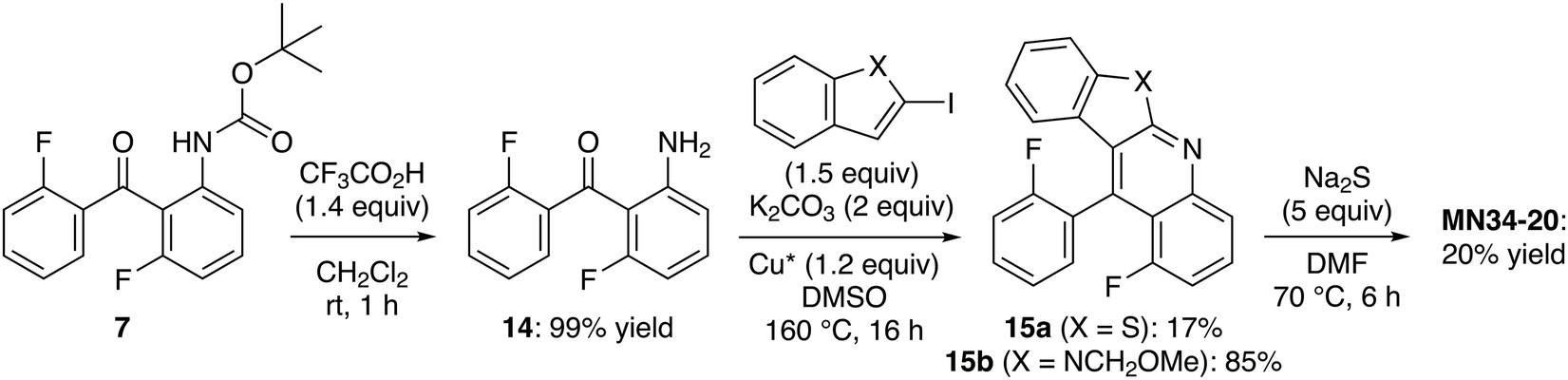 | ||
| Scheme 5 Alternative route to benzothieno[2,3-b]benzothiopyrano[4,3,2-de]quinoline (MN34-20). | ||
Cyclization to MN34-20 was finally attempted by treating 15a with excess sodium sulfide in dimethylformamide (DMF) at 70 °C for 6 h; however, the expected product was isolated in a low yield. From the substrate 15b, the corresponding hexacycle was only detected in the crude by NMR (∼10% estimated yield) after 5 h at 120 °C.
In search of aza analogs of 10-methylbenzothiopyrano[4,3,2-de]indolo[2,3-b]quinoline, benzothieno[2,3-b]benzothiopyrano[4,3,2-de]quinoline and benzofuro[2,3-b]benzothiopyrano[4,3,2-de]quinoline
For comparison, NM-049 was tested50 against the proto-oncogene protein kinases PIM1 and PIM2. However, while MN34-20 has promising activity, the IC50 of NM-049 were found to be surprisingly low (Fig. 2), highlighting the importance of the tetrahydrothiopyran ring to achieve good affinity. In addition, the inhibitory properties of benzothieno[2,3-b]1,8-naphthyridine (MN65-45) which is an aza analog of NM-049 were also evaluated, leading to IC50 values close to those of MN34-20. Given this preliminary structure–activity analysis, we thought it would be interesting to synthesize compounds with both a sulfur ring and this extra nitrogen in order to evaluate them.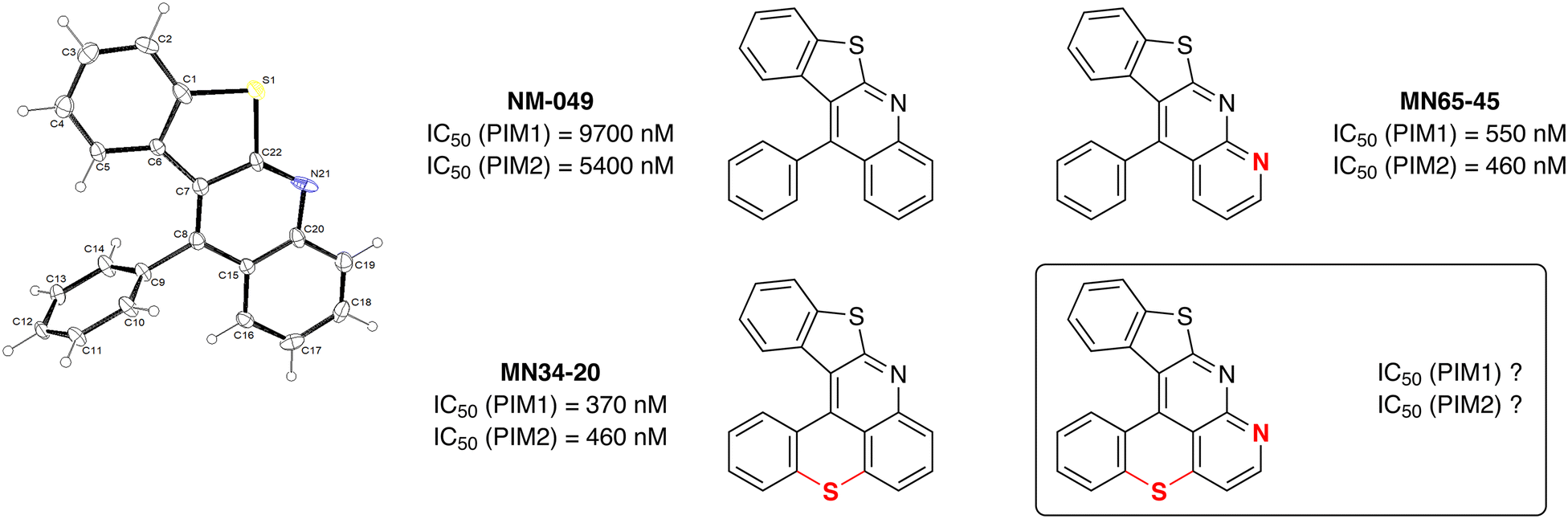 | ||
| Fig. 2 Structure-activity relationships leading to benzothieno[2,3-b]benzothiopyrano[4,3,2-de]1,8-naphthyridine, and ORTEP diagram (30% probability) of the compound NM-049. | ||
Our first approach started from 2-chloropyridine, from which deprotolithiation using LiTMP in THF at low temperature51 followed by trapping with 2-bromobenzaldehyde furnished the alcohol 16 in high yield (Scheme 6, top left). While its oxidation using Dess–Martin periodinane52 (1.4 equiv.) in THF at room temperature for 3 h afforded the ketone 17 in 62% yield (not shown), better results were obtained by using 2-iodoxybenzoic acid (IBX) in ethyl acetate at 60 °C for 6 h.53 However, our preferred method of accessing the ketone 17 remained the Swern oxidation54 which worked similarly (98% yield; Scheme 6, top middle). A direct synthesis of 17 from 2-chloropyridine was also investigated by using a lithium cuprate base derived from LiTMP,55 but it gave the product in lower yields.
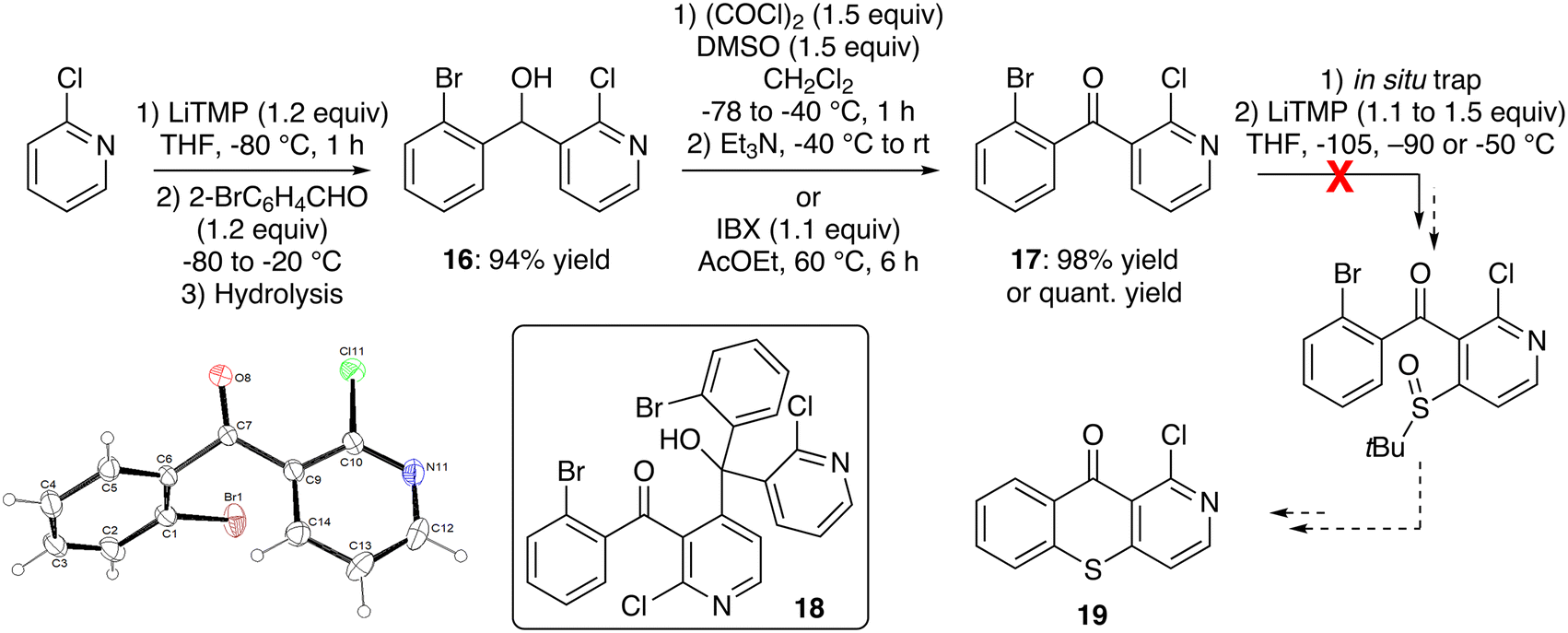 | ||
| Scheme 6 Unsuccessful attempt toward 1-chlorobenzothiopyrano[3,2-c]pyridin-10-one (19) and ORTEP diagram (30% probability) of the compound 17. | ||
As coupling reactions have been reported between aryl sulfoxides and aryl bromides,56 our next step was to introduce a tert-butylsulfinyl group at the 4-position of the pyridine ring in order to attempt cyclization. We thus studied the deprotolithiation of the ketone 17 in the presence of an in situ trap.11,13 Different electrophiles (tert-butylsulfinyl chloride, S-tert-butyl-tert-butanethiosulfinate and di-tert-butyl disulfide) were tested for their ability to intercept the aryllithiums generated by LiTMP-mediated deprotolithiation in THF. While the first electrophile led to the recovery of starting material at temperatures between −105 and −50 °C, the second gave only traces of the expected sulfoxide at −90 °C. With the third, we only obtained the tertiary alcohol 18 resulting from an attack of the aryllithium formed on the ketone 17. This last result tends to indicate that the di-tert-butyl disulfide does not trap the lithiated intermediate quickly enough, probably due to high steric hindrance (Scheme 6, right and bottom).
Another potential precursor of our targets is 2-amino-3-benzoyl-4-(methylthio)pyridine (20) which could be prepared from 2-chloro-4-(methylthio)pyridine. The synthesis of the latter was attempted from 2-chloro-4-fluoropyridine at room temperature, treating it successively with sodium hydrosulfide (1.5 equiv.) in methanol for 0.5 h and iodomethane (2 equiv.) for 1 h.57 However, under these conditions, we only recovered the starting material. Furthermore, when the more reactive sodium thiomethoxide (1 equiv.) was employed in methanol at 30 °C for 2.5 h, the expected sulfide was only isolated in a low 16% yield (Scheme 7, top) due to the recovery of starting material but also the competitive formation of a product that could result from the substitution of chlorine. Using CH2Cl2, acetone and THF as the solvent, extending the reaction time or using 2,4-difluoropyridine as the substrate did not give better results. Note that replacing sodium thiomethoxide with sodium thio-tert-butoxide, either in THF at 40 °C or in MeOH at room temperature, also did not furnish the expected 4-(tert-butylthio)pyridine. Furthermore, our attempts to convert this methyl sulfide into the expected ketone 21, by using successively the lithium cuprate base (TMP)2CuLi·LiCl58 and the benzoyl chloride, failed. Faced with these disappointing results, we turned to another path to obtain 20.
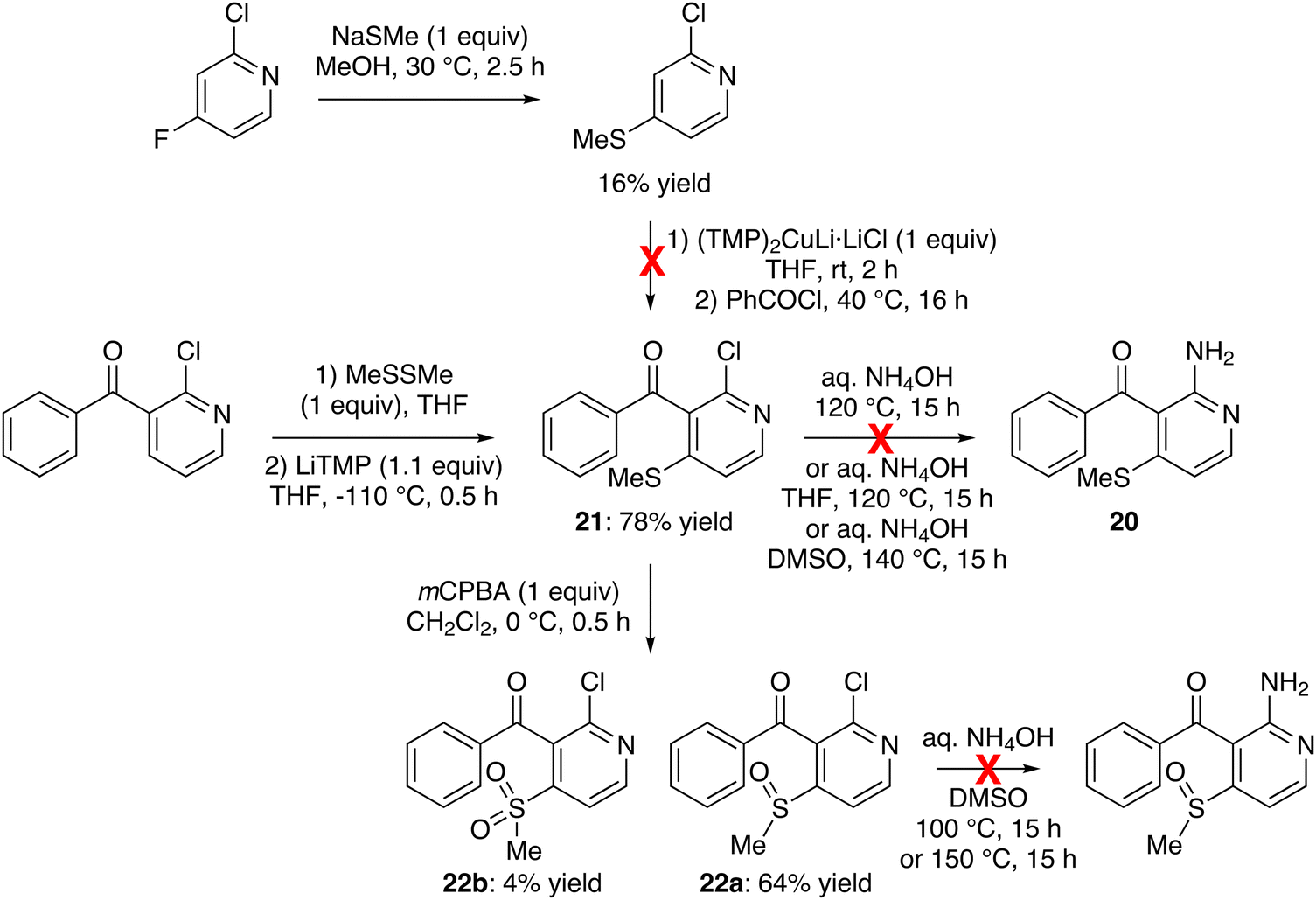 | ||
| Scheme 7 Unsuccessful attempts toward 2-amino-3-benzoyl-4-(methylthio)pyridine (20) and 2-amino-3-benzoyl-4-(methylsulfinyl)pyridine. | ||
Thus, we explored the use of dimethyl disulfide as an in situ trap for the LiTMP-mediated deprotometallation of 3-benzoyl-2-chloropyridine.59 Pleasingly, the reaction performed at −110 °C furnished the expected sulfide 21 in 78% yield. However, all attempts to replace its chloro group with an amino,60 whether using aqueous ammonia on overnight heating,61 with THF61 at 120 °C,62 or with DMSO at 140 °C, did not provide 20 (starting material recovered; Scheme 7, middle).
Since we had succeeded in converting 2-chlorothioxanthone into the corresponding Boc-protected amine (Scheme 4), we also attempted the coupling between the chloride 21 and tert-butyl carbamate (5.0 equiv.) in the presence of palladium(0) bis(dibenzylideneacetone) (Pd2(dba)3; 0.02 equiv.), XPhos (0.05 equiv.) and cesium carbonate (7.6 equiv.) in THF at 70 °C for 24 h.63 However, we only recovered the starting material (not shown).
As a substitution from an analog of 21 lacking the methylthio group is known,60 we suspected that this substituent disfavors the reaction, probably for electronic reasons. Therefore, we decided to oxidize6421 into the corresponding sulfoxide. The use of meta-chloroperbenzoic acid (mCPBA; 1 equiv.) led to the expected product 22a, isolated in 64% yield, but also to the sulfone 22b (4% yield; Scheme 7, bottom). By using cerium ammonium nitrate (CAN; 4 equiv.) in acetonitrile at room temperature,65 the sulfoxide 22a was formed selectively, but the conversion proved incomplete after 24 h (not shown). Disappointingly, all attempts to replace the chloro group in aqueous ammonia upon heating overnight with DMSO at 100 °C or 150 °C led to degradation (Scheme 7, bottom).
