
How reduction temperature affects the properties of carbon nanotubes growing over the Fe–MgO/Al2O3 catalyst
Chao
Yan
 ,
Yinrui
Yu
,
Tianle
Zhang
,
Yan
Lin
*,
Fei
Wang
and
Yan
He
*
,
Yinrui
Yu
,
Tianle
Zhang
,
Yan
Lin
*,
Fei
Wang
and
Yan
He
*
College of Mechanical and Electrical Engineering, Qingdao University of Science and Technology, Qingdao, Shandong Province 266061, China. E-mail: heyan@qust.edu.cn
First published on 23rd November 2022
Abstract
Fe2O3–MgO/Al2O3 catalyst precursors were prepared by a co-precipitation method for the growth of carbon nanotubes (CNTs), and the materials were characterized by various means. The effects of reduction temperature on the surface morphology, texture properties and reduction degree of the catalysts were systematically discussed, and the effects of the catalysts on the diameter uniformity, defect degree and carbon yield of carbon nanotubes were further investigated. The results showed that the reduction temperature strongly affected the reduction degree and morphology of Fe-containing species. As the reduction temperature increased from 560 °C to 760 °C, the reduction products obtained were (Fe2O3, Fe3O4), (Fe3O4, FexO), (FexO, Fe), and (FexO, Fe), and the specific surface area tended to decrease while the pore size gradually increased, which would have a great influence on further reduction of the catalyst after C3H6 was introduced. The results showed that the best catalyst performance was achieved when the reduction temperature was 660 °C, and carbon nanotubes with a carbon yield of 1800%, a graphitization degree of 0.98, a diameter distribution in the range of 6–20 nm, a specific surface area of 260.55 m2 g−1 and a resistivity of 52.6 mΩ cm could be obtained, which was because at this temperature, the composition and structure of the active material in the catalyst were favorable for further diffusion of carbon.
1. Introduction
Carbon nanotubes are a new material with excellent properties (high thermal conductivity, high elastic modulus and high current density), and have broad application prospects and great commercial value in catalyst support, adsorption, composite materials and other fields.1,2 After more than two decades of development, the more developed preparation methods mainly include arc discharge, laser ablation and chemical vapor deposition. Among these preparation methods, chemical vapor deposition is considered to be the most promising preparation method due to its advantages of lower synthesis temperature, a simple operation method, and high yield.3,4 In a typical CVD reactor, the carbon source gas decomposes continuously on the catalyst surface under the protection of inert gas. When carbon solubility reaches saturation, it precipitates and assembles into carbon nanotubes. Similar to other methods, the CVD method also uses transition metals as catalysts, and many studies have shown that both the active material and support in the catalyst can affect the performance of the catalyst. Carbon has high solubility and a diffusion rate in Fe at high temperatures, and has unparalleled superiority over other active materials. The influence of a support on the catalyst cannot be ignored; Al2O3 shows strong metal–support interaction and has a large specific surface area,5,6 and MgO can reduce the generation of amorphous carbon and is easier to remove by acid washing.7,8 Mixing these two supports in a certain ratio can produce a synergistic effect between both of the supports, enhancing the catalyst activity.Calcination and reduction are the key steps of catalyst activation. The importance of the catalyst reduction process for the synthesis of carbon nanotubes has been well recognized, and many papers have focused on the parameters of the reduction process. Amama et al. investigated the effect of H2O on the reduction process of catalysts and found that the average diameter of nanoparticles obtained by annealing in H2/H2O was smaller compared to that of the samples annealed in H2, and suggested that the effective lifetime of catalysts was prolonged because water vapor hindered the Ostwald ripening of catalysts.9 Kim et al. analyzed the effect of different reduction durations on the catalyst by in situ TEM and investigated the relationship between the termination of carbon nanotube array growth and the evolution of the catalyst morphology, demonstrating that Ostwald ripening and Fe atom subsurface diffusion were the causes of growth termination.10 Sakurai et al. adjusted the effect of Ostwald ripening on the diameter and density of single-walled carbon nanotubes by decoupling the catalyst formation and growth processes and found that the single-walled carbon nanotube density was inversely and positively related to the catalyst reduction temperature and total H2 flow, respectively.11 All the mentioned literature studies discussed the effect of Ostwald ripening on the catalyst during the reduction process from the perspective of catalyst morphology evolution. However, different reduction conditions, especially different reduction temperatures, lead to significant differences in the reduction rate, which strongly affect the form in which the active substances exist, the pore structure and surface properties, which in turn determine the catalyst activity,12 and the effect of this on the growth of carbon nanotubes still needs to be determined.
Generally speaking, Fe2O3 on undergoing H2 reduction does not directly turn into metal Fe. In the iron making industry, if the reduction temperature is higher than 570 °C, the reduction process is Fe2O3 → Fe3O4 → Fe(1−x)O → Fe, and if the reduction is carried out at a temperature lower than 570 °C, the reduction process of Fe2O3 is Fe2O3 → Fe3O4 → Fe, and the intermediate oxide Fe(1−x)O cannot exist stably below 570 °C.13
Reduction activation is the final step in catalyst preparation and its influence on catalyst performance cannot be ignored. In this work, the catalyst precursors prepared by co-precipitation were reduced at different temperatures to investigate the effects of reduction temperature on the microscopic morphology and pore structure of the catalysts and the relationship between these factors and the catalytic performance was determined from the perspective of the Fe form which was present, and the effects of catalysts reduced at different temperatures on the carbon yield, diameter distribution, resistivity and defect degree of multi-walled carbon nanotubes synthesized by the CVD method were further investigated. Finally, the whole reduction process undergone by the catalysts at different reduction temperatures was discussed in detail.
