DOI:
10.1039/D2NJ03661D
(Paper)
New J. Chem., 2023,
47, 284-296
‘Optimized route’ to synthesize isoelectronic and isostructural Au(III)- and Pt(II)-NHC complexes: synthesis, structure, spectral properties, electrochemistry, and molecular docking studies†
Received
24th July 2022
, Accepted 18th November 2022
First published on 21st November 2022
Abstract
We report the synthesis, structures, luminescent properties, and electrochemistry of gold(III) and platinum(II)-N-heterocyclic carbene complexes. The proligand 1-methyl-2-(pyridylmethyl)imidazo[1,5-a]pyridin-4-ylium hexafluorophosphate, (1·HPF6), and two novel isoelectronic and isostructural complexes, viz. [Au(1)2][PF6]3 (2), [Pt(1)2][PF6]2 (3), were synthesized and characterized by several spectroscopic studies. Finally, the single crystal X-ray diffraction studies established the square planar geometry of both complexes 2 and 3. The solid state structures of both the square planar complexes revealed two Ccarbene and two Npyridine present in trans fashion around the central metal ions. The complexes showed strong absorbance at ∼350 nm and DFT studies demonstrated the ILCT transition. Both complexes are luminescent at ∼400 nm. DFT and TDDFT studies were performed to gain insights into the experimental findings of solid-state structures and electronic properties. Electrochemical studies of Au(III)-NHC complex 2 showed two irreversible Au(III) → Au(I) and Au(I) →Au(0) reductions at −0.75 V and −1.28 V, whereas Pt(II)-NHC complex 3 revealed Pt(II/IV) two-electron reversible oxidation potential at 0.53 V. Molecular docking analysis revealed the highest free binding energy for complex 3 with human-DNA topoisomerase as compared to complex 2 and proligand 1·HPF6. Pharmacokinetics studies revealed the scope of the complexes to be potent drugs.
Introduction
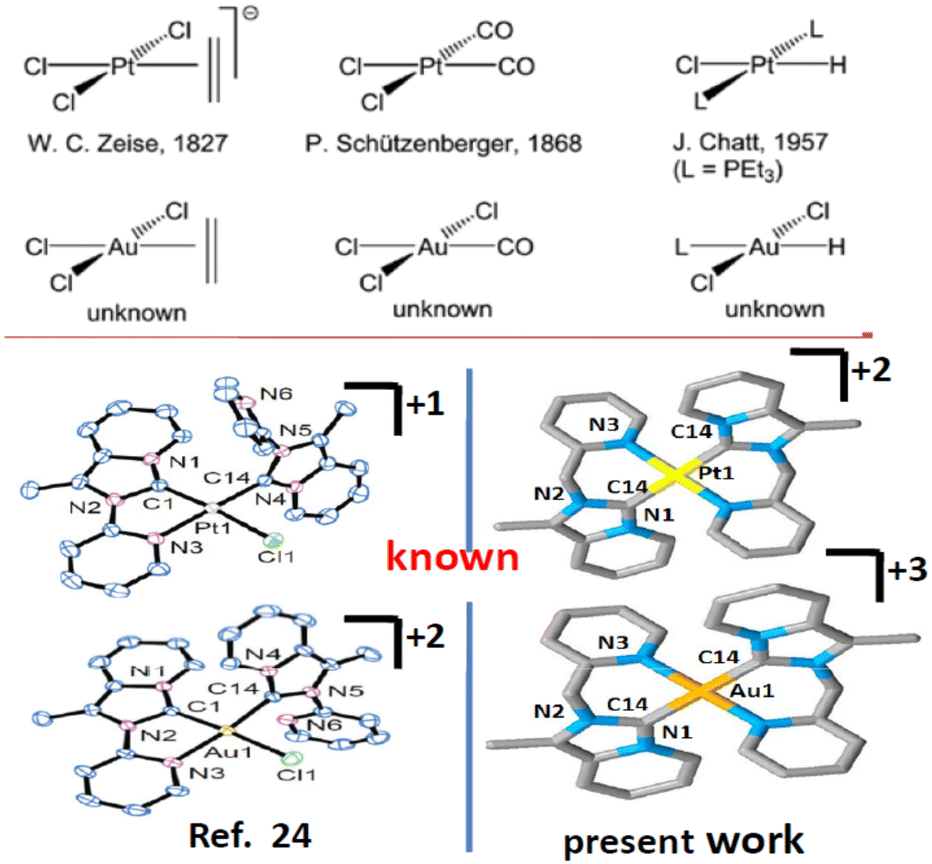
Imidazole or benzimidazole-based N-heterocyclic carbene (NHC) is now recognized as well-accepted ligand in organometallic chemistry. N-Heterocyclic carbenes (NHCs) have gained increasing research interest due to their more versatile properties1 than traditional phosphine and nitrogen donor ligands. NHC complexes provide extensive stability towards air, moisture, and heat and provide the desired product, which makes them more robust than other types of complexes.2 Additionally, easy fine-tuning of the NHC ligand provides tunable electronic properties that help in catalytic and luminescence studies. Various functional groups such as pyridine,3 pyrimidine,4 pyrazole,5 and phenanthroline6 can be easily incorporated as ‘wingtip’ to tune the electronic properties. The incorporation of pyridine or picolyl into main frame NHC systems to produce bischelating,7 pincer,8 tripodal,9 or bridging NHC ligands10 can show better catalytic,11 photophysical12 and biological activities13 than the normal NHC ligand. Besides this, chelating NHC ligands can offer a multinuclear structural motif.14 One more advantage of NHC is that it can form stable σ-bonds with a broad spectrum of transition metals from the periodic table in low to high oxidation states.15 The optoelectronic properties displayed by transition/lanthanide metal complexes can be utilized in sensors, photocatalysis, organic light-emitting diodes (OLEDs), and light-emitting electrochemical cells (LECs), etc.16 Intense research has been focused on Ir(III), Pt(II), and other cyclometalated complexes.17,18 In the literature Ir(III), Pt(II), Au(III)-NHC cyclometalated complexes are less studied19–21 compared to other metals (Chart 1).
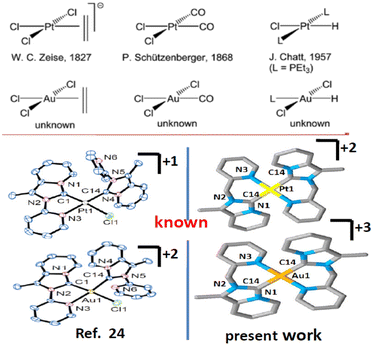 |
| | Chart 1 Isoelectronic and isostructuralgold(III) and platinum(II)-NHC. | |
In recent times cyclometalated Au(III)- and Pt(II)-NHC complexes have been gaining interest as luminescent substances used in OLEDs22,23 and are deployed in several catalytic reactions, medicinal chemistry,24,25 and other optoelectronic devices.26 Recently tridentate (C∩N∩C) or (C∩C∩N)-donor type ligands and bidentate (C∩N)-donor type N-heterocyclic carbene ligands27 have become an attractive family of highly stable cyclometalated organometallic compounds with interesting chemical and photophysical properties. Due to their isoelectronic nature (d8 system), it was expected that both Pt(II) and Au(III) may form isostructural complexes. However, due to the different inherent properties of Pt(II) and Au(III), the photophysical, electrochemical behaviour of organogold, organoplatinum complexes bearing the same ligand may be different and would be an important area of research. Thus, we confined our present studies to the synthesis, structural characterization, photophysical, and electrochemical studies of Pt(II) and Au(III) complexes of chelating C∩N donor NHC ligand. For this, we have designed 1-methyl-2-(pyridylmethyl)imidazo[1,5-a]pyridin-4-ylium hexafluorophosphate, (1·HPF6), a precursor to the annelated C∩N donor NHC, and we synthesized and characterized [Au(1)2][PF6]3, 2 and [Pt(1)2][PF6]2, 3 complexes.
Results and discussion
Synthesis and characterization
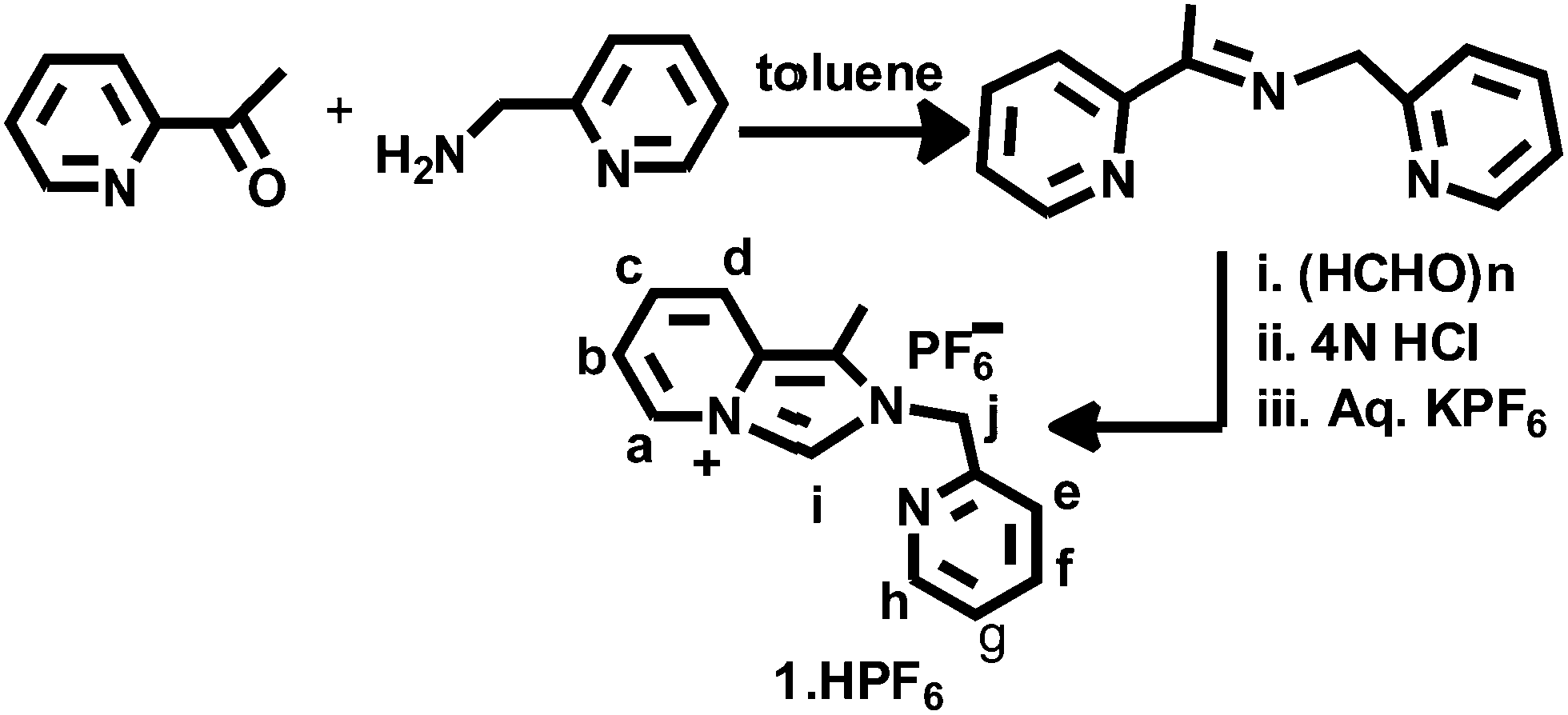
The imidazolium salt 1-methyl-2-(pyridylmethyl)imidazo[1,5-a]pyridin-4-ylium hexafluorophosphate, 1·HPF6, was synthesized through the formylative cyclization reaction of the corresponding Schiff base 2-pyridylmethyl-N-(2-acetylpyridyl)methylamine, as illustrated in Scheme 1 and following a reported procedure.28 The condensation reaction of 2-acetylpyridine and 2-picolylamine in toluene results in the Schiff base and 1-methyl-2-(pyridylmethyl)imidazo[1,5-a]pyridin-4-ylium chloride (1·HCl) was synthesized following the reported procedure. This chloride salt was converted to colorless hexafluorophosphate, 1·HPF6 by the metathesis reaction with KPF6. The formation of the proligand 1·HPF6 was confirmed by the appearance of the diagnostic imidazolium singlet NCHN signal at δ = 9.77 ppm and the methylene protons appeared as a singlet at 5.83 ppm in the 1H NMR spectrum, as shown in Fig. S1 (ESI†). Additionally, the formation of proligand was established by the presence of NCN resonance at δ = 153.19 ppm in the 13C NMR spectrum, as shown in Fig. S2 (ESI†). Imidazolium salt proligands are common precursors of N-heterocyclic carbene ligands. Indeed the transfer of imidazolium salt-derived proligand to transition metal complexes can be easily achieved through silver(I) carbene complexes.29
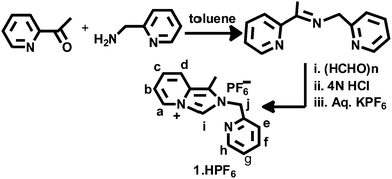 |
| | Scheme 1 Synthesis of proligand (1·HPF6). | |
The Au(III)-NHC complex 2 and Pt(II)-NHC complex 3 were obtained by the transmetallation process, as illustrated in Scheme 2. Proligand, 1·HPF6, and excess silver oxide were stirred in the CH3CN solvent in dark for 4 h, and then the solution was filtered through a plug of Celite. After that, the filtrate was added to a stoichiometric amount of KAuCl4 in CH3CN and further stirred for 2 h. Then, AgCl was removed through a plug of Celite, and the volume of the filtrate was reduced and recrystallized from CH3CN/Et2O. An orange-red compound [Au(1)2][PF6]3(2) was obtained in 82% yield. Similarly, [Pt(1)2][PF6]2(3) was prepared by the transmetallation method; pre-prepared Ag(I)-NHC was added to a stoichiometric amount of K2PtCl4 in CH3CN and further stirred for 16 h followed by reflux for 2 h at 90 °C. Then, the mixture was cooled and filtered through a plug of Celite and the volume of the filtrate was reduced. Analytically pure colourless compound [Pt(1)2][PF6]2(3) was recrystallized from CH3CN/Et2O and obtained as 84% yield.
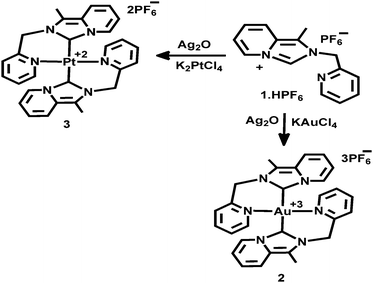 |
| | Scheme 2 Synthesis of [Au(1)2][PF6]3, 2 and [Pt(1)2][PF6]2, 3 complexes. | |
The formation of complexes 2 and 3 was confirmed as the disappearance of the diagnostic NCHN signal associated with the imidazopyridine on 1H NMR spectroscopy (shown in Fig. S3 and S5 respectively, ESI†). Additionally, the downfield shifts of carbene carbon (NCN) signals were observed at 156.69, 166.59 ppm for complex 2 and 3, respectively (free ligand 153.19 ppm) in 13C NMR spectroscopy (shown in Fig. S4 and S6, ESI†) to support the ligation of Ccarbene to Au(III)/Pt(II). The carbenic carbon signal of Pt(II)-NHC complex 3 appeared slightly downfield compared to that of the Au(III)-NHC complex 2. Additionally, the bridging methylene protons display a characteristic AB-type doublet (at 5.97 and 6.17 ppm for complex 2, and 5.96 and 6.18 ppm for complex 3), revealing the diastereotopic nature of the protons as the N-heterocyclic carbene ligand is coordinated to Au(III)/Pt(II), which is a known phenomenon.21 Additionally, both complexes 2 and 3 were characterized by the downfield shift of the aromatic and aliphatic protons, as compared 1·HPF6 using 1H NMR spectroscopy. The splitting nature of complex 3 in 1H and 13C NMR spectra was almost similar to those of complex 2, but in the case of complex 3 more downfield shift was observed for most of the protons and carbon resonance with respect to complex 2. This may be due to the presence of a more acidic Au(III) ion in complex 2 than an isoelectronic Pt(II) ion in complex 3. Strong chelation that occurs through the pyridine N-atom is supported by the more downfield shift of doublet of o-proton of the pyridine ring in both the complex 2 (8.60 ppm) and 3 (8.70 ppm), as compared to 1·HPF6 (8.57 ppm). HRMass spectral analysis of complex 2 revealed a signal at m/z 643.1891, which corresponds to [Au(NHC)2]3+, and complex 3 displayed a signal at m/z 641.1861, which corresponds to [Pt(NHC)2]2+(as shown in Fig. S7 and S8, ESI†). Additional support for the solid-state structure of complexes 2 and 3 was obtained through the single crystal X-ray diffraction analysis.
X-ray crystal structure description of complex 2 and 3
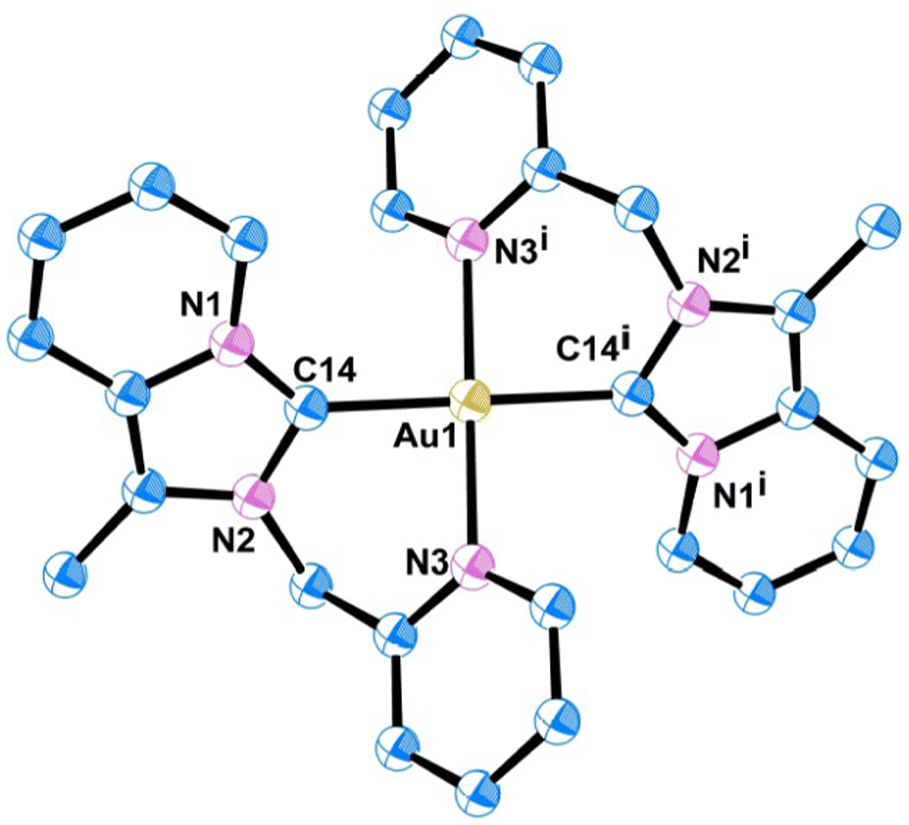
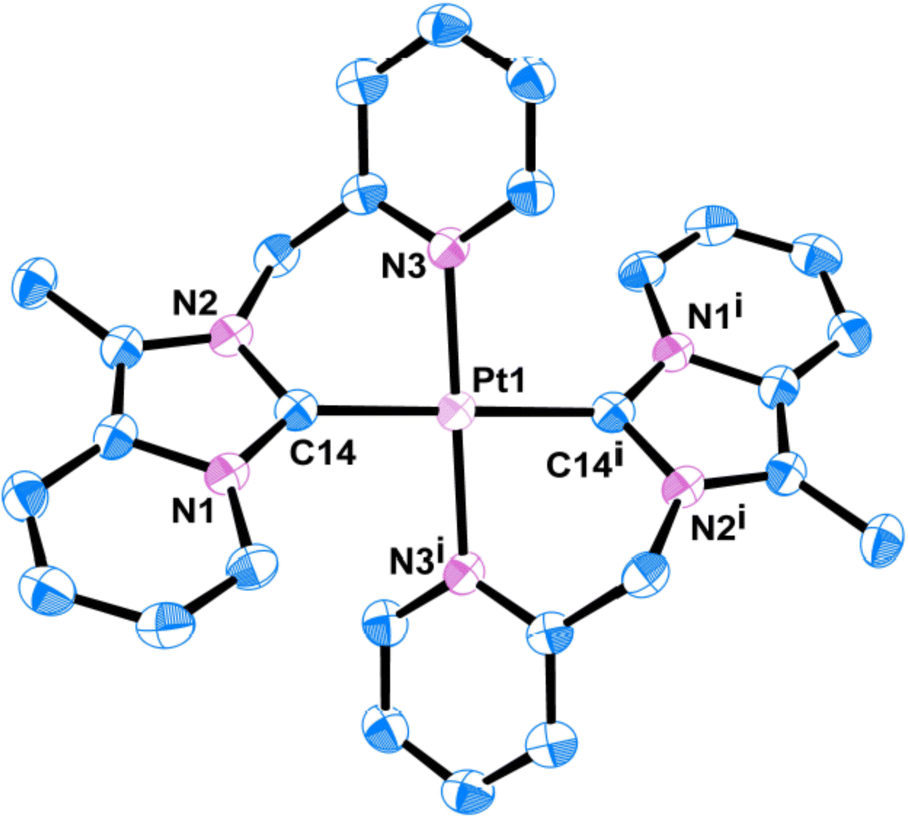
Quality single crystals of complexes 2 and 3 were obtained for X-ray diffraction studies by slow diffusion of Et2O into the saturated CH3CN solutions of the respective complexes 2 and 3. The ORTEP views of the molecular structures of 2 and 3 are shown in Fig. 1 and 3, respectively. Selected bond distances and bond angles are given in Tables 1 and 2, respectively. Key crystallographic parameters are listed in Table S1 (ESI†). Molecule 2 was crystallized with triclinic symmetry with the ‘P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ’ space group, whereas molecule 3 was crystallized with monoclinic symmetry with the C2/c space group. In both cases, the counter anionic charges of the metal ions were balanced by PF6. The geometries of both the molecules 2 and 3 around d8-metal ions Au(III) and Pt(II), respectively, are essentially square planar and tetra-coordinated square planar geometry was satisfied by the two Npy and two Ccarbene. Considering packing, molecule 2 essentially looked distorted octahedral; where the bidentate (C∩N) donor two NHC ligand comprises a biclometallated square planar array around the Au(III) centre; whereas, nitrogen atom from the two CH3CN occupy two axial coordination sites (as shown in Fig. S9, ESI†) bearing the Au(III)–NCH3CN bond distances 3.046 Å. The bidentate (C∩N)-type two NHC ligands were oriented in a cis fashion around Au(III) and Pt(II) ions in both complex 2 and 3, respectively, presumably due to a relatively large trans effect. The asymmetric units in [Au(NHC)2]3+ and [Pt(NHC)2]2+ both look just like butterfly geometry (shown in Fig. S10, ESI†). The observed Au(III)–Ccarbene bond distance (2.043 Å) in the case of complex 2 is slightly longer than the Pt(II)–Ccarbene bond distances (2.017 Å) in 3; similarly, Au(III)–Npy bond distance (2.041 Å) in complex 2 is slightly longer than Pt(II)–Npy bond distances (2.033 Å) of complex 3; which are comparable to other reported for Au(III)-NHC complexes30,31 and Pt(II)-NHC complexes.25,32,33
’ space group, whereas molecule 3 was crystallized with monoclinic symmetry with the C2/c space group. In both cases, the counter anionic charges of the metal ions were balanced by PF6. The geometries of both the molecules 2 and 3 around d8-metal ions Au(III) and Pt(II), respectively, are essentially square planar and tetra-coordinated square planar geometry was satisfied by the two Npy and two Ccarbene. Considering packing, molecule 2 essentially looked distorted octahedral; where the bidentate (C∩N) donor two NHC ligand comprises a biclometallated square planar array around the Au(III) centre; whereas, nitrogen atom from the two CH3CN occupy two axial coordination sites (as shown in Fig. S9, ESI†) bearing the Au(III)–NCH3CN bond distances 3.046 Å. The bidentate (C∩N)-type two NHC ligands were oriented in a cis fashion around Au(III) and Pt(II) ions in both complex 2 and 3, respectively, presumably due to a relatively large trans effect. The asymmetric units in [Au(NHC)2]3+ and [Pt(NHC)2]2+ both look just like butterfly geometry (shown in Fig. S10, ESI†). The observed Au(III)–Ccarbene bond distance (2.043 Å) in the case of complex 2 is slightly longer than the Pt(II)–Ccarbene bond distances (2.017 Å) in 3; similarly, Au(III)–Npy bond distance (2.041 Å) in complex 2 is slightly longer than Pt(II)–Npy bond distances (2.033 Å) of complex 3; which are comparable to other reported for Au(III)-NHC complexes30,31 and Pt(II)-NHC complexes.25,32,33
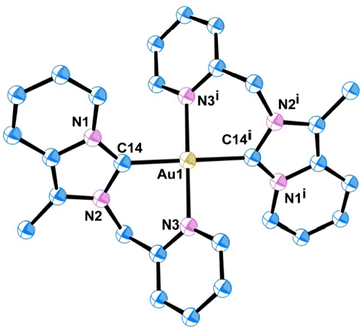 |
| | Fig. 1 ORTEP view of single crystal X-ray structure of 2; H atoms and PF6 have been omitted for clarity. | |
Table 1 Selected bond lengths (Å) of complexes 2 and 3
| Bond |
2
|
3
|
| Exp. |
Theo |
Exp. |
Theo |
| M–Ccarbene |
2.043(4) |
2.070 |
2.017(4) |
2.054 |
| M–N(3) |
2.041(3) |
2.082 |
2.033(3) |
2.081 |
| N(1)–C(14) |
1.329(5) |
1.359 |
1.355(5) |
1.361 |
| N(2)–C(14) |
1.355(5) |
1.360 |
1.357(5) |
1.360 |
Table 2 Selected bond angles (°) of complexes 2 and 3
| Bond angles |
2
|
3
|
| Exp. |
Theo |
Exp. |
Theo |
| C(14)–M(1)–C(14i) |
180.0(3) |
180.0 |
180.0(3) |
180.0 |
| N(3)–M(1)–N(3i) |
180.0(3) |
180.0 |
180.0(3) |
180.0 |
| N(3)–M(1)–C(14) |
86.52(14) |
86.06 |
85.70(14) |
85.55 |
| N(3)–M(1)–C(14i) |
93.50(14) |
93.94 |
94.31(14) |
94.45 |
| N(1)–C(14)–N(2) |
106.2(3) |
105.6 |
104.1(3) |
104.3 |
In both complexes 2 and 3, the d8-metal ion [M = Au(III)/Pt(II)] is coordinated by two Ccarbene and two Npy in a linear fashion having 180° bond angle of both C(14)–M(1)–C(14i) and N(3)–M(1)–N(3i) angle. The average C–N distance in the imidazolydene moiety is 1.36 Å for complex 2, which is slightly shorter than that in complex 3 (1.377 Å) and the imidazolydene N–C–N angle (106.17°) of complex 2 is slightly larger than that in complex 3 (104.1°), which are comparable to those found in our previous report25,33 and the other reported Au(III)/Pt(II)-NHC complexes.32–34 The dihedral angle between the NHC plane (containing C1–C2–C3–C4–C5–C6–C7–N1–N2–C14 atoms of annulated imidazopyridine moiety) and the pyridyl ring plane of picolyl wingtip is found to be 56.19° in Pt(II)-NHC complex 3, which is a little lower than that in Au(III)-NHC complex 2 (62.15°).
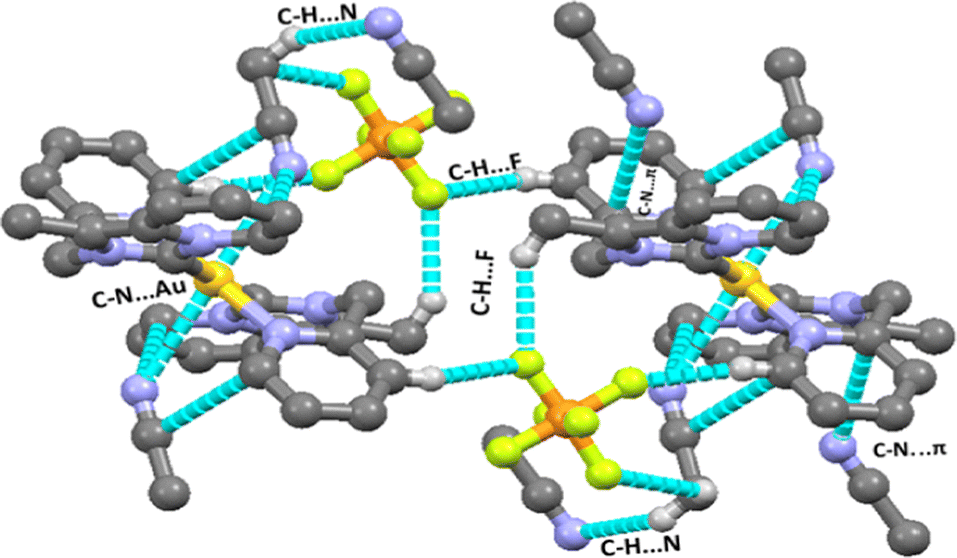
There are several types of intermolecular interactions present in both complexes 2 and 3 in the crystal packing view of complexes. The anion⋯π(d(F⋯π) = 3.559 Å), C–H⋯F (d(C–H⋯F) = 2.410–2.561 Å), C–H⋯N (d(C–H⋯N) = 2.730 Å) and C–N⋯π (3.403 Å) interactions are observed in the crystal packing view of complex 2 (shown in Fig. 2). The anion⋯π(d(F⋯π) = 3.580 Å), C–H⋯F (d(C–H⋯F) = 2.418–2.554 Å), C–H⋯N (d(C–H⋯N) = 2.698 Å) and C–H⋯π (3.464 Å) interactions are also observed in the crystal packing view of complex 3 (shown in Fig. 4).
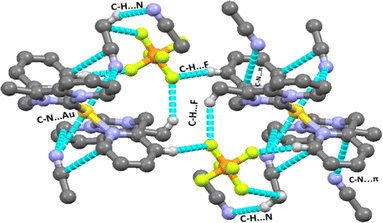 |
| | Fig. 2 Various intermolecular interactions (C–H⋯F, C–N⋯π, C–H⋯N, Au⋯N) observed in complex 2. | |
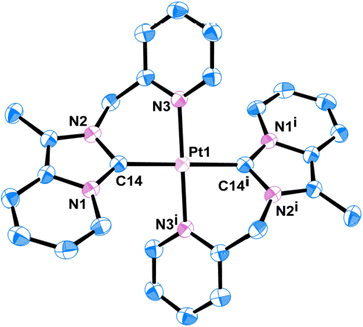 |
| | Fig. 3 ORTEP view of single crystal X-ray structure of 3; the H atoms and PF6 have been omitted for clarity. | |
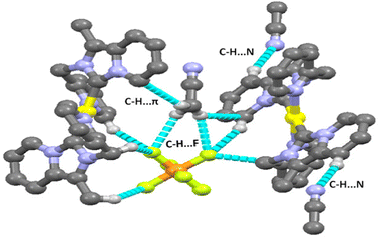 |
| | Fig. 4 Various intermolecular interactions (C–H⋯F, C–H⋯π, C–H⋯N) observed in complex 3. | |
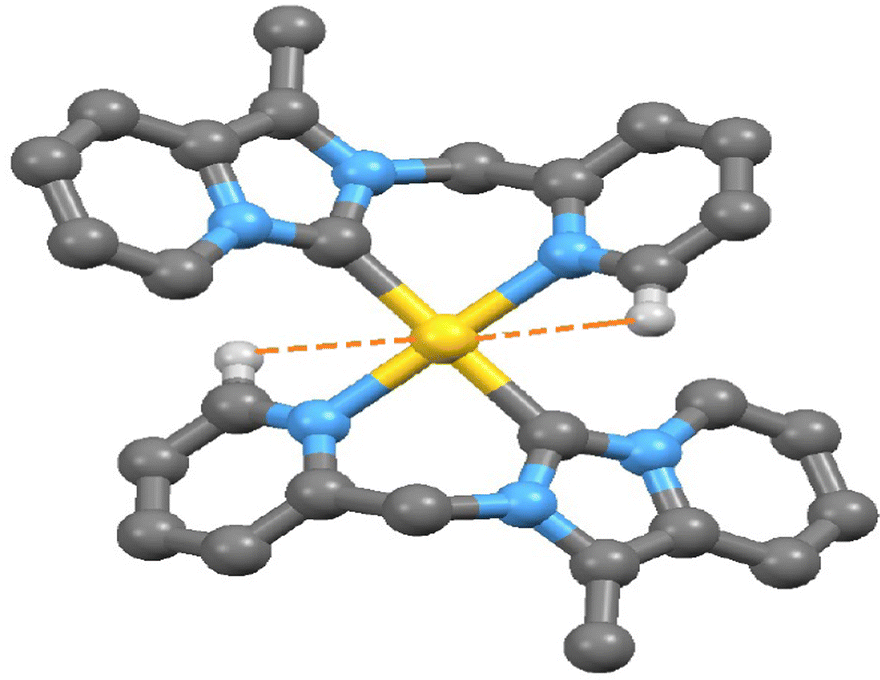
Electron-deficient transition metal-based M⋯H–C interactions now have attracted interest in organometallic compounds.35 Since the discovery of agostic interaction in early 1970, the M⋯H hydrogen bond involving N–H/O–H/C–H donor groups with various transition metal elements is well documented.36,37 Indeed, there are very few reports in the literature of M⋯H–C interactions observed in the transition metal-NHC complex.38 Here, a close look at the packing feature of 2 and 3 revealed the presence of rare intramolecular C(sp2)–H⋯Au(III)/Pt(II) non-covalent interactions. The observed shortest C(sp2)–H⋯Au(III) interaction (2.939 Å) between the metal ion and a hydrogen atom (H13) in complex 2 (shown in Fig. 5) is slightly shorter than C(sp2)–H⋯Pt(II) interaction (2.969 Å) in complex 3 (shown in Fig. 6). This observed shortest C(sp2)–H⋯Au(III) separation in complex 2 is 0.079 Å longer than the van der Waals radii of the gold and hydrogen atoms (2.86 Å), but in complex 3 C(sp2)–H⋯Pt(II) separation is very close to the van der Waals radii of the platinum and hydrogen atoms (2.95 Å). Theoretically, Au(III)–H/Pt(II)–H separation at 2.975 Å was calculated at B3LYP-D3/6-31G**, the def2TZVP level of the theory. In complex 2, the Au(1) and C(13) separation (2.925 Å, shown in Fig. S11, ESI†) is very shorter than the van der Waals Au–C distance (3.95 Å).
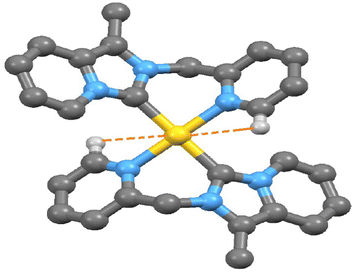 |
| | Fig. 5 The observed shortest Au(III)⋯H–C (sp2) interaction in complex 2. | |
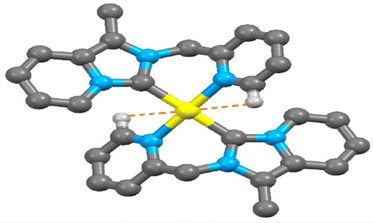 |
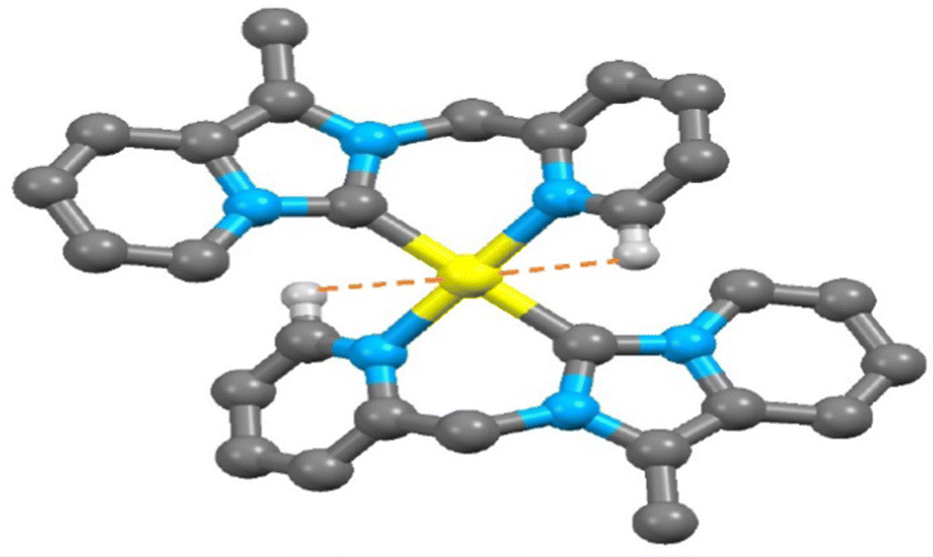
| | Fig. 6 The observed shortest Pt(II)⋯H–C (sp2) interaction in 3. | |
Similarly in complex 3, the separation between Pt(1) and C(13) atoms (2.947 Å, as shown in Fig. S12, ESI†) is very shorter than the van der Waals Pt–C distance (3.45 Å) but slightly larger than that in complex 2 (2.925 Å). The M⋯H(13)–C(sp2) bond angle in complex 3 [M = Pt(II)] is 79.46° which is a little lower than that of complex 2 [80.04°, M = Au(III)] but the observed H(13)–C(13)⋯M bond angle (82.05°) in complex 3 [M = Pt(II)] is slightly longer than complex 2 [81.71°, M = Au(III)]. At the same time, a rare intramolecular C(sp3)–H⋯Au(III)/Pt(II) interaction was observed in the packing view of 2 and 3. The C(sp3)–H⋯Pt(II) interaction (3.066 Å) in complex 3 (shown in Fig. S12, ESI†) is slightly longer than C(sp3)–H⋯Au(III) interaction (3.044 Å) as seen in complex 2 (shown in Fig. S11, ESI†). Both the separations are longer than their respective van der Waals radii of the metal and hydrogen atoms of the methylene group (dAu⋯H = 2.86 Å and dPt⋯H = 2.95 Å). The M⋯H(8B)–C(sp3) bond angle in complex 3 [M = Pt(II)] is 86.28° which is comparatively lower than Au(III)-NHC complex 2 (87.22°). Selected bond lengths (Å) and bond angle (°) of various M⋯H–C interactions of complex 2 and 3 are given in Tables S2 and S3 (ESI†), respectively.
Density functional theory studies
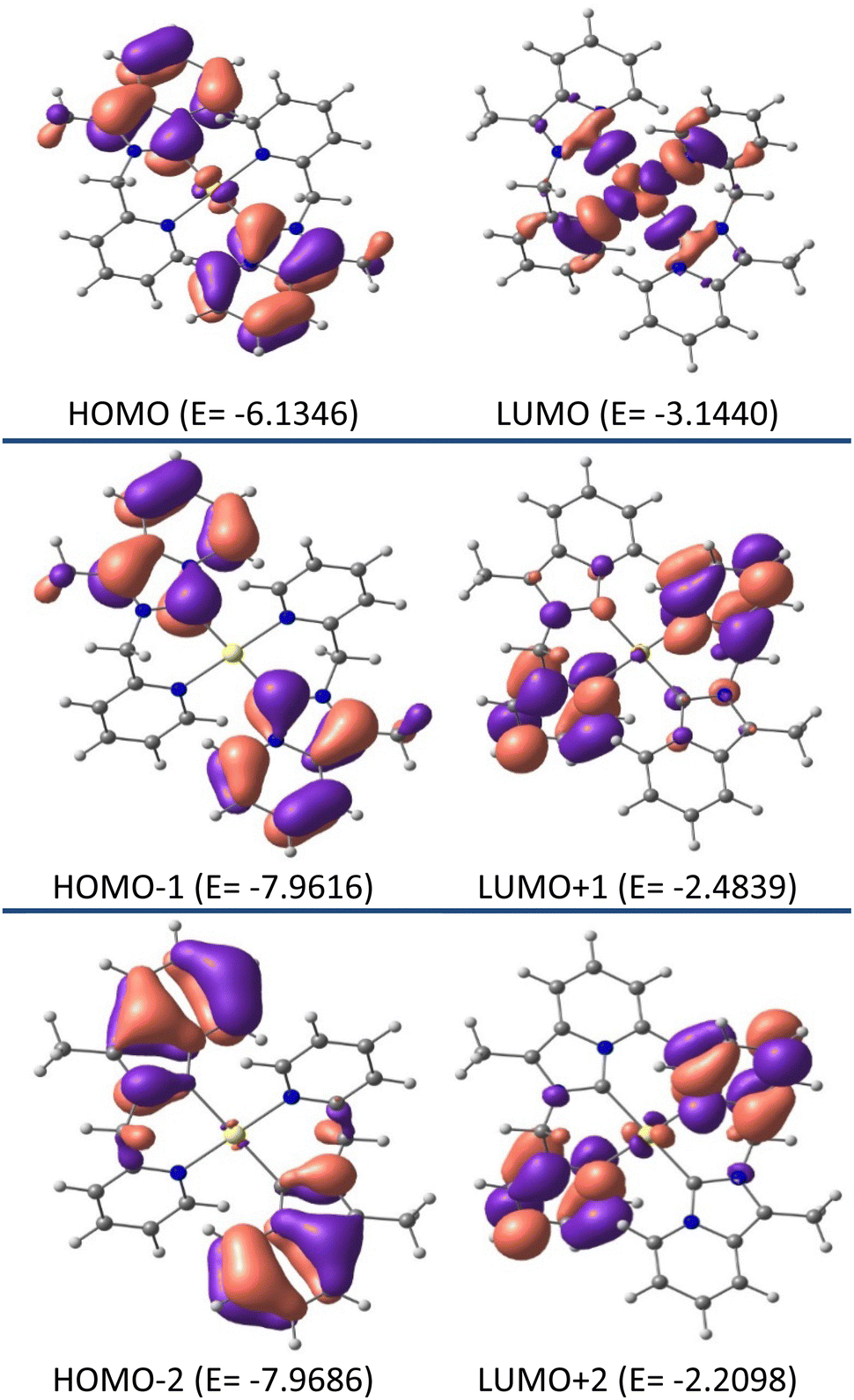
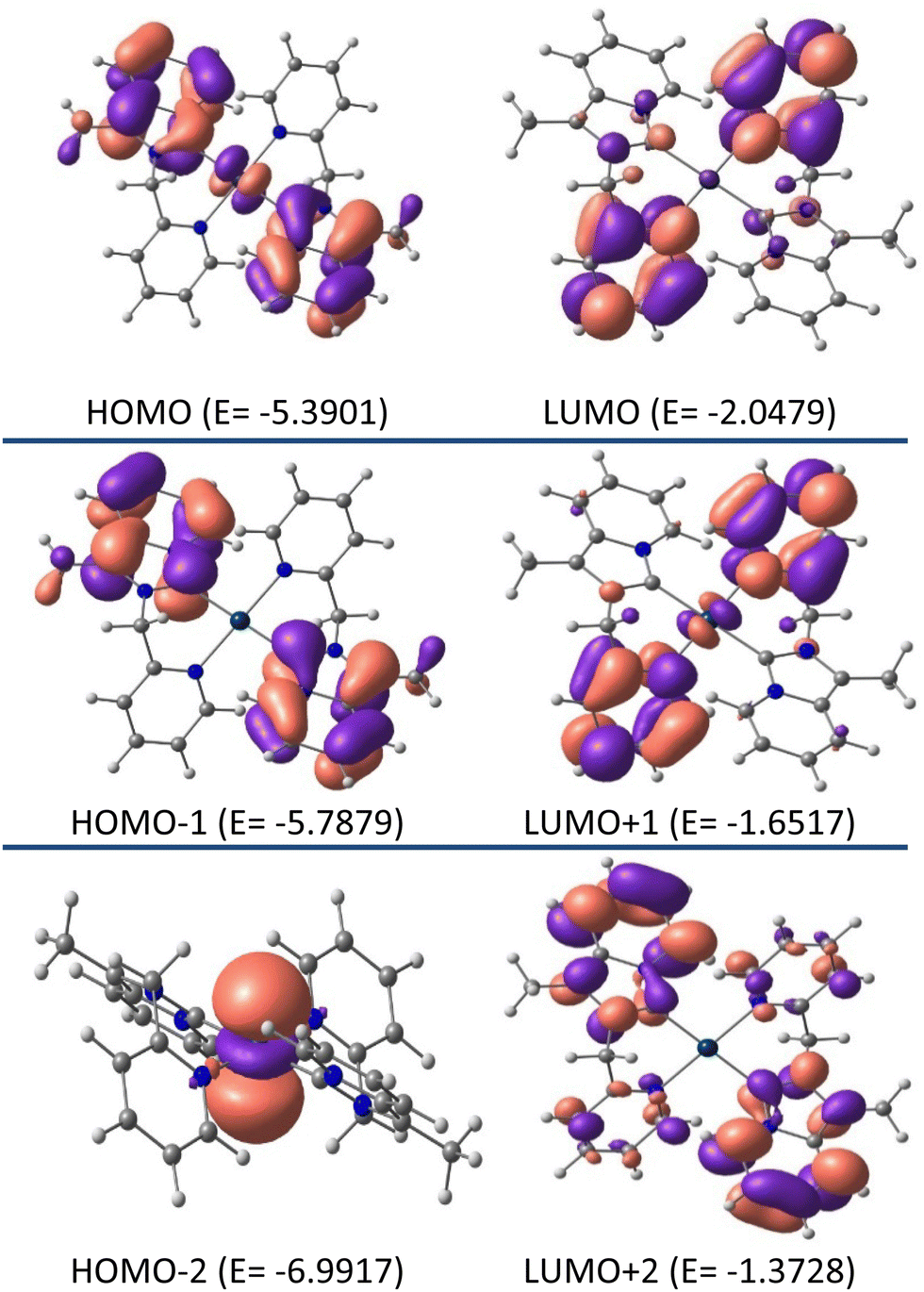
Additional insight into the structural and electronic properties of complexes 2 and 3 was gained from a series of theoretical calculations performed using DFT and TDDFT studies. The calculated geometries and bond parameters were found to be in good agreement with the crystallographic data, as represented in Tables 1 and 2. Fig. 7 and 8 show the contour plots of the selected molecular orbitals of 2 and 3.
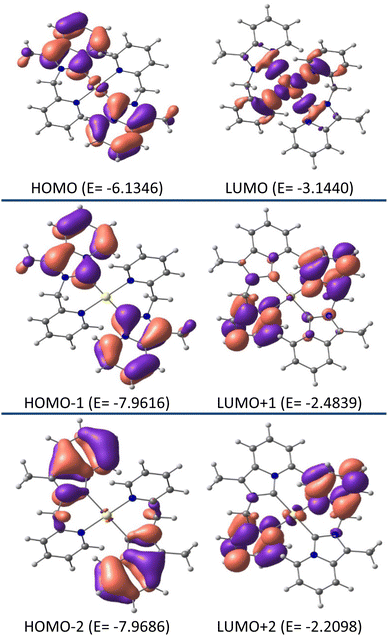 |
| | Fig. 7 Contour plots of selected molecular orbitals of 2 and energy are considered in eV. | |
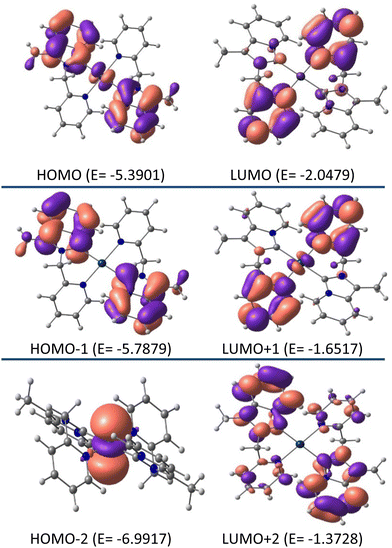 |
| | Fig. 8 Contour plots of selected molecular orbitals of 3 and energy are considered in eV. | |
Theoretical studies (B3LYP-D3/6-31G**, def2TZVP) on complex 2 show that the HOMO (E0 = −6.1346 eV) is mainly centered on the annelated imidazoline part with a minimum contribution from gold metal (98% ligand, 2% metal), whereas, in LUMO (E0 = −3.1440 eV) the electron is essentially located on the pyridyl wingtip part and gold metal (41% metal, 59% ligand character). As such, HOMO → LUMO charge transfer is assumed as mixed ILCT/LMCT. Close inspection of the FMOs for complex 2 revealed the fact that higher energy LUMOs, i.e., LUMO +½, etc. are electronically populated by the pyridyl wingtip moiety, and lower energy HOMOs i.e. HOMO −½, etc. are electronically populated by the annelated imidazoline moiety.
Unlike complex 2, similar studies on complex 3 revealed 93% ligand and 7% metal character by both HOMO and LUMO. Where the HOMO (E0 = −5.3901 eV) is mainly centered on the annelated imidazoline part, and in the LUMO (E0 = −2.0479 eV) electron is essentially concentrated on the pyridyl wingtip part. As such, the charge transfer from HOMO → LUMO in complex 3 is assumed to be ILCT. Unlike complex 2, close inspection of the FMOs for complex 3 revealed the fact that higher energy LUMOs, i.e. LUMO+1, are mainly electronically populated by the pyridyl wingtip moiety and LUMO+2 is mainly populated by the whole NHC ligand. Whereas the lower energy HOMOs, i.e. HOMO−1 are electronically populated by the annelated imidazoline moiety, and HOMO−2 is electronically concentrated by the platinum metal ion.
The chemical hardness (η) values for the complexes were also calculated to predict the energy gap (ΔE) between the corresponding frontier molecular orbitals according to the following equation: η = [(ELUMO − EHOMO)/2].39 The Chemical hardness (η) are 1.4953 and 1.6711, respectively, for complexes 2 and 3, which suggests that Pt-NHC complex 3 is harder than Au-NHC complex 2. The calculated HOMO–LUMO differences were in good agreement with the chemical hardness values.
The nature of the binding interaction in [(C∩N)–Au–(C∩N)]3+, 2 and [(C∩N)–Pt–(C∩N)]2+, 3 were probed by post-wave function analysis using the natural bond orbital (NBO) method.40 As summarized in Table 3, the natural charges on the Au and Pt atoms were calculated to be +0.93406 and +0.37407 in complexes 2 and 3, respectively. Moreover, the Wiberg Bond Index (WBI) calculations were consistent with the X-ray data, which is effectively explained by 2 and 3; each one showed distinct M-C and M–N bond distances and was consistent with reported values.41
Table 3 Calculated chemical hardness (η) and WBI values
| Complex |
Chemical hardness (η) |
WBI (bond order) |
| Complex 2 |
1.4953 |
WBI [Au(1)–C(14)]: 0.5630 |
| WBI [Au(1)–N(3)]: 0.4563 |
| Complex 3 |
1.6711 |
WBI [Pt(1)–C(1)]: 0.6419 |
| WBI [Pt(14)–N(3)]: 0.4787 |
For example; the WBI bond order for Au–Ccarbene values of 0.5630 was calculated for 2, whereas Pt–Ccarbene WBI bond order 0.6419 was found for 3. The results indicate that the Pt–Ccarbene bond is stronger than Au–Ccarbene, which was also found from single crystal X-ray data.
UV-Visible electronic absorption spectroscopy, luminescence, and lifetime studies
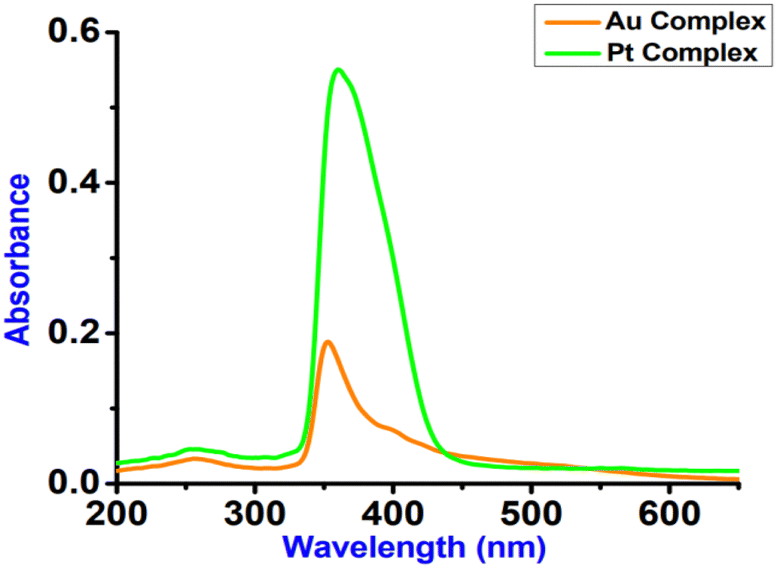
The UV-Vis absorption spectra of both complexes 2 and 3 were recorded in dry CH3CN solution at approximately 50 μM concentrations, shown in Fig. 9 and the relevant data are summarized in Table 4. The absorption spectrum of complex 2 in CH3CN exhibited strong absorption at 352 nm with a shoulder at 400, 421 nm, and a weaker absorption band at 530 nm, whereas complex 3 displayed strong absorption at 360 nm and a weaker absorption band at 570 nm. We tentatively attribute the low energy absorption band (400–570 nm) to be the ligand-to-metal charge-transfer (LMCT), similar to the LMCT bands observed in other similar systems42 and observed in platinum-tetra-N-heterocyclic carbene complex.43 In addition, both complexes 2 and 3 display high energy absorption ∼258 nm that is assigned to the intraligand π–π* transitions. Moreover, other strong absorption bands (e.g. 352, 360 nm) were assigned as ILCT transition.
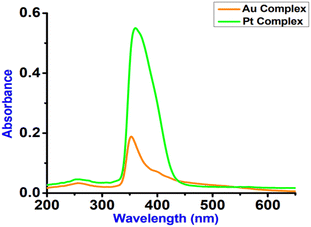 |
| | Fig. 9 Absorption spectra of complexes 2 (Au-complex) and 3 (Pt-complex) studied in dry acetonitrile at room temperature at 50 μM concentration. | |
Table 4 Summary of the photophysical data of complexes 2 and 3 studied in dry acetonitrile
| Complex |
λ
abs/nm (ε/M−1 cm−1) |
λ
em/nm (λex/nm) |
| 2 |
258, 352, 400, 421, 530, (820, 3860, 1480, 815, 600) |
412 (340) |
| 3 |
258, 360, 570 (1100, 11200, 590) |
425 (340) |
The UV-Vis absorption spectra of both complexes 2, 3, and proligand 1·HPF6, were also recorded in less polar DCM solutions at approximately 50 μM concentrations (shown in Fig. S13, ESI†), where complex 2 exhibited a strong absorption at 220 nm with a shoulder at 284, 323 nm and a weaker absorption band at 340, 391 nm. Whereas, complex 3 displayed a strong absorption at 224 nm with shoulders at 272, 290, 323, and 370 nm and a weaker absorption band at 404 nm, in comparison to the absorption bands at 219, 281, 320, 339, and 378 nm for proligand 1·HPF6. Time-dependent DFT studies displayed that complexes 2 and 3 show comparable vertical electronic transitions (shown in Fig. S14 and S15, ESI†). Theoretically, we observed high energy absorptions near 292–332 nm, whereas, low energy absorption near 370–432 nm, in addition to one very low energy absorption transition at 549 nm for complex 2 (shown in Table 5).
Table 5 Selected vertical electronic transitions of complex 2 calculated using the TDDFT/CPCM method
|
λ (nm) |
E (eV) |
Osc. strength, f |
Key excitations |
| 549.16 |
2.2577 |
0.0146 |
HOMO−1 → LUMO |
| 432.06 |
2.8696 |
0.0547 |
HOMO → LUMO+1 |
| 370.02 |
3.3507 |
0.0104 |
HOMO−1 → LUMO+2 |
| 332.22 |
3.7320 |
0.2074 |
HOMO → LUMO+3 |
| 305.50 |
4.0584 |
0.0986 |
HOMO → LUMO+6 |
| 302.88 |
4.0934 |
0.0398 |
HOMO−3 → LUMO |
| 300.95 |
4.1198 |
0.1029 |
HOMO−1 → LUMO+4 |
| 295.68 |
4.1932 |
0.1669 |
HOMO → LUMO+7 |
| 293.36 |
4.2263 |
0.0114 |
HOMO−1 → LUMO+5 |
| 292.53 |
4.2384 |
0.0484 |
HOMO−5 → LUMO |
A band at 432.06 nm is assigned for HOMO → LUMO+1 and a band at 370.02 nm for HOMO−1 → LUMO+2 transition as ILCT according to theoretical calculations. The low energy band at 549.16 nm is assigned as HOMO−1 → LUMO transition as LMCT according to theoretical calculations. Other high energy bands (292–332 nm) are assigned from HOMO/lower HOMO to LUMO/higher LUMO, which are assigned as π(L) → π*(L) ILCT transitions. Similarly, for complex 3, theoretically calculated absorption bands are observed at 276.26, 278.23, 303.74, 304.15, 309.16, 318.79, 347.18, 353.94, and 450.25 nm (as shown in Table 6). The lowest energy band at 450.25 nm is assigned to the HOMO → LUMO transition, which is assigned as the ILCT according to theoretical calculation. Other high energy bands are involved in the maximum contribution from HOMO or lower HOMO to LUMO or higher LUMO, which are also assigned as ILCT transitions. Only the band at 303.74 nm is assigned to HOMO−2 → LUMO transition, which is assigned as MLCT according to theoretical calculations. The theoretically calculated absorption wavelengths are very close to the experimentally measured values.
Table 6 Selected vertical electronic transitions to complex 3 calculated by the TDDFT/PCM method
|
λ (nm) |
E (eV) |
Osc. strength, f |
Key excitations |
| 450.25 |
2.7537 |
0.0635 |
HOMO → LUMO |
| 353.94 |
3.5030 |
0.1454 |
HOMO → LUMO+3 |
| 347.18 |
3.5712 |
0.0195 |
HOMO−1 → LUMO+1 |
| 318.79 |
3.8893 |
0.0381 |
HOMO → LUMO+5 |
| 309.16 |
4.0104 |
0.2713 |
HOMO → LUMO+6 |
| 304.15 |
4.0764 |
0.1681 |
HOMO−1 → LUMO+3 |
| 303.74 |
4.0820 |
0.0282 |
HOMO−2 → LUMO |
| 278.23 |
4.4563 |
0.0107 |
HOMO−2 → LUMO+7 |
| 276.26 |
4.4879 |
0.2365 |
HOMO−3 → LUMO |
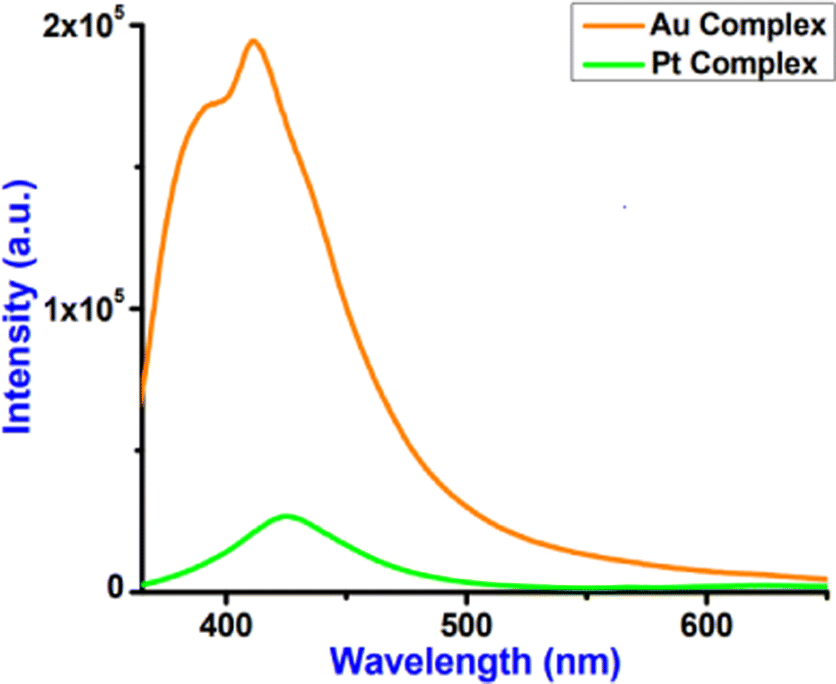
Fig. 10 represents the luminescence spectra of complexes 2 and 3. Both complexes 2 and 3 displayed relatively strong emission maxima at 412 and 425 nm, respectively, upon excitation at 340 nm. The emission spectra of these complexes are structured in dry CH3CN solution with the lifetimes of the nanosecond time scale. Fig. S16 (ESI†) provides the fluorescence decay of complexes 2 and 3 with 340 nm laser excitation, and the decay parameters are shown in Table 7.
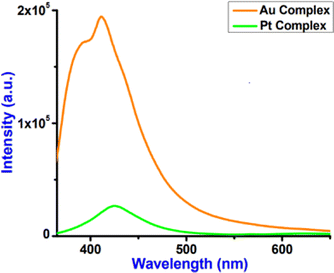 |
| | Fig. 10 Luminescence spectra of complexes 2 and 3 (λex = 340 nm) studied in dry acetonitrile at room temperature. | |
Table 7 Lifetime and quantum yield data of complexes 2 and 3 in CH3CN
|
|
τ
1(α1) |
τ
2(α2) |
τ
3(α3) |
τ
avg (ns) |
χ
2
|
ϕ
em (%) |
|
2
|
0.13 (0.34) |
2.17 (0.53) |
6.52 (0.13) |
2.10 |
1.12 |
4.1 |
|
3
|
0.42 (0.35) |
1.92 (0.61) |
7.76 (0.04) |
1.63 |
1.15 |
10.2 |
Complex 2 displays a higher lifetime (2.10 ns) compared to complex 3 (1.63 ns). Luminescence quantum yields (ϕem) in dry CH3CN solution obtained from spectra [the reference was quinine sulphate in H2SO4 (0.5 M) quantum yield (ϕF) = 0.546 at λex = 340 nm] on a wavelength scale (nm) were measured according to the approach described by Demas and Crosby44 and reported in Table 7. Complex 3 displays a higher quantum yield (10.2%) compared to complex 2 (4.1%).
Electrochemistry studies
Cyclic voltammogram studies of both complexes were conducted in acetonitrile using a Pt working electrode, Ag/AgCl as a reference electrode, and 0.1 M solutions of tetrabutylammoniumhexafluorophosphate as the supporting electrolyte. Cyclic voltammograms of Au(III)-NHC complex 2 (shown in Fig. S17, ESI†) exhibit two irreversible reduction peak potentials at −0.75 V [R1] and −1.28 V [R2] and one irreversible oxidation peak [O1] at +1.19 V. It is expected that the first reduction potential R1 is due to Au(III) → Au(I) and the second reduction potential R2 is due to Au(I) → Au(0) and one oxidation O1 is due to Au(0) → Au(I) oxidation. These redox potential values are also comparable with other reported Au(III)-NHC systems.45–47 According to Raubenheimer, Au(III) reduction is a 2e process, i.e. Au(III) → Au(I).47 According to that, the reduction potential at −0.75 V indicates the transformation of the four-coordinated square planar Au(III)-NHC complex to a two-coordinated linear Au(I) complex, and hence no existence of Au(II) intermediate for the overall two-electron reduction. Indeed, in this transformation, both +I/III oxidation states of gold are stabilized by the same NHC ligand, as reported by our group in several articles.48 It is also true that we cannot completely exclude the existence of an Au(II) intermediate, as it could react extremely fast. Here, we could not observe the Au(II) intermediate wave in cyclic voltammetry studies.
Cyclic voltammogram of complex 3 (shown in Fig. S18, ESI†) shows a bielectronic reversible process Pt(II)/Pt(IV) at 0.53 V, which is less positive than the similar C∩N donor complex [Pt(pbpy)Cl]Cl49 and the lower potential is expected due to the presence of strong σ donor NHC ligand. The lower redox potential value of Pt(II)-NHC, 3 is comparable to that of the other Pt(II)-NHC systems reported by our group.50 We have reported Pt(II)/Pt(IV) redox potential at 0.58 V for a NCN donor pincer ligand where the HOMO–LUMO energy gap is 4.102 eV; but in the present case the HOMO–LUMO energy gap is 3.342 eV, therefore a lower Pt(II)/Pt(IV) redox potential is expected and observed in comparison to the previous case. Chemical hardness may be co-related to the redox potential value, the more hard is the system more will be the Pt(II)/Pt(IV) oxidation potential; the present complex is less hard than the NCN pincer-Pt(II) complex reported by our group.50 The present studies are very consistent with the chemical hardness and redox potential.
Molecular docking studies
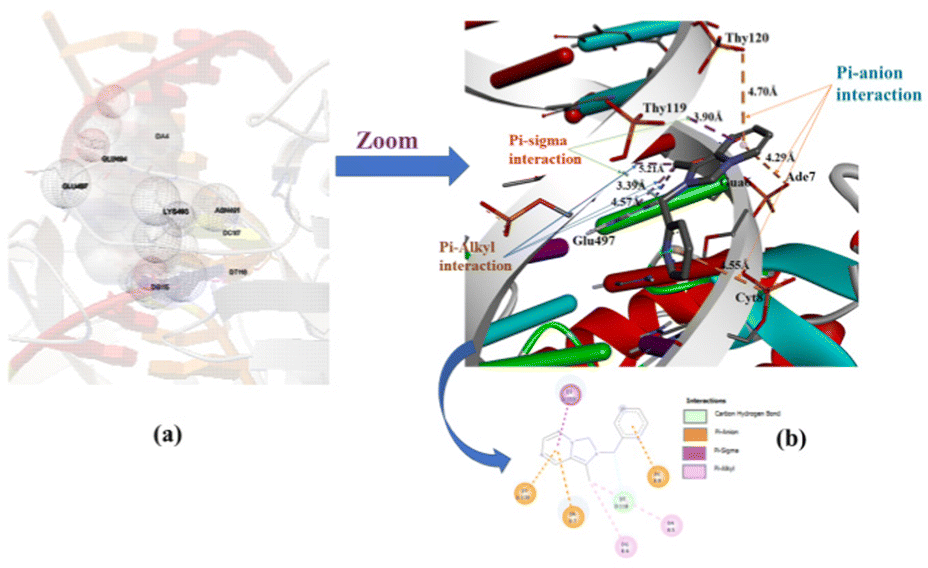
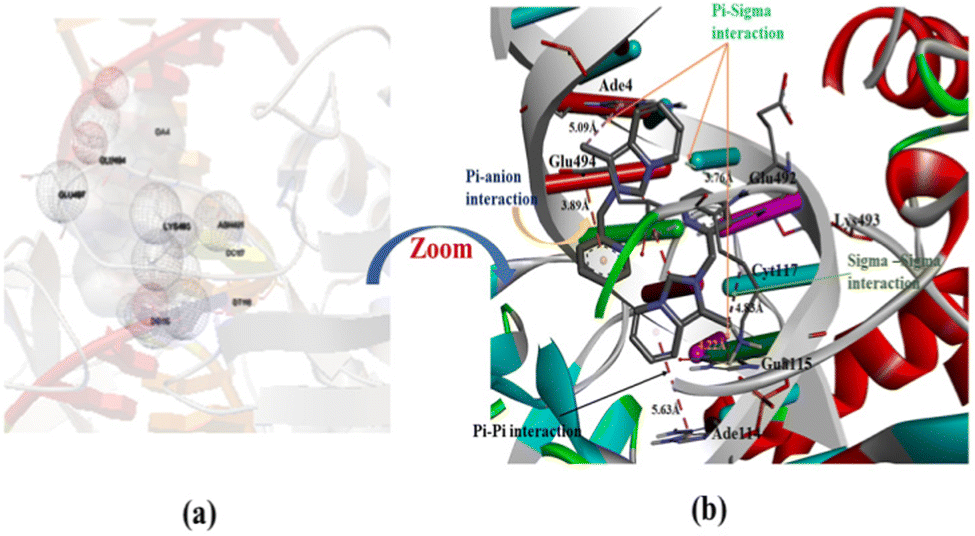
Molecular docking analysis has now appeared as an emerging tool in drug discovery for exploring the binding mode and interaction between small molecules as an organic compound and its metal complexes with double-strand DNA. The energetically most favorable docked conformations of the N-heterocyclic ligand precursor (1·HPF6) with human-DNA topoisomerase are displayed in Fig. 11(a) and (b). The ligand is encapsulated in double-strand DNA with several interactions like carbon–hydrogen bond, π–anion, π–σ, and π–alkyl type. The estimated free binding energy of the ligand is −5.73 kcal mol−1 and other energies are given in Table S4 (ESI†). The π–anion type interaction is observed between the π–electrons of the annulated imidazopyridine moiety of the N-heterocyclic ligand with the acetate group of Thy120 and Ade7 base pair at a distance of 4.70 Å and 4.29 Å respectively. The other π–σ, π–alkyl type interactions were also observed between the substituted methyl group of the annulated imidazopyridine moiety of N-heterocyclic ligand with Glu-497, Ade6, and Thy119 base pairs of DNAs. The docking result displays the NHC ligand binding to DNA with a major binding side.
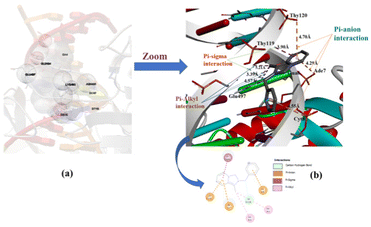 |
| | Fig. 11 Capped and stick model and 2D view of the different binding modes of the NHC ligand precursor (1·HPF6) with Human-DNA topoisomerase. | |
The energetically most favorable docked picture of Au(III)-NHC complex 2 with human-DNA topoisomerase is given in Fig. 12(a) and (b). The free binding energy of complex 2 increased with respect to the NHC ligand precursor, due to the incorporation of Au3+ ion in the NHC ligand. Mainly π–σ, π–anion, σ–σ, and π–π interactions are observed between complex 2 and the base pair of the DNA.
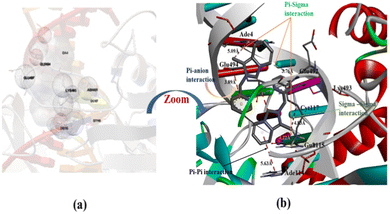 |
| | Fig. 12 Capped and stick model and 2D view into the interaction mode of complex 2 with human-DNA topoisomerase. | |
We observed the highest free binding energy for complex 3 with 1t8i DNA (−7.26 kcal mol−1) than the corresponding NHC ligand precursor and complex 2.
Complex 3 interacts with DNA at the major binding sides and inserts hydrophobic pockets in double-stranded DNA. In complex 3, the metal ion [Pt(II)] directly interacts with the negative part of the base pair of DNA. The most favorable docking picture in Fig. 13(a) and (b) implies several interactions present between DNA and the targeted molecule, complex 3. The order of estimated free binding energy is complex 3 > complex 2> NHC proligand (1·HPF6). On account of the higher effective nuclear charge and a special distribution of d-orbitals of heavy transition metal ions like Au(III) and Pt(II) shows effecting binding or interacting properties than corresponding NHC ligand molecule.
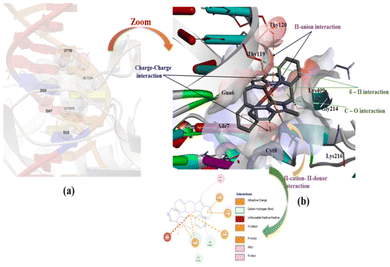 |
| | Fig. 13 Capped and stick model and 2D view into the different binding modes of complex 3 with human-DNA ttopoisomerase. | |
Pharmacokinetic, drug-likeness, and scope of medicinal chemistry studies
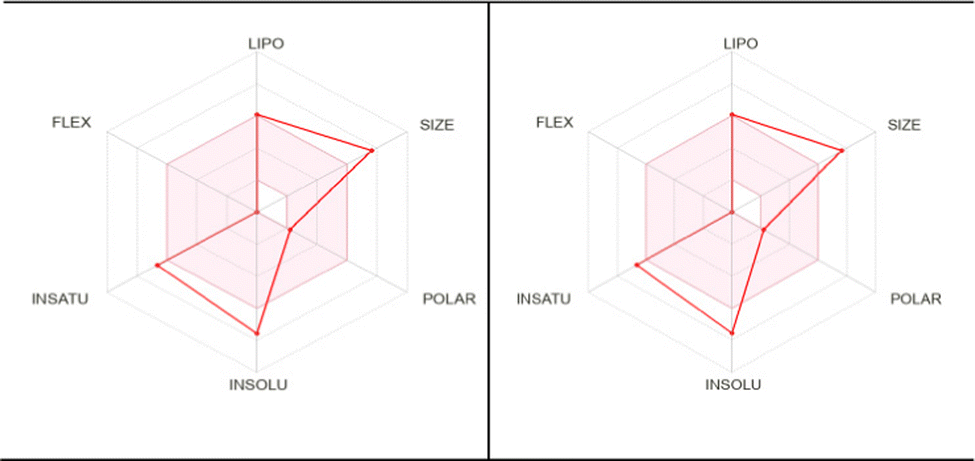
To be used as a drug, a potent molecule must reach in target in sufficient concentration, and remain in bioactive form for sufficient time for biological events to occur. Drug designing and development involves the assessment of absorption, distribution, metabolism, and excretion (ADME); Swiss ADME is a web tool that offers robust predictive models for physicochemical properties, pharmacokinetics, drug-likeness, and medicinal chemistry friendliness.51 Pioneering work of Lipinski indicates that to be considered a compound to be used as a drug it must be defined by some physicochemical parameters; the so-called ‘Rule-of-five’ establishes the relationship between pharmacokinetics and physicochemical parameters. In this context, six physicochemical properties are taken into consideration: size, solubility, lipophilicity, polarity, flexibility, and saturation. Considering the physicochemical range, a pink area is obtained in which the radar plot of the molecule has to fall entirely to be considered drug-like. Out of six properties, three properties remain within the pink area in the case of our present complexes, 2 and 3, as shown in Fig. 14 (data are plotted in Tables S5–S7, ESI†). Another important observation noted that both complexes 2 and 3 fall in the same range of the pink area, which supports the similar potency. More detailed studies, such as in vitro and in vivo studies, are needed to establish this same fact.
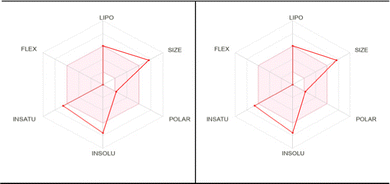 |
| | Fig. 14 Pharmacokinetic studies of complex 2 (left) and complex 3 (right) show the similar potency of the complexes to be used as a drug. | |
Conclusion
Capitalizing on the C∩N donor NHC, two Isoelectronic square planar Au(III) and Pt(II) complexes were synthesized and characterized. Finally, the solid-state X-ray structure established the isostructural geometry of both Au(III)NHC, 2 and Pt(II)-NHC, 3. As both belong to the 5d group (d8 system) they bear comparable bond parameters; e.g. Au–Ccarbene and Pt–Ccarbene distances 2.043(4) and 2.017(4) Å respectively. X-ray crystallography revealed the square planar geometry of both complexes 2 and 3. Both complexes possess several intermolecular interactions viz. C–H⋯F, C–H⋯N, anion⋯π interactions and rare intramolecular C(sp2)–H⋯Au(III)/Pt(II) non-covalent interactions in the solid-state structure. Such interactions are expected to enrich the crystal engineering design parameters, which are used to create higher-order structures. Both complexes are luminescent. Complex 2 possesses a higher a lifetime than complex 3. Cyclic voltammetric studies of complex 2 revealed irreversible Au(III) → Au(I) and Au(I) → Au(0) reductions and complex 3 displayed a reversible Pt(II)/Pt(IV) bielectronic process. Molecular docking studies revealed that complex 3 is more susceptible to binding with human-DNA topoisomerase than complex 2 and even with the NHC proligand.
Experimental section
General procedures
The reagents silver oxide, KPF6, 2-acetylpyridine, 2-picolylamine, paraformaldeyde, KAuCl4, K2PtCl4 triethylorthoformate, were purchased from Sigma Aldrich and used without further purification. All manipulations were carried out in an open atmosphere. NMR spectra were measured on a Bruker 400 MHz spectrometer at 25 °C with tetramethylsilane as an internal standard. The absorption and steady-state fluorescence spectra of the samples were recorded using the Cary 100 Bio spectrophotometer and Carry eclipse fluorescence spectrophotometer (Agilent technologies), respectively. Time correlated single photon counting (TCSPC) spectrometer (Life Spec II) was used for time-resolved fluorescence measurements along with a 330 nm picoseconds diodes laser (EPL) as an excitation source. Fluorescence spectra were recorded in dry CH3CN. Quantum yield was measured in an Agilent Cary eclipse fluorescence spectrophotometer.
Synthesis of proligand (1·HPF6)
Proligand 1·HPF6 was synthesized according to the reported procedure;28 when the Schiff base 2-pyridylmethyl N-(2-acetylpyridyl)methylamine (1.5 g, 0.0071 mol), 2–3 drops formic acid, 0.5 mL triethylorthoformate and 91% crushed paraformaldehyde powder (0.235 g, 0.0071 mol) were taken in 20 mL dioxane, stirred for 6 h, then refluxed for 4 h; 5 mL 4(N) HCl in Et2O was added slowly to the cold slurry suspension; layers separated. The lower layer of the round bottom occupied a brownish viscous liquid and a colourless liquid was in the upper part. Water (5 mL) was added to the solution and the lower aqueous phase was separated using a separating funnel. Then, the excess aqueous saturated potassium hexafluorophosphate solution was added dropwise to the solution; the immediate white precipitate of hexafluorophosphate salt of proligand was obtained. The precipitate was filtered and recrystallized from CH3CN–Et2O. The solid mass was dried under a vacuum. The yield was 2.01 g (5.45 mmol, 77%). 1H NMR (DMSO-d6, 25 °C, 400 MHz): δ = 9.77 (s, 1H, Hi), 8.57 (d, J = 6.0 Hz, 2H, Ha,h), 7.95 (t, J = 6.0 Hz, 1H, Hc), 7.91 (d, J = 6.0 Hz, 1H, Hd), 7.59 (d, J = 6.0 Hz, 1H,He), 7.45 (d, J = 6.0 Hz, 1H, Hf), 7.18 (m, 2H, Hb,g), 5.83 (s, 2H, Hj), 2.16 (s, 3H, Hk), 13C NMR (DMSO-d6, 25 °C, 400 MHz): 153.19, 149.82, 138.57, 126.98, 126.57, 124.33, 123.45, 123.18, 122.59, 120.52, 118.38, 118.11, 51.79, 8.48. Anal. Calc. for C14H14N3PF6: C, 49.32; H, 3.79; N, 11.38. Found: 49.28; H, 3.77; N, 11.21%.
Synthesis of [Au(1)2][PF6]3 (2)
The proligand 1·HPF6 (300 mg, 0.81 mmol) and Ag2O (100 mg, 0.43 mmol) were taken in 10 mL dry CH3CN and stirred in dark at room temperature for 4 h. Then, the unreacted Ag2O was separated by filtration through a plug of Celite; the filtrate was added dropwise to a solution of KAuCl4 (153 mg, 0.405 mmol taken in 5 mL CH3CN) at the stirring condition in dark at room temperature, and then stirring was continued for 2 h. The colourless solution changed to an orange-red colour, over the course of the reaction. After filtration through a plug of Celite, the solvent was evaporated at low temperature and high pressure to obtain the solid mass. An analytically pure compound was obtained by diffusion of diethyl ether into saturated acetonitrile solution of the complex 2. The yield was 82% (355 mg, 0.33 mmol). 1H NMR (DMSO-d6, 25 °C, 400 MHz): δ = 8.67 (d, J = 4.0 Hz, 1H, Ha), 8.60 (d, J = 4.0 Hz, 1H, Hh), 8.27 (d, J = 4.0 Hz, 1H, Hd), 7.91 (t, J = 8.0 Hz, 1H, Hc), 7.69 (d, J = 4.0 Hz, 1H, He), 7.57 (t, J = 8.0 Hz, 1H, Hf), 7.19 (t, J = 4.0 Hz, 1H, Hb), 7.16 (t, J = 4.0 Hz, 1H, Hg), 6.17 (d, 1H, J = 8.0 Hz, N-CH2-py, Hj), 5.97 (d, 1H, J = 12.0 Hz, N-CH2-py, Hj), 2.41 (s, 3H, Hk).13C NMR (DMSO-d6, 25 °C, 400 MHz): 156.69, 150.15, 139.56, 127.34, 126.97, 125.29, 124.17, 123.23, 122.59, 121.12, 118.53, 118.11, 51.92, 8.66. Anal. Calcd for C28H26N6AuP3F18: C, 31.16; H, 2.41; N, 7.79. Found: C, 31.11; H, 2.37; N, 7.76%.
Synthesis of [Pt(1)2][PF6]2(3)
The proligand 1·HPF6 (300 mg, 0.81 mmol) and Ag2O (100 mg, 0.43 mmol) were taken in 10 mL dry acetonitrile and stirred in dark at room temperature for 4 h. Then, the unreacted Ag2O was separated by filtration through a plug of Celite; the filtrate was added dropwise to a solution of K2PtCl4 (168 mg, 0.405 mmol taken in 5 mL CH3CN) at the stirring condition in dark at room temperature, and then stirring was continued for 16 h. Then, the resulting solution was heated at 90 °C for 2 h. During this time period, the colour changedfrom faint yellow to colourless. The solution was kept for some time at room temperature and then filtered through a plug of Celite. The volume of the solution was reduced under high pressure and at low temperatures to obtain a solid mass. An analytically pure compound was obtained by diffusion of diethyl ether into a saturated acetonitrile solution of complex 3. The yield was 84% (315 mg, 0.34 mmol). 1H NMR (DMSO-d6, 25 °C, 400 MHz): δ = 8.76 (d, J = 4.0 Hz, 1H, Ha), 8.70 (d, J = 8.0 Hz, 1H, Hh), 8.37 (d, J = 8.0 Hz, 1H, Hd), 7.89 (t, J = 8.0 Hz, 1H, Hc), 7.67 (d, J = 8.0 Hz, 1H, He), 7.40 (t, J = 8.0 Hz, 1H, Hf), 7.29 (t, J = 8.0 Hz, 1H, Hb), 7.16 (t, J = 8.0 Hz, 1H, Hg), 6.18 (d, 1H, J = 16.0 Hz, N-CH2-py, Hj), 5.96 (d, 1H, J = 16.0 Hz, N-CH2-py, Hj), 2.78 (s, 3H, Hk).13C NMR (DMSO-d6, 25 °C, 400 MHz): 166.59, 153.35, 142.16, 128.19, 127.95, 126.59, 124.25, 123.44, 123.01,121.35, 119.02, 118.34, 52.02, 8.80. Anal. Calc. for C28H26N6PtP2F12: C, 36.09; H, 2.79; N, 9.02. Found: C, 36.04; H, 2.73; N, 9.01%.
Crystal structure determination
Single crystals of complexes 2 and 3 suitable for X-ray data collection were grown by slow diffusion of diethyl ether into a saturated acetonitrile solution of the respective complexes. The crystal data and details of the data collected for 2 and 3 are given in Table 1. X-ray data were collected on a Bruker diffractometer (ImuS-Diamond source, Cu Kα radiation, λ = 1.54178 Å, Photon III detector). The structures were solved with SHELXT52 and refined with SHELXL53 programs using Olex2 as GUI.54
Computational methodology
The geometries of Au(III) and Pt(II) complexes were optimized using the DFT, TD-DFT/CPCM(acetonitrile)/B3LYP-D3/6-31G**, def2TZVP(Au,Pt), 6-31G**(C,H,N) level of theory. All DFT/B3LYP calculations were performed using the GAUSSIAN09 package program.55 During optimization, metal was considered by the quasi relativistic LANL2DZ basis set56 while the other elements were considered with the 6-31+G(d,p) basis set.57 Using the optimized ground state geometry, absorption characteristics in acetonitrile (CH3CN) were calculated using time-dependent density functional theory (TDDFT) in conjunction with the conductor-like polarizable continuum model (CPCM). Gauss Sum 2.1 was used to calculate the fractional contributions.
Cyclic voltammetric studies
Electrochemical data were acquired on a PAR model 273A electrochemistry system at a scan rate 100 mV s−1. Platinum electrode as working electrode, platinum wire as auxiliary electrodes and an aqueous saturated calomel reference electrode were used in a three-electrode configuration. The sample was dissolved in acetonitrile (Merck, p.a.) to give a concentration of 5 × 10−4 M of the analyte and 0.1 M [Bu4N]PF6. The solutions were purged with nitrogen prior to measurement and then stored under an argon blanket.
Molecular docking instrumentation and methods
Molecular docking was carried out using AUTODOCK 4.0 software as accomplished by the graphical user interface AUTODOCK TOOLS 4.0 (AD4.1_bound.dat). The macrocyclic receptor was chosen as PDB formed of a three-dimensional X-ray crystal structure of a human-DNA topoisomerase (ID: 1t8i). The graphical user interface AUTODOCK TOOLS was devoted to setting up the protein: water molecule was deleted from the crystal of protein, only polar hydrogen was added, computed gasteiger charge was calculated as −16.983 and non-polar hydrogen was merged with the carbon atom. The desired structures of the compounds were saved in the PDB format with the aid of the program MERCURY. The AUTODOCK TOOLS program was used to create a docking input file. A grid box size for the NHC ligand, complex 2 and complex 3 were 38 × 76 × 62; 54 × 64 × 56, and 48 × 46 × 50, respectively, with coordinates of the Central Grid Point of Maps equal to (22.477, 0.766, 30.352) and grid spacing 1 Å. Both receptors and the drug molecules were saved in the pdbqt format.58 Distances-dependent functions of the dielectric constant were used for the calculation of the energetic map. Fifty runs were generated by using Lamarckian genetic algorithm searches. Default settings were used with a maximum number of 2.5 × 106 energy evaluations, an initial population of 50 randomly placed individuals.59 Final docking was run with autogrid4.exe and autodock4.exe functions to generate glg and dlg files, respectively. The graphical interaction pictures were found using discovery Studio 2021 visualisation software.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
JD is grateful to SERB, New Delhi for financial support through the project EMR/2016/007022; WB-OHEPEE sponsored research project, Utkal University, and DST-FIST, Utkal University for providing infrastructural facilities.
References
-
(a) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS PubMed;
(b) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS;
(c) M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677–3707 CrossRef CAS;
(d) C. Samojłowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708–3742 CrossRef;
(e) O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445–3478 CrossRef CAS;
(f) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef.
-
(a) J. D. Egbert, A. Chartoire, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2011, 30, 4494–4496 CrossRef CAS;
(b) M. V. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef CAS PubMed;
(c) K. T. Chan, Y. H. Tsai, W. S. Lin, J. R. Wu, S. J. Chen, F. X. Liao, C. H. Hu and H. M. Lee, Organometallics, 2010, 29, 463–472 CrossRef CAS;
(d) S. DíezGonzález, E. C. Escudero-Adán, J. Benet-Buchholz, E. D. Stevens, A. M. Z. Slawin and S. P. Nolan, Dalton Trans., 2010, 39, 7595–7606 RSC;
(e) W. A. Herrmann, M. Elison, J. Fischer, C. Köcher and G. R. Artus, J. Angew. Chem., Int. Ed. Engl., 1995, 34, 2371–2374 CrossRef CAS;
(f) K. Zhang, M. Conda-Sheridan, S. R. Cooke and J. Louie, Organometallics, 2011, 30, 2546–2552 CrossRef CAS;
(g) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC.
-
(a) Z. Xi, X. Zhang, W. Chen, S. Fu and D. Wang, Organometallics, 2007, 26, 6636–6642 CrossRef CAS;
(b) F. E. Hahn, M. C. Jahnke and T. Pape, Organometallics, 2007, 26, 150–154 CrossRef CAS;
(c) B. Liu, Q. Xia and W. Chen, Angew. Chem., Int. Ed., 2009, 48, 5513–5516 CrossRef CAS;
(d) X. Liu and W. Chen, Dalton Trans., 2012, 41, 599–608 RSC.
-
(a) D. Meyer, M. A. Taige, A. Zeller, K. Hohlfeld, S. Ahrens and T. Strassner, Organometallics, 2009, 28, 2142–2149 CrossRef CAS;
(b) X. Zhang, B. Liu, A. Liu, W. Xie and W. Chen, Organometallics, 2009, 28, 1336–1349 CrossRef CAS.
-
(a) C. Chen, H. Qiu and W. Chen, Inorg. Chem., 2011, 50, 8671–8678 CrossRef CAS;
(b) U. J. Scheele, M. John, S. Dechert and F. Meyer, Eur. J. Inorg. Chem., 2008, 373–377 CrossRef CAS;
(c) Y. Zhou, Z. Xi, W. Chen and D. Wang, Organometallics, 2008, 27, 5911–5920 CrossRef CAS;
(d) B. Liu, B. Liu, Y. Zhou and W. Chen, Organometallics, 2010, 29, 1457–1464 CrossRef CAS.
-
(a) S. Gu and W. Chen, Organometallics, 2009, 28, 909–914 CrossRef CAS;
(b) S. Gu, D. Xu and W. Chen, Dalton Trans., 2011, 40, 1576–1583 RSC.
-
(a) M. Poyatos, E. Mas-Marzá, M. Sanaú and E. Peris, Inorg. Chem., 2004, 43, 1793–1798 CrossRef CAS PubMed;
(b) A. Azua, S. Sanz and E. Peris, Chem. – Eur. J., 2011, 17, 3963–3967 CrossRef CAS;
(c) P. Gigler, B. Bechlars, W. A. Herrmann and F. E. Kühn, J. Am. Chem. Soc., 2011, 133, 1589–1596 CrossRef CAS PubMed;
(d) S. Meyer, C. M. Orben, S. Demeshko, S. Dechert and F. Meyer, Organometallics, 2011, 30, 6692–6702 CrossRef CAS.
-
(a) A. A. Danopoulos, D. Pugh, H. Smith and J. Saβmannshausen, Chem. – Eur. J., 2009, 15, 5491–5502 CrossRef CAS;
(b) A. Liu, X. Zhang and W. Chen, Organometallics, 2009, 28, 4868–4871 CrossRef CAS;
(c) C. S. Lee, S. Sabiah, J. C. Wang, W. S. Hwang and I. J. B. Lin, Organometallics, 2010, 29, 286–289 CrossRef CAS;
(d) E. Fogler, E. Balaraman, Y. Ben-David, G. Leitus, L. J. W. Shimon and D. Milstein, Organometallics, 2011, 30, 3826–3833 CrossRef CAS;
(e) C. S. Lee, R. R. Zhuang, S. Sabiah, J. C. Wang, W. S. Hwang and I. J. B. Lin, Organometallics, 2011, 30, 3897–3900 CrossRef CAS.
-
(a) X. Hu and K. Meyer, J. Organomet. Chem., 2005, 690, 5474–5484 CrossRef CAS;
(b) C. Vogel, F. W. Heinemann, J. Sutter, C. Anthon and K. Meyer, Angew. Chem., Int. Ed., 2008, 47, 2681–2684 CrossRef CAS PubMed;
(c) C. E. Ellul, G. Reed, M. F. Mahon, S. I. Pascu and M. K. Whittlesey, Organometallics, 2010, 29, 4097–4104 CrossRef CAS;
(d) A. Rit, T. Pape and F. E. Hahn, J. Am. Chem. Soc., 2010, 132, 4572–4573 CrossRef CAS PubMed.
-
(a) F. E. Hahn, C. Radloff, T. Pape and A. Hepp, Organometallics, 2008, 27, 6408–6410 CrossRef CAS;
(b) C. Radloff, J. J. Weigand and F. E. Hahn, Dalton Trans., 2009, 9392–9394 RSC;
(c) A. Zanardi, J. A. Mata and E. Peris, J. Am. Chem. Soc., 2009, 131, 14531–14537 CrossRef CAS PubMed;
(d) F. M. Conrady, R. Fröhlich, C. S. Brink, T. Pape and F. E. Hahn, J. Am. Chem. Soc., 2011, 133, 11496–11499 CrossRef CAS PubMed.
-
(a) A. S. K. Hashmi and Top Organomet, Chem, 2013, 44, 143–164 Search PubMed;
(b) A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 11, 1657–1712 CrossRef PubMed;
(c) N. Marion and S. P. Nolan, Chem. Soc. Rev., 2008, 37, 1776–1782 RSC;
(d) G. Ung, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 758–761 CrossRef CAS PubMed;
(e) M. M. Hansmann, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2013, 52, 2593–2598 CrossRef CAS;
(f) S. Gaillard, C. S. J. Cazin and S. P. Nolan, Acc. Chem. Res., 2012, 45, 778–787 CrossRef CAS PubMed;
(g) K. F. Donnelly, A. Petronilho and M. Albrecht, Chem. Commun., 2013, 49, 1145–1159 RSC.
-
(a) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS PubMed;
(b) L. Mercs and M. Albrecht, Chem. Soc. Rev., 2010, 39, 1903–1912 RSC;
(c) C. Bronner and O. S. Wenger, Dalton Trans., 2011, 40, 12409–12420 RSC;
(d) T. H. T. Hsu, J. J. Naidu, B. J. Yang, M. Y. Jang and I. J. B. Lin, Inorg. Chem., 2012, 51, 98–108 CrossRef CAS PubMed;
(e) M. C. Gimeno, A. Laguna and R. Visbal, Organometallics, 2012, 31, 7146–7157 CrossRef CAS;
(f) M. Baron, C. Tubaro, A. Biffis, M. Basato, C. Graiff, A. Poater, L. Cavallo, N. Armaroli and G. Accorsi, Inorg. Chem., 2012, 51, 1778–1784 CrossRef CAS.
-
(a) W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755–773 RSC;
(b) L. Oehninger, R. Rubbiani and I. Ott, Dalton Trans., 2013, 42, 3269–3284 RSC;
(c) A. Gautier and F. Cisnetti, Metallomics, 2012, 4, 23–32 CrossRef CAS PubMed;
(d) C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS;
(e) T. Scattolina, G. Andreettaa, M. Mauceri, F. Rizzolioa, N. Demitri, V. Canzonieri and F. Visentina, J. Org. Chem., 2021, 952, 122014 CrossRef.
- V. J. Catalano and A. O. Etogo, Inorg. Chem., 2007, 46, 5608–5615 CrossRef CAS PubMed.
-
(a) X. Zeng, R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 122, 954–957 CrossRef ; Angew. Chem., Int. Ed. 2010, 49, 942;
(b) A. S. K. Hashmi, A. M. Schuster and F. Rominger, Angew. Chem., Int. Ed., 2009, 121, 8396–8398 CrossRef ; Angew. Chem.Int. Ed. 2009, 48, 8247.
-
(a) S. W. Thomas, K. Venkatesan, P. Muller and T. M. Swager, J. Am. Chem. Soc., 2006, 128, 16641–16648 CrossRef CAS PubMed;
(b) W. P. To, G. S. M. Tong, W. Lu, C. S. Ma, J. Liu, A. L. F. Chow and C. M. Che, Angew. Chem., Int. Ed., 2012, 51, 2654–2657 CrossRef CAS;
(c) G. E. Schneider, A. Pertegas, E. C. Constable, C. E. Housecroft, N. Hostettler, C. D. Morris, J. A. Zampese, H. J. Bolink, J. M. Junquera-Hernandez, E. Orti and M. Sessolo, J. Mater. Chem. C, 2014, 2, 7047–7055 RSC;
(d) A. Bonfiglio, P. Hsiao, Y. Chen, C. Gourlaouen, Q. Marchand, V. César, S. B. Laponnaz, Y. Wang, C. Lu, C. Daniel, F. Polo, H. Su and M. Mauro, Chem. Mater., 2022, 34(4), 1756–1769 CrossRef CAS.
-
(a) J. Li, P. I. Djurovich, B. D. Alleyne, M. Yousufuddin, N. N. Ho, J. C. Thomas, J. C. Peters, R. Bau and M. E. Thompson, Inorg. Chem., 2005, 44, 1713–1727 CrossRef CAS;
(b) P.-T. Chou and Y. Chi, Chem. – Eur. J., 2007, 13, 380–395 CrossRef CAS PubMed;
(c) A. R. Naziruddin, A. Galstyan, A. Iordache, C. G. Daniliuc, C. A. Strassert and L. De Cola, Dalton Trans., 2015, 44, 8467–8477 RSC.
- S. Leuthaußer, D. Schwarz and H. Plenio, Chem. – Eur. J., 2007, 13, 7195–7203 CrossRef PubMed.
- B. Liu, M. A. Jabed, J. Guo, W. Xu, S. L. Brown, A. Ugrinov, E. K. Hobbie, S. Kilina, A. Qin and W. Sun, Inorg. Chem., 2019, 58, 14377–14388 CrossRef CAS.
- V. W. W. Yam, K. M. C. Wong, L. L. Hung and N. Zhu, Angew. Chem., Int. Ed., 2005, 44, 3107–3110 CrossRef CAS PubMed.
- S. D. Adhikary, T. Samanta, G. Roymahapatra, F. Loiseau, D. Jouvenot, S. Giri, P. K. Chattaraj and J. Dinda, New J. Chem., 2010, 34, 1974–1980 RSC.
- V. K.-M. Au, K. M.-C. Wong, D. P.-K. Tsang, M.-Y. Chan, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2010, 132, 14273–14278 CrossRef CAS PubMed.
- V. K. M. Au, W. H. Lam, W. T. Wong and V. W. W. Yam, Inorg. Chem., 2012, 51, 7537–7545 CrossRef CAS.
- B. K. Rana, S. Mishra, D. Sarkar, T. K. Mondal, S. K. Seth, V. Bertolasi, K. Das Saha, C. W. Bielawski, A. A. Isab and J. Dinda, New J. Chem., 2018, 42, 10704–10711 RSC.
- S. D. Adhikary, D. Bose, P. Mitra, K. D. Saha, V. Bertolasi and J. Dinda, New J. Chem., 2012, 36, 759–767 RSC.
-
(a) A. Y.-Y. Tam and V. W.-W. Yam, Chem. Soc. Rev., 2013, 42, 1540–1567 RSC;
(b) D. A. Rosca, J. Fernandez-Cestau, J. Morris, J. A. Wright and M. Bochmann, Sci. Adv., 2015, 1, e1500761 CrossRef PubMed;
(c) H. A. Wegner and M. Auzias, Angew. Chem., Int. Ed., 2011, 50, 8236–8247 CrossRef CAS ; Angew. Chem. 2011, 123, 8386-8397;
(d) M. Joost, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 15022–15045 CrossRef CAS ; Angew. Chem. 2015, 127, 15234–15258.
-
(a) A. A. D. Tulloch, A. A. Danopoulos, R. P. Tooze, S. M. Cafferkey, S. Kleinhenz and M. B. Hursthouse, Chem. Commun., 2000, 1247–1248 RSC;
(b) F. E. Hahn, M. C. Jahnke, V. Gomez-Benitez, D. Morales-Morales and T. Pape, Organometallics, 2005, 24, 6458–6463 CrossRef CAS;
(c) F. E. Hahn, C. Holtgrewe, T. Pape, M. Martin, E. Sola and L. A. Oro, Organometallics, 2005, 24, 2203–2209 CrossRef CAS;
(d) F. E. Hahn, M. C. Jahnke and T. Pape, Organometallics, 2007, 26, 150–154 CrossRef CAS;
(e) J. T. Hutt and Z. D. Aron, Org. Lett., 2011, 13(19), 5256–5259 CrossRef CAS.
-
(a) T. Samanta, B. K. Rana, G. Roymahapatra, S. Giri, P. Mitra, R. Pallepogu, P. K. Chattaraj and J. Dinda, Inorg. Chim. Acta, 2011, 375, 271–279 CrossRef CAS;
(b) T. Samanta, R. N. Munda, G. Roymahapatra, A. Nandy, K. D. Saha, S. S. Al-Deyab and J. Dinda, J. Organomet. Chem., 2015, 791, 183–191 CrossRef CAS.
- H. M. J. Wang and I. J. B. Lin, Organometallics, 1998, 17, 972–975 CrossRef CAS.
- Z. Lu, S. A. Cramer and D. M. Jenkins, Chem. Sci., 2012, 3, 3081–3087 RSC.
- H. V. Huynh, S. Guo and W. Wu, Organometallics, 2013, 32, 4591–4600 CrossRef CAS.
- S. Gaillard, A. M. Z. Slawin, A. T. Bonura, E. D. Stevens and S. P. Nolan, Organometallics, 2010, 29, 394–402 CrossRef CAS.
- J. Dinda, T. Samanta, A. Nandy, K. Das Saha, S. K. Seth, S. K. Chattopadhyay and C. W. Bielawski, New J. Chem., 2014, 38, 1218–1224 RSC.
-
(a) J. F. Britten, C. J. L. Lock and Z. Wang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 1600–1603 CrossRef;
(b) K. M. Lee, C. K. Lee and I. J. B. Lin, Angew. Chem., Int. Ed. Engl., 1997, 36, 1850–1852 CrossRef CAS;
(c) G. J. Kruger, P. J. Olivier, L. Lindeque and H. G. Raubenheimer, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 1814–1816 CrossRef;
(d) B. Bovio, A. Burini and B. R. Pietroni, J. Organomet. Chem., 1993, 452, 287–291 CrossRef CAS;
(e) X. Hu, I. Castro-Rodriguez, K. Olsen and K. Mayer, Organometallics, 2004, 23, 755–764 CrossRef CAS.
- M. A. Bakar, M. Sugiuchi, M. Iwasaki, Y. Shichibu and K. Konishi, Nat. Commun., 2017, 8, 576 CrossRef PubMed.
-
(a) L. Brammer, Dalton Trans., 2003, 3145–3157 RSC;
(b) L. Brammer, J. M. Charnock, P. L. Goggin, R. J. Goodfellow, T. F. Koetzle and A. G. Orpen, J. Chem. Soc., Chem. Commun., 1987, 443–445 RSC.
-
(a) J. M. Casas, L. R. Falvello, J. Forniés, A. Martín and A. J. Welch, Inorg. Chem., 1996, 35, 6009–6014 CrossRef CAS;
(b) S. Chatterjee, J. A. Krause, A. G. Oliver and W. B. Connick, Inorg. Chem., 2010, 49, 9798–9808 CrossRef CAS PubMed;
(c) S. Rizzato, J. Bergès, S. A. Mason, A. Albinati and J. Kozelka, Angew. Chem., Int. Ed., 2010, 49, 7440–7443 CrossRef CAS;
(d) M. Baya, Ú. Belío and A. Martín, Inorg. Chem., 2014, 53, 189–200 CrossRef CAS.
-
(a) M. Straka, E. Andris, J. Vícha, A. Růžička, J. Roithová and L. Rulíšek, Angew. Chem., Int. Ed., 2019, 58, 2011–2016 CrossRef CAS;
(b) L. Estévez, Dalton Trans., 2020, 49, 4797–4804 RSC;
(c) M. Vaddamanu, A. Sathyanarayana, Y. Masaya, S. Sugiyama, O. Kazuhisa, K. Velappan, K. Subramaniyam, K. Hisano, O. Tsutsumi and G. Prabusankar, Organometallics, 2020, 39, 2202–2206 CrossRef CAS.
- V. Rani, H. B. Singh and R. J. Butcher, Organometallics, 2017, 36, 4741–4752 CrossRef CAS.
- D. Pathak, S. Deuri and P. Phukan, J. Phys. Chem. A, 2016, 120, 128–138 CrossRef CAS PubMed.
- F. Cui, P. Yang, X. Huang, X. J. Yang and B. Wu, Organometallics, 2012, 31, 3512–3518 CrossRef CAS.
- A. H. Mageed, B. W. Skeltona and M. V. Baker, Dalton Trans., 2017, 46, 7844–7856 RSC.
- Y. Unger, A. Zeller, S. Ahrensy and T. Strassner, Chem. Commun., 2008, 3263–3265 RSC.
-
(a) J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991–1024 CrossRef;
(b) W. H. Melhuish, J. Phys. Chem., 1961, 65, 229–235 CrossRef CAS.
-
(a) M. Pazicky, A. Loos, M. J. Ferreira, D. Serra, N. Vinokurov, F. Rominger, C. Jakel, A. S. K. Hashmi and M. Limbach, Organometallics, 2010, 29, 4448 CrossRef CAS;
(b) H. V. Huynh, S. Guo and W. Wu, Organometallics, 2013, 32, 4591–4600 CrossRef CAS;
(c) C. Tubaro, M. Baron, M. Costante, M. Basato, A. Biffis, A. Gennaro, A. A. Isse, C. Graiff and G. Accorsi, Dalton Trans., 2013, 42, 10952–10963 RSC.
- P. Kuhlkamp, H. G. Raubenheimer, J. S. Field and M. Desmet, J. Organomet. Chem., 1998, 552, 69–74 CrossRef CAS.
- A. H. Mageed, B. W. Skelton and M. V. Baker, Dalton Trans., 2017, 46, 7844 RSC.
-
(a) J. Dinda, S. D. Adhikary, S. K. Seth and A. Mahapatra, New J. Chem., 2013, 37, 431 RSC;
(b) T. Samanta, R. N. Munda, G. Roymahapatra, A. Nandy, K. D. Saha, S. S. Al-Deyab and J. Dinda, J. Organomet. Chem., 2015, 791, 183–191 CrossRef CAS;
(c) J. Dinda, T. Samanta, A. Nandy, K. D. Saha, S. K. Seth, S. K. Chattopadhyay and C. W. Bielawski, New J. Chem., 2014, 38, 1218–1224 RSC.
-
(a) S.-W. Lai, M. C.-W. Chan, K.-K. Cheung and C.-M. Che, Organometallics, 1999, 18, 3327–3336 CrossRef CAS;
(b) S.-W. Lai, H.-W. Lam, W. Lu, K.-K. Cheung and C.-M. Che, Organometallics, 2002, 21, 226–234 CrossRef CAS.
- S. D. Adhikary, T. Samanta, G. Roymahapatra, F. Loiseau, D. Jouvenot, S. Giri, P. K. Chattaraj and J. Dinda, New J. Chem., 2010, 34, 1974–1980 RSC.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., 2015, A71, 3 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
-
(a)
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, Jr., J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2013 Search PubMed;
(b) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
-
(a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC;
(b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
-
(a) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS;
(b) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- A. Jana, A. Aher, P. Brandao, S. Sharda, P. Bera, U. Phadikar, S. K. Manna, A. K. Mahapatra and P. Bera, J. Mol. Struct., 2022, 1256, 132479 CrossRef CAS.
- J. M. Brenchley and D. C. Douek, Method Cell Biol., 2004, 75, 481–496 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. additional crystallographic data, calculated energies, compositions of selected molecular orbitals, calculated vertical electronic excitations, and spectral data (NMR and UV-vis). CCDC 2183850 and 2183851 for 2 and 3 and contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2nj03661d |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 d and
Joydev
Dinda
d and
Joydev
Dinda