Since substitution of the chloro group with an amino turned out to be an issue for the methylthio- and methylsulfinyl-containing 2-chloropyridines 21 and 22a, we attempted the LiTMP-mediated deprotometallation of 3-benzoyl-2-fluoropyridine59 in the presence of dimethyl disulfide, aiming to achieve a more easily substitutable fluorinated analog of 21. Unfortunately, the reaction only gave a complex mixture (not shown). This led us to consider the synthesis of 2-amino-3-(2-methylthio)benzoylpyridine (23), an isomer of 2-amino-3-benzoyl-4-(methylthio)pyridine (20), as another precursor of our targets. To this purpose, 2-fluoropyridine was easily deprotocuprated at its 3-position by using (TMP)2CuLi·LiCl, in situ prepared from LiTMP and CuCl, in THF at room temperature for 2 h;55 subsequent trapping with 2-(methylthio)benzoyl chloride66 afforded the ketone 24a in 80% yield (Scheme 8, top left). As expected, the replacement of the fluoro group using aqueous ammonia at 50 °C for 16 h67 provided the 2-aminopyridine 23 in a quantitative yield. It is interesting to note that the methylthio group cannot be introduced from a 2-amino-3-(2-fluorobenzoyl)pyridine (e.g.25; Scheme 8, right). The amine 23 was then involved in the tandem N-arylation-cyclization with 2-iodobenzofuran as before,47 to furnish 11-(2-(methylthio)phenyl)benzofuro[2,3-b]1,8-naphthyridine (26) in 50% yield (Scheme 8, middle). It was oxidized64 to the sulfoxide 27 in 79% yield by using mCPBA. NMR analysis seemed to show that 27 exists as a mixture of diastereomers, even at 100 °C (see experimental part), possibly due to impeded rotation around the biaryl bond.
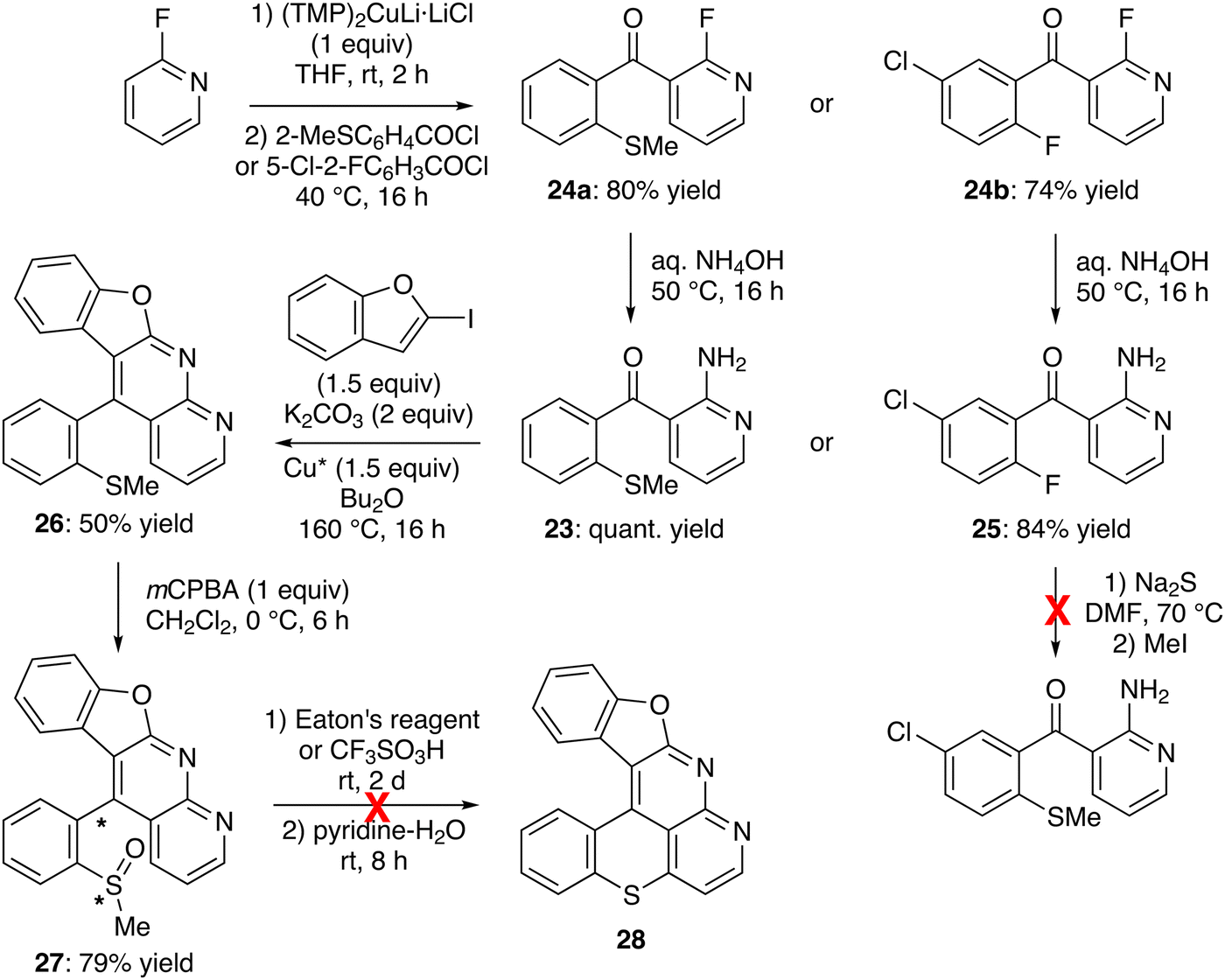 | ||
| Scheme 8 Unsuccessful attempt toward benzofuro[2,3-b]benzothiopyrano[4,3,2-de]1,8-naphthyridine (28). | ||
Unfortunately, all attempts to convert 27 into the hexacycle 28 have failed, whether in the presence of Eaton's reagent and then pyridine,28 or using trifluoromethanesulfonic (triflic) acid before demethylation,29 due to degradation (Scheme 8, bottom). Softer conditions using triflic acid in dichloromethane30 or pyridinium chloride at 220 °C for 15 min68 have also been tested, but without greater success (not shown).
Even though intramolecular reactions of pyridines with remote (methylsulfinyl)aryl moieties have been claimed,69 electrophilic aromatic substitutions are generally less favored from π-deficient heteroaromatic compounds. Therefore, we decided to undertake the synthesis of a substrate in which the methylsulfinyl group would be at the 4-position of the pyridine ring, and designed another synthesis of 2-amino-3-benzoyl-4-(methylthio)pyridine (20). To discard issues related to nucleophilic aromatic substitution, we started from Boc-protected 2-aminopyridine, prepared from 2-aminopyridine by modifying a reported procedure.20 This substrate can also be obtained, albeit in a lower 32% yield, by coupling 2-chloropyridine with tert-butyl carbamate in the presence of cesium carbonate, catalytic palladium(II) acetate and 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (Xantphos) in dioxane at 80 °C.70 However, no product was formed by replacing Xantphos by XPhos.40
Deprotolithiation of Boc-protected 2-aminopyridine was carried out as reported, using tert-butyllithium (2.4 equiv.) in THF at −20 °C for 2.5 h.71 Subsequent trapping with benzaldehyde led to the expected alcohol 29 in a moderate 31% yield (Scheme 9, top left). Alternatively, we attempted to deprotolithiate 2-aminopyridine doubly protected by Boc and CH2OMe (MOM) groups, this by using tert-butyllithium in THF at −80 °C. However, the expected alcohol was not formed at all and only 10% of the substrate was recovered (not shown).
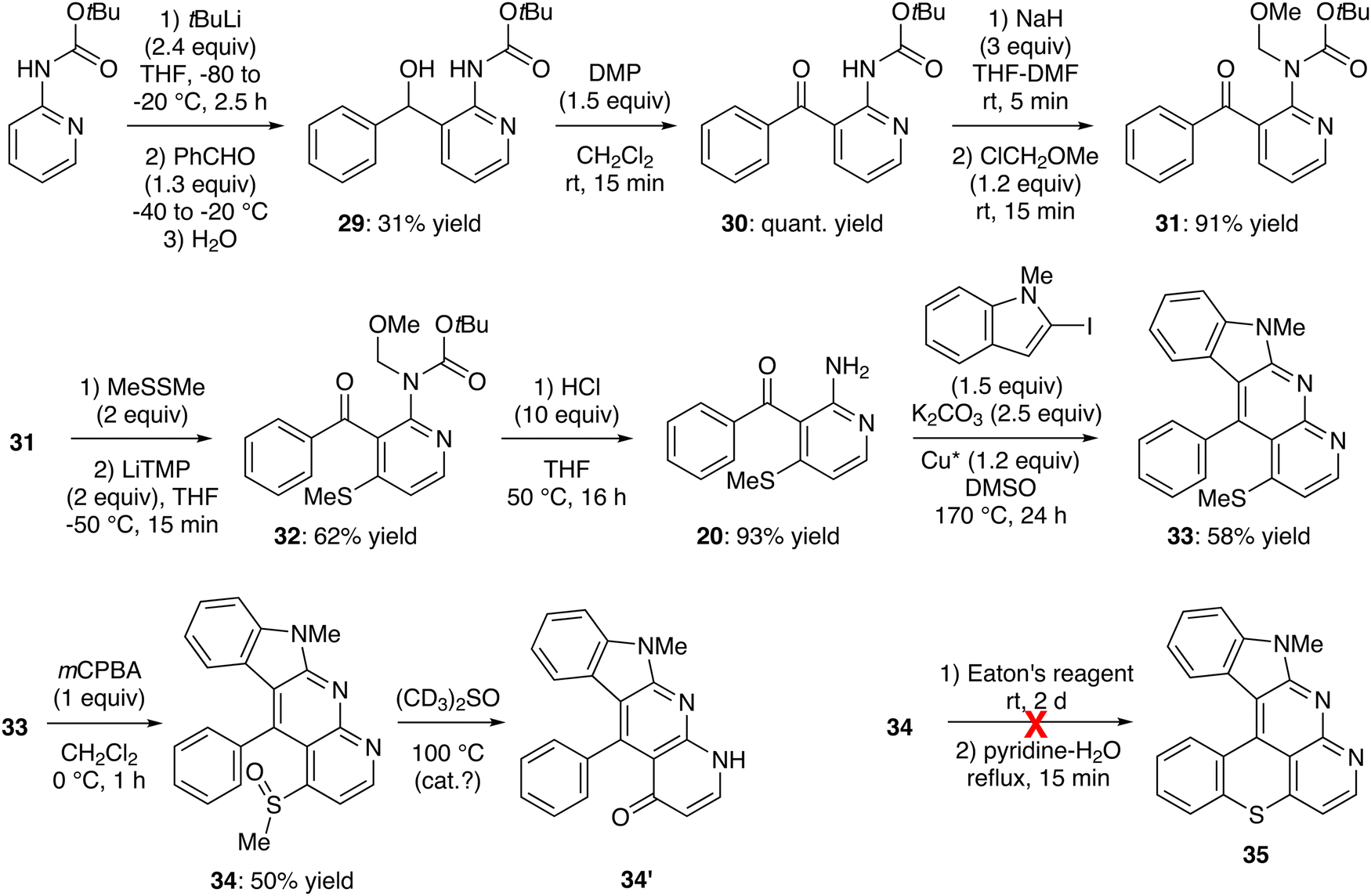 | ||
| Scheme 9 Unsuccessful attempt toward 10-methylbenzothiopyrano[4,3,2-de]indolo[2,3-b]1,8-naphthyridine (35). | ||
While the Swern oxidation failed to deliver the ketone 30 due to a competitive intramolecular reaction of the alcohol function with the carbamate, the use of Dess–Martin periodinane in dichloromethane52 led to 30 in quantitative yield (Scheme 9, top middle). In order to introduce the methylthio group at the 4-position of the pyridine ring, 30 was treated with LiTMP (2 or 3 equiv.) at different temperatures between −90 and −50 °C in the presence of dimethyl disulfide as an in situ trap.59 Unfortunately, regardless of the conditions used, only an unidentified side product formed in addition to the recovered starting material (not shown). However, after MOM-protection, functionalization of the ketone 31 was found to be much easier, affording the 4-methylthio derivative 32 in 62% yield (Scheme 9, middle left). The amine was finally deprotected to deliver 2-amino-3-benzoyl-4-(methylthio)pyridine (20). A few attempts were necessary to optimize the reaction and revealed the superiority of hydrochloric acid (10 equiv.) in THF at 50 °C over trifluoroacetic acid (5 equiv.) in dichloromethane under the same conditions (93% vs. 65% yield). Tandem N-arylation-cyclization on the amine 20 was performed with 2-iodo-N-methylindole as usual,47 to give 11-phenyl-10-(methylthio)indolo[2,3-b]1,8-naphthyridine (33) in 58% yield. However, while the latter was easily oxidized64 to the sulfoxide 34 (50% yield), an attempt to cyclize to the hexacycle 35 using Eaton's reagent gave only a complex mixture from which the product has not been identified (Scheme 9, bottom).
During the NMR experiment (several hours in deuterated DMSO at 375 K), the sulfoxide 34 was quantitatively converted into what could the pyridone 34′,72 on the basis of its NMR and high-resolution mass spectra. However, when these NMR experiments were repeated on a repurified sample, no reaction occurred, suggesting it was catalyzed by traces of an impurity.
With evidence that electrophilic aromatic substitution with a remote sulfoxide will hardly deliver the target hexacycles, we reasoned that the presence of the pyridine ring would favor the nucleophilic aromatic substitution and reverted to using a difluorinated diarylketone as in Schemes 1 and 5. Thus, 2-chloro-4-fluoropyridine was deprotolithiated by using LiTMP (1.2 equiv.) in THF at low temperature, before trapping with 2-fluorobenzaldehyde. The expected alcohol 36, isolated in 76% yield, was then engaged in a Swern oxidation54 to deliver the ketone 37 in high yield (Scheme 10, top). Successive treatment of 2-chloro-4-fluoropyridine with (TMP)2CuLi·LiCl in THF at room temperature for 2 h and 2-fluorobenzoyl chloride also afforded 37, but with lower yields (not shown).
 | ||
| Scheme 10 Synthesis of benzofuro[2,3-b]benzothiopyrano[4,3,2-de]1,8-naphthyridine (28), 10-methylbenzothiopyrano[4,3,2-de]indolo[2,3-b]1,8-naphthyridine (35) and benzothieno[2,3-b]benzothiopyrano[4,3,2-de]1,8-naphthyridine (39), and ORTEP diagram (30% probability) of the compound 36. a0.2 equiv. of activated copper (Cu*) was used in this case. | ||
The difluoroketone 37 was then reacted with sodium sulfide in NMP,73 and the resulting 1-chlorobenzothiopyrano[3,2-c]pyridin-10-one (19) was directly involved in the substitution reaction with aqueous ammonia at 110 °C to give 38 with an overall yield of 40% for two steps. Our targets 28, 35 and 39 were finally reached from 38 through the N-arylation-cyclization tandem reaction using 2-iodobenzofuran, 2-iodo-N-methylindole and 2-iodobenzothiophene as coupling partners. Competitive deiodination is responsible for the moderate yields recorded.
Evaluation on kinases and molecular modeling experiments
Our target compounds finally in hand, their ability to act as inhibitors of PIM kinases was evaluated and compared to those of benzothieno[2,3-b]benzothiopyrano[4,3,2-de]quinoline (MN34-20) and benzofuro[2,3-b]benzothiopyrano[4,3,2-de]quinoline (MN42-62) previously studied14 (Fig. 1). The results collected in Fig. 3 show that, if there is a certain interest in introducing an additional nitrogen on structures devoid of a sulfur bridge (top), its presence in the hexacycles has a deleterious effect on the affinity for the PIM kinases (bottom).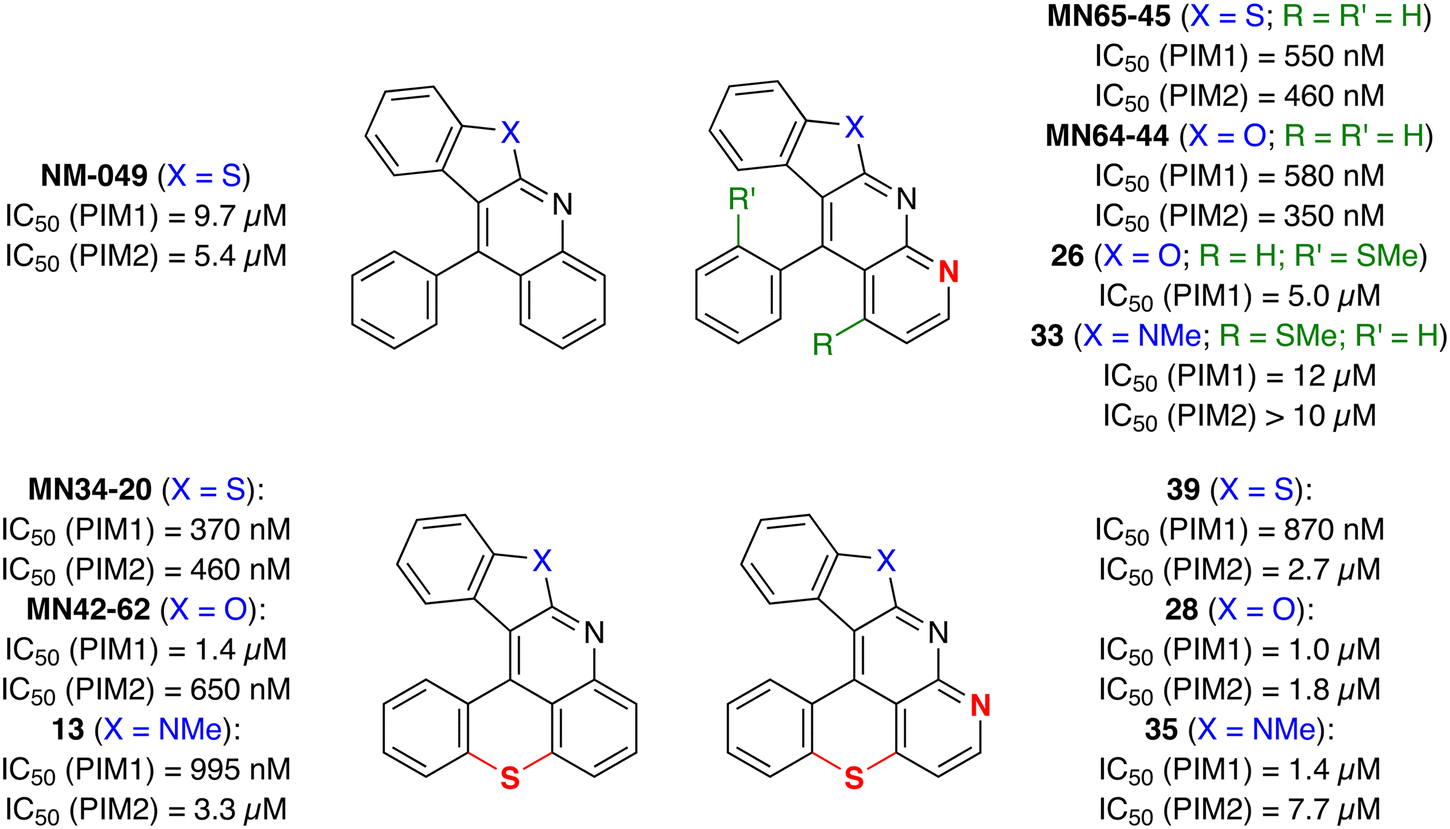 | ||
| Fig. 3 IC50 values for some of the synthesized products and related compounds. ATP concentration used in the kinase assays was 10 μM. | ||
To better understand these results and highlight the structural features essential to the inhibition of PIM1/2 kinases in these series, molecular modeling studies on the hexacycle 39 and the compounds NM-049 and MN65-45 were undertaken. After geometry optimization, docking experiments were performed with PIM1 model generated from 3JPV structure74 using AutoDock Vina.75,76 The putative binding mode of MN34-20 with the hexacyclic scaffold highly stabilized in the ATP-binding pocket via several hydrophobic interactions involving ILE104, LEU120, VAL126, LEU174 and ILE185 residues as well as LEU44, PHE49 and VAL52 residues from the P-loop flexible domain was previously described (see compound 6b in ref. 14 and Fig. 4A). The presence of an additional nitrogen group in the analogous structure 39 leads to a sliding of the molecule into the binding pocket which has the consequence of selecting the P helix. The interaction with PHE49 then becomes impossible leading to a much lower binding score. Indeed, the region delimited by residues LYS67, LEU93, ASP186 and GLU89 seems unable to accept the presence of a nitrogen group (Fig. 4B).
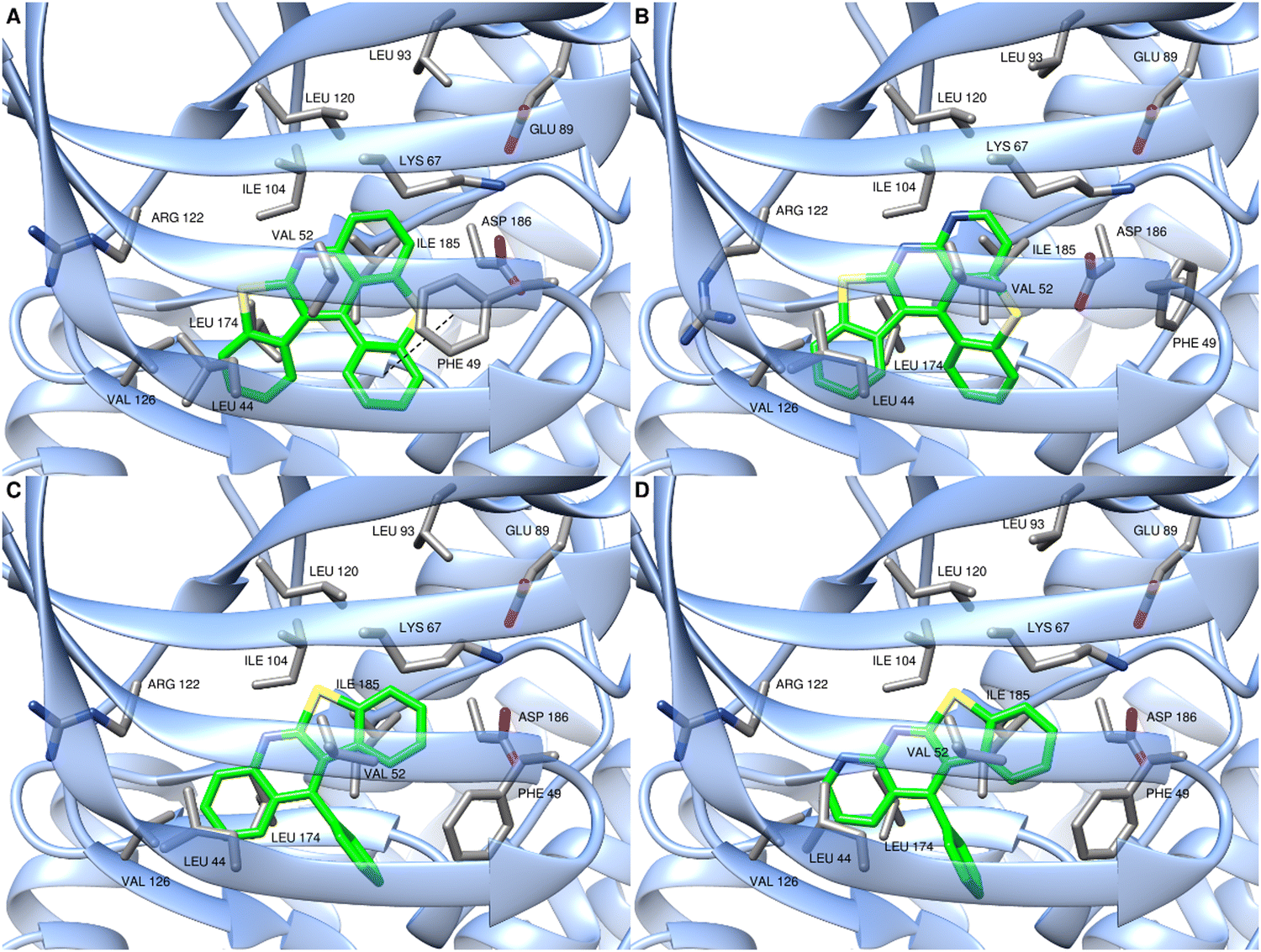 | ||
| Fig. 4 Plausible binding mode of MN34-20 (A), 39 (B), NM-049 (C) and MN65-45 (D) within PIM1 ATP-binding site. The images were produced using UCSF Chimera.77 | ||
As described in Fig. 4, similar binding modes were observed for the compounds NM-049 and MN65-45 with the planar tetracyclic moiety inserted into the ATP-binding pocket, and also stabilized via hydrophobic interactions with VAL52, ILE104, LEU174 and LEU44 residues (Fig. 4C and D). Compared to MN34-20, these two compounds did not fit into the pocket with the same orientation but with a 180° flip due to the presence of the phenyl group. Thus, NM-049 and MN65-45 were not able to interact anymore with the PHE49 residue while this interaction seems crucial in this series. Altogether, the results obtained only explain partially the differences in PIM1 inhibitory potency observed between these compounds. This may result from different solubilities in testing media or solvation networks for the studied compounds.
Biological evaluation on melanoma cells
For six compounds produced, 3, 13, 28, 35, 39 and NM-049, the properties were evaluated against A2058 human melanoma cells. Moderate growth inhibitions of 33.2% ± 3.2%, 32.8% ± 9.7%, 18.7% ± 1.5%, 25.9% ± 2.0%, 32.5% ± 2.6% and 16.9% ± 9.7%, respectively, were observed at 10−5 M after 72 h (Fig. 5). Thus, the introduction of an additional nitrogen to 13 (32.8% ± 9.7%) and MN34-20 (38.5% ± 4.2%)14 led to compounds possessing reduced antiproliferative activity (25.9% ± 2.0% for 35 and 32.5% ± 2.6% for 39, respectively).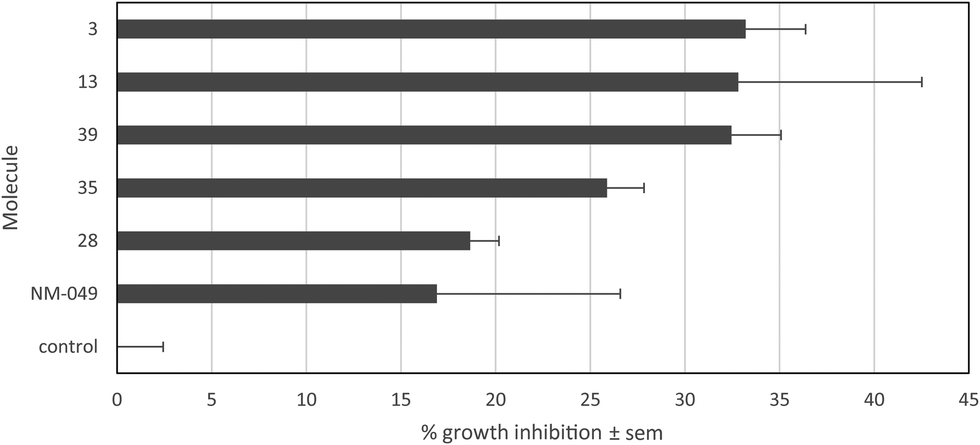 | ||
| Fig. 5 Antiproliferative activity (% growth inhibition) of the compounds 3, 13, 28, 35, 39 and NM-049. 2000 A2058 human melanoma cells were treated for 72 h with 10−5 M of compound. Control cells were treated with an equivalent dose of DMSO (solvent of the tested compounds). | ||
Photophysical properties
As the hexacycles MN34-20 and MN42-62 are strongly fluorescent (Fig. 1), it was of interest to examine the photophysical properties of their analog 13 (X = NMe), as well as those of the corresponding hexacycles with an additional nitrogen 39, 28 and 35, respectively (Fig. 3).The UV-visible absorption and fluorescence emission properties of these six hexacycles are collected in Table 2. All compounds exhibit a lower energy band in the violet-blue part of the visible region (Fig. 6), with maximum wavelengths ranging between 426 and 470 nm. This value increases depending on X (Fig. 3) in the order O < S < NMe, and introduction of an extra nitrogen atom leads to a hypsochromic shift of about 10 nm. The three compounds bearing this extra nitrogen (39, 28 and 35) emit in the blue region, whereas the three other ones (MN34-20, MN42-62 and 13) emit at higher wavelengths in the green (Fig. 7), the introduction of the heteroatom leading to a marked hypsochromic shift of more than 40 nm, and therefore to a decrease of the Stokes shifts of more than 1000 cm−1.
| Compound | λ abs (nm) | ε max (M−1 cm−1) | λ em (nm) | Stokes shiftd (cm−1) | Φ F | τ (ns) | k r (s−1) | k nr (s−1) |
|---|---|---|---|---|---|---|---|---|
| a Absorption maximum. b Molar extinction coefficient at λabs. c Emission maximum. d Stokes shift = (1/λabs − 1/λem). e Fluorescence quantum yield using quinine bisulfate in 0.5 M H2SO4 as a standard. f Fluorescence lifetime. g Radiative (kr) and nonradiative (knr) decay rates derived from fluorescence quantum yield and lifetime values: kr = ΦF/τ and knr = (1 − ΦF)/τ. | ||||||||
| MN34-20 | 449 | 6800 | 532 | 3470 | 0.50 | 16.5 | 3.0 × 107 | 3.0 × 107 |
| MN42-62 | 437 | 8300 | 512 | 3350 | 0.49 | 13.6 | 3.6 × 107 | 3.8 × 107 |
| 13 | 470 | 6800 | 519 | 2010 | 0.61 | 14.1 | 4.3 × 107 | 2.8 × 107 |
| 39 | 441 | 3100 | 483 | 1970 | 0.76 | 9.7 | 7.8 × 107 | 2.5 × 107 |
| 28 | 426 | 11100 |
460 | 1740 | 0.86 | 7.8 | 11.0 × 107 | 1.8 × 107 |
| 35 | 457 | 3900 | 479 | 1010 | 0.50 | 10.7 | 4.7 × 107 | 4.7 × 107 |
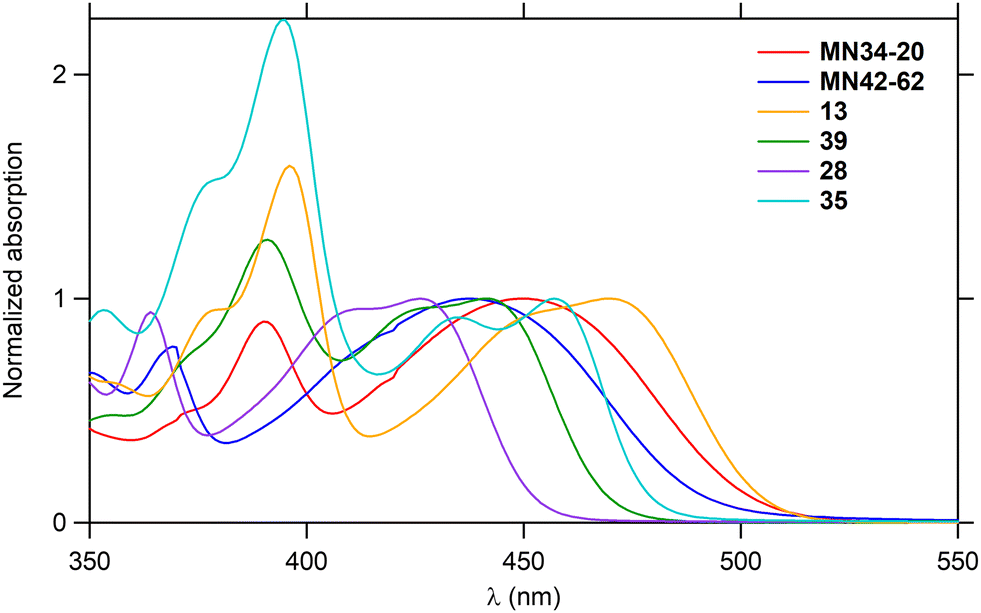 | ||
| Fig. 6 Absorption spectra of the hexacycles in toluene. | ||
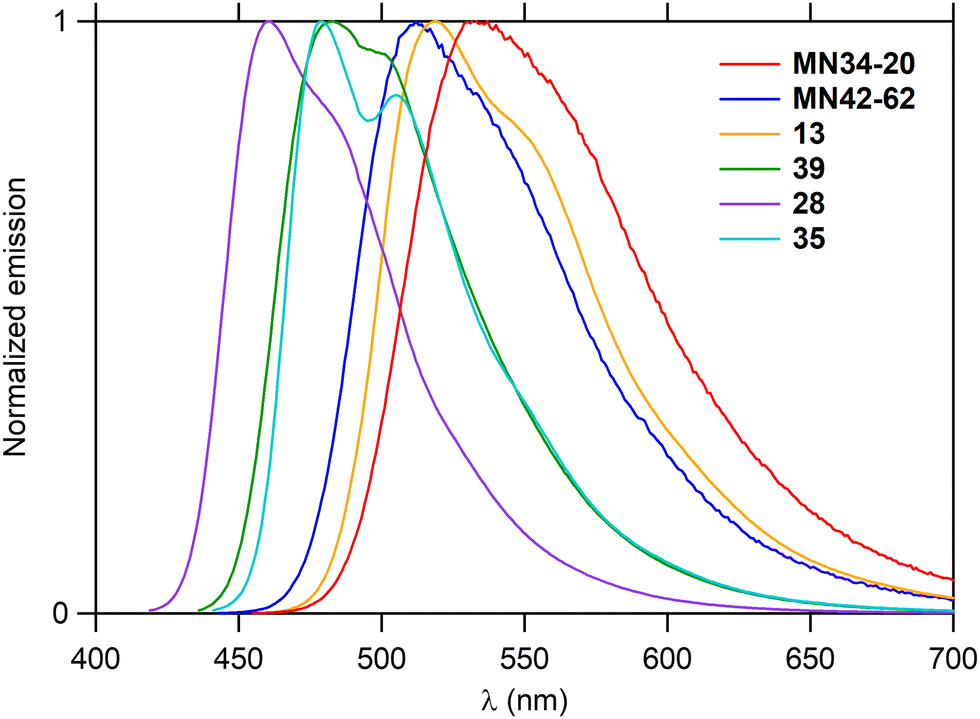 | ||
| Fig. 7 Emission spectra of the hexacycles in toluene. | ||
All compounds are highly fluorescent, with quantum yields ranging between 49% and 86%. The lifetimes vary between 8 and 16 ns, in agreement with a fluorescence emission. It should be noticed that the most fluorescent compounds are 28 and 39 with the extra nitrogen, which is consistent with their much higher radiative decay rate, but also with their slightly lower nonradiative decay rate, in comparison with their analogs MN42-62 and MN34-20 lacking this nitrogen, respectively.