2. Experimental
2.1. Catalyst preparation
Fe2O3–MgO/Al2O3 was prepared as catalyst precursors by a co-precipitation method. A certain amount of Fe(NO3)3·9H2O, Mg(NO3)2·6H2O and Al(NO3)3·9H2O was dissolved in deionized water to form an acid solution and the molar ratio of Mg, Fe and Al was ensured to be 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.8:0.2, and then a certain amount of (NH4)2CO3 and NaOH was dissolved in deionized water to form a basic solution. The acid solution was transferred to a magnetic stirrer and the basic solution was added drop by drop with a rubber tipped dropper until the pH reached 9.5. Then the suspension was aged at 60 °C for 10 h, and the whole process should be carried out under constant agitation. After aging, the sediment was filtered until the filtrate was neutral, and the filter cake was dried in a drying oven at 70 °C for 10 h. Then it was calcined in a muffle furnace at 500 °C for 2 h. The calcined solids were ground and sieved with a 120-mesh sieve. The powder was placed in a fixed-bed reactor with 200 mL min−1 nitrogen as the protective gas and reduced by passing 150 mL min−1 hydrogen gas for 10 min at 560 °C, 660 °C, 690 °C and 760 °C and the obtained catalysts were labeled RT-X (X = 560, 660, 690, 760), respectively.
:0.8:0.2, and then a certain amount of (NH4)2CO3 and NaOH was dissolved in deionized water to form a basic solution. The acid solution was transferred to a magnetic stirrer and the basic solution was added drop by drop with a rubber tipped dropper until the pH reached 9.5. Then the suspension was aged at 60 °C for 10 h, and the whole process should be carried out under constant agitation. After aging, the sediment was filtered until the filtrate was neutral, and the filter cake was dried in a drying oven at 70 °C for 10 h. Then it was calcined in a muffle furnace at 500 °C for 2 h. The calcined solids were ground and sieved with a 120-mesh sieve. The powder was placed in a fixed-bed reactor with 200 mL min−1 nitrogen as the protective gas and reduced by passing 150 mL min−1 hydrogen gas for 10 min at 560 °C, 660 °C, 690 °C and 760 °C and the obtained catalysts were labeled RT-X (X = 560, 660, 690, 760), respectively.
2.2. Growth of CNTs
The carbon nanotubes were prepared by a chemical vapor deposition method. In a fixed-bed reactor, 0.1 g of catalyst powder was added to a quartz boat before the reaction, and the reactor temperature was raised to 660 °C in a N2 atmosphere with a heating rate of 10 °C min−1; then C3H6 was introduced and the temperature was kept at 660 °C for 45 min, and C3H6 was turned off after the reaction, and N2 was turned off when the reaction mixture cooled to room temperature. The black powder obtained on the quartz boat was the prepared carbon nanotubes. The catalytic ratio, namely, carbon yield, was defined as:M1 is the total mass of carbon nanotubes and RT-X, M2 is the initial mass of RT-X.

2.3. Characterization
Powder X-ray diffraction (XRD) data were collected from a MiniFlex 600-C X diffractometer, and the samples were step-scanned in steps of 0.01° 2θ using a count time of 10° min−1.Scanning electron microscopy (SEM) and energy dispersive spectrometry (EDS) observations were recorded using a Hitachi SU-8000 apparatus with an applied voltage of 10 kV.
Low-temperature N2 adsorption–desorption isotherms of the samples were obtained on a BSD-3H-2000PS4 sorptometer apparatus. Before the test, the samples were degassed at room temperature for 4 h, and then isothermal adsorption and desorption were carried out at liquid nitrogen temperature. The total specific surface areas were evaluated by the multipoint Brunauer–Emmett–Teller (BET) method; the size distribution and average pore diameter were determined by the Barrett–Joyner–Halenda (BJH) method.
X-ray photoelectron spectroscopy (XPS) was performed on a Thermo Scientific K-Alpha X-ray photoelectron spectrometer at a base pressure of 5.0 × 10−7 mBar using Al Kα X-ray (1486.6 eV) as the excitation source. The binding energy calibration of all spectra was referenced to the C1s signal at 284.8 eV.
Raman spectra were recorded on a confocal Raman spectrometer (Renishaw inVia) at room temperature using a 532 nm Ar+ laser as an excitation source.
Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) observations were carried out on a JEM-2100PLUS electron microscope operating at 200 kV.
An RTS-8 four-probe resistivity meter was used to measure the resistivity of CNTs. Before the test, a certain amount of CNTs was pressed into thin discs with a mold.
3. Results and discussion
3.1. Characterization of catalysts
The surface morphology of the catalysts after reduction was observed using a scanning electron microscope, and the images are shown in Fig. 1. The morphology of the catalysts was also significantly different at different reduction temperatures; RT-560 and RT-660 (Fig. 1a and b) catalysts could still maintain the lamellar structure and the surface was regular and dense; however, as the reduction temperature increased to 690 °C (Fig. 1c), the lamellar structure was destroyed. It can be seen that the smaller lamellar material after fragmentation was scattered in the field of vision, and the surface particle agglomeration increased significantly. When the reduction temperature reached 760 °C, the lamellar structure basically disappeared, and the catalyst sintered and agglomerated into large particles. It can be determined that the specific surface area would decrease sharply at this time. In the growth process of carbon nanotubes, it is crucial for the carbon source gas to transfer and diffuse inside the porous catalyst, and a larger specific surface area can provide more active sites and a larger pore volume is beneficial for the diffusion of gas to the interior, both of which are beneficial for the preparation of carbon nanotubes.14Fig. 1e–h shows the elemental mapping of RT-660; as expected, the catalyst consisted of four elements, Mg, O, Al and Fe, and the atomic ratio of Mg, Fe and Al was 0.73:0.21:0.06, which was close to the molar ratio of the three elements.
 | ||
| Fig. 1 SEM images of the catalyst (a) RT-560, (b) RT-660, (c) RT-690, and (d) RT-760. (e–h) Elemental mapping of Mg, O, Al and Fe of RT-660. | ||
N2-Physisorption was performed to characterize the physical properties of the catalysts, and the N2 adsorption–desorption isotherms are shown in Fig. 2. All four isotherms are type IV with H3-type hysteresis loops, illustrating that the catalysts have a mesoporous structure.15,16 At lower pressures, the adsorption volume was very small, while in the range of P/P0 = 0.8–1, the adsorption volume increased significantly. This was due to the capillary condensation of mesopores by the formation of multilayer films at higher relative pressures,17 which also showed that the catalysts had a large mesopore after reduction. As the reduction temperature gradually increased, the closure point moved toward a higher relative pressure direction, indicating an increasing catalyst pore size.18 The BJH pore size distribution is shown in the inset of Fig. 2, and it can be observed that the pore size of RT-560 mainly ranged from 10–30 nm, while pores in the range of 30–100 nm in RT-660, RT-690 and RT-760 also occupied a considerable portion of the pore volume, suggesting that a certain number of macropores appeared in the catalyst. The mesopores can provide a larger specific surface area with more active sites, and macropores are conducive to the diffusion of gas and the deposition of carbon atoms, which are both beneficial for the reaction. The specific surface area, pore volume, and average pore size of the samples are summarized in Table 1. Obviously, with an increase in reduction temperature, the specific surface area and pore volume of catalyst generally decrease while the average pore size increases. As can be seen from Table 1, the specific surface areas calculated by the BET method were 89.69 m2 g−1, 87.71 m2 g−1, 72.03 m2 g−1, and 49.95 m2 g−1, respectively, with increasing reduction temperature, indicating a gradual decrease in the N2 adsorption capacity of the catalyst, which is consistent with the state of the particles in the SEM micrographs discussed earlier. When the reduction temperature was lower than 660 °C, the effect of increasing the reduction temperature on the specific surface area was almost negligible. However, when the reduction temperature was higher than 660 °C, the specific surface area of the catalyst decreased greatly if the reduction temperature continued to increase. This may be due to the sintering of the iron phase at high temperature, which blocked the pore structure of the support.19,20 Compared with RT-690 and RT-760, RT-660 had a larger specific surface area and pore volume, which was consistent with higher catalytic activity. In addition, although the specific surface areas of RT-560 and RT-660 were similar, their carbon yields were significantly different, which also indicated that the specific surface area was not the decisive factor affecting the catalytic activity in this catalytic system, and the products obtained at this stage of reductive activation had a greater influence on the catalytic performance (Table 2).
 | ||
| Fig. 2 N2 adsorption–desorption isotherms of catalysts (a) RT-560, (b) RT-660, (c) RT-690, and (d) RT-760. The inset shows the pore size distributions. | ||
| Sample | RT-560 | RT-660 | RT-690 | RT-760 |
|---|---|---|---|---|
| Surface area (m2 g−1) | 89.69 | 87.71 | 72.03 | 49.95 |
| Pore volume (cm3 g−1) | 0.43 | 0.41 | 0.39 | 0.30 |
| Average pore diameter (nm) | 12.35 | 12.88 | 13.04 | 15.32 |
| Yield of carbon (%) | 1330 | 1800 | 1670 | 530 |
| Sample | MgO | RT-560 | RT-660 | Mg1−xFexO |
|---|---|---|---|---|
| Crystallite size in a/b/c direction (nm) | 0.42114 | 0.421135 | 0.423542 | 0.42646 |
Fig. 3 shows the XRD patterns of the catalysts treated at different reduction temperatures. It can be seen from the figure that the phase of the catalyst obtained was different when the catalyst was reduced at different temperatures. The reduction temperature changes the form in which the active metal Fe species exists, thus affecting the performance of the catalyst. The three diffraction peaks near 36.7°, 42.7°, 62.1°, 74.4° and 78.4° observed in the RT-560 catalyst can be ascribed to (111), (200), (220), (311) and (222) crystal planes representing MgO (JCPDS no: 75-0447), and the catalyst powder obtained at this time is black in color and can be considered to have a small amount of Fe3O4. Due to the small size and high dispersion of Fe3O4 nanoparticles, the diffraction peaks of Fe3O4 nanoparticles were not observed in the XRD pattern. With the increase in reduction temperature from 560 °C to 660 °C, the reduced species of iron reduction products gradually appeared, and a characteristic diffraction peak (JCPDS no: 82-1533) of the (311) crystal plane of Fe3O4 appeared near 35.7°, implying that a large amount of Fe3O4 had appeared at this time. At the same time, the intensity of the (200) crystal plane diffraction peak enhanced, which may be attributed to the reduction of some FexO.21 Due to the similar structure of FexO and MgO, which both have a face-centered cubic structure and similar lattice parameters, it is also easy to form a solid solution.22,23 Most FexO peaks overlapped with MgO peaks, so it is difficult to distinguish the diffraction peaks of FexO, MgO and Mg1−xFexO.24 The strongest diffraction peak of FexO was the diffraction peak corresponding to the crystal plane of FexO (200) near 42.7°, but it overlapped with the diffraction peak of the MgO support. In order to verify this conjecture, the catalyst crystal parameters were calculated and the results are shown in Table 1. Obviously, the crystal parameters of RT-560 are close to those of MgO, while the crystal parameters of RT-660 are between those of RT-560 and Mg1−xFexO, which can be ascribed to the reduction of FexO. For RT-560 and RT-660 samples (Fig. 3a and b), the diffraction peak of metal Fe was not visible. Continuing to increase the reduction temperature to 690 °C, the characteristic diffraction peak of the Fe (110) crystal plane began to appear (JCPDS no: 85-1410), implying that a considerable amount of Fe2+ was reduced and migrated out of Mg(Fe, Al)O and that Fe metal particles were still being generated. When the reduction temperature increased to 760 °C, the characteristic diffraction peak of the (110) crystal plane of Fe became stronger, and the (200) and (211) crystal planes of metal Fe appeared, suggesting that more crystalline Fe can be formed under high temperature reduction.
 | ||
| Fig. 3 XRD patterns of the catalysts (a) RT-560, (b) RT-660, (c) RT-690, and (d) RT-760. | ||
Fig. 4 shows the high-resolution XPS spectrum of Fe 2p of the catalyst. As shown in Fig. 4a, the Fe 2p3/2 and Fe 2p1/2 characteristic peaks appeared near 710.9 eV and 724.4 eV, respectively,25 which are typical peak positions of Fe2O3. As the reduction temperature was raised to 660 °C (Fig. 4b), the Fe 2p3/2 and Fe 2p1/2 peaks became wider and tended to move toward lower binding energy, corresponding to the formation of Fe3O4, meaning that Fe2O3 was first reduced to Fe3O4.26 For the RT-690 catalyst, (Fig. 4c), a weak peak was observed at 708.9 eV, which may be related to Fe2+, showing a further reduction of Fe3O4 to Fe2+.27 When the reduction temperature was increased to 760 °C (Fig. 4d), no obvious diffraction peak of Fe0 was found near 706.8 eV,28 but Fig. 1 shows a more intense diffraction peak of Fe, suggesting that the reduction process of FeO to Fe was in line with the nuclear growth model of oxygen outward migration, rather than hydrogen inward diffusion. As nano-Fe is easily oxidized,29 an oxide layer of 2–3 nm consisting of γ-Fe2O3 and Fe3O4 is present on the outer surface of the catalyst during the test,30,31 which explains why Fe3+ has also been detected in the XPS spectra of both RT-690 and RT-760 catalysts. And due to a low percentage of this oxide layer, the corresponding diffraction peaks did not appear in the XRD pattern.
 | ||
| Fig. 4 High-resolution XPS spectra of Fe 2p in catalysts (a) RT-560, (b) RT-660, (c) RT-690, and (d) RT-760. | ||
The possible pathways that the iron oxides in the catalyst may undergo during the H2 reduction are shown in eqn (1)–(6). The change in thermodynamic parameter ΔG with reaction temperature is calculated by using the reaction equation module in HSC Chemistry 6.0. The calculated results are shown in Fig. 5
| 3Fe2O3 + H2(g) = 2Fe3O4 + H2O | (1) |
| Fe2O3 + H2(g) = 2FeO + H2O | (2) |
| Fe2O3 + 3H2(g) = 2Fe + 3H2O | (3) |
| Fe3O4 + 4H2(g) = 3Fe + 4H2O | (4) |
| Fe3O4 + H2(g) = 3FeO + H2O | (5) |
| FeO + H2(g) = Fe + H2O | (6) |
 | ||
| Fig. 5 Gibbs free energy–temperature relation diagram in the reduction process. | ||
Fig. 5 shows the ΔG–T diagram of the six reactions. It can be seen from the figure that in the range of 0–1000 °C, only the ΔG value of eqn (1) was less than 0, suggesting that eqn (1) can proceed spontaneously in the standard state, or at least it is thermodynamically allowed. In other words, the reduction reaction from Fe2O3 to Fe3O4 can easily proceed when the kinetic conditions are also allowed, which corresponded to the diffraction peak of Fe3O4 observed in the XRD pattern at a reduction temperature of 660 °C. The ΔG value of eqn (2)–(6) are always higher than 0 in the range of 0–1000 °C, indicating that the reduction activation of catalysts cannot be carried out under standard conditions. But under experimental conditions, the reduction activation of catalysts was carried out under non-standard conditions. Suppose that P(H2O′) and  were the experimental pressures of water vapor and hydrogen, then
were the experimental pressures of water vapor and hydrogen, then  , and P(H2O) and P(H2) were the standard pressures of water vapor and hydrogen, then
, and P(H2O) and P(H2) were the standard pressures of water vapor and hydrogen, then  . For eqn (2)–(6) to proceed to the right side of the equation, the following thermodynamic conditions must be reached ΔG = ΔG0 + RTlnK′ = RT(lnK′ − lnK) < 0, that is,
. For eqn (2)–(6) to proceed to the right side of the equation, the following thermodynamic conditions must be reached ΔG = ΔG0 + RTlnK′ = RT(lnK′ − lnK) < 0, that is,  . In the experiment, the high purity hydrogen produced through reduction activation contains almost no water, and the water vapor produced in the reaction in the tubular furnace is also discharged, so the above equation is valid, and reactions (2)–(6) can also be carried out.
. In the experiment, the high purity hydrogen produced through reduction activation contains almost no water, and the water vapor produced in the reaction in the tubular furnace is also discharged, so the above equation is valid, and reactions (2)–(6) can also be carried out.
According to Fig. 5, the Gibbs free energy of reaction (1) is always less than zero under standard conditions, and there is a great driving force for the reaction to proceed in the forward direction, so the reaction easily proceeds after the dynamic conditions are reached. In contrast, the Gibbs free energies of reactions (2)–(6) are all positive under the standard conditions, and the reaction can proceed only by increasing the partial pressure of hydrogen. Therefore, reactions (2)–(6) are the main segments limiting the reduction reaction, so the reduction reaction is closely related to the reduction temperature and gas pressure.
3.2. Characterization of CNTs
The XRD patterns of the carbon products were recorded to determine the crystal phases. Although the XRD pattern was not suitable to distinguish the microstructure of carbon nanotubes and graphite,32 it provided the main evidence of graphite deposition because the diffraction peaks of carbon nanotubes and graphite were close.33 Different RT-X (X = 560, 660, 690, and 760) compounds were used as catalysts in the CVD process for the preparation of carbon nanotubes. Fig. 6 shows their XRD patterns with an intense and a weaker characteristic peak in each diffraction line, and the characteristic peaks appearing around 2θ = 26°, 42.8° correspond to the (002) and (100) crystal planes of graphite (JCPDS no: 41-1487), respectively, indicating that carbon nanotubes were formed from crimped multilayered graphite. The intensity of the (002) diffraction peak is related to the degree of graphitization of the synthesized product, and this peak appears usually due to the presence of multi-walled carbon tubes.34 With the increase in reduction temperature from 560 °C to 660 °C, the diffraction peak of the (002) crystal plane became stronger, revealing that the crystallinity of carbon nanotubes became higher, and as the reduction temperature continued to rise to 760 °C, the (002) diffraction peak became weaker instead; it was concluded that the catalyst performance deteriorates and the crystallinity of carbon nanotubes decreases significantly after the reduction temperature exceeds 660 °C. Therefore, the carbon nanotubes prepared with RT-660 as the catalyst had the highest crystallinity. | ||
| Fig. 6 XRD patterns of carbon nanotubes (a) CNTRT-560, (b) CNTRT-660, (c) CNTRT-690, and (d) CNTRT-760. | ||
Scanning electron microscopy was used to observe the microscopic morphology of carbon nanotubes, and Fig. 7 shows the SEM image of the synthesized carbon nanotube samples. SEM images shows a large number of dense, randomly distributed carbon nanotubes, whose morphology varies with catalyst parameters, and no visible amorphous carbon or catalytic metal particles were detected in the samples. Obviously, a small number of coarser carbon nanotubes with very non-uniform tube diameters appear in Fig. 7a and d, showing poor catalytic performance of the catalysts. The reason may be that the activity of the main components of the catalyst is low or the active substances are agglomerated, which leads to an increase in the diameter of carbon nanotubes. As shown in Fig. 7b and c, the diameter of the carbon nanotubes was uniform, and the outer surface was smooth, suggesting that the catalyst performance was fine at this time. In summary, it can be seen that the reduction temperature of the catalyst affects the surface morphology of carbon nanotubes. Therefore, high quality carbon nanotubes can only be obtained when RT-X is used as the catalyst at an appropriate reduction temperature.
 | ||
| Fig. 7 SEM images of carbon nanotubes (a) CNTRT-560, (b) CNTRT-660, (c) CNTRT-690, and (d) CNTRT-760. | ||
In order to obtain more details about the microstructure of the carbon nanotubes, transmission electron microscopy (TEM) and high resolution transmission electron microscopy (HRTEM), are employed to observe the morphologies of these samples. As shown in Fig. 8a–d, the prepared carbon nanotubes have smooth surfaces and a uniform diameter distribution. The carbon nanotubes showed an obvious hollow tubular structure, and it was difficult to observe their actual lengths due to their agglomeration and entanglement, but they were at least a few microns in length, and their diameters were about ten nanometers. In Fig. 8d, it could be observed that iron nanoparticles were located in the carbon nanotubes and their diameters were similar to the inner diameters of the carbon nanotubes, which also confirmed that the particle size of the catalyst affects the diameter of carbon nanotubes.35Fig. 9 shows the locally enlarged images of the carbon nanotubes, and the HRTEM image shows that the carbon nanotubes are composed of multilayer parallel lamellar structures with a layer spacing of about 0.35 nm, which is similar to the (002) crystal plane distance of graphite. Fifty carbon nanotubes from HRTEM images were selected for the wall layer count. The average wall layer numbers were 10.3, 10.8, 11.3 and 12.2 as the reduction temperature increased from 560 °C to 760 °C, respectively. Therefore, the wall layer numbers of the prepared carbon nanotubes tended to increase gradually as the reduction temperature increased.
 | ||
| Fig. 8 TEM images of carbon nanotubes (a) CNTRT-560, (b) CNTRT-660, (c) CNTRT-690, and (d) CNTRT-760. | ||
 | ||
| Fig. 9 HRTEM images of carbon nanotubes (a) CNTRT-560, (b) CNTRT-660, (c) CNTRT-690, and (d) CNTRT-760. | ||
In order to predict the effect of RT-X on the diameter change trend of carbon nanotubes, 100 carbon nanotubes were randomly selected from TEM images for measurement and the diameter of carbon nanotubes was calculated and the histogram showing their diameter distribution is shown in Fig. 10. As can be seen from the figure, with the increase in reduction temperature from 560 °C to 760 °C, the diameter distribution range and average diameter of carbon nanotubes also increase. The diameter distribution of the carbon nanotubes grown on RT-560 ranged from 6 to 20 nm, and the average diameter was 11.79 nm, while the diameter distribution range of the carbon nanotubes grown on RT-760 increased to 6–24 nm, and the average diameter increased to 13.30 nm. The increase in carbon nanotube diameter also indicates that the size of active Fe nanoparticles enlarged accordingly.
 | ||
| Fig. 10 Histogram showing the diameter distribution of carbon nanotubes (a) CNTRT-560, (b) CNTRT-660, (c) CNTRT-690, and (d) CNTRT-760. | ||
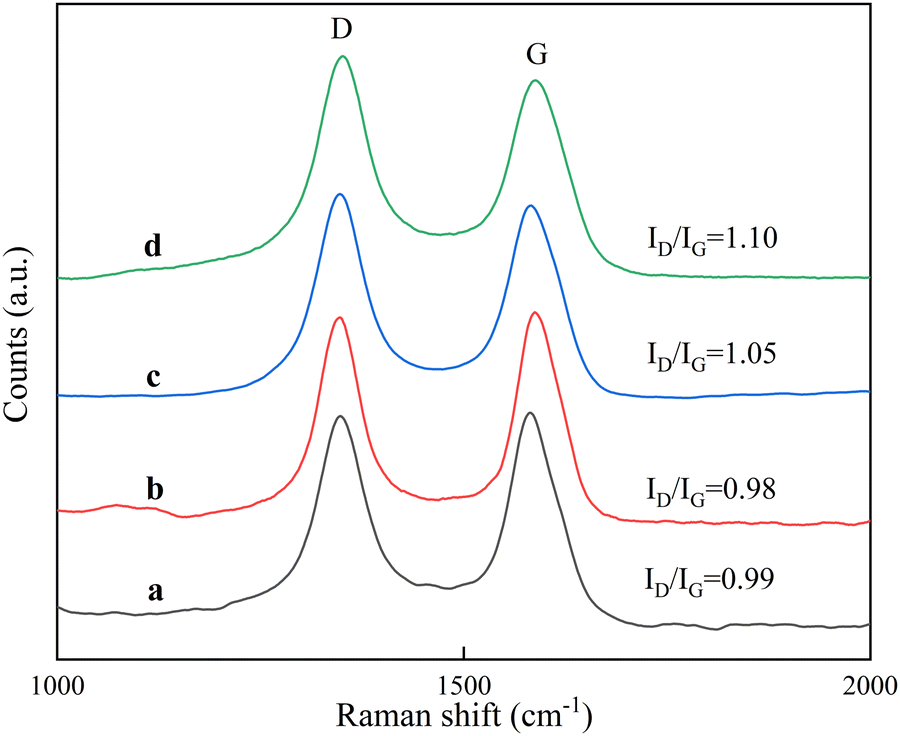
Raman spectrometry can be used to detect the lattice vibration of ordered carbon materials, and to further confirm the quality of the formed carbon material, the carbon material was characterized using Raman spectroscopy, and the results are shown in Fig. 11. A strong peak near 1580 cm−1 (designated as the G-band) is due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C tangential vibration of sp2 hybridized carbon, representing the formation of graphitized CNTs.36 Another strong peak near 1350 cm−1 (designated as the D-band) represents the vibration of other hybrid carbon atoms,37 such as amorphous carbon and lattice defects.38 The D band and G band of all four spectra were similar, and the Raman intensity ratio (ID/IG) of the D band and G band can be used to measure the structural regularity of carbon nanotubes, and ID/IG gives the degree of surface defects and lattice distortion of the graphite layer within the carbon material. It can be seen that the ID/IG ratios of carbon nanotubes grown on RT-560 and RT-660 were 0.99 and 0.98, respectively, showing a high degree of graphitization. When the reduction temperature was higher than 660 °C, the ID/IG ratio of the prepared carbon nanotubes increased rapidly, and the ID/IG ratio of the carbon nanotubes grown on RT-690 and RT-760 reached 1.05 and 1.10, respectively. The results were consistent with SEM and TEM results. The above results indicate that carbon nanotubes have a high number multiwall crystalline structure and the lower reduction temperature is favorable for the synthesis of carbon nanotubes with more ordered arrangement of graphite flakes and fewer defects on the wall surface.
C tangential vibration of sp2 hybridized carbon, representing the formation of graphitized CNTs.36 Another strong peak near 1350 cm−1 (designated as the D-band) represents the vibration of other hybrid carbon atoms,37 such as amorphous carbon and lattice defects.38 The D band and G band of all four spectra were similar, and the Raman intensity ratio (ID/IG) of the D band and G band can be used to measure the structural regularity of carbon nanotubes, and ID/IG gives the degree of surface defects and lattice distortion of the graphite layer within the carbon material. It can be seen that the ID/IG ratios of carbon nanotubes grown on RT-560 and RT-660 were 0.99 and 0.98, respectively, showing a high degree of graphitization. When the reduction temperature was higher than 660 °C, the ID/IG ratio of the prepared carbon nanotubes increased rapidly, and the ID/IG ratio of the carbon nanotubes grown on RT-690 and RT-760 reached 1.05 and 1.10, respectively. The results were consistent with SEM and TEM results. The above results indicate that carbon nanotubes have a high number multiwall crystalline structure and the lower reduction temperature is favorable for the synthesis of carbon nanotubes with more ordered arrangement of graphite flakes and fewer defects on the wall surface.
 | ||
| Fig. 11 Raman patterns of carbon nanotubes (a) CNTRT-560, (b) CNTRT-660, (c) CNTRT-690, and (d) CNTRT-760. | ||
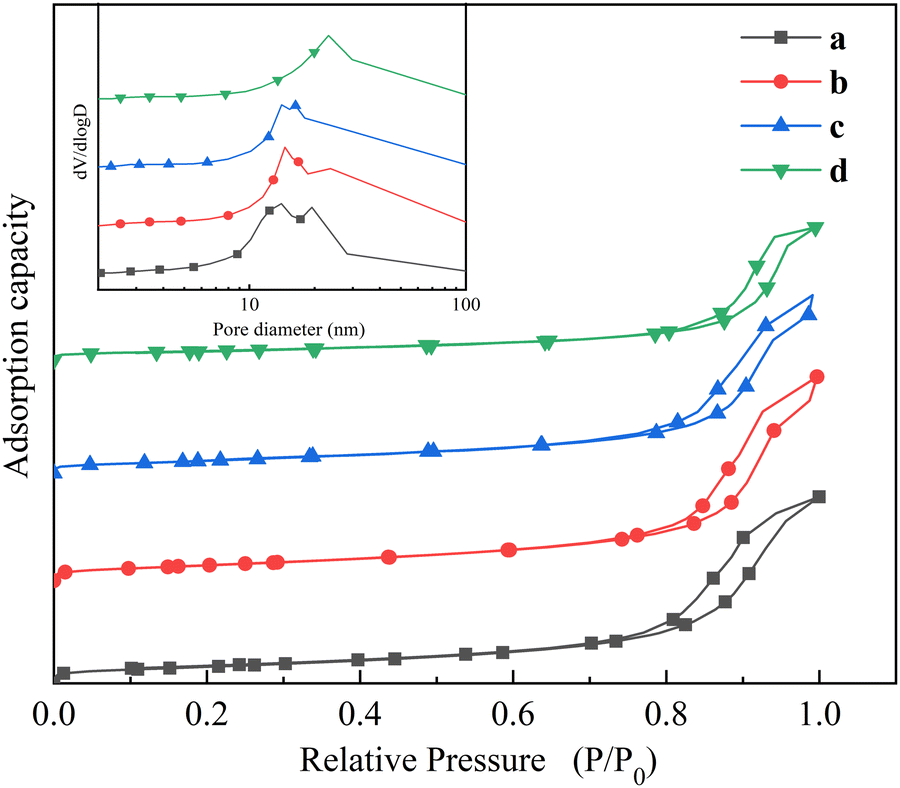
Fig. 12 shows the N2 adsorption–desorption isotherms of CNTRT-X and their pore size distributions. All four isotherms are type IV with H3-type hysteresis loops, illustrating that the carbon nanotubes have a mesoporous structure. According to the image showing the pore size distribution, the pore sizes of the prepared carbon nanotubes were mainly concentrated in the range of 2–4 nm, which might be affected by the roughness of the carbon nanotubes, while the pore sizes larger than 4 nm might be related to the voids between the wound carbon nanotubes.39Table 3 shows the test results of the specific surface area, pore volume, average pore size and resistivity of carbon nanotubes. It can be seen that the specific surface area and pore volume of CNTRT-690 and CNTRT-760 were significantly smaller than those of CNTRT-560 and CNTRT-660, and the reason might be that the defects in CNTRT-690 and CNTRT-760 were larger, and there were more impurities such as amorphous carbon on the surface, which blocked the pore structure. The trend of resistivity of carbon nanotubes was similar to that of the diameter, and both increased with an increase in the reduction temperature. The reason for this phenomenon might be that the thin discs pressed from the smaller diameter carbon nanotubes contain more carbon nanotubes with the same mass and pressure, which could form more conductive networks and therefore have better electrical conductivity and lower resistivity.
 | ||
| Fig. 12 N2 adsorption–desorption isotherms of carbon nanotubes (a) CNTRT-560, (b) CNTRT-660, (c) CNTRT-690, and (d) CNTRT-760. The inset shows the pore size distributions. | ||
| Simple | CNTRT-560 | CNTRT-660 | CNTRT-690 | CNTRT-760 |
|---|---|---|---|---|
| Surface area (m2 g−1) | 265.14 | 260.55 | 237.71 | 229.34 |
| Pore volume (cm3 g−1) | 1.06 | 1.08 | 1.02 | 0.98 |
| Average pore diameter (nm) | 18.85 | 22.97 | 23.76 | 26.87 |
| Electrical resistivity (mΩ cm) | 50.2 | 52.6 | 55.2 | 63.7 |
In summary, under ideal conditions, the influence of reduction temperature on the reduction process of RT-X and the growth of carbon nanotubes can be explained as follows (Fig. 13): according to the Arrhenius formula, the temperature is exponentially related to the reaction rate. At 560 °C, the reduction reaction rate was slow, and after 10 minutes of H2 reduction, only a small amount of Fe3O4 was generated, and a great deal of Fe2O3 still remained unreduced. When C3H6 was introduced for the growth of carbon nanotubes, C3H6 cleaved on the catalyst surface to produce carbon atoms and hydrogen, and at this time, the reduction of Fe2O3 followed the core-shrinking model,40 and the surface layer was gradually reduced to Fe3O4 and FexO. However, due to the poor carburizing ability of Fe3O4, the inward diffusion of carbon atoms was hindered, so the yield of carbon nanotubes was not high. With the increase in reduction temperature to 660 °C, the reaction rate of reactions (1) and (2) was accelerated, and a large amount of Fe3O4 and FexO appeared within 10 min of reduction. The reduction process of Fe3O4 → FexO → Fe followed the nuclear growth model;41 oxygen migrated outward, the Fe3O4 layer became thinner, and the carburizing capacity of FexO and Fe is better than that of Fe3O4,42 so the resistance to carbon diffusion became smaller. At this time, the specific surface area and pore diameter of RT-660 were larger, which was conducive to the adsorption of carbon atoms, internal diffusion of hydrogen and subsequent reduction reaction, so the production and graphitization degree of carbon nanotubes reached the highest. At 690 °C, the reaction rate was further accelerated; combined with Fig. 1, it was not difficult to infer that FexO and Fe were the main components of the active substances after hydrogen reduction for 10 min; when C3H6 was introduced, the reduction reaction shown in eqn (6) was dominant. Although the carbon diffusion rate of FexO was higher than that of Fe3O4, the specific surface area of RT-690 significantly reduced due to the higher reduction temperature, resulting in fewer active sites and a slight decrease in carbon yield. When the reduction temperature was increased to 760 °C, the reaction rate of the reduction process was highly accelerated and more Fe appeared. But due to the high reduction temperature, the specific surface area reduced drastically, the iron nanoparticles were heavily agglomerated, and the active sites were reduced, all of which are unfavorable for the growth of carbon nanotubes.
 | ||
| Fig. 13 Effect of reduction temperature on the RT-X reduction process. | ||
4. Conclusions
The catalyst was prepared by a co-precipitation method and carbon nanotubes were synthesized by a chemical vapor deposition method using C3H6 as the carbon source. It was found that the reduction temperature had a significant effect on the reduction degree of Fe3+, Fe particle size and catalytic performance. The phase composition of the products obtained from the reduction of the catalysts at different temperatures for 10 min also differs, which would have a large effect on the reaction after the introduction of C3H6. With an increase of reduction temperature, the reduction process was accelerated and the particle size of metallic Fe increased gradually. The sample reduced at 660 °C had a larger specific surface area, and the phase structure of the outer layer of FexO and the inner layer of Fe3O4 was also conducive to further reduction and carbon diffusion, so the activity was the highest. However, a higher reduction temperature led to a lower dispersion of Fe and a larger particle size, which had an adverse effect on the catalytic performance. In addition, the analysis results of carbon nanotubes showed that with an increase of RT-X reduction temperature, the wall number and diameter of the carbon nanotubes gradually increased, and the diameter distribution range became wider. When the reduction temperature was 660 °C, the carbon nanotubes with high yield, less defects and a concentrated diameter distribution could be obtained.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 52176076 and 51676103) and the Taishan Scholar Project of Shandong Province (China) (no. ts20190937)References
- K. Z. Li, H. J. Zhang, Y. F. Zheng, G. Q. Yuan, Q. L. Jia and S. W. Zhang, Nanomaterials, 2020, 10, 1517 CrossRef CAS.
- A. Oyewemi, A. S. Abdulkareem, J. O. Tijani, M. T. Bankole, O. K. Abubakre, A. S. Afolabi and W. D. Roos, Arabian J. Sci. Eng., 2019, 44, 5411–5432 CrossRef CAS.
- K. A. Shah and B. A. Tali, Mater. Sci. Semicond. Process., 2016, 41, 67–82 CrossRef CAS.
- S. U. Rather, Int. J. Hydrogen Energy, 2020, 45, 4653–4672 CrossRef.
- S. Adamu, A. Bawah, O. Muraza, Z. Malaibari and M. M. Hossain, Can. J. Chem. Eng., 2020, 98, 2425–2434 CrossRef CAS.
- G. A. Queiroz and C. M. M. Barbosa, Materia, 2019, 24, e12322 Search PubMed.
- S. A. Shokry, A. K. El Morsi, M. S. Sabaa, R. R. Mohamed and H. E. El Sorogy, Egypt. J. Pet., 2014, 23, 183–189 CrossRef.
- E. Maccallini, T. Tsoufis, A. Policicchio, S. L. Rosa, T. Caruso, G. Chiarello, E. Colavita, V. Formoso, D. Gournis and R. G. Agostino, Carbon, 2010, 48, 3434–3445 CrossRef CAS.
- P. B. Amama, C. L. Pint, L. McJilton, S. M. Kim, E. A. Stach, P. T. Murray, R. H. Hauge and B. Maruyama, Nano Lett., 2009, 9, 44–49 CrossRef CAS.
- S. M. Kim, C. L. Pint, P. B. Amama, D. N. Zakharov, R. H. Hauge, B. Maruyama and E. A. Stach, J. Phys. Chem. Lett., 2010, 1, 918–922 CrossRef CAS.
- S. Sakurai, M. Inaguma, D. N. Futaba, M. Yumura and K. Hata, Small, 2013, 9, 3584–3592 CrossRef CAS.
- F. Barzegari, F. Farhadi, M. Rezaei, M. Kazemeini and A. Keshavarz, J. Energy Inst., 2021, 96, 38–51 CrossRef CAS.
- D. Spreitzer and J. Schenk, Steel Res. Int., 2019, 99, 1900108 CrossRef.
- Z. X. Song, Y. Xing, T. J. Zhang, J. G. Zhao, J. K. Wang, Y. L. Mao, B. L. Zhao, X. J. Zhang, M. Zhao and Z. A. Ma, Appl. Organomet. Chem., 2019, 34, e5446 Search PubMed.
- A. Shukla, R. K. Singha, T. Sasaki, S. Adak, S. Bhandari, V. V. D. N. Prasad, A. Bordoloi and R. Bal, Mol. Catal., 2020, 49, 110943 CrossRef.
- G. Leofanti, M. Padovan, G. Tozzola and B. Venturelli, Catal. Today, 1998, 41, 207–219 CrossRef CAS.
- B. Guan, H. Lin, L. Zhu, B. Tian and Z. Huang, Chem. Eng. J., 2012, 181, 307–322 CrossRef.
- O. M. El-Ahwany, A. E. Awadallah, A. A. Aboul-Enein, S. M. Abdel-Azim, N. A. K. Aboul-Gheit and S. A. Abo-EL-Enein, Fullerenes, Nanotubes, Carbon Nanostruct., 2019, 28, 435–445 CrossRef.
- A. Montesinos-Castellanos, T. A. Zepeda, B. Pawelec, E. Lima, J. L. G. Fierro, A. Olivas and J. A. de los Reyes H, Appl. Catal., A, 2008, 334, 330–338 CrossRef CAS.
- M. W. Tan, X. G. Wang, Y. Hu, X. J. Zou, W. Z. Ding and X. G. Lu, Int. J. Hydrogen Energy, 2015, 40, 16202–16214 CrossRef CAS.
- N. Zhang, G. Y. Qian, Z. Wang, K. X. Wei, W. H. Ma and W. Gong, Silicon, 2019, 12, 1145–1156 CrossRef.
- K. Takehira, T. Shishido, P. Wang, T. Kosaka and K. Takaki, J. Catal., 2004, 221, 43–54 CrossRef CAS.
- Y. Y. Zhan, K. Song, Z. M. Shi, C. S. Wan, J. H. Pan, D. L. Li, C. Au and L. L. Jiang, Int. J. Hydrogen Energy, 2019, 45, 2794–2807 CrossRef.
- M. U. Rashid and W. M. A. Wan Daud, RSC Adv., 2016, 6, 91603–91616 RSC.
- Q. L. Li, Q. C. Zhang, C. L. Liu, W. B. Gong, Z. Y. Zhou, P. Man, J. B. Guo, B. He, K. Zhang, W. B. Lu and Y. G. Yao, Energy Storage Mater., 2020, 27, 316–326 CrossRef.
- D. B. Bukur, L. Nowicki, R. K. Manne and X. S. Lang, J. Catal., 1995, 155, 366–375 CrossRef CAS.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- Y. J. Cao, H. Y. Peng, S. Q. Chu, Y. T. Tang, C. J. Huang, Z. L. Wang, F. Liu, J. S. Wu, B. Shan and R. Chen, Chem. Eng. J., 2021, 42, 129713 CrossRef.
- S. Sakurai, M. Yamada, J. P. He, K. Hata and D. N. Futaba, J. Phys. Chem. Lett., 2022, 13, 1879–1885 CrossRef CAS.
- L. Signorini, L. Pasquini, L. Savini, R. Carboni, F. Boscherini, E. Bonetti, A. Giglia, M. Pedio, N. Mahne and S. Nannarone, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 195423 CrossRef.
- S. Linderoth, S. Mørup and M. D. Bentzon, J. Mater. Sci., 1995, 30, 3142–3148 CrossRef CAS.
- W. Z. Zhu, D. E. Miser, W. G. Chan and M. R. Hajaligol, Mater. Chem. Phys., 2003, 82, 638–647 CrossRef CAS.
- E. Maccallin I, T. Tsoufis, A. Policicchio, S. L. Rosa, T. Caruso, G. Chiarello, E. Colavita, V. Formoso, D. Gournis and R. G. Agostino, Carbon, 2010, 48, 3434–3445 CrossRef CAS.
- A. Gamboa, L. M. Marques and E. C. Fernandes, Diamond Relat. Mater., 2021, 113, 108274 CrossRef CAS.
- X. T. Liu, Y. S. Zhang, M. A. Nahil, P. T. Williams and C. F. Wu, J. Anal. Appl. Pyrolysis, 2017, 125, 32–39 CrossRef CAS.
- Y. Jin, G. W. Wang and Y. D. Li, Appl. Catal., A, 2012, 445, 121–127 CrossRef.
- G. T. T. Le, P. Mala, S. Ratchahat and T. Charinpanitkul, J. Anal. Appl. Pyrolysis, 2021, 158, 105257 CrossRef CAS.
- M. S. Shamsudin, N. A. Asli, S. Abdullah, S. Y. S. Yahya and M. Rusop, Adv. Condens. Matter Phys., 2012, 2012, 186–192 Search PubMed.
- L. M. Esteves, H. A. Oliveira, Y. T. Xing and F. B. Passos, New J. Chem., 2021, 45, 14218–14226 RSC.
- J. Li, H. D. Zhang, Z. P. Gao, J. Fu, W. Y. Ao and J. J. Dai, Energy Fuels, 2017, 31, 3475–3524 CrossRef CAS.
- H. A. Alalwan, S. E. Mason, V. H. Grassian and D. M. Cwiertny, Energy Fuels, 2018, 32, 7959–7970 CrossRef CAS.
- M. Y. Ding, Y. Yang, B. S. Wu, Y. W. Li, T. J. Wang and L. L. Ma, Appl. Energy, 2015, 160, 982–989 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |