
Pt single atoms meet metal–organic frameworks to enhance electrocatalytic hydrogen evolution activity†
Jingting
Zhu
 abc,
Yingqian
Cen
ab,
Haibin
Ma
c,
Weiguang
Lian
ab,
Jidong
Liu
ab,
Haohui
Ou
ab,
Fangping
Ouyang
d,
Lifu
Zhang
*ab and
Wenjing
Zhang
*ab
abc,
Yingqian
Cen
ab,
Haibin
Ma
c,
Weiguang
Lian
ab,
Jidong
Liu
ab,
Haohui
Ou
ab,
Fangping
Ouyang
d,
Lifu
Zhang
*ab and
Wenjing
Zhang
*ab
aState Key Laboratory of Radio Frequency Heterogeneous Integration, Shenzhen University, Shenzhen 518060, China. E-mail: wjzhang@szu.edu.cn; zhanglifu@szu.edu.cn
bInternational Collaborative Laboratory of 2D Materials for Optoelectronics Science and Technology of Ministry of Education, Institute of Microscale Optoelectronics, Shenzhen University, Shenzhen 518060, China
cSIAMC Advanced Materials Co., Ltd, 313100, Huzhou, China
dSchool of Physics and Electronics, Central South University, Changsha 410083, China
First published on 19th July 2023
Abstract
The electrochemical hydrogen evolution reaction (HER) effectively produces clean, renewable, and sustainable hydrogen; however, the development of efficient electrocatalysts is required to reduce the high energy barrier of the HER. Herein, we report two excellent single-atom (SA)/metal–organic framework (MOF) composite electrocatalysts (PtSA-MIL100(Fe) and PtSA-MIL101(Cr)) for HER. The obtained PtSA-MIL100(Fe) and PtSA-MIL101(Cr) electrocatalysts exhibit overpotentials of 60 and 61 mV at 10 mA cm−2, respectively, which are close to that of commercial Pt/C (38 mV); they exhibit overpotentials of 310 and 288 mV at 200 mA cm−2, respectively, which are comparable to that of commercial Pt/C (270 mV). Theoretical simulations reveal that Pt SAs modulate the electronic structures of the MOFs, leading to the optimization of the binding strength for H* and significant enhancement of the HER activity. This study describes a novel strategy for preparing desirable HER electrocatalysts based on the synergy between SAs and MIL-series MOFs. Using MIL-series MOFs to support SAs could be valuable for future catalyst design.
New conceptsThe aim of this study is to develop new inexpensive and high-performance single-atom catalysts (SACs) for the hydrogen evolution reaction (HER) based on low loadings of Pt decorated on high-surface-area metal–organic framework (MOF) supports. We believe that our study makes a significant contribution to the HER literature because we firstly prepared two Pt SA/MIL-series MOF composite catalysts (PtSA-MIL100(Fe) and PtSA-MIL101(Cr)) which show performances similar to that of a commercial Pt/C catalyst, but with a much lower Pt loading. Furthermore, these catalysts were prepared using a simple and inexpensive route that is considered viable for upscaling. Using MIL-series MOFs to support SAs could be valuable for future catalyst design. |
1. Introduction
Global environmental problems are inspiring the development of clean, renewable, and sustainable energy sources as alternatives to traditional fossil fuels. Hydrogen is considered one of the most promising alternatives owing to its eco-friendly production method and high energy density.1,2 The electrocatalytic hydrogen evolution reaction (HER) has attracted considerable interest because it produces high-purity hydrogen gas.3,4 The HER needs to be accelerated by appropriate catalysts, and platinum (Pt) is currently considered the most efficient catalyst for this reaction. However, the high cost and scarcity of Pt limit the widespread application of Pt-based catalysts, and researchers are exploring effective strategies to reduce catalyst costs.5,6Single-atom catalysts (SACs) have attracted tremendous interest owing to their maximum atomic utilization efficiency and well-defined active centres.7–9 Preparing Pt SACs can significantly reduce the amount of Pt while maintaining the excellent catalytic activity of Pt, which is an effective strategy to reduce catalyst costs. The catalytic activity of SACs results from the synergistic effect between the single-atoms (SAs) and supports. An ideal support is expected to provide abundant anchoring sites to stabilize the SAs. Moreover, the support should interact with the SAs and modulate their coordination environment, facilitating the activity, selectivity, and stability of the SAs.10–13
Metal–organic frameworks (MOFs) are a class of porous crystalline materials constructed from metal centres and organic ligands. In recent years, electrocatalysts based on MOFs with good catalytic activity have attracted extensive attention.14 Micheroni et al. prepared a Hf12-CoDBP/CNT hybrid with an overpotential of 650 mV at 10 mA cm−2 and a Tafel slope of 178 mV dec−1,15 Sun et al. prepared NiMn-MOF aerogels with an overpotential of 243 mV at 10 mA cm−2 and a Tafel slope of 126 mV dec−1.16 Additionally, owing to their high porosity, structural diversity, abundant anchoring sites, and tailorability for specific applications, MOFs are considered promising supports for SAs.17–19 Although SACs composed of SAs and MOFs have been frequently used as high-performance photocatalysts,20–23 SA–MOF composite electrocatalysts appropriate for HER applications have been rarely reported.24
The aim of this study was to develop inexpensive and efficient catalyst systems for the HER. Here, we propose two MOFs, MIL100(Fe) and MIL101(Cr), as new supports for anchoring Pt SAs to produce SACs for the electrocatalytic HER. MIL100(Fe) and MIL101(Cr) MOFs were chosen because they are inexpensive and can be fabricated by simple methods that are suitable for upscaling, i.e., solution agitation and hydrothermal treatment, respectively. Subsequently, the composite catalysts composed of Pt SAs and MOFs (named PtSA-MIL100(Fe) and PtSA-MIL101(Cr)) were prepared under simulated solar irradiation. The preparation processes for the two SA/MOF catalysts were previously shown to be cost-effective and simple,25,26 facilitating their industrial application. The HER activities of as-prepared PtSA-MIL100(Fe) and PtSA-MIL101(Cr) were significantly enhanced compared with those of pristine MIL100(Fe) and MIL101(Cr) and comparable to those of Pt/C compounds, i.e., ultralow overpotentials of 60 and 61 mV, respectively, at 10 mA cm−2 (close to that of Pt/C; 38 mV) and low overpotentials of 310 and 288 mV, respectively, at 200 mA cm−2 (close to that of Pt/C; 270 mV). Density functional theory (DFT) calculations confirmed that the electronic structures of both MIL100(Fe) and MIL101(Cr) were regulated by the introduction of Pt atoms, thereby optimizing the adsorption strength for H* and enhancing HER performance.
2. Results and discussion
2.1. Characterization of PtSA-MIL100(Fe) and PtSA-MIL101(Cr)
First, the MIL100(Fe) and MIL101(Cr) MOFs were synthesized by two different methods, i.e., solution agitation and hydrothermal treatment, respectively. Subsequently, Pt SAs were anchored on the MIL100(Fe) and MIL101(Cr) by exposing mixtures of H2PtCl6·6H2O and one of the MOFs to simulated solar irradiation (Scheme 1). By varying the concentration of H2PtCl6·6H2O, a series of composite catalysts with different Pt loadings were obtained, determined by inductively coupled plasma optical emission spectrometry (ICP-OES) (Table S1, ESI†). Hereafter, these samples are denoted as PtX–MIL100(Fe) and PtX–MIL101(Cr), where X is the molar ratio of Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe and Pt:Cr, respectively; further details are provided in the Experimental section. The molar ratio is used to describe the metal loadings to reflect the amount of metal atoms replaced by Pt, to be consistent with previous literature.24 The structures of these samples were studied using X-ray diffraction (XRD). As shown in Fig. S1a (ESI†), the main diffraction peaks of the pristine and Pt-decorated MIL100(Fe) were similar and matched well with the simulated spectrum for model MIL100(Fe),27 indicating that decoration with Pt did not change the crystal structure. Similarly, the pristine and Pt-decorated MIL101(Cr) had the same crystal structure (Fig. S1b, ESI†), which was consistent with that of the simulated MIL101(Cr).25
:Fe and Pt:Cr, respectively; further details are provided in the Experimental section. The molar ratio is used to describe the metal loadings to reflect the amount of metal atoms replaced by Pt, to be consistent with previous literature.24 The structures of these samples were studied using X-ray diffraction (XRD). As shown in Fig. S1a (ESI†), the main diffraction peaks of the pristine and Pt-decorated MIL100(Fe) were similar and matched well with the simulated spectrum for model MIL100(Fe),27 indicating that decoration with Pt did not change the crystal structure. Similarly, the pristine and Pt-decorated MIL101(Cr) had the same crystal structure (Fig. S1b, ESI†), which was consistent with that of the simulated MIL101(Cr).25
 | ||
| Scheme 1 Experimental flow chart for the synthesis processes of PtSA-MIL100(Fe) and PtSA-MIL101(Cr). | ||
The morphologies and microstructures of the catalyst products were investigated using scanning electron microscopy (SEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). As shown in Fig. 1a and Fig. S2a–c (ESI†), Pt0.009-MIL100(Fe), Pt0.015-MIL100(Fe), and Pt0.023-MIL100(Fe) have nonuniform faceted morphologies similar to those of pristine MIL100(Fe), with particle sizes ranging from 20 nm to 1.2 μm. The HAADF-STEM images in Fig. 1b, c and Fig. S2d–i (ESI†) reveal that Pt0.015-MIL100(Fe) has numerous Pt SAs (bright dots) and a few Pt atomic clusters. The energy-dispersive X-ray spectroscopy (EDS) maps of Pt0.015-MIL100(Fe) in Fig. 1d further indicate a uniform distribution of Pt atoms without obvious aggregation. The pristine and Pt-decorated MIL101(Cr) samples have similar uniform octahedral morphologies, with a particle size of approximately 90 nm (Fig. 1e and Fig. S3a–c, ESI†). The HAADF-STEM images (Fig. 1f, g and Fig. S3d–i, ESI†) and EDS maps (Fig. 1h) of Pt0.019-MIL101(Cr) also indicate uniformly dispersed Pt atoms without aggregation throughout the sample, similar to Pt0.015-MIL100(Fe).
 | ||
| Fig. 1 (a) SEM, (b) and (c) HAADF-STEM images, and (d) STEM-EDS maps of Pt0.015-MIL100(Fe); (e) SEM, (f) and (g) HAADF-STEM images, and (h) STEM-EDS maps of Pt0.019-MIL101(Cr). | ||
To further investigate the local electronic structure and coordination environment of Pt in Pt0.015-MIL100(Fe) and Pt0.019-MIL101(Cr), X-ray absorption fine structure spectroscopy (XAFS) was performed including the X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analyses. According to the XANES results (Fig. 2a), the Pt L3-edge white-line (WL) intensities of Pt0.015-MIL100(Fe) and Pt0.019-MIL101(Cr) both lie between those of Pt foil and PtO2, suggesting a positively charged state of Pt in Pt0.015-MIL100(Fe) and Pt0.019-MIL101(Cr).6Fig. 2b and Table S2 (ESI†) present the oxidation states estimated from the integrated areas under the Pt L3-edge WL intensities, which indicate a higher oxidation state of Pt in Pt0.019-MIL101(Cr) (+2.98) than that in Pt0.015-MIL100(Fe) (+2.10). The quantitative WL peak intensity results indicate that the Pt atoms in Pt0.015-MIL100(Fe) and Pt0.019-MIL101(Cr) contain a higher number of unoccupied 5d-electron states than Pt foil, which is considered favourable for enhancing the HER activity.28–30 Furthermore, the peaks in the k2-weighted Fourier transformed (FT) EXAFS spectra (Fig. 2c) for Pt0.015-MIL100(Fe) and Pt0.019-MIL101(Cr) are similar to those for PtO2 and correspond to Pt–O bonds, whereas they are significantly different from those measured for Pt foil, suggesting that Pt aggregation did not occur in the catalysts.31 The fitting results from FT EXAFS in Fig. S4 and Table S3 (ESI†) show that the first shells of the central Pt atom display a coordination number of 5 with one C/O atom (with fitted bond length of 2.07 Å) in Pt0.015-MIL100(Fe), and a coordination number of 6 with one C/O atom (with fitted bond length of 2.14 Å) in Pt0.019-MIL101(Cr) respectively. Contrarily, the Pt foil displays a high coordination number of 12 and corresponding Pt–Pt bond length of 2.76 Å, further disclosing the fact of atomic dispersion of Pt in Pt0.015-MIL100(Fe) and Pt0.019-MIL101(Cr). Fig. S5 and S6 (ESI†) show the X-ray photoelectron spectroscopy (XPS) spectra of Pt-MIL100(Fe) and Pt-MIL101(Cr) samples compared with those of pristine MIL100(Fe) and MIL101(Cr). These spectra reveal the coexistence of Fe, C, O, Pt and Cr, C, O, Pt elements in Pt0.015-0.023-MIL100(Fe) and Pt0.019-0.034-MIL101(Cr) samples respectively, whereas Pt was not detected in Pt0.009-MIL100(Fe) and Pt0.011-MIL101(Cr) because the Pt content was below the detection limit of the XPS instrument. Additionally, the normalized Pt 4f XPS spectra (Fig. S5e and S6e, ESI†) show that the peaks measured for Pt0.015–0.023-MIL100(Fe) and Pt0.019–0.034-MIL101(Cr) are considerably red-shifted compared with those of metallic Pt in Pt/C, confirming that no Pt particles were formed in PtSA-MIL100(Fe) and PtSA-MIL101(Cr).32,33
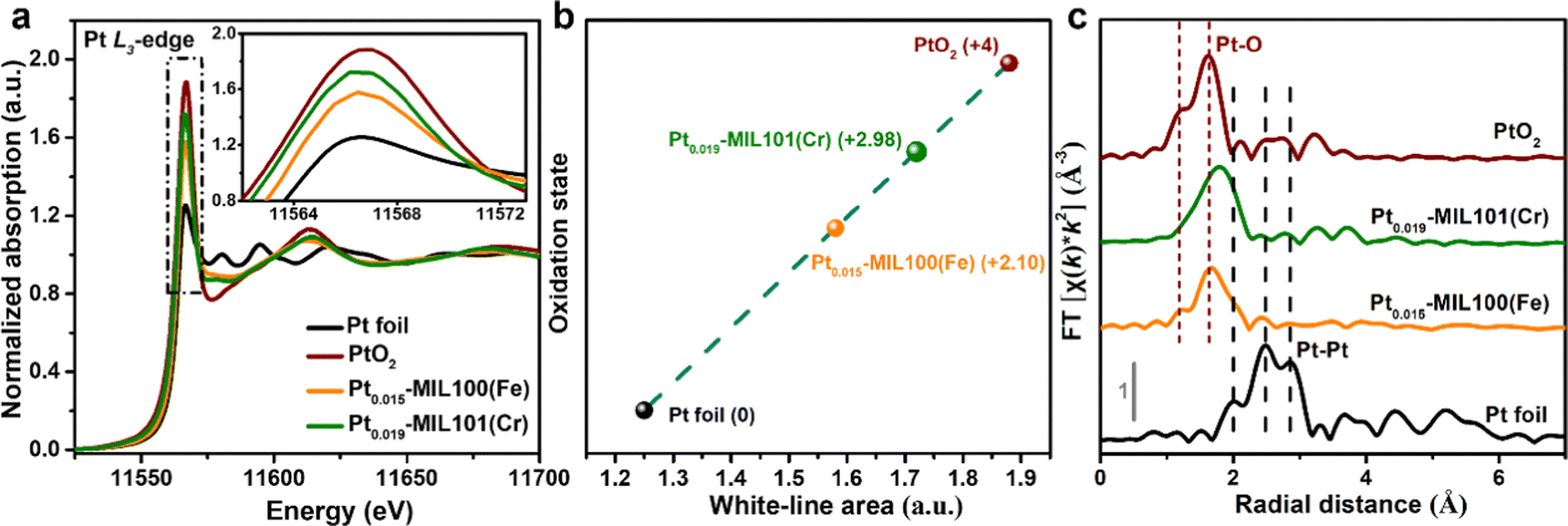 | ||
| Fig. 2 XAFS results. (a) Normalized XANES spectra of the Pt L3-edge, (b) fitted curve correlating the average oxidation state of Pt, and (c) k2-weighted FT EXAFS spectra of the Pt0.015-MIL100(Fe) and Pt0.019-MIL101(Cr) compared to Pt foil and PtO2 reference spectra. | ||
2.2. HER Performance of PtSA-MIL100(Fe) and PtSA-MIL101(Cr)
The HER performance of the SACs was measured using a three-electrode cell system in an acidic medium. Fig. 3a shows the linear sweep voltammetry (LSV) curves of pristine and Pt-decorated MIL100(Fe) compared with those of the 20 wt% Pt/C catalyst. Pristine MIL100(Fe) shows poor HER activity with an η10 (overpotential at a current density of 10 mA cm−2) of 418 mV. In contrast, after introducing Pt SAs into MIL100(Fe), the HER activity was significantly enhanced. Compared with the η10 value of pristine MIL100(Fe) (418 mV), the addition of Pt resulted in a significant decrease in η10, with values of 128, 60, and 63 mV measured for Pt0.009-MIL100(Fe), Pt0.015-MIL100(Fe), and Pt0.023-MIL100(Fe), respectively, with Pt loadings of 0.55, 0.91, and 1.41 wt%, respectively. Remarkably, the HER performance of Pt0.015-MIL100(Fe) with the lowest η10 but Pt loading of only 0.91 wt% is close to that of commercial Pt/C (38 mV) with a 20 wt% Pt loading. Additionally, Pt0.015-MIL100(Fe) exhibits a low overpotential of 310 mV to reach a current density of 200 mA cm−2 comparable to that of Pt/C (270 mV). The Tafel slope (Fig. 3b) of Pt0.015-MIL100(Fe) is 31.16 mV dec−1, which is lower than those of MIL100(Fe) (139.55 mV dec−1), Pt0.009-MIL100(Fe) (82.71 mV dec−1), and Pt0.023-MIL100(Fe) (33.71 mV dec−1), and comparable to that of Pt/C (28.87 mV dec−1). Both the LSV and Tafel results indicate that Pt0.015-MIL100(Fe) is the best-performing catalyst in this series. The LSV curves (Fig. 3c) and Tafel slopes (Fig. 3d) of the pristine and Pt-decorated MIL101(Cr) were also obtained. Pt0.019-MIL101(Cr) exhibits the best HER activity among these samples, with η10 of 61 mV and Tafel slope of 27.02 mV dec−1, which are much lower than those of pristine MIL101(Cr) (η10 of 356 mV and Tafel slope of 183.9 mV dec−1) and comparable to those of Pt/C (η10 of 38 mV and Tafel slope of 28.87 mV dec−1). Similarly, Pt0.019-MIL101(Cr) exhibits a low overpotential at 200 mA cm−2 (288 mV) closed to that of commercial Pt/C (270 mV), indicating its superiority for HER applications. For reference, the Pt loadings of Pt0.011-MIL101, Pt0.019-MIL101, and Pt0.034-MIL101 were 0.44, 0.84, and 1.52 wt%, respectively. Compared with other Pt SA-based catalysts reported from previous studies (Table S4, ESI†), the PtSA–MIL100(Fe) and PtSA-MIL101(Fe) samples in this study have good HER activity in acidic media.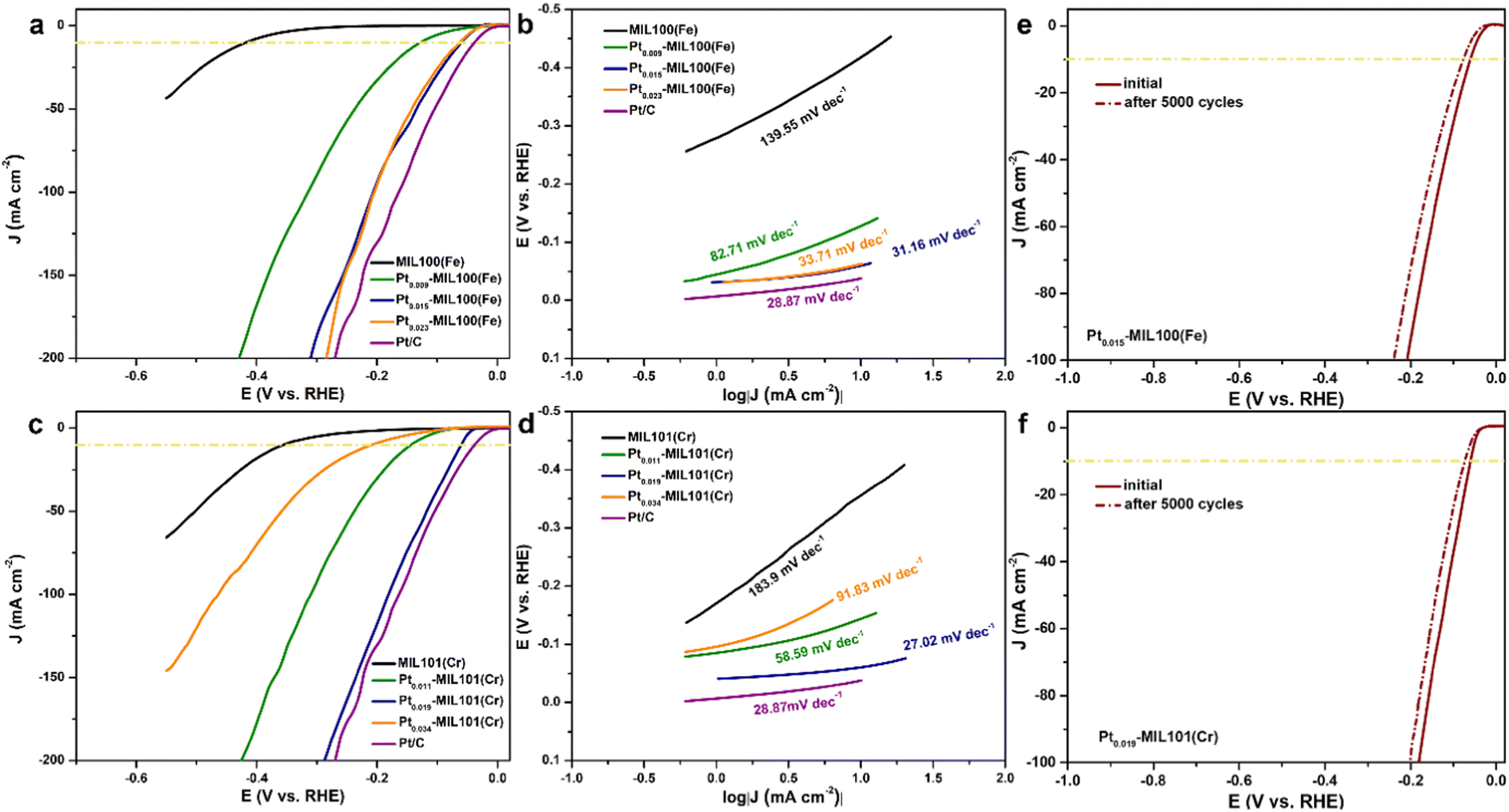 | ||
| Fig. 3 HER electrochemical performance for catalysts in 0.5 M H2SO4. (a) LSV curves and (b) Tafel plots of MIL100(Fe), Pt0.009-MIL100(Fe), Pt0.015-MIL100(Fe), Pt0.023-MIL100(Fe), and 20 wt% Pt/C for comparison. (c) LSV curves and (d) Tafel plots of MIL101(Cr), Pt0.011-MIL101(Cr), Pt0.019-MIL101(Cr), Pt0.034-MIL101(Cr), and 20 wt% Pt/C for comparison. Long-term stability test: polarization curves of (e) Pt0.015-MIL100(Fe) and (f) Pt0.019-MIL101(Cr) recorded before and after 5000 cycles from 0.05 to −0.60 V vs. RHE. | ||
The double-layer capacitance (Cdl) is an important parameter for a catalyst because it is directly proportional to the electrochemical surface area (ECSA).34–36 As shown in Fig. S7 (ESI†), all of the Pt-decorated MOFs samples exhibit a higher Cdl than the pristine MOFs. The Cdl was calculated to be 36.8 and 54.2 mF cm−2 for Pt0.015-MIL100(Fe) and Pt0.019-MIL101(Cr), respectively, which are approximately 28 and 25 times higher than that of pristine MIL100(Fe) (1.3 mF cm−2) and MIL101(Cr) (2.2 mF cm−2), respectively. The increase in the ECSA indicates an increase in the number of active sites provided by the Pt SAs and atomic clusters, which should enhance anion accessibility and HER activity. The enhanced anion accessibility was verified by measuring the electron-transfer resistance (Fig. S8, ESI†), where the resistance decreases with the introduction of Pt. Because the cycling stability is critical for evaluating the industrial potential of a catalyst, polarization curves were measured for Pt0.015-MIL100(Fe) and Pt0.019-MIL101(Cr) over 5000 sweeps (Fig. 3e and f). The curves for both catalysts shifted only slightly during cycling compared with those of the initial states, indicating excellent stability. In addition, their current densities at an overpotential of 80 mV were stable after 12 h (Fig. S9, ESI†). The excellent durability of the Pt-decorated MOFs indicates a strong interaction between the Pt SAs and MOF supports. Furthermore, considering the results presented so far, PtSA-MIL100(Fe) and PtSA-MIL101(Cr) seem to be promising substitutes for commercial Pt/C catalysts for the HER.
2.3. DFT results
DFT calculations were performed to understand the mechanism through which Pt SAs incorporated into MIL100(Fe) and MIL101(Cr) participate in the HER catalytic reaction (Fig. 4). The DFT-optimized geometric structures of H* on Fe in MIL100(Fe) and Pt-MIL100(Fe), Pt in Pt-MIL100(Fe), Cr in MIL101(Cr) and Pt-MIL101(Cr), and Pt in Pt-MIL101(Cr) are shown in Fig. S10 (ESI†). All structures in the calculation were modelled using the cluster model based on previous studies because DFT calculations based on periodic cells cannot be performed for MIL-series MOFs.37–40 The calculated adsorption free energies of H* (ΔGH*) are shown in Fig. 4a and b, i.e. 1.91, 1.67, and −0.66 eV for Fe in MIL100(Fe), Fe in Pt-MIL100(Fe), and Pt in Pt-MIL100(Fe), respectively, and 2.18, 0.97, and −0.62 eV for Cr in MIL101(Cr), Cr in Pt-MIL101(Cr), and Pt in Pt-MIL101(Cr), respectively. Moderate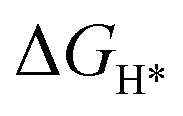 values (close to zero) indicate feasible H* adsorption and satisfactory H2 desorption.41–43 Therefore, the
values (close to zero) indicate feasible H* adsorption and satisfactory H2 desorption.41–43 Therefore, the 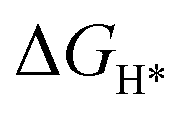 values for Fe and Pt in Pt-MIL100(Fe) are closer to the thermoneutral value than that of MIL100(Fe), indicating enhanced HER activity. The same conclusions were drawn for Cr and Pt in Pt-MIL101(Cr). The differential charge density diagrams (Fig. 4c and d) indicate that after the introduction of Pt atoms, the local charges in both MIL100(Fe) and MIL101(Cr) are redistributed, which is beneficial for H* adsorption.
values for Fe and Pt in Pt-MIL100(Fe) are closer to the thermoneutral value than that of MIL100(Fe), indicating enhanced HER activity. The same conclusions were drawn for Cr and Pt in Pt-MIL101(Cr). The differential charge density diagrams (Fig. 4c and d) indicate that after the introduction of Pt atoms, the local charges in both MIL100(Fe) and MIL101(Cr) are redistributed, which is beneficial for H* adsorption.
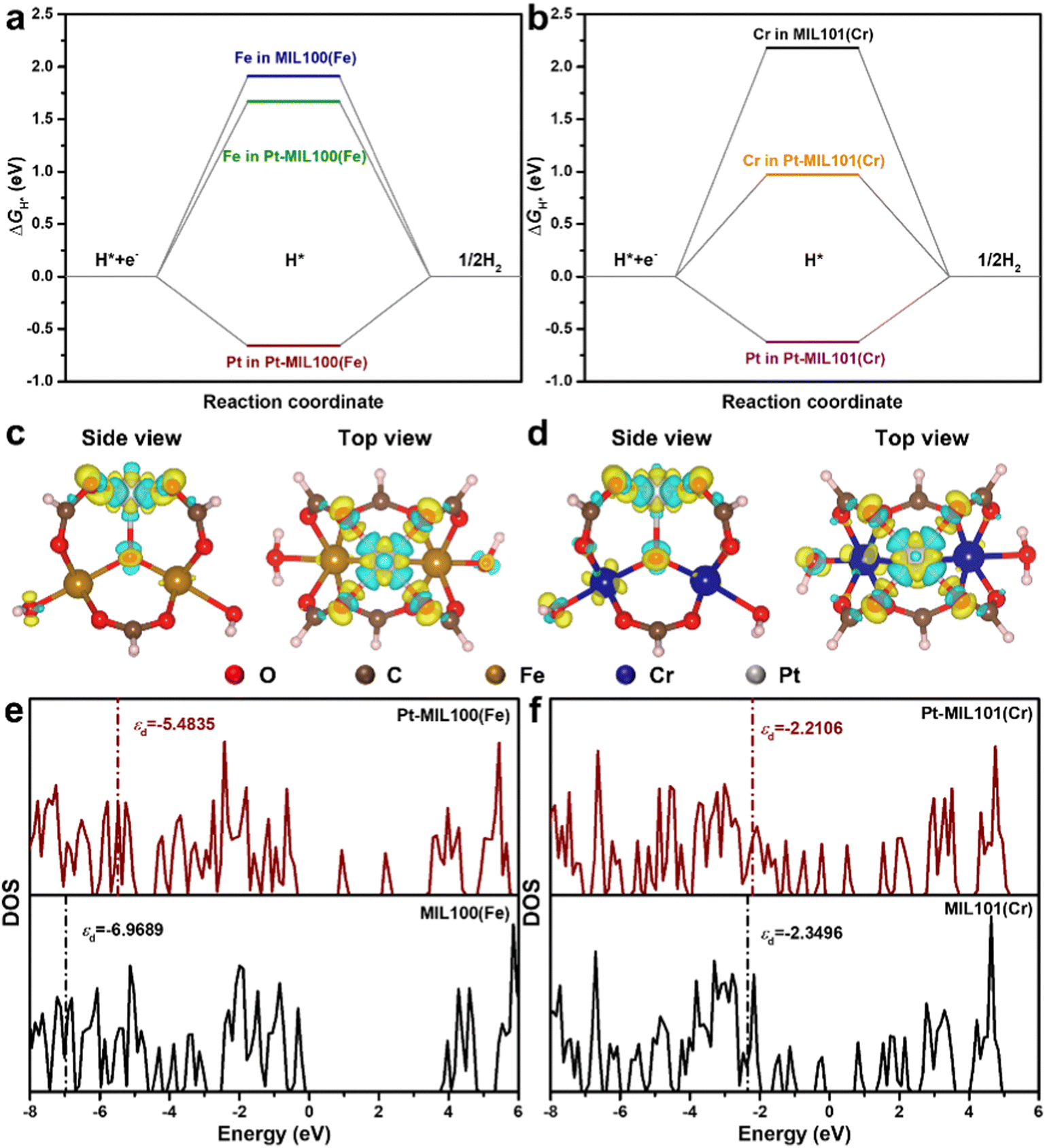 | ||
| Fig. 4 Free-energy diagram for the HER on (a) Fe in MIL100(Fe), Fe in Pt-MIL100(Fe), and Pt in Pt-MIL100(Fe); (b) Cr in MIL101(Cr), Cr in Pt-MIL101(Cr), and Pt in Pt-MIL101(Cr). Difference charge density of (c) Pt-MIL100(Fe) and (d) Pt-MIL101(Cr). Charge accumulation and depletion are shown in yellow and blue, respectively. Comparison of calculated DOS of (e) MIL100(Fe) and Pt-MIL100(Fe), and f MIL101(Cr) and Pt-MIL101(Cr). | ||
The calculated density of states (DOS) of MIL100(Fe), Pt-MIL100(Fe), MIL101(Cr), and Pt-MIL101(Cr) and their corresponding partial DOS are presented in Fig. 4e, f and Fig. S11 (ESI†). This reveals that after the introduction of Pt atoms, the electronic structures of MIL100(Fe) and MIL101(Cr) significantly changed. In addition, some localized states are observed in the bandgap of Pt-MIL100(Fe) and Pt-MIL101(Cr) that are not present in pristine MIL100(Fe) and MIL101(Cr), implying that more carriers can participate in H* adsorption and H2 formation.10 It should be noted that a d-band centre (εd) closer to the Fermi level indicates stronger H* adsorption.44,45 Therefore, as shown in Fig. 4e, f and Table S4 (ESI†), the εd values of −5.4835 and −2.2106 eV for Pt-MIL100(Fe) and Pt-MIL101(Cr), respectively, further prove that these catalysts have stronger H* adsorption than MIL100(Fe) (εd = −6.9689 eV) and MIL101(Cr) (εd = −2.3496 eV), respectively.
3. Conclusion
In this study, we designed and prepared two new electrocatalysts with enhanced HER performance by introducing Pt SAs into MOFs (MIL100(Fe) and MIL101(Cr)). The as-prepared PSA-MIL100(Fe) and PtSA-MIL101(Cr) SACs exhibited low overpotentials over a wide range of current densities from 10 to 200 mA cm−2, which are comparable to those of commercial Pt/C catalysts. The XAFS and XPS results confirmed the strong electron interactions between the Pt atoms and MOFs, as well as the atomic dispersion of the Pt atoms. Theoretical investigations demonstrated that the absorption behaviour of H* and charge-transfer kinetics can be enhanced by Pt decoration, which improves the HER performance. This study provides two efficient HER catalysts based on Pt SAs and MIL-series MOFs, which provide similar performance to that of commercial Pt/C but require <1 wt% of the Pt and are produced by inexpensive methods. Therefore, these findings significantly contribute to the field by providing strategies for decreasing the cost of catalysts, which could support the industrial-scale application of hydrogen energy.4. Experimental section
Synthesis of MIL100(Fe)
The MIL100(Fe) was prepared refer to reported sustainable method.27 A typical reaction started with the preparation of two different solutions. Solution 1 was prepared by dissolving 8 mmol H3BTC (Adamas Reagent, Ltd) in 23 mL 1 M NaOH (Sinopharm Chemical Reagent Co., Ltd) aqueous solution. Solution 2 was prepared by dissolving 11.5 mmol of FeCl2·4H2O (Dieckmann, Ltd) in 98 mL deionized (DI) water. After magnetically stirring for 30 min, Solution 1 was added dropwise into Solution 2 under stirring, then the mixture was continuously stirred for 24 h at room temperature. The product was collected by centrifugation and washed with DI water and ethanol, and then dried at 60 °C for further use.Synthesis of MIL101(Cr)
The MIL101(Cr) was prepared refer to reported hydrothermal method.26 In a typical reaction, 2 mmol Cr(NO3)3·9H2O (Adamas Reagent, Ltd) and 2 mmol H2BDC (Adamas Reagent, Ltd) were dissolved in 30 mL DI water. After magnetically stirring for 30 min, the above solution was added into a 50 mL Teflon-lined autoclave. After 24 h reaction in the sealed autoclave at 180 °C, the product was obtained by centrifugation and washed with DI water and ethanol, and then dried at 60 °C for further use.Synthesis of Pt-MIL100(Fe) and Pt-MIL101(Cr)
Solar irradiation strategy was applied to introduce Pt SAs into MIL100(Fe) and MIL101(Cr) structure. 0.03 g as-prepared MIL100(Fe) and MIL101(Cr) powders were respectively dispersed into 20 mL H2PtCl6·6H2O (99.9%, Adamas Reagent, Ltd) of different concentration by ultrasonic, which was then irradiated with a Xe lamp (NBET Solar-500) at the standard solar AM1.5G intensity of 100 mW cm−2. After irradiating for 20 min, the Pt-MIL100(Fe) and Pt-MIL101(Cr) were obtained, followed by washing with DI water and ethanol and drying at 60 °C for 2 h. The molar ratio of Pt:Fe in Pt-MIL100(Fe) samples prepared in 0.1, 0.2 and 0.3 mM H2PtCl6·6H2O were 0.009, 0.015 and 0.023, respectively. The molar ratio of Pt:Cr in Pt-MIL101(Cr) samples prepared in 0.05, 0.1 and 0.2 mM H2PtCl6·6H2O were 0.011, 0.019 and 0.034, respectively.
Materials characterization
The Pt content was detected by ICP-OES (Agilent 720ES). XRD (Model D/Max 2550 V, Rigaku) was conducted to identify the crystal phases of the products with Cu Kα radiation (λ = 1.5418 Å) at the current of 40 mA and the voltage of 40 kV. The morphology of the product was characterized by field-emission SEM (GeminiSEM 560). HAADF-STEM and EDS mapping were performed by using Thermo Fisher Scientific Themis Z TEM/STEM with probe and image correctors. XAFS was measured at the 4B9A beamline of BSRF (Beijing Synchrotron Radiation Facility) under ring conditions of 2.2 GeV and about 80 mA. A Si (111) double-crystal monochromator was used for energy selection, and samples data were collected in fluorescence mode, while standards (i.e., Pt foil and PtO2) data were collected in transmission mode. XPS was performed using a Thermo Scientific K-Alpha+ with monochromatic Al Kα radiation.Electrochemical measurements.
All electrochemical measurements were conducted by a typical three-electrode configuration with CHI 760E electrochemical workstation, where a graphite rod was used as the counter electrode and a saturated calomel electrode (SCE) was used as the reference electrode. The working electrodes were prepared by dropping different catalyst inks onto glassy carbon electrodes. The catalyst ink was prepared by 30 min sonication of a mixture containing 3 mg catalyst, 80 μL Nafion solution (5 wt%, Dupont), 300 μL isopropanol and 700 μL DI water. Then, 5 μL catalyst ink was transferred onto the glassy carbon electrode with a catalyst loading of ∼0.2 mg cm−2. The electrolyte was 0.5 M H2SO4 solution. LSV curves were characterized from 0.05 to −0.60 V vs. reversible hydrogen electrode (RHE) at a scan rate of 5 mV s−1. Cyclic voltammograms (CV) taken at various scan rates (30, 50, 70, 90, and 110 mV s−1) were recorded in the potential range of 0.35–0.45 V vs. RHE and were used to estimate the Cdl. The stability was tested by measuring accelerated 5000 loops of LSV tests at a scan rate of 20 mV s−1, and the current-time response was monitored at a static potential of −0.08 V vs. RHE for up to 12 h additionally. All the potentials reported in this work were referenced to a RHE according to the following equation:| E(RHE) = E(SCE) + 0.241 + 0.0591 × PH. |
DFT Calculations
DFT calculations were performed by using Vienna ab initio simulation package (VASP) based on the plane-wave basis sets with the projector augmented-wave method.46,47 The exchange–correlation potential was treated by using a generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) parametrization.48 The van der Waals correction of Grimme's DFT-D3 model was also adopted.49 Following previous works,50 an effective Hubbard Ueff = U − J = 5.3/3.7 eV is used for MIL100(Fe)/MIL101(Cr). Given the structural complexity, it is impossible to complete the structural optimization of the two MOFs at the DFT level. Therefore, refer to previous literature,37–40 the cluster models were used to investigate their reaction processes at the atomic scale. For Pt-decorated structures, we assumed that Pt atom replaces the corresponding unsaturated metal position of the MOF metal node. The energy cutoff was set to be 520 eV. The Brillouin-zone integration was sampled with a Γ-centred Monkhorst-Pack mesh of 1 × 1 × 1.51 The structures were fully relaxed until the maximum force on each atom was less than 0.01 eV Å−1, and the energy convergent standard was 10−5 eV.The configuration with hydrogen atom adsorbed on OMS site (with slight structural distortion) was used to study the HER performance. Under standard conditions, the HER pathway can be described by:52,53
| H+(aq) + e− → 1/2H2(g). |
An initial state H+(aq) and an e− form an intermediate adsorbed H*, and two H* form H2. Thus, the Gibbs free energy of the adsorption of the H* on the catalyst (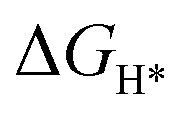 ) is a key descriptor for the HER activity.
) is a key descriptor for the HER activity. 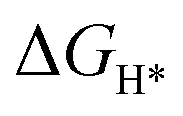 is obtained by:
is obtained by:
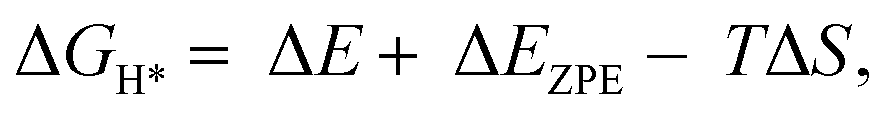
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Shenzhen University Outstanding Young Teacher Training Program, National Natural Science Foundation of China (NSFC)-Research Grant Council of Hong Kong (RGC) Key International (Regional) Joint Research Program (NSFC Grant No. 62261160574 and RGC Grant No. CRS_PolyU502/22), the Educational Commission of Guangdong Province project (Key program, Grant no. 2020ZDZX3041), the Guangdong Basic and Applied Basic Research Foundation (Grant No. 2020A1515110333), the Science, Technology and Innovation Commission of Shenzhen Municipality (Grant No. JCYJ20200109105422876). The authors would like to thank Shiyanjia Lab (www.shiyanjia.com) for ICP-OES, XAFS, XPS measurements and DFT calculations.References
- S.-Y. Bae, J. Mahmood, I.-Y. Jeon and J.-B. Baek, Nanoscale Horiz., 2020, 5, 43–56 RSC.
- T. E. Mallouk, Nat. Chem., 2013, 5, 362–363 CrossRef CAS PubMed.
- Z. Wu, J. Yang, W. Shao, M. Cheng, X. Luo, M. Zhou, S. Li, T. Ma, C. Cheng and C. Zhao, Adv. Fiber Mater., 2022, 4, 774–785 CrossRef CAS.
- Y. Zhu, Q. Lin, Y. Zhong, H. A. Tahini, Z. Shao and H. Wang, Energy Environ. Sci., 2020, 13, 3361–3392 RSC.
- M. Li, K. Duanmu, C. Wan, T. Cheng, L. Zhang, S. Dai, W. Chen, Z. Zhao, P. Li, H. Fei, Y. Zhu, R. Yu, J. Luo, K. Zang, Z. Lin, M. Ding, J. Huang, H. Sun, J. Guo, X. Pan, W. A. Goddard, P. Sautet, Y. Huang and X. Duan, Nat. Catal., 2019, 2, 495–503 CrossRef CAS.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985–992 CrossRef CAS.
- D. Li, X. Li, S. Chen, H. Yang, C. Wang, C. Wu, Y. A. Haleem, S. Duan, J. Lu, B. Ge, P. M. Ajayan, Y. Luo, J. Jiang and L. Song, Nat. Energy, 2019, 4, 512–518 CrossRef.
- Y. Zhao, T. Ling, S. Chen, B. Jin, A. Vasileff, Y. Jiao, L. Song, J. Luo and S.-Z. Qiao, Angew. Chem., Int. Ed., 2019, 58, 12252–12257 CrossRef CAS PubMed.
- L. Zhang, K. Doyle-Davis and X. Sun, Energy Environ. Sci., 2019, 12, 492–517 RSC.
- J. Zhu, Y. Tu, L. Cai, H. Ma, Y. Chai, L. Zhang and W. Zhang, Small, 2022, 18, 2104824 CrossRef CAS PubMed.
- J. Zhu, L. Cai, Y. Tu, L. Zhang and W. Zhang, J. Mater. Chem. A, 2022, 10, 15370–15389 RSC.
- J. Zhu, L. Cai, X. Yin, Z. Wang, L. Zhang, H. Ma, Y. Ke, Y. Du, S. Xi, A. T. S. Wee, Y. Chai and W. Zhang, ACS Nano, 2020, 14, 5600–5608 CrossRef CAS PubMed.
- C. Cui, R. Cheng, H. Zhang, C. Zhang, Y. Ma, C. Shi, B. Fan, H. Wang and X. Wang, Adv. Funct. Mater., 2020, 30, 202000693 Search PubMed.
- N. Lv, Q. Li, H. Zhu, S. Mu, X. Luo, X. Ren, X. Liu, S. Li, C. Cheng and T. Ma, Adv. Sci., 2023, 10, 2206239 CrossRef CAS PubMed.
- D. Micheroni, G. Lan and W. Lin, J. Am. Chem. Soc., 2018, 140, 15591–15595 CrossRef CAS PubMed.
- Y. Sun, S. Xu, C. A. Ortíz-Ledón, J. Zhu, S. Chen and J. Duan, Exploration, 2021, 1, 20210021 CrossRef PubMed.
- C.-C. Hou, H.-F. Wang, C. Li and Q. Xu, Energy Environ. Sci., 2020, 13, 1658–1693 RSC.
- Y.-S. Wei, M. Zhang, R. Zou and Q. Xu, Chem. Rev., 2020, 120, 12089–12174 CrossRef CAS PubMed.
- H. D. Liu, M. Cheng, Y. Liu, J. Wang, G. X. Zhang, L. Li, L. Du, G. F. Wang, S. Z. Yang and X. Y. Wang, Energy Environ. Sci., 2022, 15, 3722–3749 RSC.
- X. Ma, H. Liu, W. Yang, G. Mao, L. Zheng and H.-L. Jiang, J. Am. Chem. Soc., 2021, 143, 12220–12229 CrossRef CAS PubMed.
- Q. Zuo, T. Liu, C. Chen, Y. Ji, X. Gong, Y. Mai and Y. Zhou, Angew. Chem., Int. Ed., 2019, 58, 10198–10203 CrossRef CAS PubMed.
- J. Li, H. Huang, P. Liu, X. Song, D. Mei, Y. Tang, X. Wang and C. Zhong, J. Catal., 2019, 375, 351–360 CrossRef CAS.
- X. Fang, Q. Shang, Y. Wang, L. Jiao, T. Yao, Y. Li, Q. Zhang, Y. Luo and H.-L. Jiang, Adv. Mater., 2018, 30, 1705112 CrossRef PubMed.
- Y. Sun, Z. Xue, Q. Liu, Y. Jia, Y. Li, K. Liu, Y. Lin, M. Liu, G. Li and C.-Y. Su, Nat. Commun., 2021, 12, 1369 CrossRef CAS PubMed.
- F. Guo, S. Yang, Y. Liu, P. Wang, J. Huang and W.-Y. Sun, ACS Catal., 2019, 9, 8464–8470 CrossRef CAS.
- S. Su, X. Li, X. Zhang, J. Zhu, G. Liu, M. Tan, Y. Wang and M. Luo, J. Colloid Interface Sci., 2022, 621, 406–415 CrossRef CAS PubMed.
- K. Guesh, C. A. D. Caiuby, A. Mayoral, M. Diaz-Garcia, I. Diaz and M. Sanchez-Sanchez, Cryst. Growth Des., 2017, 17, 1806–1813 CrossRef CAS.
- L. Zhang, R. Si, H. Liu, N. Chen, Q. Wang, K. Adair, Z. Wang, J. Chen, Z. Song, J. Li, M. N. Banis, R. Li, T.-K. Sham, M. Gu, L.-M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2019, 10, 4936 CrossRef PubMed.
- H. Yoshida, S. Nonoyama, Y. Yazawa and T. Hattori, Phys. Scr., 2005, 2005, 813–815 CrossRef.
- P. Kuang, Y. Wang, B. Zhu, F. Xia, C.-W. Tung, J. Wu, H. M. Chen and J. Yu, Adv. Mater., 2021, 2008599 CrossRef CAS PubMed.
- S. Ye, F. Luo, Q. Zhang, P. Zhang, T. Xu, Q. Wang, D. He, L. Guo, Y. Zhang, C. He, X. Ouyang, M. Gu, J. Liu and X. Sun, Energy Environ. Sci., 2019, 12, 1000–1007 RSC.
- K. Jiang, B. Liu, M. Luo, S. Ning, M. Peng, Y. Zhao, Y.-R. Lu, T.-S. Chan, F. M. F. de Groot and Y. Tan, Nat. Commun., 2019, 10, 1743 CrossRef PubMed.
- Y. Qu, B. Chen, Z. Li, X. Duan, L. Wang, Y. Lin, T. Yuan, F. Zhou, Y. Hu, Z. Yang, C. Zhao, J. Wang, C. Zhao, Y. Hu, G. Wu, Q. Zhang, Q. Xu, B. Liu, P. Gao, R. You, W. Huang, L. Zheng, L. Gu, Y. Wu and Y. Li, J. Am. Chem. Soc., 2019, 141, 4505–4509 CrossRef CAS PubMed.
- Q. Ma, C. Hu, K. Liu, S.-F. Hung, D. Ou, H. M. Chen, G. Fu and N. Zheng, Nano Energy, 2017, 41, 148–153 CrossRef CAS.
- Y. Chen, Z. Ren, H. Fu, X. Zhang, G. Tian and H. Fu, Small, 2018, 14, 1800763 CrossRef PubMed.
- T. Liu, P. Diao, Z. Lin and H. Wang, Nano Energy, 2020, 74, 104787 CrossRef CAS.
- U. Kokcam-Demir, A. Goldman, L. Esrafili, M. Gharib, A. Morsali, O. Weingart and C. Janiak, Chem. Soc. Rev., 2020, 49, 2751–2798 RSC.
- K. Gopalsamy and R. Babarao, Ind. Eng. Chem. Res., 2020, 59, 15718–15731 CrossRef CAS.
- T. Wei, Z. Wang, Q. Zhang, Y. Zhou, C. Sun, M. Wang, Y. Liu, S. Wang, Z. Yu, X. Qiu, S. Xu and S. Qin, CrystEngComm, 2022, 24, 5014–5030 RSC.
- J. N. Hall and P. Bollini, React. Chem. Eng., 2019, 4, 207–222 RSC.
- H. Duan, C. Wang, G. Li, H. Tan, W. Hu, L. Cai, W. Liu, N. Li, Q. Ji, Y. Wang, Y. Lu, W. Yan, F. Hu, W. Zhang, Z. Sun, Z. Qi, L. Song and S. Wei, Angew. Chem., Int. Ed., 2021, 60, 7251–7258 CrossRef CAS PubMed.
- J. Yu, A. Wang, W. Yu, X. Liu, X. Li, H. Liu, Y. Hu, Y. Wu and W. Zhou, Appl. Catal., B, 2020, 277, 119236 CrossRef CAS.
- S. Ye, W. Xiong, P. Liao, L. Zheng, X. Ren, C. He, Q. Zhang and J. Liu, J. Mater. Chem. A, 2020, 8, 11246–11254 RSC.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- B. Hammer and J. K. Norskov, Nature, 1995, 376, 238–240 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Zhou, M. Cococcioni, C. A. Marianetti, D. Morgan and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 235121 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- X. Liao, R. Lu, L. Xia, Q. Liu, H. Wang, K. Zhao, Z. Wang and Y. Zhao, Energy Environ. Mater., 2022, 5, 157–185 CrossRef CAS.
- Q. Tang and D.-E. Jiang, ACS Catal., 2016, 6, 4953–4961 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Including ICP-OES, XRD, SEM, HAADF-STEM, WL parameters, XPS, CV, Nyquist, stability results; DFT results comprising optimized atomic configurations, and calculated PDOS, d-band centre, ΔEZPE and ΔS. See DOI: https://doi.org/10.1039/d3nh00159h |
| This journal is © The Royal Society of Chemistry 2023 |