Conclusions
Our objective was to develop syntheses of analogs of MN34-20 and MN42-62 and to evaluate their biological and photophysical properties. Through this study, we were confronted with the difficulty of reaching sulfur aromatic compounds and functionalizing them in a controlled way toward helical structures. Different approaches have thus been successively designed and carefully evaluated, taking advantage of successes and failures. Not only did this work ultimately lead to original hexacycles as the first fluorescent helical PIM kinase inhibitors, but we also established dimethyl disulfide as an original in situ trap to functionalize aromatic ketones using LiTMP. Given the usefulness of sulfides in chemistry, the ability to perform such a simple transformation will greatly enhance the interest of in situ traps in organic synthesis.Experimental
General details
THF and dioxane were freshly distilled from sodium-benzophenone. Acetonitrile, dichloromethane, dibutyl ether and pyridine were distilled over CaH2 under argon. Dimethylformamide and dimethylsulfoxide were distilled over CaH2 under vacuum. All alkyllithiums were titrated before use.78 2,2,6,6-Tetramethylpiperidine (TMPH) was distilled over CaH2 and stored under argon. The other commercially available starting materials were used without further purification. 1-Iodo-9-thioxanthone,14tert-butyl N-(4-fluorophenyl)carbamate,20 2-iodo-N-methylindole,42 2-iodobenzothiophene,43,44 2-iodo-N-(methoxymethyl)indole,49 3-benzoyl-2-chloropyridine59 and 2-(methylthio)benzoyl chloride66 were prepared as described. Column chromatography separations were achieved on silica gel (40–63 μm). Melting points were measured on a Kofler apparatus. IR spectra were taken on a PerkinElmer Spectrum 100 spectrometer. The 1H, 13C{1H} and 19F{1H} Nuclear Magnetic Resonance (NMR) spectra were recorded on Bruker Avance III spectrometers equipped with a BBFO probe (the solvent, temperature and frequency are specified below, in the product description). 1H chemical shifts (δ) are given in ppm relative to the solvent residual peak and 13C chemical shifts are relative to the central peak of the solvent signal.79 High resolution mass spectra measurements were performed at the CRMPO in Rennes (Centre Régional de Mesures Physiques de l’Ouest).General crystallographic details
The X-ray diffraction data were collected at T = 150(2) K either on an APEXII Kappa-CCD (Bruker-AXS) diffractometer equipped with a CDD plate detector (graphite monochromator; 3, 6a, 9, 13 and NM-049), or on a D8 Venture (Bruker-AXS) diffractometer equipped with a CMOS-PHOTON70 detector (multilayer monochromator; 17 and 36), using Mo-Kα radiation (λ = 0.71073 Å). The crystal structure was solved by dual-space algorithm using the SHELXT program,80 and then refined with full-matrix least-squares methods based on F2 (SHELXL program).81 All non-hydrogen atoms were refined with anisotropic atomic displacement parameters. Except those linked to nitrogen and oxygen that were introduced in the structural model through Fourier difference maps analysis in the case of 6a and 36, H atoms were finally included in their calculated positions and treated as riding on their parent atom with constrained thermal parameters. The molecular diagrams were generated by ORTEP-3 (version 2.02).82Synthesis of 1-aminothioxanthone (2) from 1-iodothioxanthone (1)
O), 180.0 (C, CO) ppm; HRMS (ASAP; Maxis 4G), m/z = 357.0453 (0 ppm) found (calcd for C21H11NO3S, M+˙, requires 357.04542). Crystal data for3: 2(C21H11NO3S)·CH2Cl2, M = 799.66, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 8.2771(3), b = 13.4367(6), c = 15.9360(6) Å, α = 95.3390(10), β = 93.9650(10), γ = 99.893(2) °, V = 1731.86(12) Å3, Z = 2, d = 1.533 g cm−3, μ = 0.365 mm−1. A final refinement on F2 with 7818 unique intensities and 500 parameters converged at ωR(F2) = 0.1502 (R(F) = 0.0591) for 5578 observed reflections with I > 2σ(I). CCDC 2204834.†
, a = 8.2771(3), b = 13.4367(6), c = 15.9360(6) Å, α = 95.3390(10), β = 93.9650(10), γ = 99.893(2) °, V = 1731.86(12) Å3, Z = 2, d = 1.533 g cm−3, μ = 0.365 mm−1. A final refinement on F2 with 7818 unique intensities and 500 parameters converged at ωR(F2) = 0.1502 (R(F) = 0.0591) for 5578 observed reflections with I > 2σ(I). CCDC 2204834.†
Without isolation of the hydrazinocarboxamide. To 1-(N-phthalimido)thioxanthone (3; 0.12 g, 0.30 mmol) were added EtOH (2 mL), CH2Cl2 (2 mL) and hydrazine hydrate (51% in H2O; 0.10 mL, 1.6 mmol). The reaction mixture was stirred at reflux for 3 h and then cooled down to rt. The white precipitate of phthalhydrazide was discarded by filtration, and the solvent was removed under reduced pressure to afford 1-aminothioxanthone (1) in 97% yield (66 mg) as a yellow solid. Further purification by column chromatography over silica gel (eluent: petroleum ether–CH2Cl2–AcOEt 75
:15:10) afforded the title product, but with non-reproducible yields.
With isolation of the hydrazinocarboxamide. 1-(N-phthalimido)thioxanthone (3; 1.4 g, 4.0 mmol) was dissolved in CHCl3 (30 mL) before slow addition of hydrazine hydrate (51% in H2O; 2.0 mL, 31 mmol). The stirring was continued for 2 h at rt. The yellow precipitate was then filtered, washed with CHCl3 (30 mL) and identified by NMR: 1H NMR ((CD3)2SO, 300 K, 300 MHz) δ 4.35 (br s, 2H, NH2), 7.55–7.70 (m, 5H), 7.76–7.89 (m, 4H), 8.45 (d, 1H, J = 8.3 Hz), 8.81 (d, 1H, J = 8.2 Hz), 9.70 (br s, 1H, NH), 13.42 (s, 1H, NH) ppm. The solid was next dissolved in CH2Cl2 (15 mL) and EtOH (15 mL), and the mixture was stirred for 2 h at reflux and cooled to rt. After filtration of the phthalhydrazide, the solvent was removed under reduced pressure to afford pure 1-aminothioxanthone (1) in 74% overall yield (0.67 g) as a yellow solid. Its 1H NMR spectrum was found similar to that reported previously:141H NMR (CDCl3, 300 K, 300 MHz) δ 6.55 (dd, 1H, J = 8.2 and 1.1 Hz), 6.76 (dd, 1H, J = 7.7 and 1.1 Hz), 7.00 (br s, 2H, NH2), 7.25 (t, 1H, J = 8.0 Hz), 7.37–7.46 (m, 2H), 7.50–7.55 (m, 1H), 8.50 (dd, 1H, J = 8.1 and 1.5 Hz).
Synthesis of 1-aminothioxanthone (2) from 3-fluoroaniline
O), 160.1 (d, C, J = 246 Hz, C3 or C2′, C–F), 160.6 (d, C, J = 247 Hz, C3 or C2′, C–F) ppm; 19F{1H} NMR (CDCl3, 300 K, 471 MHz) δ −117.3 (d, J = 9.3 Hz), −117.1 (d, J = 9.2 Hz) ppm. Crystal data for6a: C18H19F2NO3, M = 335.34, monoclinic, P21/n, a = 11.5846(9), b = 7.0406(5), c = 21.0857(13) Å, β = 101.424(2) °, V = 1685.7(2) Å3, Z = 4, d = 1.321 g cm−3, μ = 0.104 mm−1. A final refinement on F2 with 3823 unique intensities and 226 parameters converged at ωR(F2) = 0.1244 (RF = 0.0430) for 2812 observed reflections with (I > 2σ). CCDC 2204836.†
Replacing 2-fluorobenzaldehyde with 5-chloro-2-fluorobenzaldehyde in the present procedure only furnished the expected alcohol 6b in 6% yield (oxidation of 6b worked in ∼60% conversion using Dess–Martin periodinane while using pyridinium chlorochromate (PCC) proved much less efficient).
:10) led to the title product in 34% yield (0.11 g) as a white solid: mp 81 °C; IR (ATR) ν 667, 682, 729, 757, 780, 802, 829, 887, 929, 966, 1035, 1086, 1103, 1150, 1222, 1248, 1277, 1311, 1368, 1393, 1419, 1454, 1467, 1517, 1579, 1611, 1639, 1732, 2980, 3354 cm−1; 1H NMR (CDCl3, 300 K, 300 MHz) δ 1.51 (s, 9H, tBu), 6.71 (ddd, 1H, J = 10.7, 8.3 and 1.0 Hz), 7.10 (ddd, 1H, J = 10.7, 8.3 and 1.1 Hz), 7.24 (td, 1H, J = 7.5 and 1.0 Hz), 7.46 (td, 1H, J = 8.3 and 6.2 Hz), 7.49–7.56 (m, 1H), 7.64 (td, 1H, J = 7.5 and 1.8 Hz), 8.18 (d, 1H, J = 8.6 Hz), 9.37 (br s, 1H, NH) ppm; 13C{1H} NMR (CDCl3, 300 K, 75 MHz) δ 28.4 (3CH3, CMe3), 81.2 (C, CMe3), 109.1 (d, CH, J = 22.8 Hz, C4 or C3′), 114.9 (d, C, J = 14.3 Hz, C2 or C1′), 115.8 (d, CH, J = 3.1 Hz), 116.4 (d, CH, J = 22.0 Hz, C4 or C3′), 124.4 (d, CH, J = 3.7 Hz), 129.0 (d, C, J = 2.4 Hz), 129.1 (d, C, J = 2.5 Hz), 130.5 (C), 134.3 (d, CH, J = 8.9 Hz), 134.8 (d, CH, J = 11.1 Hz), 141.2 (d, CH, J = 4.1 Hz), 152.8 (C), 160.7 (d, C, J = 253 Hz, C3 or C2′, C–F), 162.4 (d, C, J = 251 Hz, C3 or C2′, C–F), 191.9 (d, C, J = 1.7 Hz, CO) ppm; 19F{1H} NMR (CDCl3, 300 K, 282 MHz) δ −113.3, −106.6 ppm.
:20) = 0.40; IR (ATR) ν 692, 762, 785, 852, 901, 943, 1057, 1099, 1123, 1151, 1185, 1251, 1296, 1451, 1524, 1576, 1602, 1647, 1747, 2857, 2928, 2960, 3095 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 4.77 (dd, 1H, J = 6.2 and 2.0 Hz, OCH = CHH), 5.07 (dd, 1H, J = 13.9 and 2.0 Hz, OCH = CHH), 7.07 (td, 1H, J = 8.7 and 1.1 Hz, H5), 7.24 (d, 1H, J = 8.1 Hz, H3), 7.37 (td, 1H, J = 8.3 and 5.9 Hz, H4), 7.47 (dd, 1H, J = 13.9 and 6.2 Hz, OCHCH2) ppm; 13C{1H} NMR (CDCl3, 300 K, 126 MHz) δ 99.8 (CH2), 114.7 (d, CH, J = 21.5 Hz, C5), 121.2 (d, C, J = 19.3 Hz, C1), 125.8 (d, CH, J = 3.3 Hz, C3), 132.4 (d, CH, J = 9.1 Hz, C4), 133.1 (d, C, J = 4.4 Hz, C2), 141.1 (CH, OCHCH2), 160.2 (d, C, J = 255 Hz, C6, C–F), 160.3 (C, CO) ppm; 19F{1H} NMR (CDCl3, 300 K, 471 MHz) δ −111.0 ppm.
:5 to 50:50), the title product was isolated in 75% yield (0.50 g) as white crystals: Rf (CHCl3–petroleum ether 65:35) = 0.26; mp 102 °C; IR (ATR) ν 732, 752, 770, 802, 819, 841, 854, 913, 937, 1006, 1030, 1053, 1078, 1103, 1145, 1240, 1279, 1352, 1370, 1395, 1455, 1489, 1598, 1612, 1682, 1741, 2852, 2923 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 1.25 (s, 9H, tBu), 7.03 (dt, 1H, J = 9.3 and 2.3 Hz), 7.07–7.13 (m, 3H), 7.25 (td, 1H, J = 7.6 and 1.1 Hz), 7.41 (td, 1H, J = 8.1 and 6.2 Hz), 7.46–7.52 (m, 1H), 7.63 (td, 1H, J = 7.2 and 1.8 Hz) ppm; 13C{1H} NMR (CDCl3, 300 K, 126 MHz) δ 27.5 (3CH3, CMe3), 84.3 (C, CMe3), 115.4 (d, CH, J = 10.4 Hz), 115.6 (d, CH, J = 11.0 Hz), 116.2 (d, CH, J = 22.9 Hz), 124.4 (d, CH, J = 3.2 Hz), 124.6 (d, CH, J = 3.5 Hz), 125.7 (d, C, J = 13.9 Hz), 130.2 (d, CH, J = 2.5 Hz), 130.3 (d, CH, J = 8.8 Hz), 132.9 (d, CH, J = 8.3 Hz), 139.9 (d, C, J = 10.0 Hz), 152.2 (C), 159.2 (d, C, J = 250 Hz), 162.9 (d, C, J = 247.1 Hz), 167.8 (C) ppm; 19F{1H} NMR (CDCl3, 300 K, 282 MHz) δ −115.25, −112.0 ppm. Crystal data for9: C18H17F2NO3, M = 333.33, triclinic, P, a = 9.0170(11), b = 11.7369(15), c = 17.176(2) Å, α = 70.454(4), β = 81.368(4), γ = 75.963(4) °, V = 1657.2(4) Å3, Z = 4, d = 1.336 g cm−3, μ = 0.106 mm−1. A final refinement on F2 with 7335 unique intensities and 443 parameters converged at ωR(F2) = 0.1316 (RF = 0.0468) for 5203 observed reflections with (I > 2σ). CCDC 2204831.†
Unsuccessful attempt to prepare 2-(5-chloro-2-methylthiobenzoyl)aniline
:5 to 90:100) led to the title product which was isolated in 50% yield (0.28 g) as a white solid: Rf (petroleum ether–AcOEt 80:20) = 0.34; mp 70 °C; IR (ATR) ν 675, 698, 730, 818, 876, 973, 1060, 1077, 1105, 1155, 1187, 1252, 1293, 1372, 1425, 1437, 1457, 1541, 1582, 1682, 2734, 2839, 3047 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 2.50 (s, 3H, Me), 7.29 (d, 1H, J = 8.6 Hz, H3), 7.49 (dd, 1H, J = 8.6 and 2.4 Hz, H4), 7.78 (d, 1H, J = 2.4 Hz, H6), 10.24 (s, 1H, CHO) ppm; 13C{1H} NMR (CDCl3, 300 K, 126 MHz) δ 16.0 (CH3, SMe), 127.6 (CH, C3), 131.1 (C), 132.2 (CH, C6), 134.0 (CH, C4), 134.1 (C), 141.9 (C), 190.0 (CH, CHO) ppm.
Synthesis of Boc-protected 2-aminothioxanthone
:40 to 100:0), the title product was isolated in 72% yield (0.24 g) as a yellow solid: Rf (CH2Cl2) = 0.29; mp 225 °C; IR (ATR) ν 732, 744, 778, 811, 908, 922, 1007, 1058, 1086, 1122, 1158, 1184, 1245, 1309, 1368, 1482, 1519, 1572, 1591, 1604, 1624, 1702, 2979, 3315 cm−1; 1H NMR (CDCl3, 300 K, 300 MHz) δ 1.54 (d, 9H, J = 4.8 Hz, tBu), 6.70 (br s, 1H), 7.48 (ddd, 1H, J = 8.2, 6.2 and 2.0 Hz), 7.55 (d, 1H, J = 8.9 Hz), 7.59–7.60 (m, 2H), 8.11 (br d, 1H, J = 9.1 Hz), 8.26 (d, 1H, J = 2.5 Hz), 8.61 (d, 1H, J = 8.1 Hz) ppm; 1H NMR ((CD3)2SO, 300 K, 500 MHz) δ 1.51 (s, 9H, tBu), 7.56 (ddd, 1H, J = 8.1, 7.5 and 1.3 Hz, H7), 7.73–7.76 (m, 2H, H5 and H6), 7.81 (d, 1H, J = 8.9 Hz, H4), 7.83 (dd, 1H, J = 8.9 and 2.3 Hz, H3), 8.46 (dd, 1H, J = 8.1 and 0.8 Hz, H8), 8.66 (d, 1H, J = 2.3 Hz, H1), 9.75 (br s, 1H, NH) ppm; 13C{1H} NMR (CDCl3, 300 K, 75 MHz) δ 28.5 (3CH3, CMe3), 81.3 (C, CMe3), 118.3 (CH), 124.1 (CH), 126.2 (CH), 126.3 (CH), 127.0 (CH), 128.9 (C), 129.8 (C), 130.1 (CH), 131.2 (C), 132.3 (CH), 137.5 (C), 137.7 (C), 152.8 (C), 179.8 (C, CO) ppm; 13C{1H} NMR ((CD3)2SO, 300 K, 126 MHz) δ 28.1 (3CH3, CMe3), 79.6 (C, CMe3), 116.9 (CH), 123.9 (CH), 126.5 (CH), 126.5 (CH), 127.0 (CH), 128.0 (C), 128.8 (C), 129.1 (CH), 129.3 (C), 132.7 (CH), 136.7 (C), 138.6 (C), 152.7 (C), 178.6 (C, CO) ppm.
N-Arylation-cyclization from 1-aminothioxanthone using 2-iodo-N-methylindole
:39:1) to give the title product in 94% yield (0.16 g) as an orange solid: Rf (CHCl3–petroleum ether 65:35) = 0.14; mp 190 °C; IR (ATR) ν 803, 811, 839, 870, 972, 1125, 1160, 1245, 1268, 1288, 1337, 1392, 1425, 1475, 1554, 1583, 1616, 2166, 2921 cm−1; 1H NMR (CDCl3, 300 K, 300 MHz) δ 4.00 (s, 3H, Me), 7.17 (ddd, 1H, J = 8.2, 7.1 and 1.1 Hz), 7.31 (d, 1H, J = 7.3 Hz), 7.34 (dd, 1H, J = 7.3 and 1.6 Hz), 7.39 (dd, 1H, J = 7.8 and 1.7 Hz), 7.41 (d, 1H, J = 7.3 Hz), 7.49 (dd, 1H, J = 7.8 and 1.7 Hz), 7.50–7.57 (m, 2H), 7.83 (d, 1H, J = 8.4 Hz), 8.33 (d, 1H, J = 8.0 Hz), 8.48 (dd, 1H, J = 7.8 and 1.7 Hz) ppm; 13C{1H} NMR (CDCl3, 300 K, 75 MHz) δ 28.0 (CH3), 108.9 (CH), 112.8 (C), 117.8 (CH), 119.5 (CH), 120.8 (C), 122.0 (C), 123.8 (CH), 124.1 (CH), 126.1 (CH), 127.3 (CH), 128.0 (CH), 128.5 (CH), 129.6 (C), 129.9 (C), 130.0 (CH), 130.6 (CH), 135.7 (C), 136.4 (C), 143.2 (C), 147.7 (C), 154.6 (C) ppm; HRMS (ASAP; Maxis 4G), m/z = 338.0865 (2 ppm) found (calcd for C22H14N2S, M+·, requires 338.0872). Crystal data for13: C22H14N2S, M = 338.41, monoclinic, P21/c, a = 13.8094(10), b = 12.1986(9), c = 19.1371(15) Å, β = 99.043(3)°, V = 3183.7(4) Å3, Z = 8, d = 1.412 g cm−3, μ = 0.209 mm−1. A final refinement on F2 with 7274 unique intensities and 453 parameters converged at ωR(F2) = 0.0990 (R(F) = 0.0477) for 4807 observed reflections with I > 2σ(I). CCDC 2204832.†
11-Phenylbenzothieno[2,3-b]quinoline (NM-049) was similarly prepared in 30% yield. Its analyses proved similar to those reported.50Crystal data forNM-049: C21H13NS, M = 311.38, triclinic, P, a = 8.8473(9), b = 9.7021(9), c = 10.1837(10) Å, α = 66.774(3), β = 69.333(4), γ = 77.281(3)°, V = 748.26(13) Å3, Z = 2, d = 1.382 g cm−3, μ = 0.214 mm−1. A final refinement on F2 with 3338 unique intensities and 208 parameters converged at ωR(F2) = 0.2687 (R(F) = 0.0991) for 2620 observed reflections with I > 2σ(I). CCDC 2204837.†
Synthesis of 2-amino-6,2′-difluorobenzophenone and its N-arylation-cyclization
:20) = 0.24; mp 99 °C; 1H NMR (CDCl3, 300 K, 400 MHz) δ 6.03 (bs s, 2H, NH2), 6.29 (ddd, 1H, J = 11.5, 8.0 and 0.64 Hz), 6.48 (d, 1H, J = 8.3 Hz), 7.08 (ddd, 1H, J = 10.5, 8.4 and 1.0 Hz), 7.18 (td, 1H, J = 8.2 and 5.9 Hz), 7.20 (td, 1H, J = 7.5 and 1.1 Hz), 7.44 (tdd, 1H, J = 7.2, 5.1 and 1.9 Hz), 7.55 (td, 1H, J = 7.4 and 1.7 Hz) ppm; 19F{1H} NMR (CDCl3, 300 K, 282 MHz) δ −115.3 (d, J = 2.6 Hz), −106.5 (d, J = 3.5 Hz) ppm.
:35) 10-fluoro-11-(2-fluorophenyl)benzothieno[2,3-b]quinoline (15a) in 17% yield (29 mg). It was identified by NMR: 1H NMR (CDCl3, 300 K, 400 MHz) δ 6.65 (d, 1H, J = 8.3 Hz), 7.09–7.17 (m, 2H), 7.33–7.40 (m, 3H), 7.43 (td, 1H, J = 7.7 and 1.2 Hz), 7.60–7.66 (m, 1H), 7.70 (ddd, 1H, J = 8.6, 7.7 and 5.4 Hz), 7.82 (d, 1H, J = 7.8 Hz), 8.03 (d, 1H, J = 8.6 Hz) ppm; 19F{1H} NMR (CDCl3, 300 K, 376 MHz) δ −114.3, −111.0 ppm. This compound was directly involved in the cyclization step to the hexacycle as follows. To 15a (29 mg, 0.80 mmol) in dry DMF (5 mL) under argon, was added Na2S (0.34 g, 4.3 mmol). The mixture was stirred at 70 °C for 6 h and poured onto ice-water (20 mL). The precipitate was collected by filtration. It was then dissolved in Et2O (40 mL), and washed with brine (20 mL). The organic layer was dried over MgSO4 before removal of the solvents under reduced pressure. Benzothieno[2,3-b]benzothiopyrano[4,3,2-de]quinoline (MN34-20) was obtained in 20% yield (6 mg) and identified by comparison of its 1H NMR spectrum with the one reported:141H NMR (CDCl3, 300 K, 400 MHz) δ 7.22–7.27 (m, 2H), 7.38–7.44 (m, 2H), 7.46 (dd, 1H, J = 7.3, 1.0 Hz), 7.54 (dd, 1H, J = 7.8 and 1.2 Hz), 7.60 (dd, 1H, J = 8.4 and 7.2 Hz), 7.82 (d, 1H, J = 7.9 Hz), 7.85 (dd, 1H, J = 8.4 and 1.1 Hz), 8.20 (dd, 1H, J = 8.1 and 1.0 Hz), 8.24 (d, 1H, J = 8.2 Hz) ppm.
:15; Rf = 0.14) 10-fluoro-11-(2-fluorophenyl)-N-(methoxymethyl)indolo[2,3-b]quinoline (15b) in 85% yield (62 mg) as a brown solid: mp 168 °C; IR (ATR) ν 695, 752, 830, 882, 918, 995, 1076, 1177, 1233, 1268, 1284, 1315, 1396, 1487, 1595, 1716, 2149, 2863, 2943 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 3.45 (s, 3H, Me), 5.97 (d, 1H, J = 11.0 Hz, CHH), 6.00 (d, 1H, J = 11.0 Hz, CHH), 6.84 (d, 1H, J = 7.8 Hz), 7.04 (ddd, 1H, J = 7.6, 2.7 and 1.1 Hz), 7.05 (t, 1H, J = 7.6 Hz), 7.34 (t, 1H, J = 8.9 Hz), 7.38 (td, 1H, J = 7.5 and 1.1 Hz), 7.48 (td, 1H, J = 7.4 and 1.8 Hz), 7.50 (t, 1H, J = 7.1 Hz), 7.58–7.60 (m, 2H), 7.63 (ddd, 1H, J = 8.7, 7.6 and 5.5 Hz), 7.99 (d, 1H, J = 8.5 Hz) ppm; 13C{1H} NMR (CDCl3, 300 K, 126 MHz) δ 56.8 (CH3), 72.7 (CH2), 108.7 (d, CH, J = 21.7 Hz), 110.1 (CH), 114.8 (d, C, J = 9.6 Hz), 116.0 (d, CH, J = 21.5 Hz), 117.9 (C), 120.9 (C), 121.3 (CH), 123.2 (CH), 124.5 (CH), 124.5 (d, CH, J = 7.9 Hz), 126.6 (dd, C, J = 17.4 and 3.3 Hz), 128.4 (d, CH, J = 10.1 Hz), 128.7 (CH), 130.4 (t, CH, J = 2.9 Hz), 130.5 (d, CH, J = 7.7 Hz), 133.3 (C), 142.3 (C), 148.3 (C), 152.8 (C), 159.5 (d, C, J = 243 Hz, C–F), 159.8 (d, C, J = 256 Hz, C–F) ppm; 19F{1H} NMR (CDCl3, 300 K, 471 MHz) δ −114.5, −112.4 ppm. This compound was directly involved in the cyclization step to the hexacycle as follows. To 15b (40 mg, 0.11 mmol) in dry DMF (1 mL) under argon, was added Na2S (52 mg, 0.60 mmol). The mixture was stirred at 120 °C for 5 h and poured onto ice-water (5 mL). The product was extracted with AcOEt (3 × 10 mL), and the organic layer washed with brine (10 mL). The organic layer was dried over MgSO4 before removal of the solvents under reduced pressure. The expected product was only detected in the crude by NMR (∼10% yield).
Attempts to prepare 1-chlorobenzothiopyrano[3,2-c]pyridin-10-one (19) from 2-chloropyridine
:20; Rf = 0.11), the title product in 94% yield (0.28 g) as a white solid: mp 114 °C; IR (ATR) ν 738, 750, 816, 854, 949, 1018, 1072, 1132, 1196, 1228, 1313, 1414, 1440, 1467, 1569, 3265 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 2.77 (d, 1H, J = 4.1 Hz, OH), 6.43 (d, 1H, J = 4.1 Hz, CH(OH)), 7.21 (ddd, 1H, J = 9.1, 7.4 and 2.1 Hz, H4′ or H5′), 7.27 (dd, 1H, J = 7.6 and 4.7 Hz, H5), 7.30 (dd, 1H, J = 7.8 and 2.1 Hz, H3′ or H6′), 7.33 (td, 1H, J = 7.4 and 0.95 Hz, H4′ or H5′), 7.60 (dd, 1H, J = 8.0 and 0.9 Hz, H3′ or H6′), 7.73 (dd, 1H, J = 7.6 and 1.7 Hz, H4), 8.35 (dd, 1H, J = 4.7 and 1.9 Hz, H6) ppm; JMOD{1H} NMR (CDCl3, 300 K, 126 MHz) δ 71.7 (CH, CH(OH)), 122.8 (CH, C5), 123.8 (C), 127.9 (CH, C4′ or C5′), 128.6 (CH, C3′ or C6′), 129.9 (CH, C4′ or C5′), 133.3 (CH, C3′ or C6′), 136.3 (C), 137.4 (CH, C4), 140.1 (C), 148.9 (CH, C6), 150.7 (C) ppm.
With Dess–Martin periodinane. To the alcohol 16 (0.88 g, 2.9 mmol) in THF (30 mL) under argon, was added Dess–Martin periodinane (DMP; 1.8 g, 4.2 mmol). The reaction mixture was stirred at rt for 3 h before being quenched with a saturated aqueous solution of Na2S2O3 (9 mL). The product was extracted with AcOEt (3 × 20 mL). The organic layer was washed with a saturated aqueous solution of Na2CO3 (10 mL) and dried over MgSO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography over silica gel (eluent: petroleum ether–AcOEt 75
:25) to give the title product in 62% yield (0.53 g). It was identified by NMR: 1H NMR (CDCl3, 300 K, 300 MHz) δ 7.37 (dd, 1H, J = 7.6 and 4.8 Hz, H5), 7.39 (td, 1H, J = 7.5 and 2.1 Hz, H4′ or H5′), 7.43 (td, 1H, J = 7.4 and 1.9 Hz, H4′ or H5′), 7.51–7.54 (m, 1H, H3′ or H6′), 7.64–7.67 (m, 1H, H3′ or H6′), 7.90 (dd, 1H, J = 7.6 and 2.0 Hz, H4), 8.54 (dd, 1H, J = 4.8 and 2.0 Hz, H6) ppm; JMOD{1H} NMR (CDCl3, 300 K, 126 MHz) δ 119.7 (C), 121.5 (CH), 123.8 (CH), 127.7 (CH), 129.6 (CH), 132.5 (CH), 133.7 (CH), 138.5 (C), 145.4 (C), 150.9 (CH), 152.8 (C), 193.7 (C, CO) ppm. These data are close to those already reported.55Crystal data for17: C12H7BrClNO, M = 296.55, orthorhombic, Pna21, a = 13.664(2), b = 8.7402(15), c = 18.815(4) Å, V = 2247.0(7) Å3, Z = 8, d = 1.753 g cm−3, μ = 3.871 mm−1. A final refinement on F2 with 4810 unique intensities and 293 parameters converged at ωR(F2) = 0.1482 (RF = 0.0564) for 3506 observed reflections with (I > 2σ). CCDC 2204833.†
With 2-iodoxybenzoic acid. 2-Iodoxybenzoic acid (IBX; 0.84 g, 3.0 mmol) was added to a solution of 16 (0.30 g, 1.0 mmol) in AcOEt (7 mL) and the reaction mixture was heated in a preheated oil bath at 80 °C for 6 h. The reaction mixture was cooled to rt, filtrated over silica and the filter cake was thoroughly washed with AcOEt. The combined filtrates were concentrated under vacuum to give the crude product. This was purified by column chromatography over silica gel (eluent: petroleum ether–AcOEt 80
:20 to 70:30) to give the title product in 95% yield (0.28 g).
By Swern oxidation. To a solution of oxalyl chloride (0.32 mL, 3.8 mmol) in dry CH2Cl2 (10 mL) at −80 °C under argon, was added dry DMSO (0.27 mL, 3.8 mmol), and the mixture was stirred for 15 min at this temperature. A solution of the alcohol 16 (0.76 g, 2.5 mmol) in dry CH2Cl2 (5 mL) was then added dropwise, and the mixture was allowed to reach −40 °C in 0.5 h and kept at this temperature for an additional hour. Et3N (1.7 mL, 12.5 mmol) was next added slowly, and the mixture was warmed up to rt before addition of H2O (20 mL). The product was extracted with CH2Cl2 (2 × 20 mL). The organic layer was dried over MgSO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography over silica gel (eluent: CH2Cl2) to give the title product in 98% yield (1.3 g).
Synthesis of 3-benzoyl-2-chloro-4-(methylthio)pyridine (21) and 3-benzoyl-2-chloro-4-(methylsulfinyl)pyridine (22a)
:18:2), the title product was obtained in 16% yield as a white solid: IR (ATR) ν 685, 726, 791, 807, 821, 858, 950, 984, 1081, 1113, 1130, 1148, 1230, 1281, 1318, 1369, 1437, 1454, 1522, 1568, 1920, 2927, 3052 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 2.48 (s, 3H, Me), 7.00 (dd, 1H, J = 5.4 and 1.6 Hz, H5), 7.08 (d, 1H, J = 1.4 Hz, H3), 8.15 (d, 1H, J = 5.4 Hz, H6) ppm; JMOD{1H} NMR (CDCl3, 300 K, 126 MHz) δ 13.9 (CH3), 118.9 (CH, C5), 119.3 (CH, C3), 148.7 (CH, C6), 151.9 (C, C2), 153.7 (C, C4) ppm.
O) ppm.
O) ppm. Note that the sulfone 22b was similarly isolated in 4% yield (21 mg) as a white solid: IR (ATR) ν 736, 748, 780, 818, 847, 924, 962, 1001, 1070, 1111, 1147, 1201, 1265, 1317, 1359, 1450, 1560, 1596, 1670, 2925 cm−1; 1H NMR (CDCl3, 300 K, 300 MHz) δ 3.16 (s, 3H, Me), 7.43–7.49 (m, 2H, H3′ and H5′), 7.68 (tt, 1H, J = 7.4 and 1.3 Hz, H4′), 7.73–7.76 (m, 2H, H2′ and H6′), 7.93 (d, 1H, J = 5.1 Hz, H5), 8.76 (d, 1H, J = 5.1 Hz, H6) ppm.
Synthesis of 11-(2-(methylsulfinyl)phenyl)benzofuro[2,3-b]1,8-naphthyridine (27)
:20; Rf = 0.14), the title ketone in 80% yield (0.51 g) as a yellow solid: mp 176 °C; IR (ATR) ν 669, 688, 741, 760, 784, 815, 849, 927, 963, 1047, 1098, 1130, 1171, 1210, 1261, 1313, 1429, 1458, 1567, 1586, 1597, 1647, 1736, 2927, 2984, 3063 cm−1; 1H NMR (CDCl3, 300 K, 300 MHz) δ 2.29 (s, 3H, Me), 7.03 (ddd, 1H, J = 8.0, 7.2 and 1.2 Hz, H4′ or H5′), 7.21 (ddd, 1H, J = 7.4, 4.9 and 1.9 Hz, H5), 7.23 (dd, 1H, J = 8.1 and 1.2 Hz, H3′ or H6′), 7.29 (dtd, 1H, J = 7.8, 1.5 and 0.5 Hz, H3′ or H6′), 7.34 (ddd, 1H, J = 8.1, 7.2 and 1.5 Hz, H4′ or H5′), 7.90 (ddd, 1H, J = 9.4, 7.4 and 2.0 Hz, H4), 8.22 (ddd, 1H, J = 4.9, 2.1 and 1.1 Hz, H6) ppm; 13C{1H} NMR (CDCl3, 300 K, 75 MHz) δ 15.8 (CH3), 121.5 (d, CH, J = 4.6 Hz, C5), 121.6 (d, C, J = 28.6 Hz, C3), 123.7 (CH), 125.8 (CH), 131.4 (d, CH, J = 1.7 Hz, C5), 132.4 (CH), 134.7 (C), 141.6 (d, CH, J = 3.0 Hz, C4), 141.7 (C), 150.3 (d, CH, J = 15.0 Hz, C6), 159.8 (d, C, J = 244 Hz, C2, C–F), 191.2 (d, C, J = 4.9 Hz, CO) ppm; 19F{1H} NMR (CDCl3, 300 K, 282 MHz) δ −63.95 (d, J = 9.1 Hz) ppm.
:20), in 74% yield (0.94 g) as a yellow oil: IR (ATR) ν 702, 743, 776, 818, 860, 895, 960, 1102, 1117, 1158, 1202, 1225, 1251, 1273, 1302, 1401, 1427, 1458, 1476, 1571, 1598, 1667, 3064 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 7.10 (dd, 1H, J = 9.7 and 8.8 Hz, H3′), 7.37 (ddd, 1H, J = 7.5, 4.9 and 1.9 Hz, H5), 7.53 (ddd, 1H, J = 8.8, 4.3 and 2.7 Hz, H4′), 7.72 (dd, 1H, J = 6.0 and 2.7 Hz, H6′), 8.18 (ddd, 1H, J = 9.3, 7.4 and 2.0 Hz, H4), 8.42 (ddd, 1H, J = 4.9, 2.1 and 1.1 Hz, H6) ppm; 13C{1H} NMR (CDCl3, 300 K, 126 MHz) δ 118.0 (d, CH, J = 24.3 Hz, C3′), 121.8 (d, C, J = 1.0 Hz), 122.0 (d, CH, J = 4.5 Hz, C5), 127.7 (d, C, J = 13.9 Hz), 130.3 (d, C, J = 3.3 Hz), 130.5 (d, CH, J = 2.4 Hz, C6′), 134.7 (d, CH, J = 9.0 Hz, C4′), 141.8 (d, CH, J = 2.7 Hz, C4), 151.7 (d, CH, J = 15.5 Hz, C6), 159.7 (d, C, J = 256 Hz, C–F, C2′), 160.7 (d, C, J = 245 Hz, C–F, C2), 187.1 (d, C, J = 5.2 Hz, CO) ppm; 19F{1H} NMR (CDCl3, 300 K, 471 MHz) δ −114.5 (d, J = 2.5 Hz), −64.3 ppm.
:20) = 0.32; mp not measured due to decomposition; IR (ATR) ν 740, 761, 844, 932, 971, 1066, 1126, 1240, 1302, 1354, 1430, 1549, 1585, 1618, 2974, 3142, 3406 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 2.42 (s, 3H, Me), 6.54 (dd, 1H, J = 7.8 and 4.7 Hz, H5), 6.98 (br s, 2H, NH2), 7.23 (dd, 1H, J = 7.7 and 1.7 Hz, H3′ or H6′), 7.25 (td, 1H, J = 7.5 and 1.2 Hz, H4′ or H5′), 7.40 (d, 1H, J = 7.6 Hz, H3′ or H6′), 7.44 (ddd, 1H, J = 8.1, 6.4 and 2.2 Hz, H4′ or H5′), 7.52 (dd, 1H, J = 7.8 and 1.9 Hz, H4), 8.24 (dd, 1H, J = 4.7 and 1.9 Hz, H6) ppm; 13C{1H} NMR (CDCl3, 300 K, 126 MHz) δ 17.1 (CH3), 112.5 (CH, C5), 113.3 (C, C3), 125.3 (CH, C4′ or C5′), 127.9 (CH, C3′ or C6′), 128.1 (CH, C3′ or C6′), 130.5 (CH, C4′ or C5′), 136.8 (C), 139.6 (C), 143.3 (CH, C4), 154.7 (CH, C6), 159.6 (C, C2), 198.1 (C, CO) ppm.
:20) = 0.11; mp 174 °C; IR (ATR) ν 704, 782, 810, 880, 954, 1080, 1106, 1140, 1158, 1221, 1256, 1299, 1348, 1398, 1433, 1474, 1549, 1618, 3127, 3398 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 6.61 (dd, 1H, J = 7.9 and 4.7 Hz, H5), 7.06 (br s, 2H, NH2), 7.11 (t, 1H, J = 8.8 Hz, H3′), 7.39 (dd, 1H, J = 5.8 and 2.7 Hz, H6′), 7.45 (ddd, 1H, J = 8.5, 4.4 and 2.8 Hz, H4′), 7.61 (dt, 1H, J = 8.1 and 2.4 Hz, H4), 8.28 (dd, 1H, J = 4.8 and 1.8 Hz, H6) ppm; 13C{1H} NMR (CDCl3, 300 K, 126 MHz) δ 112.7 (CH, C5), 112.8 (C, C3), 117.8 (d, CH, J = 23.5 Hz, C3′), 129.0 (d, C, J = 18.6 Hz, C1′), 129.6 (d, CH, J = 3.8 Hz, C6′), 130.0 (d, C, J = 3.4 Hz, C5′), 132.2 (d, CH, J = 8.2 Hz, C4′), 143.0 (d, CH, J = 2.3 Hz, C4), 155.3 (CH, C6), 157.5 (d, C, J = 250 Hz, C–F, C2′), 159.5 (C, C2), 192.4 (C, CO) ppm; 19F NMR (CDCl3, 300 K, 471 MHz) δ −115.7 ppm.
:50) to give the title product in 50% yield (0.255 g). This compound was identified by NMR: 1H NMR (CDCl3, 300 K, 500 MHz) δ 2.29 (s, 3H, Me), 6.94 (d, 1H, J = 7.5 Hz), 7.15 (t, 1H, J = 7.6 Hz), 7.30 (dd, 1H, J = 7.5 and 1.5 Hz), 7.41 (td, 1H, J = 7.5 and 1.3 Hz), 7.49 (ddd, 1H, J = 8.3, 7.8 and 1.0 Hz), 7.52 (d, 1H, J = 8.2 Hz), 7.46 (br s, 1H, H9), 7.62 (d, 1H, J = 7.1 Hz, H4), 7.64 (td, 1H, J = 7.7 and 1.25 Hz), 8.04 (br d, 1H, J = 7.3 Hz, H10), 9.17 (br s, 1H, H8) ppm; JMOD{1H} NMR (CDCl3, 300 K, 126 MHz) δ 15.7 (CH3, Me), 112.1 (CH, C4), 117.5 (C), 121.8 (C), 123.0 (CH), 123.8 (CH), 125.6 (CH, C9), 126.0 (CH), 129.7 (CH), 129.9 (CH), 130.1 (C), 130.3 (CH), 132.6 (C), 135.4 (CH, C10), 138.5 (C), 142.5 (C), 153.2 (CH), 154.2 (C), 156.3 (C), 164.3 (C) ppm (1CH not seen).
Attempt to prepare benzofuro[2,3-b]benzothiopyrano[4,3,2-de]1,8-naphthyridine (28) from 27
Synthesis of 5-methyl-10-(methylsulfinyl)-11-phenylindolo[2,3-b]1,8-naphthyridine (34)
:20 to 70:30) to give the title product in 60% yield as a white solid (11.6 g): Rf (petroleum ether–AcOEt 70:30) = 0.89; mp 95–96 °C; IR (ATR) ν 735, 753, 773, 804, 840, 902, 993, 1027, 1049, 1063, 1111, 1152, 1167, 1234, 1256, 1267, 1292, 1308, 1365, 1390, 1468, 1531, 1586, 1715, 2982, 3183 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 1.54 (s, 9H, tBu), 6.93 (ddd, 1H, J = 7.4, 5.0 and 1.1 Hz, H5), 7.65 (ddd, 1H, J = 8.9, 7.3 and 2.0 Hz, H4), 8.00 (d, 1H, J = 8.5 Hz, H3), 8.35 (ddd, 1H, J = 5.0, 2.1 and 0.9 Hz, H6), 9.54 (br s, 1H, NH) ppm; JMOD{1H} NMR (CDCl3, 300 K, 126 MHz) δ 28.5 (3CH3, CMe3), 80.8 (C, CMe3), 112.7 (CH, C3), 118.1 (CH, C5), 138.4 (CH, C4), 147.8 (CH, C6), 152.9 (C, C2 or CO), 153.0 (C, C2 or CO) ppm. These data are as described previously.84
:50; Rf = 0.36) to give the title product in 31% yield (0.93 g) as a white solid: mp 168–169 °C; IR (ATR) ν 727, 756, 773, 793, 821, 843, 879, 920, 1023, 1046, 1148, 1229, 1254, 1322, 1369, 1392, 1445, 1460, 1515, 1591, 1731, 2974, 3223 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 1.49 (s, 9H, tBu), 4.31 (br s, 1H, OH), 5.96 (s, 1H, CH(OH)), 7.02 (dd, 1H, J = 7.7 and 4.8 Hz, H5), 7.25–7.29 (m, 1H, H4′), 7.31–7.37 (m, 4H, H2′, H3′, H5′ and H6′), 7.45 (dd, 1H, J = 7.7 and 1.8 Hz, H4), 7.81 (br s, 1H, NH), 8.32 (dd, 1H, J = 4.8 and 1.8 Hz, H6) ppm; JMOD{1H} NMR (CDCl3, 300 K, 126 MHz) δ 28.4 (3CH3, CMe3), 72.0 (CH, CH(OH)), 81.4 (C, CMe3), 120.8 (CH, C5), 126.4 (2CH, C2′ and C6′), 127.6 (CH, C4′), 128.5 (2CH, C3′ and C5′), 131.7 (C, C3), 138.7 (CH, C4), 141.9 (C, C1′), 147.8 (CH, C6), 149.3 (C, C2), 154.2 (C, CO) ppm.
:20 to 70:30) to give the title product in 88% yield (2.1 g) as a colorless oil: Rf (petroleum ether–AcOEt 80:20) = 0.48; IR (ATR) ν 744, 757, 786, 813, 857, 913, 976, 993, 1073, 1094, 1143, 1253, 1281, 1310, 1367, 1402, 1435, 1472, 1573, 1589, 1707, 2935, 2978 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 1.49 (s, 9H, tBu), 3.38 (s, 3H, CH2OMe), 5.32 (s, 2H, CH2OMe), 7.06 (ddd, 1H, J = 7.5, 4.9 and 1.1 Hz, H5), 7.47 (d, 1H, J = 8.2 Hz, H3), 7.64 (ddd, 1H, J = 8.2, 7.4 and 2.0 Hz, H4), 8.41 (dd, 1H, J = 4.9 and 1.3 Hz, H6) ppm; JMOD{1H} NMR (CDCl3, 300 K, 126 MHz) δ 28.3 (3CH3, CMe3), 56.5 (CH3, OMe), 78.5 (CH2), 81.8 (C, CMe3), 120.6 (CH, C5), 120.9 (CH, C3), 137.3 (CH, C4), 148.2 (CH, C6), 154.0 (C, C2 or CO), 154.1 (C, C2 or CO) ppm.
:50; Rf = 0.57) to give the title product in a quantitative yield (745 mg) as a white solid: mp 175–176 °C; IR (ATR) ν 708, 745, 763, 771, 788, 814, 839, 902, 931, 939, 960, 1028, 1051, 1066, 1087, 1148, 1221, 1272, 1308, 1367, 1395, 1444, 1517, 1579, 1593, 1670, 1726, 2989, 3069, 3172 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 1.46 (s, 9H, tBu), 7.02 (dd, 1H, J = 7.8 and 4.8 Hz, H5), 7.47 (t, 2H, J = 7.7 Hz, H3′ and H5′), 7.58 (tt, 1H, J = 7.5 and 1.3 Hz, H4′), 7.68 (dd, 2H, J = 8.3 and 1.4 Hz, H2′ and H6′), 7.86 (dd, 1H, J = 7.8 and 1.9 Hz, H4), 8.60 (dd, 1H, J = 4.8 and 1.9 Hz, H6), 9.69 (br s, 1H, NH) ppm; JMOD{1H} NMR (CDCl3, 300 K, 126 MHz) δ 28.3 (3CH3, CMe3), 81.3 (C, CMe3), 117.3 (CH, C5), 119.5 (C, C3), 128.5 (2CH, C3′ and C5′), 129.8 (2CH, C2′ and C6′), 132.8 (CH, C4′), 137.8 (C, C1′), 141.6 (CH, C4), 151.3 (C, C2 or N–CO), 152.1 (C, C2 or N–CO), 152.5 (CH, C6), 197.0 (C, CO) ppm. Swern oxidation gave 30 in 7% yield due to competitive cyclization between the alcohol and the carbamate of 27 giving 1,4-dihydro-4-phenylpyrido[2,3-d][1,3]2-oxazinone: 1H NMR (CDCl3, 300 K, 300 MHz) δ 6.36 (s, 1H, CH-Ph), 7.00 (dd, 1H, J = 7.6 and 5.0 Hz, H5), 7.19 (d, 1H, J = 7.5 Hz, H4), 7.35–7.38 (m, 2H, Ph), 7.41–7.45 (m, 3H, Ph), 8.36 (dd, 1H, J = 5.2 and 1.7 Hz, H6), 9.55 (br s, 1H, NH) ppm.
:40) to give the title product in 91% yield (2.35 g) as a colorless oil: Rf (petroleum ether–AcOEt 50:50) = 0.58; IR (ATR) ν 705, 764, 787, 824, 853, 923, 977, 1001, 1082, 1111, 1151, 1199, 1254, 1271, 1295, 1367, 1400, 1431, 1449, 1581, 1598, 1669, 1704, 2976 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 1.30 (s, 9H, tBu), 3.41 (s, 3H, CH2OMe), 5.20 (br s, 2H, CH2OMe), 7.24 (dd, 1H, J = 7.6 and 4.8 Hz, H5), 7.44 (t, 2H, J = 7.8 Hz, H3′ and H5′), 7.57 (tt, 1H, J = 7.4 and 1.3 Hz, H4′), 7.76 (dd, 1H, J = 7.7 and 1.9 Hz, H4), 7.80 (dd, 2H, J = 8.0 and 1.5 Hz, H2′ and H6′), 8.59 (dd, 1H, J = 5.0 and 1.9 Hz, H6) ppm; JMOD{1H} NMR (CDCl3, 300 K, 126 MHz) δ 28.1 (3CH3, CMe3), 57.2 (CH3, OMe), 79.7 (CH2), 82.2 (C, CMe3), 120.9 (CH, C5), 128.4 (2CH, C3′ and C5′), 130.2 (2CH, C2′ and C6′), 131.0 (C, C3), 133.2 (CH, C4′), 138.4 (CH, C4), 150.2 (CH, C6), 136.5 (C, C1’), 152.1 (C, C2), 154.0 (C, N–CO), 194.1 (C, CO) ppm.
:50 to 60:40) to give the title product in 62% yield (0.24 g) as a white solid: Rf (AcOEt–petroleum ether 50:50) = 0.27; mp 121–122 °C; IR (ATR) ν 709, 773, 794, 805, 858, 914, 927, 991, 1036, 1081, 1110, 1147, 1208, 1221, 1269, 1290, 1302, 1364, 1400, 1430, 1537, 1559, 1582, 1594, 1663, 1703, 2930, 2981 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 1.27 (s, 9H, tBu), 2.44 (s, 3H, SMe), 3.24 (s, 3H, CH2OMe), 4.86 (br s, 2H, CH2OMe), 7.15 (d, 1H, J = 5.4 Hz, H5), 7.42 (t, 2H, J = 7.8 Hz, H3′ and H5′), 7.56 (tt, 1H, J = 7.4 and 1.3 Hz, H4′), 7.78 (d, 2H, J = 7.7 Hz, H2′ and H6′), 8.48 (d, 1H, J = 5.4 Hz, H6) ppm; JMOD{1H} NMR (CDCl3, 300 K, 126 MHz) δ 15.2 (CH3, SMe), 28.0 (3CH3, CMe3), 56.7 (CH3, OMe), 80.5 (CH2), 81.8 (C, CMe3), 118.1 (CH, C5), 128.6 (2CH, C3′ and C5′), 129.9 (2CH, C2′ and C6′), 131.2 (C, C3), 134.0 (CH, C4′), 136.2 (C, C1′), 149.2 (CH, C6), 150.8 (C, C4), 151.1 (C, C2), 154.0 (C, N–CO), 193.9 (C, CO) ppm.
:30; Rf = 0.36) to give the title product in 93% yield (0.39 mg) as a white solid: mp 140–141 °C; IR (ATR) ν 752, 776, 794, 921, 969, 1001, 1025, 1070, 1123, 1149, 1174, 1208, 1243, 1265, 1310, 1357, 1436, 1459, 1540, 1558, 1594, 1636, 1659, 2928, 3133, 3303, 3420 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 2.35 (s, 3H, SMe), 4.86 (br s, 2H, NH2), 6.60 (d, 1H, J = 5.5 Hz, H5), 7.47 (t, 2H, J = 7.9 Hz, H3′ and H5′), 7.61 (tt, 1H, J = 7.1 and 1.3 Hz, H4′), 7.83 (dd, 2H, J = 8.3 and 1.4 Hz, H2′ and H6′), 8.05 (d, 1H, J = 5.6 Hz, H6) ppm; JMOD{1H} NMR (CDCl3, 300 K, 126 MHz) δ 15.6 (CH3, SMe), 110.5 (CH, C5), 116.0 (C, C3), 129.0 (2CH, C3′ and C5′), 129.7 (2CH, C2′ and C6′), 134.0 (CH, C4′), 137.4 (C, C1′), 149.5 (CH, C6), 150.9 (C, C4), 156.2 (C, C2), 197.0 (C, CO) ppm. Upon storage with or without solvent, it was observed that this compound could be converted to a tautomeric form 20′. This tautomer was separated by chromatography over silica gel (eluent: AcOEt–petroleum ether 70:30) and identified as being 3-benzoyl-2-imino-4-(methylthio)-1,2-dihydropyridine on the basis of its NMR data: 1H NMR (CDCl3, 300 K, 300 MHz) δ 2.33 (s, 3H, SMe), 4.92 (t, 1H, J = 6.4 Hz, NH), 6.04 (t, 1H, J = 6.5 Hz, =NH), 6.50 (d, 1H, J = 5.6 Hz, H5), 7.37 (tt, 2H, J = 7.6 and 1.7 Hz, H3′ and H5′), 7.53 (tt, 1H, J = 7.4 and 1.4 Hz, H4′), 7.73–7.77 (m, 2H, H2′ and H6′), 7.91 (d, 1H, J = 5.5 Hz, H6) ppm; JMOD{1H} NMR (CDCl3, 300 K, 126 MHz) δ 15.5 (CH3, SMe), 109.9 (CH, C5), 117.2 (C, C3), 128.9 (2CH, C3′ and C5′), 129.6 (2CH, C2′ and C6′), 133.9 (CH, C4′), 137.1 (C, C1′), 148.9 (CH, C6), 149.8 (C, C4), 155.1 (C, C2), 196.6 (C, CO) ppm.
:30 to 100:0) to give the title product in 58% yield (0.25 g) as a brown solid: Rf (AcOEt) = 0.09; mp 190 °C (dec.); IR (ATR) ν 666, 702, 744, 799, 814, 867, 981, 1023, 1110, 1160, 1243, 1272, 1315, 1345, 1366, 1394, 1424, 1473, 1506, 1552, 1571, 1594, 1626, 1732, 2919, 3049 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 2.34 (s, 3H, SMe), 4.07 (s, 3H, NMe), 6.55 (d, 1H, J = 7.8 Hz, H1), 6.96 (t, J = 7.4 Hz, 1H, H2), 7.05 (br s, 1H, H9), 7.41 (d, 1H, J = 7.5 Hz, H4), 7.48 (t, 1H, J = 7.5 Hz, H3), 7.50 (d, 2H, J = 7.2 Hz, H2′ and H6′), 7.59 (t, 2H, J = 7.4 Hz, H3′ and H5′), 7.66 (t, J = 7.4 Hz, 1H, H4′), 8.84 (br s, 1H, H8) ppm; JMOD{1H} NMR (CDCl3, 300 K, 126 MHz) δ 17.3 (CH3, SMe), 27.9 (CH3, NMe), 108.7 (CH, C4), 120.3 (CH, C2), 120.7 (C), 123.4 (CH, C1), 126.7 (CH), 128.3 (CH), 128.9 (4CH, C2′, C3′, C5′ and C6′), 129.4 (CH), 129.8 (CH), 139.9 (C), 143.2 (C), 148.1 (C), 153.2 (C) ppm (4C not seen).
:5): Rf (AcOEt) = 0.05; mp 250 °C (dec.); IR (ATR) ν 663, 682, 707, 727, 746, 773, 798, 814, 841, 876, 954, 1023, 1044, 1087, 1134, 1161, 1205, 1275, 1316, 1341, 1394, 1427, 1473, 1505, 1564, 1595, 1624, 2921, 3050, 3273 cm−1; 1H NMR (CDCl3, 300 K, 300 MHz) δ 2.42 (s, 3H, S(O)Me), 4.10 (s, 3H, NMe), 6.75 (d, 1H, J = 7.6 Hz), 7.02 (ddd, 1H, J = 8.0, 7.6 and 1.1 Hz), 7.44–7.61 (m, 4H), 7.64–7.76 (m, 3H, H3′, H4′ and H5′), 8.29 (d, 1H, J = 4.5 Hz, H9), 9.29 (d, 1H, J = 4.1 Hz, H8) ppm; 1H NMR ((CD3)2SO, 300 K, 500 MHz) δ 2.44 (s, 3H, S(O)Me), 3.98 (s, 3H, NMe), 6.60 (d, 1H, J = 7.8 Hz), 7.04 (td, 1H, J = 7.6 and 1.0 Hz), 7.54 (d, 1H, J = 7.5 Hz), 7.58 (td, 1H, J = 7.6 and 1.2 Hz), 7.68–7.78 (m, 5H, H4, H2′, H6′), 8.15 (d, 1H, J = 4.4 Hz, H9), 9.26 (br s, 1H, H8) ppm; JMOD{1H} NMR ((CD3)2SO, 300 K, 126 MHz) δ 27.7 (CH3, NMe), 45.5 (CH3, S(O)Me), 109.9 (CH, C4), 113.4 (CH, C9), 113.6 (C), 117.7 (C), 119.4 (C), 120.5 (CH), 122.6 (CH), 129.0 (CH), 129.2 (CH), 129.3 (CH), 129.6 (CH), 130.0 (CH), 130.2 (CH), 136.5 (C), 141.1 (C), 143.0 (C), 151.8 (CH), 152.9 (C), 154.0 (C), 159.3 (C) ppm; 1H NMR ((CD3)2SO, 375 K, 500 MHz) δ 2.43 (s, 3H, S(O)Me), 4.02 (s, 3H, NMe), 6.69 (d, 1H, J = 7.8 Hz, H1), 7.03 (t, 1H, J = 7.6 Hz, H2), 7.52 (d, 1H, J = 7.7 Hz, H4), 7.57 (td, 1H, J = 7.6 and 1.2 Hz), 7.66 (d, 1H, J = 8.1 Hz), 7.67–7.72 (m, 2H), 7.73–7.78 (m, 2H), 8.18 (d, 1H, J = 4.5 Hz, H9), 9.26 (d, 1H, J = 4.5 Hz, H8) ppm; JMOD{1H} NMR ((CD3)2SO, 375 K, 126 MHz) δ 27.1 (CH3, NMe), 45.2 (CH3, S(O)Me), 109.2 (CH, C4), 112.9 (CH, C9), 113.3 (C), 119.1 (C), 119.9 (CH), 122.2 (CH, C1), 128.4 (CH), 128.6 (CH), 129.0 (CH), 129.1 (CH), 129.4 (CH), 129.8 (CH), 136.1 (C), 140.5 (C), 142.8 (C), 151.1 (CH, C8), 152.8 (C), 159.3 (C), 163.2 (C) ppm (1C not seen). During the NMR experiment (several hours in deuterated DMSO at 375 K), the product was quantitatively converted into what could be 10-methyl-5-phenyl-1,10-dihydro-4H-indolo[2,3-b]1,8-naphthyridin-4-one (34′), on the basis of its NMR spectra and HRMS: 1H NMR ((CD3)2SO, 375 K, 500 MHz) δ 3.95 (s, 3H, NMe), 6.37 (d, 1H, J = 7.0 Hz), 6.56 (d, 1H, J = 7.9 Hz), 6.99 (t, 1H, J = 7.6 Hz), 7.36–7.38 (m, 2H), 7.49 (t, 1H, J = 7.7 Hz), 7.54–7.55 (m, 3H), 7.62 (d, 1H, J = 8.1 Hz), 8.10 (d, 1H, J = 7.1 Hz) ppm (NH not unambiguously seen); 1H NMR ((CD3)2SO, 300 K, 500 MHz) δ 3.96 (s, 3H, NMe), 6.48 (d, 1H, J = 7.8 Hz, H6), 6.63 (d, 1H, J = 7.0 Hz, H3), 6.99 (td, 1H, J = 7.6 and 1.0 Hz, H7), 7.39–7.40 (m, 2H, H2′ and H6′), 7.54 (ddd, 1H, J = 8.2, 7.4 and 1.3 Hz, H8), 7.56–7.59 (m, 3H, H3′, H4′ and H5′), 7.69 (d, 1H, J = 8.1 Hz, H9), 8.40 (d, 1H, J = 7.0 Hz, H2) ppm; JMOD{1H} NMR (CDCl3, 300 K, 126 MHz) δ 28.0 (CH3, NMe), 106.8 (CH, C4), 109.9 (C), 110.3 (CH, C9), 116.4 (C), 119.9 (C), 121.1 (CH, C7), 122.2 (CH, C6), 127.0 (2CH, C2′ and C6′), 127.8 (CH), 128.3 (CH), 128.4 (2CH, C3′ and C5′), 138.5 (C), 141.7 (CH, C2), 141.9 (C), 144.8 (C), 149.2 (C), 152.8 (C), 174.6 (C, CO) ppm; HRMS (ESI; Maxis 4G), m/z = 326.1286 (1 ppm) found (calcd for C21H16N3O, [M + H]+, requires 326.12879).
Synthesis of benzofuro- and benzothieno[2,3-b]benzothiopyrano[4,3,2-de]1,8-naphthyridine (28 and 39), and attempt to prepare 10-methylbenzothiopyrano[4,3,2-de]indolo[2,3-b]1,8-naphthyridine, from 2-chloro-4-fluoropyridine
:20), the title product in 76% yield (1.95 g) as a white solid: Rf (CHCl3) = 0.25; Rf (petroleum ether–AcOEt 60:40) = 0.37; mp 112 °C; IR (ATR) ν 762, 804, 831, 855, 870, 918, 949, 1031, 1044, 1067, 1105, 1167, 1191, 1221, 1267, 1385, 1452, 1460, 1482, 1569, 1591, 3198 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 3.52 (d, 1H, J = 6.7 Hz, OH), 6.54 (d, 1H, J = 6.1 Hz, CH(OH)), 6.96–7.00 (m, 2H, H5 and H3′), 7.18 (td, 1H, J = 7.55 and 0.65 Hz, H4′ or H5′), 7.28 (tdd, 1H, J = 7.45, 5.4 and 1.6 Hz), 7.67 (t, 1H, J = 7.7 Hz), 8.25 (dd, 1H, J = 7.7 and 5.6 Hz, H6) ppm; JMOD{1H} NMR (CDCl3, 300 K, 126 MHz) δ 65.4 (t, CH, J = 2.4 Hz, CH(OH)), 112.2 (d, CH, J = 19.4 Hz, C5 or C3′), 115.4 (d, CH, J = 21.1 Hz, C5 or C3′), 124.1 (d, CH, J = 3.4 Hz, C4′ or C5′), 125.1 (d, C, J = 11.3 Hz, C3 or C1′), 127.7 (d, C, J = 12.8 Hz, C3 or C1′), 127.9 (t, CH, J = 3.2 Hz), 129.7 (d, CH, J = 8.2 Hz), 150.4 (d, CH, J = 9.8 Hz, C6), 151.7 (d, C, J = 6.2 Hz, C2), 159.7 (d, C, J = 247 Hz, C–F, C4 or C2′), 168.2 (d, C, J = 268 Hz, C–F, C4 or C2′) ppm; 19F{1H} NMR (CDCl3, 300 K, 471 MHz) δ −116.4 (d, J = 6.5 Hz), −100.0 (d, J = 6.5 Hz). Crystal data for36: C12H8ClF2NO, M = 255.64, monoclinic, P 21/c, a = 13.651(2), b = 5.9725(11), c = 13.013(2) Å, β = 90.997(6) °, V = 1060.8(3) Å3, Z = 4, d = 1.601 g cm−3, μ = 0.368 mm−1. A final refinement on F2 with 2387 unique intensities and 157 parameters converged at ωR(F2) = 0.0746 (R(F) = 0.0283) for 2290 observed reflections with I > 2σ(I). CCDC 2204835.†
:20; Rf = 0.29) to give the title product in 98% yield (0.58 g) as an orange oil: IR (ATR) ν 750, 828, 915, 1233, 1292, 1381, 1454, 1482, 1560, 1594, 1607, 1675 cm−1; 1H NMR (CDCl3, 300 K, 300 MHz) δ 7.08 (ddd, 1H, J = 11.1, 8.3 and 1.1 Hz, H3′), 7.10 (dd, 1H, J = 8.0, 5.7 Hz, 1H, H5), 7.27 (td, 1H, J = 7.6 and 1.1 Hz), 7.59 (dddd, 1H, J = 8.3, 7.0, 5.0 and 1.8 Hz, H4′), 7.91 (td, 1H, J = 7.7 and 1.9 Hz), 8.42 (dd, 1H, J = 8.1 and 5.7 Hz, H6) ppm; JMOD{1H} NMR (CDCl3, 300 K, 75 MHz) δ 111.3 (d, CH, J = 17.8 Hz, C5), 117.0 (d, CH, J = 22.5 Hz, C3′), 124.5 (d, C, J = 9.0 Hz, C3), 124.9 (d, CH, J = 3.7 Hz), 125.9 (dd, C, J = 17.5 and 1.4 Hz, C1′), 131.2 (CH), 136.6 (d, CH, J = 9.4 Hz, C4′), 148.5 (dd, C, J = 5.7 and 2.0 Hz, C2), 151.8 (d, CH, J = 8.5 Hz, C6), 162.3 (d, C, J = 259 Hz, C–F, C4 or C2′), 165.9 (dd, C, J = 267 and 2.0 Hz, C–F, C4 or C2′), 185.1 (C, CO) ppm; 19F{1H} NMR (CDCl3, 300 K, 282 MHz) δ −111.3 (d, J = 1.7 Hz), −102.7 (d, J = 2.0 Hz). This ketone was directly involved in the cyclization step below.
:10) the title product in 25% yield (35 mg) as a yellow solid: Rf (AcOEt–petroleum ether 80:20) = 0.31; mp 228 °C; IR (ATR) ν 707, 731, 749, 836, 850, 879, 890, 991, 1030, 1095, 1127, 1190, 1226, 1246, 1262, 1324, 1372, 1448, 1458, 1490, 1552, 1593, 2851, 2920 cm−1; 1H NMR (CDCl3, 300 K, 500 MHz) δ 7.32–7.35 (m, 2H, H6), 7.46–7.59 (m, 4H), 7.68 (d, 1H, J = 8.2 Hz), 8.23 (d, 1H, J = 7.8 Hz), 8.55 (dd, 1H, J = 7.6 and 1.05 Hz), 8.80 (br s, 1H, H7) ppm; JMOD{1H} NMR (CDCl3, 300 K, 126 MHz) δ 112.3 (C), 112.5 (CH), 114.6 (CH, C6), 117.6 (C), 122.2 (C), 123.3 (CH), 123.7 (CH), 127.0 (CH), 127.4 (CH), 128.3 (C), 129.7 (CH), 129.9 (CH), 131.2 (CH), 133.3 (C), 137.8 (C), 143.3 (C), 151.4 (CH, C7), 154.7 (C), 156.3 (C), 166.1 (C) ppm; HRMS (ASAP; Maxis 4G), m/z = 327.0589 (1 ppm) found (calcd for C20H11N2OS, [M + H]+, requires 327.05866).
:0 to 85:15) the title product in 5% yield (7 mg) as a yellow solid: Rf (AcOEt–petroleum ether 80:20) = 0.34; mp 232 °C; 1H NMR (CDCl3, 300 K, 500 MHz) δ 7.28 (t, 1H, J = 7.6 Hz), 7.33 (t, 1H, J = 7.6 Hz), 7.38 (br s, 1H, H6), 7.47–7.50 (m, 2H), 7.58 (dd, 1H, J = 7.9, 0.9 Hz), 7.85 (d, 1H, J = 8.0 Hz), 8.26 (d, 1H, J = 8.2 Hz), 8.34 (d, 1H, J = 7.9 Hz), 8.83 (br s, 1H, H7) ppm; JMOD{1H} NMR (CDCl3, 300 K, 126 MHz) δ 114.8 (CH), 119.3 (CH), 123.4 (CH), 124.2 (CH), 125.4 (CH), 126.9 (CH), 128.0 (CH), 128.3 (C), 128.6 (CH), 131.0 (CH), 132.6 (C), 133.3 (C), 137.1 (C), 139.1 (C), 151.4 (CH, C7), 154.3 (C), 169.0 (C) ppm (3C not seen); HRMS (ASAP; Maxis 4G), m/z = 343.0357 (0 ppm) found (calcd for C20H11N2S2, M+·, requires 343.03582).
:10) the title product in 20% yield (34 mg) as a yellow solid: Rf (CHCl3) = 0.10; mp 78 °C; IR (ATR) ν 734, 747, 803, 816, 879, 989, 1083, 1124, 1159, 1241, 1263, 1315, 1345, 1365, 1392, 1427, 1474, 1543, 1585, 1618, 2851, 2921 cm−1; 1H NMR ((CD3)2SO, 300 K, 500 MHz) δ 3.94 (br s, 3H, NMe), 7.15 (t, 1H, J = 7.6 Hz), 7.43 (t, 1H, J = 7.6 Hz), 7.54 (t, 2H, J = 7.5 Hz), 7.59 (d, 1H, J = 8.1 Hz), 7.64 (d, 1H, J = 7.8 Hz), 8.07 (d, 1H, J = 7.9 Hz), 8.37 (d, 1H, J = 7.9 Hz) ppm (H6 and H7 not seen at this temperature due to very large signals); 1H NMR ((CD3)2SO, 375 K, 500 MHz) δ 3.96 (s, 3H, NMe), 7.22 (t, 1H, J = 7.5 Hz, H12), 7.38 (br s, 1H, H6), 7.50 (t, 1H, J = 7.6 Hz), 7.57 (t, 1H, J = 7.5 Hz), 7.60 (t, 1H, J = 7.4 Hz), 7.63 (d, 1H, J = 8.0 Hz, H11), 7.67 (d, 1H, J = 7.8 Hz), 8.23 (d, 1H, J = 8.0 Hz), 8.50 (d, 1H, J = 8.0 Hz), 8.74 (br s, 1H, H7) ppm; JMOD{1H} NMR ((CD3)2SO, 375 K, 126 MHz) δ 27.3 (CH3, NMe), 109.3 (CH, C11), 111.5 (C), 112.4 (CH, C6), 114.2 (C), 119.0 (C), 119.5 (CH, C12), 122.6 (CH), 126.5 (CH), 126.8 (CH), 127.5 (C), 128.1 (CH), 129.6 (CH), 130.4 (CH), 131.8 (C), 134.6 (C), 141.4 (C), 142.6 (C), 149.7 (CH, C7), 153.4 (C), 155.0 (C) ppm; HRMS (ESI; Maxis 4G), m/z = 340.0901 (1 ppm) found (calcd for C21H14N3S, [M + H]+, requires 340.09029).
Photophysical measurements
Measurements were performed on freshly-prepared air-equilibrated solutions contained in quartz cells of 1 cm pathlength at room temperature (25 °C). UV-Vis absorption spectra were recorded on a Jasco V-770 spectrophotometer. Steady-state measurements were performed using a Jasco FP-8300 fluorimeter equipped with a 150 W Xenon lamp and a Hamamatsu R928 photomultiplier tube. Fully corrected emission spectra were obtained, for each compound, after excitation at the wavelength of the absorption maximum, with Aλex < 0.1 to minimize internal absorption. Quinine bisulfate in 0.5 M H2SO4 (Φ = 0.546 at λex = 346 nm) was used as a standard. Fluorescence lifetimes were measured by time correlated single-photon counting (TCSPC) by using an Edinburgh Instrument (FLS920) fluorimeter. Excitation at 375 nm was achieved by a pulsed laser diode EDEPL375. The instrument response (FWHM ca. 1 ns) was determined by measuring the light scattered by a toluene suspension. The TCSPC traces were analyzed by standard iterative reconvolution methods implemented in the software of the fluorimeter. All compounds displayed strictly monoexponential fluorescence decays.Protein kinase assays
Kinase enzymatic activities were assayed in 384-well plates using the ADP-GloTM assay kit (Promega, Madison, WI) according to the recommendations of the manufacturer. In order to determine the half maximal inhibitory concentration (IC50), the assays were performed in duplicate in the absence or presence of increasing doses of the tested compounds. ATP concentration used in the kinase assays was 10 μM. PIM1 (human proto-oncogene, recombinant, expressed in bacteria) and PIM2 (human proto-oncogene, recombinant, expressed by baculovirus in Sf9 insect cells) were assayed in kinase buffer “A” with 630 μM of PIMtide peptide (ARKRRRHPSGPPTA) as substrate. Peptide substrates were obtained from Proteogenix (Schiltigheim, France). Kinase buffer “A”: 10 mM MgCl2, 1 mM EGTA, 1 mM DTT, 25 mM Tris-HCl pH 7.5, 50 μg mL−1 heparin.Molecular modeling experiments
After geometric optimization of the compounds MN34-20, 39, NM-049 and NM65-45 structures using Gaussian 16 at DFT B3LYP level and 6-31g orbital basis sets,85 the docking studies were performed with AutoDock Vina version 1.1.2.76 Files for the docking were prepared from 3JPV PIM1 structure after removing of water molecules and pdbqt files prepared with Chimera software.77 Apolar hydrogen atoms were removed and Gasteiger charges were added. Docking experiments were performed using the default AutoDock Vina parameters.Author contributions
Conceptualization, F. M., W. E., J. P. H., N. M.; investigation, N. M., W. E., M. M., T. R., L. N., O. M., L. P., T. R.; supervision, F. M., W. E., J. P. H.; writing initial draft, F. M., P. M., O. M.; writing – review and editing, W. E., N. M., S. B., P. M., L. N., J. P. H., T. R., L. P., V. T.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge the financial support by the Université de Rennes 1 for a PhD grant (to N. M.). We also thank Rennes Métropole (Spectrum 100 PerkinElmer spectrometer) and the Fonds Européen de Développement Régional (FEDER; D8 Venture Bruker-AXS diffractometer). Thermofisher is also acknowledged for generous gift of 2,2,6,6-tetramethylpiperidine. S. B. and T. R. thank IBiSA (French Infrastructures en sciences du vivant: Biologie, Santé et Agronomie) and Biogenouest (Western France life science and environment core facility network supported by the Conseil Régional de Bretagne) for supporting the KISSf screening facility (FR2424, CNRS and Sorbonne Université). L. P. and V. T. are grateful to the French Cancer League (Ligue contre le cancer Comité 17 Charente-Maritime) for financial support and to the Cancéropôle Grand Ouest Marine Molecules, Metabolism and Cancer network (3MC) for scientific support. We thank Nicolas Richy for his contribution to this study.References
- M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed.
- A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Żyła-Karwowska and M. Stępień, Chem. Rev., 2022, 122, 565–788 CrossRef CAS.
- W. Niu, Y. Fu, H. Komber, J. Ma, X. Feng, Y. Mai and J. Liu, Org. Lett., 2021, 23, 2069–2073 CrossRef CAS PubMed.
- A. Palmeira, M. H. Vasconcelos, A. Paiva, M. X. Fernandes, M. Pinto and E. Sousa, Biochem. Pharmacol., 2012, 83, 57–68 CrossRef CAS.
- J. Barbosa, R. T. Lima, D. Sousa, A. S. Gomes, A. Palmeira, H. Seca, K. Choosang, P. Pakkong, H. Bousbaa, M. M. Pinto, E. Sousa, M. H. Vasconcelos and M. Pedro, Molecules, 2016, 21, 81 CrossRef.
- V. Silva, E. Gil-Martins, B. Silva, C. Rocha-Pereira, M. E. Sousa, F. Remiao and R. Silva, Expert Opin. Drug Metab. Toxicol., 2021, 17, 441–482 CrossRef CAS.
- N. F. Nikitas, P. L. Gkizis and C. G. Kokotos, Org. Biomol. Chem., 2021, 19, 5237–5253 RSC.
- D. I. S. P. Resende, F. Duraes, M. Maia, E. Sousa and M. M. M. Pinto, Org. Chem. Front., 2020, 7, 3027–3066 RSC.
- A. M. Paiva, M. M. Pinto and E. Sousa, Curr. Med. Chem., 2013, 20, 2438–2457 CrossRef CAS PubMed.
- N. Mokhtari Brikci-Nigassa, G. Bentabed-Ababsa, W. Erb and F. Mongin, Synthesis, 2018, 3615–3633 Search PubMed.
- M. Hedidi, W. Erb, F. Lassagne, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, T. Roisnel, G. Bentabed-Ababsa and F. Mongin, RSC Adv., 2016, 6, 63185–63189 RSC.
- R. Amara, G. Bentabed-Ababsa, M. Hedidi, J. Khoury, H. Awad, E. Nassar, T. Roisnel, V. Dorcet, F. Chevallier, Z. Fajloun and F. Mongin, Synthesis, 2017, 4500–4516 CAS.
- M. Hedidi, J. Maillard, W. Erb, F. Lassagne, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, T. Roisnel, V. Dorcet, M. Hamzé, Z. Fajloun, B. Baratte, S. Ruchaud, S. Bach, G. Bentabed-Ababsa and F. Mongin, Eur. J. Org. Chem., 2017, 5903–5915 CrossRef CAS.
- N. Mokhtari Brikci-Nigassa, L. Nauton, P. Moreau, O. Mongin, R. E. Duval, L. Picot, V. Thiery, M. Souab, B. Baratte, S. Ruchaud, S. Bach, R. Le Guevel, G. Bentabed-Ababsa, W. Erb, T. Roisnel, V. Dorcet and F. Mongin, Bioorg. Chem., 2020, 94, 103347 CrossRef CAS.
- H. S. Jung, T. Yun, Y. Cho and H. B. Jeon, Tetrahedron, 2016, 72, 5988–5993 CrossRef CAS.
- K. Xiang and X. Wang, CN Pat., CN111087494A, 2020.
- M. Sato, S. Ebine and S. Akabori, Synthesis, 1981, 472–473 CrossRef CAS.
- R. A. Smits, M. Adami, E. P. Istyastono, O. P. Zuiderveld, C. M. E. van Dam, F. J. J. de Kanter, A. Jongejan, G. Coruzzi, R. Leurs and I. J. P. de Esch, J. Med. Chem., 2010, 53, 2390–2400 CrossRef CAS.
- O. M. Sinead, J. E. McCormick, R. McElhinney and T. B. H. McMurry, ARKIVOC, 2003, 180–189 Search PubMed.
- P. R. K. Murthia, G. P. Kumar, D. Rambabua, M. V. B. Rao and M. Pal, Curr. Catal., 2014, 3, 47–52 Search PubMed.
- E. J. Warawa, B. M. Migler, C. J. Ohnmacht, A. L. Needles, G. C. Gatos, F. M. McLaren, C. L. Nelson and K. M. Kirkland, J. Med. Chem., 2001, 44, 372–389 CrossRef CAS PubMed.
- K. Kobayashi and T. Suzuki, Heterocycles, 2012, 85, 403–411 CrossRef CAS.
- Z. R. Woydziak, L. Fu and B. R. Peterson, J. Org. Chem., 2012, 77, 473–481 CrossRef CAS.
- A. El Alaoui, F. Schmidt, M. Sarr, D. Decaudin, J.-C. Florent and L. Johannes, ChemMedChem, 2008, 3, 1687–1695 CrossRef CAS.
- J. Clayden, Organolithiums: Selectivity for Synthesis, Pergamon: Oxford, 2002 Search PubMed.
- J. M. Fourquez, A. Godard, F. Marsais and G. Queguiner, J. Heterocycl. Chem., 1995, 32, 1165–1170 CrossRef CAS.
- S. Horne and R. Rodrigo, J. Chem. Soc., Chem. Commun., 1991, 1991, 1046–1048 RSC.
- C. H. E. Chow, H. Phan, X. Zhang and J. Wu, J. Org. Chem., 2020, 85, 234–240 CrossRef CAS PubMed.
- H. Sirringhaus, R. H. Friend, C. Wang, J. Leuninger and K. Mullen, J. Mater. Chem., 1999, 9, 2095–2101 RSC.
- S. Zhang, X. Qiao, Y. Chen, Y. Wang, R. M. Edkins, Z. Liu, H. Li and Q. Fang, Org. Lett., 2014, 16, 342–345 CrossRef CAS PubMed.
- L. G. Voskressensky, E. A. Sokolova, A. A. Festa and A. V. Varlamov, Tetrahedron Lett., 2013, 54, 5172–5173 CrossRef CAS.
- Y. Wu, B. Li, F. Mao, X. Li and F. Y. Kwong, Org. Lett., 2011, 13, 3258–3261 CrossRef CAS PubMed.
- W. Rauf, A. L. Thompson and J. M. Brown, Chem. Commun., 2009, 3874–3876 RSC.
- A. M. Schoevaars, W. Kruizinga, R. W. J. Zijlstra, N. Veldman, A. L. Spek and B. L. Feringa, J. Org. Chem., 1997, 62, 4943–4948 CrossRef CAS.
- C. W. Cheung, D. S. Surry and S. L. Buchwald, Org. Lett., 2013, 15, 3734–3737 CrossRef CAS.
- M. Fan, W. Zhou, Y. Jiang and D. Ma, Org. Lett., 2015, 17, 5934–5937 CrossRef CAS PubMed.
- H. Xu and C. Wolf, Chem. Commun., 2009, 3035–3037 RSC.
- R. Komati and B. S. Jursic, Tetrahedron Lett., 2014, 55, 1523–1527 CrossRef CAS.
- S.-K. Xiang, D.-X. Zhang, H. Hu, J.-L. Shi, L.-G. Liao, C. Feng, B.-Q. Wang, K.-Q. Zhao, P. Hu, H. Yang and W.-H. Yu, Adv. Synth. Catal., 2013, 355, 1495–1499 CrossRef CAS.
- L. Qin, H. Cui, D. Zou, J. Li, Y. Wu, Z. Zhu and Y. Wu, Tetrahedron Lett., 2010, 51, 4445–4448 CrossRef CAS.
- A. I. Vogel, A. R. Tatchell, B. S. Furnis, A. J. Hannaford and P. W. G. Smith, Vogel's textbook of practical organic chemistry, Prentice Hall, 5th edn, 1996 Search PubMed.
- V. Weingand, T. Wurm, V. Vethacke, M. C. Dietl, D. Ehjeij, M. Rudolph, F. Rominger, J. Xie and A. S. K. Hashmi, Chem. – Eur. J., 2018, 24, 3725–3728 CrossRef CAS.
- J. M. L'Helgoual'ch, A. Seggio, F. Chevallier, M. Yonehara, E. Jeanneau, M. Uchiyama and F. Mongin, J. Org. Chem., 2008, 73, 177–183 CrossRef.
- M. Hedidi, G. Bentabed-Ababsa, A. Derdour, T. Roisnel, V. Dorcet, F. Chevallier, L. Picot, V. Thiéry and F. Mongin, Bioorg. Med. Chem., 2014, 22, 3498–3507 CrossRef CAS.
- M. W. Hooper, M. Utsunomiya and J. F. Hartwig, J. Org. Chem., 2003, 68, 2861–2873 CrossRef CAS PubMed.
- M. W. Hooper and J. F. Hartwig, Organometallics, 2003, 22, 3394–3403 CrossRef CAS.
- S. Bouarfa, G. Bentabed-Ababsa, W. Erb, L. Picot, V. Thiery, T. Roisnel, V. Dorcet and F. Mongin, Synthesis, 2021, 1271–1284 CAS.
- L. I. Belen'kii, I. A. Suslov and N. D. Chuvylkin, Chem. Heterocycl. Compd., 2003, 39, 36–48 CrossRef.
- H. Miyamoto, T. Hirano, Y. Okawa, A. Nakazaki and S. Kobayashi, Tetrahedron, 2013, 69, 9481–9493 CrossRef CAS.
- N. Mokhtari Brikci-Nigassa, G. Bentabed-Ababsa, W. Erb, F. Chevallier, L. Picot, L. Vitek, A. Fleury, V. Thiéry, M. Souab, T. Robert, S. Ruchaud, S. Bach, T. Roisnel and F. Mongin, Tetrahedron, 2018, 74, 1785–1801 CrossRef.
- K. Snégaroff, T. T. Nguyen, N. Marquise, Y. S. Halauko, P. J. Harford, T. Roisnel, V. E. Matulis, O. A. Ivashkevich, F. Chevallier, A. E. H. Wheatley, P. C. Gros and F. Mongin, Chem. – Eur. J., 2011, 17, 13284–13297 CrossRef.
- Y. Yamazaki, Y. Kido, K. Hidaka, H. Yasui, Y. Kiso, F. Yakushiji and Y. Hayashi, Bioorg. Med. Chem., 2011, 19, 595–602 CrossRef CAS PubMed.
- J. D. More and N. S. Finney, Org. Lett., 2002, 4, 3001–3003 CrossRef CAS PubMed.
- K. Nishide, S.-i Ohsugi, T. Miyamoto, K. Kumar and M. Node, Monatsh. Chem., 2004, 135, 189–200 CrossRef CAS.
- N. Marquise, F. Chevallier, E. Nassar, M. Frédérich, A. Ledoux, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, T. Roisnel, V. Dorcet and F. Mongin, Tetrahedron, 2016, 72, 825–836 CrossRef CAS.
- F. Gelat, J.-F. Lohier, A.-C. Gaumont and S. Perrio, Adv. Synth. Catal., 2015, 357, 2011–2016 CrossRef CAS.
- L. I. Panferova, M. O. Zubkov, V. A. Kokorekin, V. V. Levin and A. D. Dilman, Angew. Chem., Int. Ed., 2021, 60, 2849–2854 CrossRef CAS PubMed.
- P. J. Harford, A. J. Peel, F. Chevallier, R. Takita, F. Mongin, M. Uchiyama and A. E. H. Wheatley, Dalton Trans., 2014, 43, 14181–14203 RSC.
- N. Marquise, P. J. Harford, F. Chevallier, T. Roisnel, V. Dorcet, A.-L. Gagez, S. Sablé, L. Picot, V. Thiéry, A. E. H. Wheatley, P. C. Gros and F. Mongin, Tetrahedron, 2013, 69, 10123–10133 CrossRef CAS.
- F. Trécourt, F. Marsais, T. Güngor and G. Queguiner, J. Chem. Soc., Perkin Trans. 1, 1990, 2409–2415 RSC.
- E. C. Ashby and A. B. Goel, J. Organomet. Chem., 1981, 204, 139–145 CrossRef CAS.
- T. Ikemoto, T. Kawamoto, H. Wada, T. Ishida, T. Ito, Y. Isogami, Y. Miyano, Y. Mizuno, K. Tomimatsu, K. Hamamura, M. Takatani and M. Wakimasu, Tetrahedron, 2002, 58, 489–493 CrossRef CAS.
- R. Nirogi, A. R. Mohammed, A. K. Shinde, S. R. Gagginapally, D. M. Kancharla, S. K. Pandey, R. Abraham and V. Jasti, WO Pat., WO2019082140A1, 2019.
- Q. Zhu, E. Yildirim, X. Wang, A. K. K. Kyaw, T. Tang, X. Y. D. Soo, Z. M. Wong, G. Wu, S.-W. Yang and J. Xu, Mol. Syst. Des. Eng., 2020, 5, 976–984 RSC.
- V. A. Loskutov and V. B. Shelkovnikov, Russ. J. Org. Chem., 2006, 42, 298–301 CrossRef CAS.
- L. Wang, H. Wang, W. Meng, X.-H. Xu and Y. Huang, Chin. Chem. Lett., 2021, 32, 389–392 CrossRef CAS.
- T. Güngor, F. Marsais and G. Queguiner, J. Organomet. Chem., 1981, 215, 139–150 CrossRef.
- F. Mongin, O. Mongin, F. Trécourt, A. Godard and G. Queguiner, Tetrahedron Lett., 1996, 37, 6695–6698 CrossRef CAS.
- A. Parham, J. Kroeber, D. Joosten, A. Ludemann, T. Grossmann, P. Stoessel and C. Eickhoff, WO Pat., WO2017-EP81292 2018104195, 2018 Search PubMed.
- J. H. Conway and D. Fiedler, Angew. Chem., Int. Ed., 2015, 54, 3941–3945 CrossRef CAS.
- M. C. Venuti, R. A. Stephenson, R. Alvarez, J. J. Bruno and A. M. Strosberg, J. Med. Chem., 1988, 31, 2136–2145 CrossRef CAS PubMed.
- M. J. Maslankiewicz, Pol. J. Chem., 1999, 73, 1477–1487 CAS.
- K. Kobayashi, T. Komatsu, K. Nakagawa, E. Hara and S. Yuba, Heterocycles, 2013, 87, 2577–2587 CrossRef CAS.
- R. Akué-Gédu, E. Rossignol, S. Azzaro, S. Knapp, P. Filippakopoulos, A. N. Bullock, J. Bain, P. Cohen, M. Prudhomme, F. Anizon and P. Moreau, J. Med. Chem., 2009, 52, 6369–6381 CrossRef.
- M. F. Sanner, J. Mol. Graphics Modell., 1999, 17, 57–61 CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS.
- A. F. Burchat, J. M. Chong and N. Nielsen, J. Organomet. Chem., 1997, 542, 281–283 CrossRef CAS.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3–8 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- J. Cao, X. Dai, Z. Wang, Q. Di, Y. Tang, W. Guo and Y. Zhao, CN Pat., CN2021-10850813 113563384, 2021 Search PubMed.
- D. M. Krein and T. L. Lowary, J. Org. Chem., 2002, 67, 4965–4967 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, GAUSSIAN 16, Revision B.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of 3, 4, 5, 6a, 7, 8, 9, 10, 11, 12-Boc, 13, 14, 15a, 15b, 16, 17, 2-chloro-4-(methylthio)pyridine, 21, 22a, 22b, 24a, 24b, 23, 25, 26, 27, tert-butyl N-(2-pyridyl)carbamate, 29, 2-((tert-butoxycarbonyl)(methoxymethyl)amino)pyridine, 1,4-dihydro-4-phenylpyrido[2,3-d][1,3]2-oxazinone, 30, 31, 32, 20, its tautomeric form 20′, 33, 34, 34′, 36, 37, 28, 39 and 35. CCDC 2204831–2204837. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2nj04567b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |