DOI:
10.1039/D2NH00348A
(Communication)
Nanoscale Horiz., 2023,
8, 270-278
Attenuating endothelial leakiness with self-assembled DNA nanostructures for pulmonary arterial hypertension†
Received
26th July 2022
, Accepted 9th December 2022
First published on 15th December 2022
Abstract
Vascular endothelium dysfunction plays an important role in oncological and pulmonary diseases. Endothelial barrier dysfunction is the initial step of pulmonary vascular remodeling (PVR) and pulmonary arterial hypertension. Upregulation of a pro-autophagy protein Atg101 in the endothelial cells triggered a cascade of intracellular events that leads to endothelial dysfunction through apoptosis. Herein, we proposed a strategy that used endothelial targeting DNA nanostructures to deliver Atg101 siRNA (siAtg101) as a safe, biocompatible “band-aid” to restore pulmonary arterial endothelial barrier integrity within the intricate milieu of pulmonary cells and the pulmonary vasculature. The siAtg101 and aptamer conjugated DNA nanostructures were found to attenuate hypoxia-induced pulmonary endothelial leakiness with surprisingly high selectivity and efficacy. Further in vivo study revealed that functionalized DNA nanostructures likewise attenuated the vascular remodeling in a monocrotaline-induced PVR mouse model. Mechanistically, functionalized DNA nanostructures suppressed PVR by knocking down Atg101, which in turn, downregulated Beclin-1 and subsequently upregulated VE-cadherin to restore endothelial cells’ adherin junctions. This work opened a new window for future nanomaterial design that directly addresses the interfacial endothelial cell layer that often stands between the blood and many diseased sites of nanotherapeutic interest.
New concepts
Vascular endothelial cells represent a key target for the treatment of neurological, cardiovascular, oncological, and pulmonary diseases. Discovering and developing new therapeutic targets and drug nanocarriers are highly desirable for nanomedicine. Endothelial leakiness under hypoxia conditions initiates aberrant pulmonary vascular remodeling (PVR) and is believed to be the initial step of this process. In contrast to our previous study that nanoparticles induce endothelial leakiness, herein, we propose using programmable DNA self-assemblies to restore endothelial barrier functions and subsequently attenuate PVR. The current proof-of-concept study provides a new strategy and nanomaterial platform for the therapy of noncancerous, vascular endothelium-related diseases.
|
Introduction
The vascular endothelium represents a major target in various vascular-related diseases in nanomedicine.1 Aberrant increased endothelial leakiness is the cause and often possible feedback dire outcome of many important diseases such as cancer, acute respiratory distress syndrome, inflammation, and atherosclerosis.2 This phenomenon is highly associated with disease progress and is the key to biomaterial and bionanotechnology interferences. One of the abnormal endothelial leakiness phenomena is the enhanced permeability and retention (EPR) effect although there are several controversial literatures that may debunk this assumed increased leakiness.3,4 However, numerous mice studies showed increased drug delivery to tumours using nanoparticles through the tumour vascular endothelium without confirming the mechanism of accessibility. One possible explanation to resolve this conundrum is that the nanoparticles themselves can induce endothelial leakiness (NanoEL).5–12 NanoEL was observed to have occurred in vitro and in vivo even without the presence of tumors.13,14 The non-essentiality of the tumor in NanoEL therefore also expands the scope of designed increased leakiness to be beyond just cancer applications. NanoEL works on the basis of the indirect effects of nanomaterials often through the use of drug or molecule or ion release to the endothelial cells to bring about that important endothelial leakiness.15 Unlike pathogenic stimuli provoked endothelial hyperpermeability, NanoEL provides an alternative way to manipulate endothelium barrier integrity for desirable intervention in many non-cancer disease treatments.
Pulmonary arterial hypertension (PAH) is a vital lung disease that will eventually lead to right ventricular failure. Pulmonary vascular remodeling and loss of small arterial vessels are the pathological basis of PAH. The apoptosis of human pulmonary arterial endothelial cells (HPAECs) and dysregulation of endothelial barrier function is the first step of PVR.16 There have been numerous studies investigating the underlying molecular mechanisms and crosstalk between the endothelial cells and arterial smooth muscle cells.17,18 Atg101 is an autophagy-related gene and plays an important role in regulating autophagy levels in HPAECs.19 It was reported that Atg101 upregulated autophagy levels under hypoxia conditions.20 Yet studies on endothelial cells as the main therapeutic target and utility of nanomaterials and nanotechnology as an intervention strategy are rarely reported for PVR.21 Considering that endothelial cells suffer from collateral damage from detrimental stimuli and are the unintended targets of nanomedicine, we reasoned that therapeutic nanomaterials that can target and restore endothelial integrity would be highly desirable. We further reckoned that if we could specifically target HPAECs and repair endothelial leakiness, we would prevent the subsequent apoptosis-resistant HPAEC proliferation, thus stemming the early progress of PVR.22
DNA nanostructures provide an excellent nanomaterial platform with remarkable structural control and great potential of incorporating multiple functionalities.23 This class of nanomaterials is nontoxic, low immuno-responsive and biocompatible, thus favorable for biomedical applications.24,25 Previously, we found that DNA nanostructures had higher cellular uptake efficiencies compared to single-stranded siRNA in vulnerable primary cells such as pulmonary arterial smooth muscle cells and microvascular endothelial cells, indicating that DNA nanomaterials are suitable drug carriers for nanomedicines targeting the vasculature.26,27 In addition, the advantage of using DNA nanostructures to recover endothelial integrity is that the density of DNA is quite low, thus avoiding the risk of side effects of high density that might be detrimental to the endothelial barrier.
In this study, we proposed that endothelial leakiness under pathological conditions such as hypoxia can be significantly attenuated with functionalized DNA nanostructures. The DNA aptamer decorated on DNA nanostructures specifically recognizes HPAECs and siAtg101 cargo is then internalized into HAPECs and inhibits cell autophagy and subsequently abnormal proliferation. The targeting ability of the functionalized DNA nanostructures was also investigated by deliberate positioning of the aptamer on the global structure. Confocal laser scanning microscopy (CLSM) and transwell assay results demonstrated that the hypoxia-induced endothelial leakiness was greatly reduced after being treated with DNA nanostructures. The current study opened a new window of using DNA nanomaterials to recover the endothelial barrier integrity that is disrupted by detrimental stimuli.
Experimental
Materials and antibodies
All short DNA strands were purchased from Sangon Biological Engineering (Shanghai, China). siRNA strands were synthesized by Genepharma. The Acryl/Bis Solution (40%, 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), Tris, EDTA-Na, acetic acid, boric acid, magnesium acetate, Hoechst 33342, and 100 bp DNA Ladder were bought from Sangon. The “Stains-all” for gel staining was purchased from Sigma Aldrich, (USA). ECM was obtained from ScienCell (USA), and 0.25% trypsin was ordered from Gibco (USA). Cytotoxicity and Cell Proliferation Kits were purchased from Beyotime Biotechnology (Shanghai, China). X-tremeGENE HP DNA Transfection Reagent and FastStart Essential DNA Green MasterMix were purchased from Roche (Basel, Switzerland). FITC-dextran was ordered from MedChemExpress (MCE, USA). All antibodies were obtained as follows: Atg101 Rabbit mAb (Cell Signaling Technology, E1Z4W), Beclin-1 (Cell Signaling Technology, D40C5), VE-cadherin Rabbit mAb (Cell Signaling Technology, D87F2), CD31 Rabbit mAb (abcam, RM1006), LC3 Rabbit mAb (Cell Signaling Technology, D11), and Cyto-Green autophagy dye kits (Enzo Life Sciences Company, ENA-510310K200).
:1), Tris, EDTA-Na, acetic acid, boric acid, magnesium acetate, Hoechst 33342, and 100 bp DNA Ladder were bought from Sangon. The “Stains-all” for gel staining was purchased from Sigma Aldrich, (USA). ECM was obtained from ScienCell (USA), and 0.25% trypsin was ordered from Gibco (USA). Cytotoxicity and Cell Proliferation Kits were purchased from Beyotime Biotechnology (Shanghai, China). X-tremeGENE HP DNA Transfection Reagent and FastStart Essential DNA Green MasterMix were purchased from Roche (Basel, Switzerland). FITC-dextran was ordered from MedChemExpress (MCE, USA). All antibodies were obtained as follows: Atg101 Rabbit mAb (Cell Signaling Technology, E1Z4W), Beclin-1 (Cell Signaling Technology, D40C5), VE-cadherin Rabbit mAb (Cell Signaling Technology, D87F2), CD31 Rabbit mAb (abcam, RM1006), LC3 Rabbit mAb (Cell Signaling Technology, D11), and Cyto-Green autophagy dye kits (Enzo Life Sciences Company, ENA-510310K200).
DNA and siRNA sequences. D1: 5′-TTCAATCTCTATCATTCCGTCCTTCAATCTCTATCATTCCGTCCTTCAATCTCTATCATTCCGTCC-3′; D2 (for DTA-V1): 5′-TGACTGCGAGGAGAAGGTGGGTGAGAAATTTTAGAGATTGAAGGACGGAATG-3′; D2′ (for DTA-V2): 5′-GAGAAGGTGGGTGAGAAATTAGAGATTGAAGGACGGAATGTGACTGCGAG-3′; D2′′ (for scrambled siAtg101 loaded DNA triangles): 5′-TGACTGCGAGTACTTTTGTGTAGTACAATTTTAGAGATTGAAGGACGGAATG-3′; DNA aptamer: 5′-GATGTGAGTGTGTGACGAGCTACGACGTCTGGTGTAATTTATAAAGACACTGTGTATATCAACAACAGAACAAGGAAAGGCTCGCAGTCA-3′; D3 (siAtg101): 5′-UUUCUCACCCACCUUCUCCCG-3′; scrambled siAtg101: 5′-UUGUACUACACAAAAGUACUG-3′.
Synthesis and characterization of DNA nanostructures
DNA strands D1, D2, and D3 were mixed at 1:3:3 (molar ratio) in Tris–acetic–EDTA–Mg2+ (TAE/Mg2+) buffer (pH 8.0) and then subjected to the following annealing process: 95 °C/5 min, 65 °C/30 min, 50 °C/30 min, 37 °C/30 min, and 22 °C/30 min. Then, DNA aptamers were added into the pre-annealed DNA triangle without aptamer (DTA) solution at 3:1 (molar ratio) at room temperature for 30 min. 6% native PAGE in an electrophoresis unit (Bio-Rad) was employed to analyze the DNA nanostructure formation (constant voltage, 120 V). The buffer used for electrophoresis was TAE/Mg2+. ‘Stains-All’ solution (0.01%) was used for gel staining. For DLS measurement, the two versions of DNA triangles with aptamers (DTA-V1/V2) were diluted to 100 nM and then measured by a Zetasizer (Malvern, UK). The solvents were TAE/Mg2+.
Cell culture and cellular uptake studies
HPAECs were cultured in ECM medium (ScienCell). The medium contained 10% FBS and 1% penicillin–streptomycin. A humidified CO2 atmosphere (5%) at 37 °C was applied for cell growth. CLSM was employed to evaluate the cellular uptake efficiencies. siAtg101 was modified with a fluorophore and used for DNA triangle synthesis in TAE/Mg2+ buffer. Cells (2 × 104 cells per cm2) were pre-cultured for 24 h on a coverslip in a 24-well plate. Thereafter, Cy3-tagged DTA-V1 (Cy3 concentration was 300 nM) was incubated with the cells for 24 h. The ECM medium and DNA sample volume ratio was kept at 9:1. Cells were then stained with Hoechst 33342. Before being observed by Leica DMRA2 CLSM, the cells were washed with PBS 5 times. 4% paraformaldehyde was used to fix cells (15 min).
Cell viability determined by MTT assay
HPAECs were pre-cultured for 12 h in a 96-well plate. The proliferation of DNA nanostructure-treated cells was evaluated by MTT. HPAECs were cultured with different sample groups for 24 h in a 96-well plate. The final concentrations of the DNA samples are: DTA-scAtg101 (scrambled siAtg101-loaded DNA triangles, 200 nM), scrambled siAtg101 (600 nM), siAtg101 (600 nM), DTA (200 nM), and DTA-V1 (200 nM). The cells were then processed with MTT before being detected by a microplate reader.
Western blot
HPAECs were cultured for 12 h in a 6 cm Petri dish, and then incubated with DTA-scAtg101 (200 nM), siAtg101 (600 nM), DTA (200 nM), and DTA-V1 (200 nM) separately for 24 h under hypoxia. For western blot analysis, the sample was washed with PBS and then incubated with 100 μL protein lysis buffer containing protease inhibitor cocktail for 15 min. After that, the protein was harvested and centrifuged at 12000 rpm for 10 min at 4 °C and the supernatant was collected. The concentration of protein was measured by ultraviolet spectrophotometer and the sample was separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis before being transferred onto a polyvinylidene difluoride membrane. After that, the membrane was blocked by 5% non-fat milk diluted in TBST for 1 h before incubating the following antibodies: Atg101 (1:1000, Cell Signaling Technology, E1Z4W), LC3 (1:1000, cell signaling technology D3U4C), Beclin-1 (1:1000, Cell Signaling Technology, D40C5), VE-cadherin (1:1000, Cell Signaling Technology, D87F2), β-actin (1:1000, cell signaling technology, 8H10D10) overnight at 4 °C. Subsequently, the blots were washed with PBS 3 times before being incubated with second antibody Peroxidase-AffiniPure Goat Anti-Rabbit IgG (H+L) (Jackson immunoresearch, #111-035-0003) for 1 h. The blots were washed and analyzed by the enhanced chemiluminescence system (Bio-Rad ChemiDoc MP). The β-actin antibody was considered as a loading control.
Scratch assay
HPAECs were cultured in a 6-well-plate and cultivated to 80% confluence, and the cells were cultured in serum-free medium before scratching with a 200 μL pipette tip. After that the cells were incubated with DTA-scAtg101 (200 nM), siAtg101 (600 nM), DTA (200 nM), and DTA-V1 (200 nM) in serum-free medium for 24 h under hypoxia. Then the microscopic view was observed under an inverted microscope at 4 × 20magnification.
Animal model
All animal experiments were performed in line with the guidelines of the “Laboratory animal-Guideline for ethical review of animal welfare” (People's Republic of China National Standard GB/T 35892-2018), and the “Animal Care and Use Committee of the Third Military Medical University”. All animal experiments were approved by the Animal Ethics Committee of Third Military Medical University, Chongqing China. 6-week-old female mice were supplied from Beijing Vital River Laboratory (Beijing, China). Monocrotaline (600 mg kg−1) was administered by subcutaneous injection using sterilized syringes with 26-gauge needles (Terumo Corporation, Tokyo, Japan) once a week 4 times. The dosage of siRNA decorated on the DNA nanostructures is 5 mg kg−1 and it was injected by vein injection. The same doses of DTA-scAtg101 and DTA were injected as negative controls.
Hematoxylin-eosin staining
Mouse lungs were removed and fixed with 4% paraformaldehyde, and the fixed tissues were embedded in paraffin and cut into 5 μm slices. Next, after the tissue slides were dewaxed by xylene and rehydrated by alcohol gradient, hematoxylin and eosin staining was used to analyze the morphometric changes of the pulmonary artery.
Immunofluorescence
Mouse lungs were excised and were frozen at −80 °C with optimal cutting temperature compound. After that, the tissues were cut into slices with a thickness of 5 μm. Next, the tissue slides were fixed with 4% formaldehyde for 15 min at room temperature. And then the specimen was blocked in a blocking buffer for 60 min at RT. After aspirating the blocking buffer, the sample was incubated with the prepared primary antibody by diluting with Antibody Dilution Buffer at 4 °C overnight. The samples were washed with PBS for 5 min three times. The samples were incubated with fluorescent secondary antibody at 37 °C for 1 hour in the dark and washed with PBS for three times, and then analyzed by CLSM.
Autophagy detection
HPAECs were pre-cultured overnight on a coverslip in a 24-well plate. Different DNA nanostructures were added to the cells followed by exposure to normal or hypoxic conditions for 48 h. The Cyto-ID® Green autophagy dye was used to detect autophagy. The dye solution was first mixed with medium at 1:1000 ratio, and then added into cells for 30 min at 37 °C. The cells were washed 3 times with PBS before CLMS imaging.
Transwell assay
A 24 well HTS Transwell permeable support (Corning; Cat No. 3396) was used to detect HPAEC permeability under hypoxic conditions. HPAECs were pre-cultured overnight in the transwell, and then HPAECs were incubated with siAtg101, DTA, and DTA-V1 for 6 h. After that, the complete medium was replaced and incubated for another 48 h under hypoxia. The upper chambers were incubated with 100 μL fluorescein isothiocyanate (FITC)-dextran for 2 h. Spectrophotometry was used to assess the intensity of the dye in the lower chamber at 485 nm excitation and 520 nm emission.
Echocardiography analysis
Echocardiography was carried out on a VisualSonics Vevo 2100 (FujiFilm VisualSonics Inc.). DTA-V1 (5 mg kg−1) was injected into the veins of PVR mice once a week 4 times. The cardiac functions of the mice were then detected.
Results and discussion
Synthesis and characterization of DNA nanostructures with aptamers
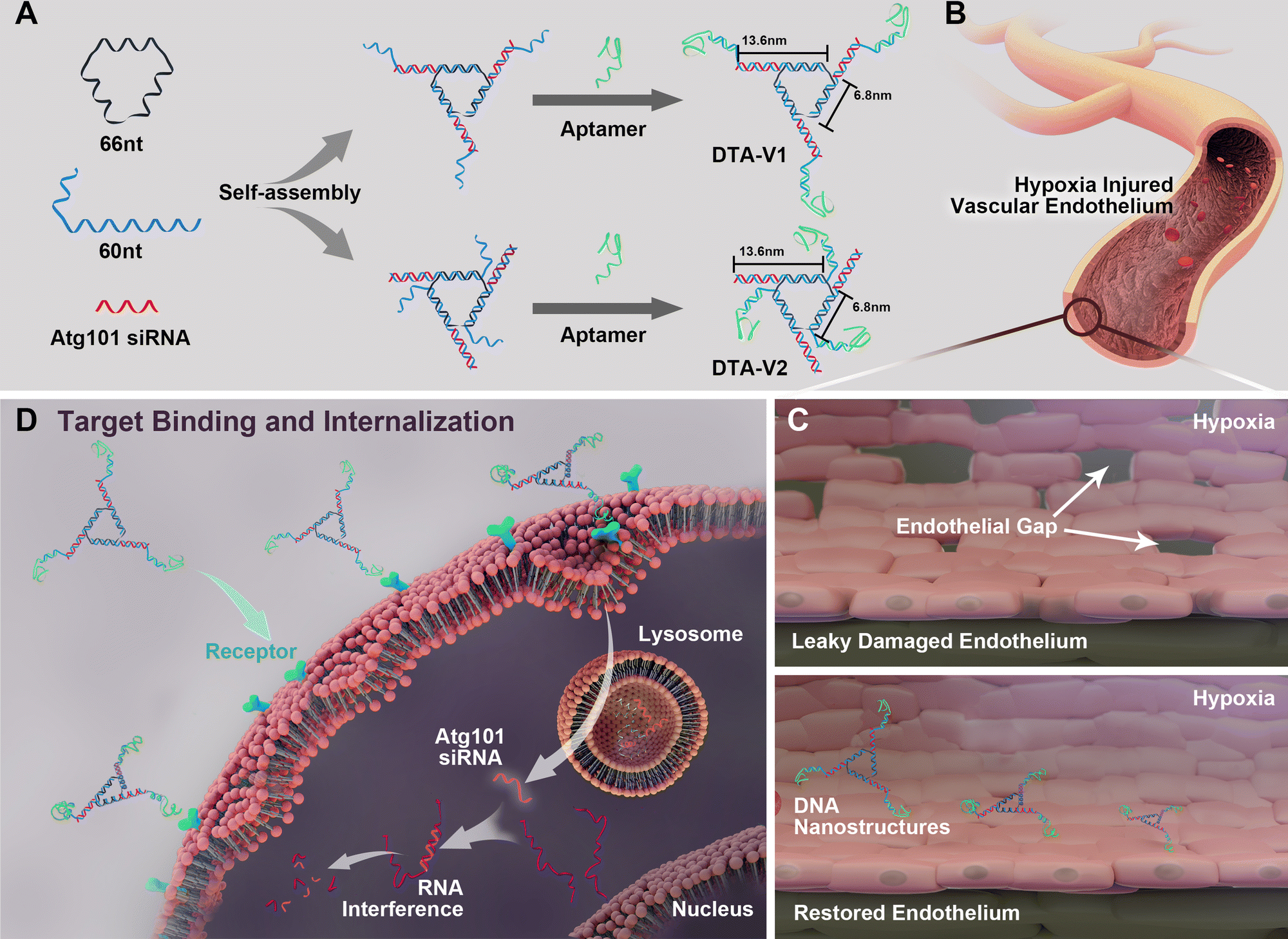
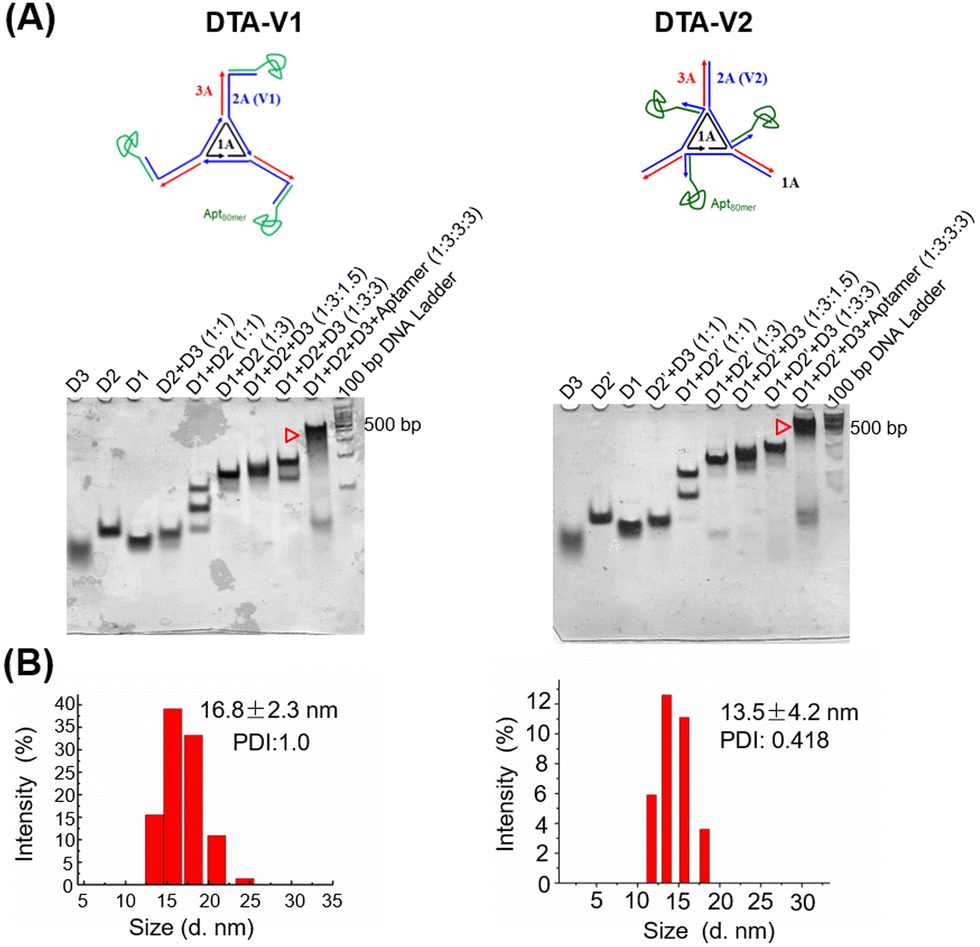
As illustrated in Scheme 1, the DNA triangle with aptamers consists of a central strand D1, three side strands D2, three D3 (siAtg101) and three DNA aptamers (detailed base pairing of the nanostructures can be seen in Fig. S1, ESI†).28 Aptamers were either positioned on the outer points of the triangle (DTA-V1) or at the corners of the structure core (DTA-V2). The secondary and higher-order structures of the aptamers are essential for their functioning. To ensure the correct folding of the aptamer, a 10-base long stem was introduced in between the triangle core and aptamer sequences to endow enough flexibility. We reasoned that three aptamers at the same plane could directly engage the cell surface at the same time for strong and specific binding. Moreover, the rationale for putting the aptamer at the edge is that it allows for easy and direct access to HPAECs, while closer to the structural center confers more protection to aptamers and probably lower binding kinetics. DTA-V1 binds to HPAECs and delivers siAtg101 into cells (Scheme 1B and C). The internalization mechanism might be the aptamer-mediated endocytosis pathway.29 Although the endosome or lysosome escape efficiency might be low, DNA nanostructures somehow could silence target gene expressions after entering into cells.30–32 siAtg101 then interferes with autophagy-related cellular machinery to inhibit the apoptosis-resistant HPAEC proliferation and attenuated endothelial leakiness. Native polyacrylamide gel electrophoresis (PAGE) results indicated that both DTA-V1 and DTA-V2 formed dominant, sharp bands on the gel with reasonable mobilities (Fig. 1A). The yields of DTA-V1 and DTA-V2 were 76.5% and 62.6%, respectively. Dynamic laser scattering (DLS) measurements further confirmed the formation of DNA triangles with aptamers (Fig. 1B). The hydrodynamic diameters of DTA-V1 and DTA-V2 were determined to be 16.8 ± 2.3 and 13.5 ± 4.2 nm, respectively. These results suggested the successful synthesis of DNA nanostructures and good correlation to the theoretical designs.
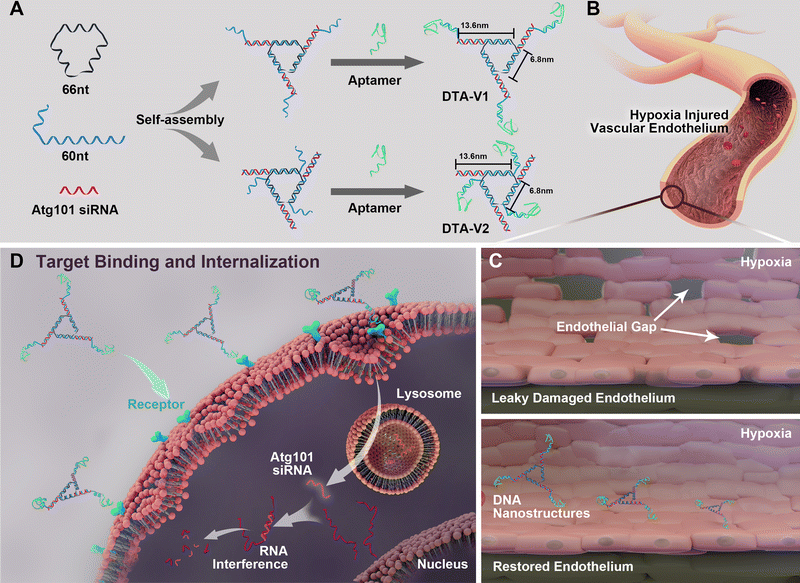 |
| | Scheme 1 (A) Design of DNA aptamer and Atg101 siRNA (siAtg101) conjugated DNA nanostructures. DNA aptamers are positioned either at the protruding points (DTA-V1) or the corners of the structure core (DTA-V2). (B) Aptamer-decorated DNA nanostructures bind to HPAECs and are subsequently internalized (C and D). The DNA nanostructures might be internalized through aptamer-mediated endocytosis.28 The embedded siRNA takes effect and restores endothelial integrity similar to the reversal of “NanoEL”. | |
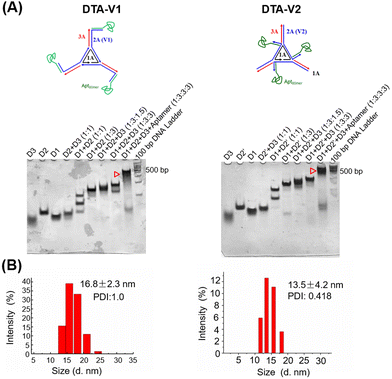 |
| | Fig. 1 Design, synthesis, and characterization of DNA triangles with aptamers. (A) Native PAGE analysis of the synthesized DNA triangles with aptamers, DTA-V1 and DTA-V2. Both designs showed high synthesis yields. (B) DLS measurements of DTA-V1 and DTA-V2. DNA nanoparticles were diluted to 100 nM before measurement. | |
Cellular uptake of DTA-V1
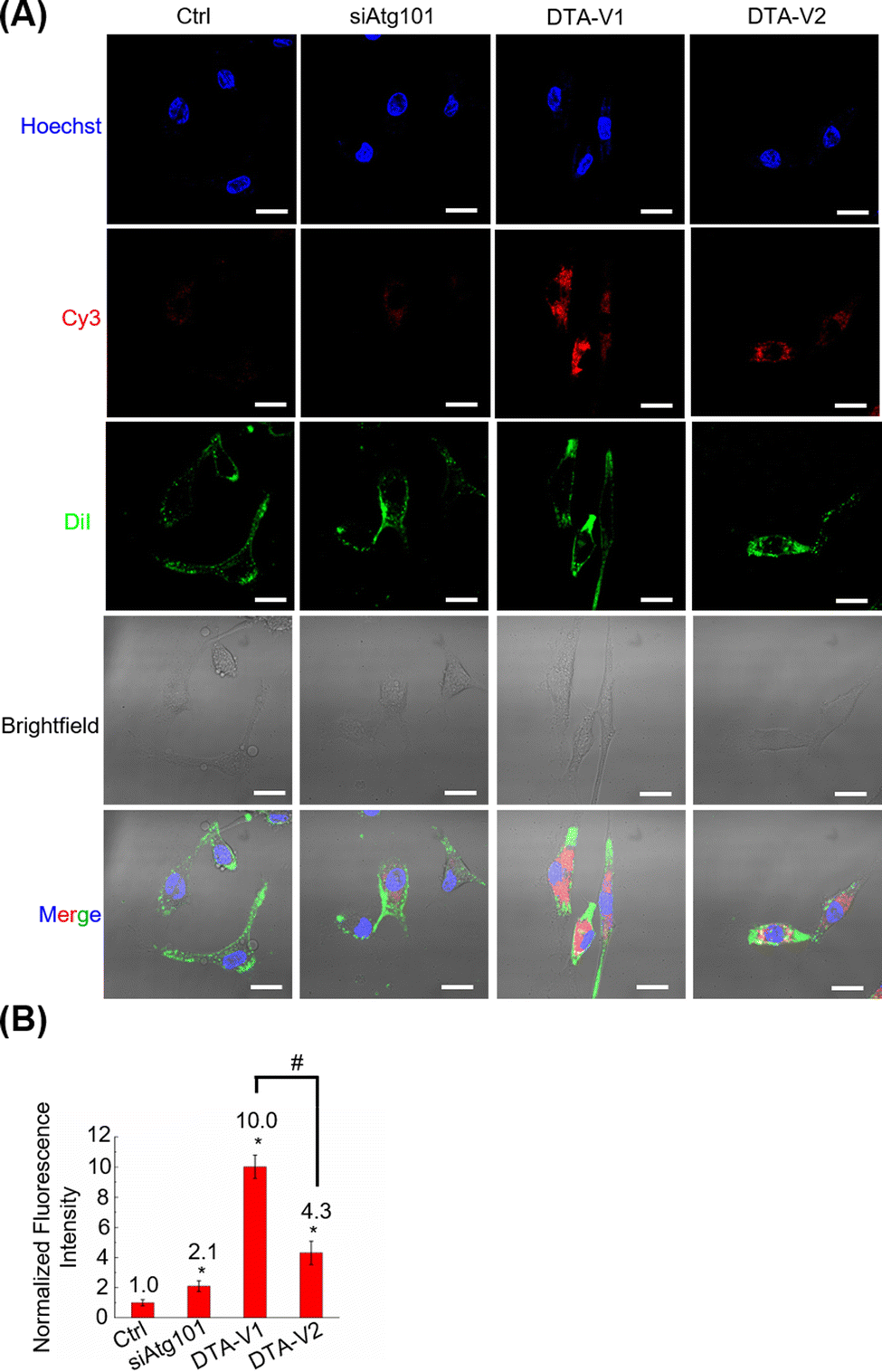
The cellular uptake and targeting capability of DTA-V1 and DTA-V2 were evaluated by CLSM. It was found that both DTA-V1 and DTA-V2 were readily taken up by HPAECs (Fig. 2A). To further confirm the internalization of DTA-V1 and DTA-V2, we performed Z-stacking during CLSM imaging. The internalized red dots were scattered in the cytoplasm at multiple focus layers (see video file in supporting materials). DTA-V1 had a much higher cellular uptake efficiency compared with DTA-V2. Quantitative analysis indicated that the cellular uptake efficiency of DTA-V1 was 2.3 times higher than that of DTA-V2 (Fig. 2B). The higher uptake efficiency of DTA-V1 was probably attributed to the fact that the aptamers were more exposed to the environment compared with DTA-V2 in a short time window of 24 hours in the current experiment setting. It is worth noting that although the fluorescence intensity of the siAtg101 group was higher than that of the control group, the naked siRNA may not function well in harsh physiological settings. Endothelial cells have high heterogeneity and complex interactions with nanoparticles. Binding and internalization to endothelial cells with high efficiency and specificity is the first step and critical for endothelial-targeted nanomedicine. In the current study, DNA aptamers that can bind to HPAECs were utilized to anchor DNA triangles onto endothelial cells and allow for subsequent functioning, thus greatly avoiding unfavourable interaction with the endothelium. More importantly, the superior structural control of the DNA nanostructure guarantees the targeting performances of the aptamer in the current design and other potential targeting strategies.
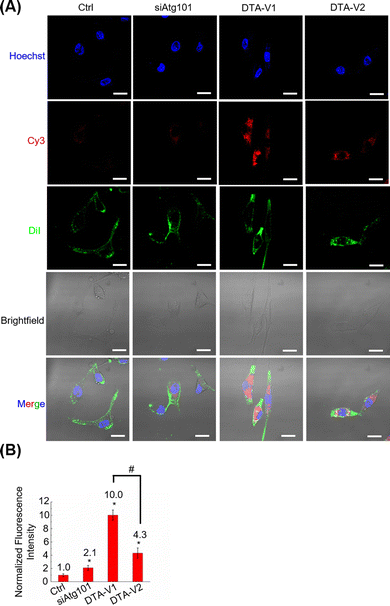 |
| | Fig. 2 CLSM imaging of the cellular uptake of DTA-V1 by HPAECs. (A) CLSM imaging of the cellular uptake of DTA-V1 and DTA-V2. Cells were treated with Cy3-tagged siAtg101, DTA-V1 and DTA-V2 for 24 h. Cells received DTA-V1/V2 and Cy3 at 100 and 300 nM, respectively. DiI was employed to stain cell membranes. Scale bar: 20 μm. (B) Quantitative analysis of the cellular uptake efficiencies of siAtg101, DTA-V1 and DTA-V2 in (A). Data represent the mean value ± SD (n = 3). p-Values were calculated using one-way ANOVA, *p < 0.05 compared with Ctrl; #p < 0.05. | |
DNA nanostructures restored endothelial barrier integrity in vitro
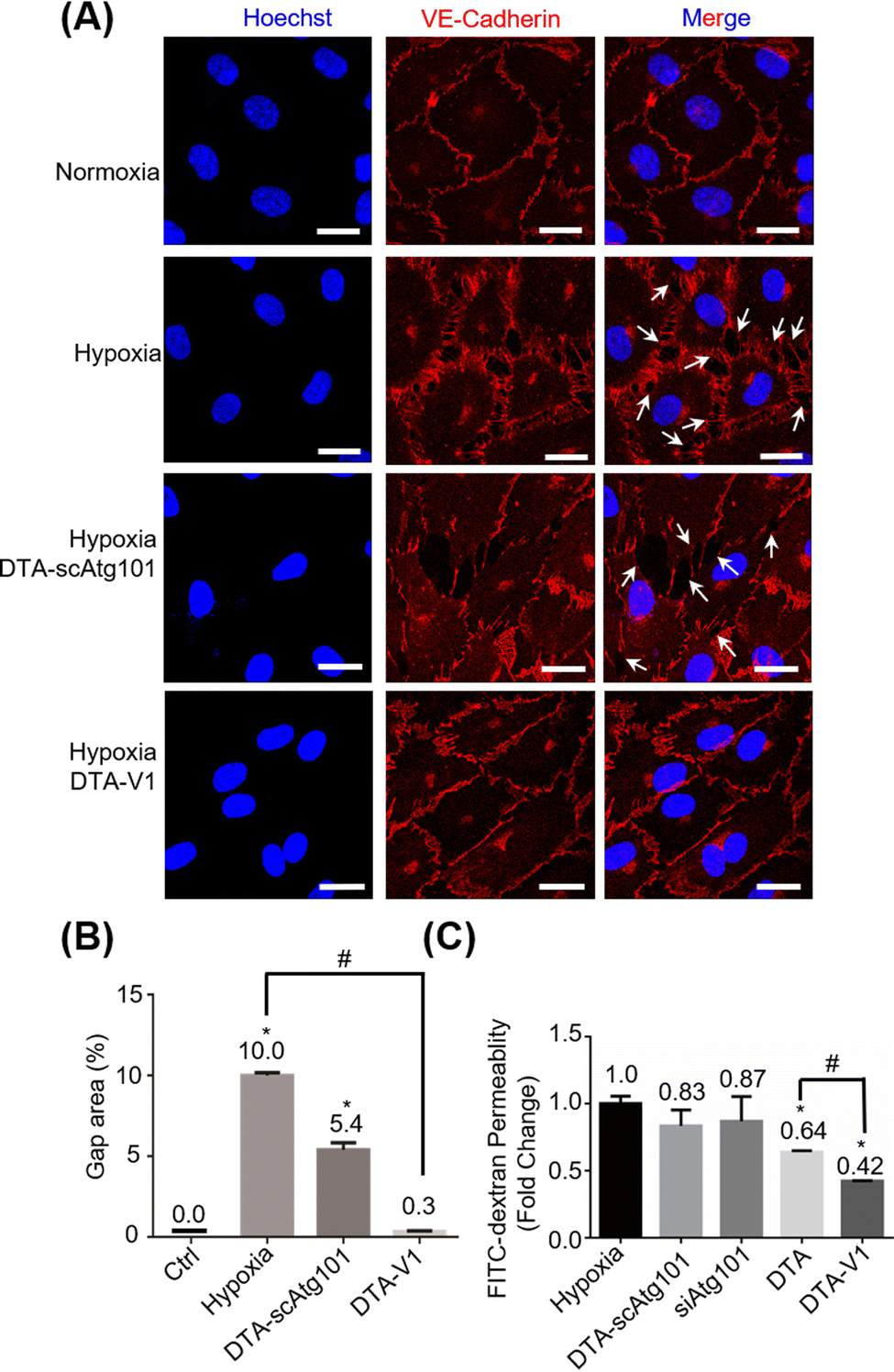
Under hypoxia and other pathological conditions, the apoptosis-resistant HPAECs formed irregular gaps and triggered PVR. To evaluate the capability of DTA-V1 to restore endothelial integrity, CLSM imaging of the endothelial integrity under different experimental conditions was performed (Fig. 3A). Under hypoxia, there were pronounced endothelial gaps found compared with the normoxia group. Under the same hypoxia conditions, after treatment with DTA-V1, the endothelial gaps were significantly reduced, while the scrambled siAtg101-loaded DNA triangles (DTA-scAtg101) group caused similar endothelial disruption as the hypoxia group. Semi-quantitative analysis of the gap area indicated that hypoxia-induced a pronounced endothelial leakiness that was ten times higher than that of normal endothelium (Fig. 3B). While for the DTA-V1 treated group, the endothelial gap area was reduced to 0.3% compared to that of 10% of the hypoxia group. These morphological results suggested that DTA-V1 indeed restored the HPAEC-formed endothelial integrity under hypoxia conditions, and the endothelial integrity restoration was probably attributed to the siAtg101 drug. We further conducted a transwell assay to examine the ability of DTA-V1 in attenuating endothelial leakiness (Fig. 3C). It was found that the hypoxia and DTA-scAtg101 group had severe endothelial leakiness as measured by the FITC fluorescence intensities. The fluorescence signal of the DTA-V1-treated group was 42% of the hypoxia group, indicating that the endothelial leakiness was greatly reduced under the hypoxia condition. This result correlated well with the morphological results and demonstrated the robust therapeutic effect of DTA-V1.
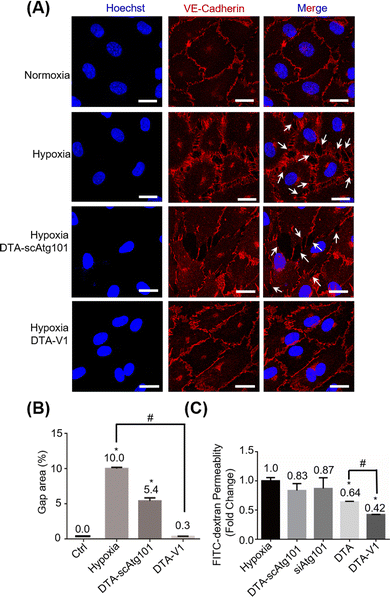 |
| | Fig. 3 Restoration of endothelial barrier function with DTA-V1 under hypoxic conditions. (A) CLSM imaging analyzed the restoration of the endothelial barrier after DTA-V1 treatment for 48 h under hypoxia conditions. The scrambled siAtg101-loaded DNA triangles (DTA-scAtg101) were also applied as a control. Scale bar: 20 μm. (B) Quantification analysis of the formed endothelial barriers in (A), p-values were calculated using one-way ANOVA, *p < 0.05 compared with Ctrl; #p < 0.05. (C) Quantitative analysis with a transwell insert permeability assay showed a significant reduction of permeability following the DTA-V1 treatment. DTA refers to siAtg101-loaded DNA triangles without aptamers. p-Values were calculated using one-way ANOVA, *p < 0.05 compared with hypoxia group; #p < 0.05. All the data of the figures represent the mean value ± SD (n = 3). | |
DNA nanostructures inhibited HPAEC growth by the Atg101/Beclin-1/VE-cadherin pathway
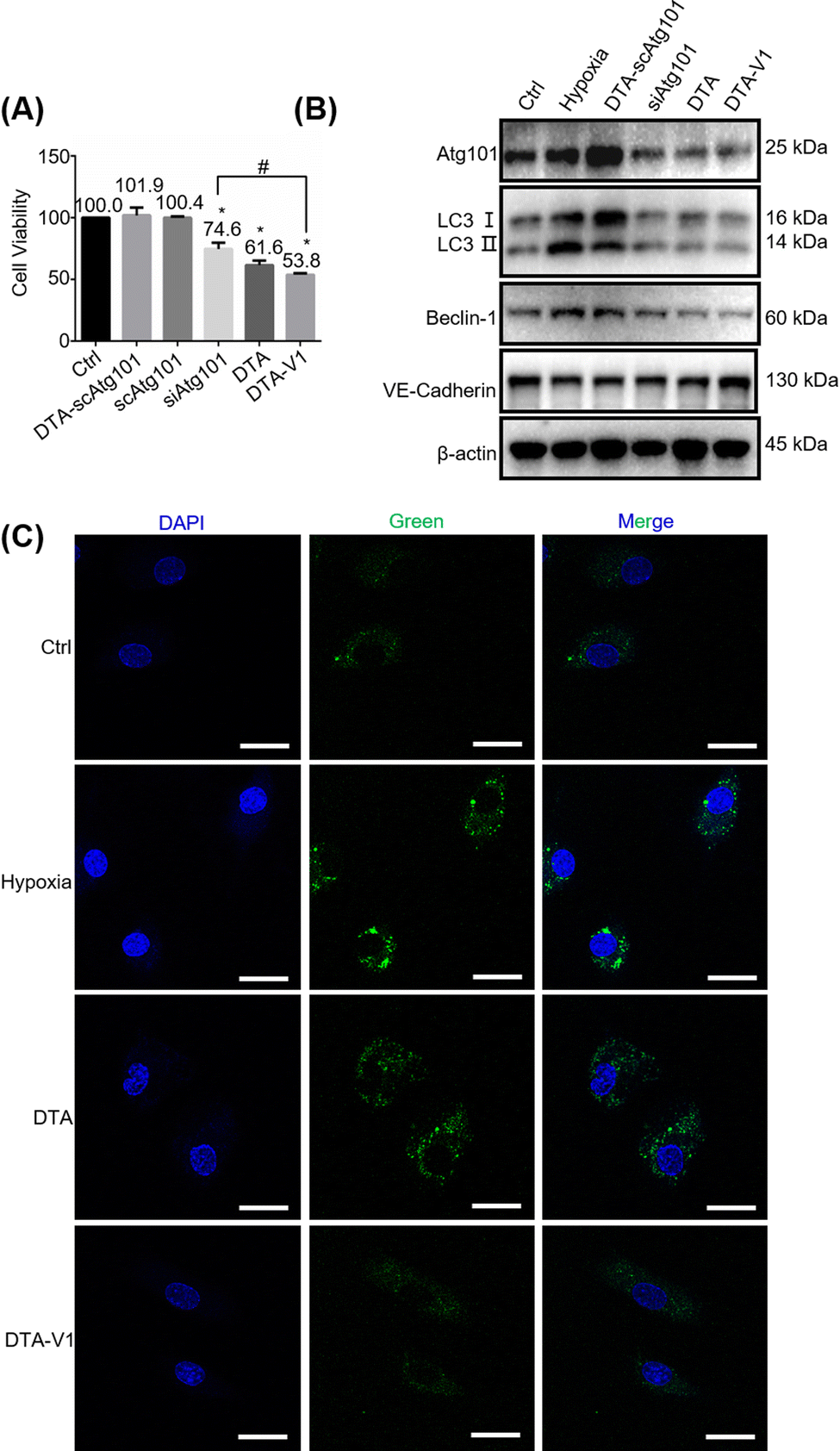
To further understand the role of DTA-V1 in protecting the endothelial barrier, we firstly investigated the HPAEC cell viability by methyl thiazolyl tetrazolium (MTT) assay. Fig. 4A shows that DTA-V1 effectively suppressed the HPAEC growth compared with the scrambled siAtg101 and siAtg101 groups. Further scratch assay indicated that DTA-V1 effectively suppressed HPAEC proliferation under hypoxia conditions (Fig. S2, ESI†). It was reported that the autophagy levels were aberrantly elevated under hypoxia conditions.33 Atg101 is an autophagy-related gene that regulates the autophagy flux by forming an autophagy complex with ULK1.34 Western blot results indicated that the Atg101 expression level was significantly knocked down upon DTA-V1 treatment (Fig. 4B and Fig. S3, ESI†). Quantification analysis indicated that Atg101 was knocked down by 46.2% for the DTA-V1 treated group compared to the hypoxia group (Fig. S4, ESI†). These results suggested that DTA-V1 inhibited HPAEC growth by successfully knocking down Atg101 expression.
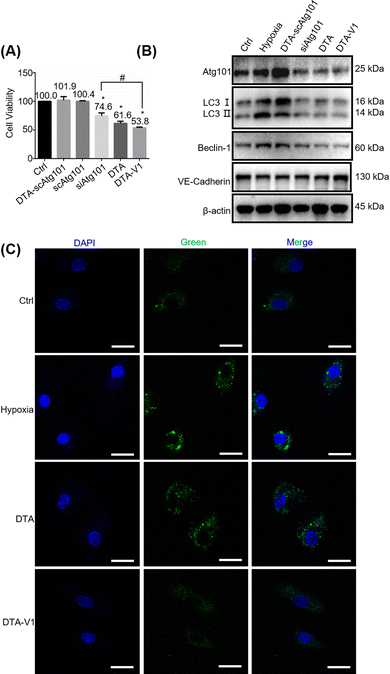 |
| | Fig. 4 DTA-V1 inhibited HPAEC growth by the Atg101/Beclin-1/VE-cadherin pathway. (A) The MTT assay showed that HPAEC cell proliferation was inhibited by DTA-V1 and DTA (siAtg101-loaded DNA triangles without aptamers). Different samples were incubated with HAPECs for 24 hours. The siAtg101 concentration was 600 nM. All the data of the figures represent the mean value ± SD (n = 5). p-Values were calculated using one-way ANOVA, *p < 0.05 compared with Ctrl group; #p < 0.05. (B) Western blot analysis of the siAtg101 knockdown efficiency and the downstream signalling pathways. Knockdown of Atg101 inhibited HPAEC cell autophagy through downregulating Beclin-1. Inhibition of Beclin-1 upregulated VE-cadherin. (C) CLSM analysis of the inhibition of cell autophagy after treatment with DTA-V1 for 48 h under hypoxia. Accumulated autophagic vacuoles were stained fluorescent green. Scale bar: 20 μm. | |
To elucidate the downstream signaling pathways triggered by Atg101 inhibition, we first evaluated the autophagy marker gene LC3. Western blot results showed that the LC3B II/I expression decreased to 25% compared with the hypoxia group (Fig. S4, ESI†), indicating a pronounced autophagy inhibition. We next detected the autophagosome using CLSM to further confirm the reduction of cell autophagy. As shown in Fig. 4C, the green fluorescence intensity of DTA-V1 treated HAPECs was greatly reduced compared with the hypoxia group, suggesting a decreased autophagy activity. It was reported that Atg101 participates in autophagy through the activation of Beclin-1.35 We thus investigated the expression of Beclin-1. As expected, Atg101 knockdown significantly downregulated Beclin-1 expression (Fig. 4B and Fig. S4, ESI†). Leonard et al. reported that silencing Beclin-1 protects the endothelial barrier by inhibiting the loss of VE-cadherin through thrombin-induced phosphorylation of Confilin-1.36 The expression of VE-cadherin was upregulated for the DTA-V1-treated group, as expected (Fig. 4B and Fig. S4, ESI†). The upregulated VE-cadherin was in agreement with the restored endothelial integrity displayed in Fig. 3. Taken together, we demonstrated that knockdown of Atg101 inhibited Beclin-1 expression and HPAEC autophagy, and subsequently upregulated VE-cadherin expression. The knockdown of Atg101 by DTA-V1 eventually inhibited HPAEC growth and restored the endothelial integrity. This process was actually a reversal of the “NanoEL” effect by interfering with the intracellular signaling pathway with DNA nanostructures.
DNA nanostructures attenuated PVR in vivo
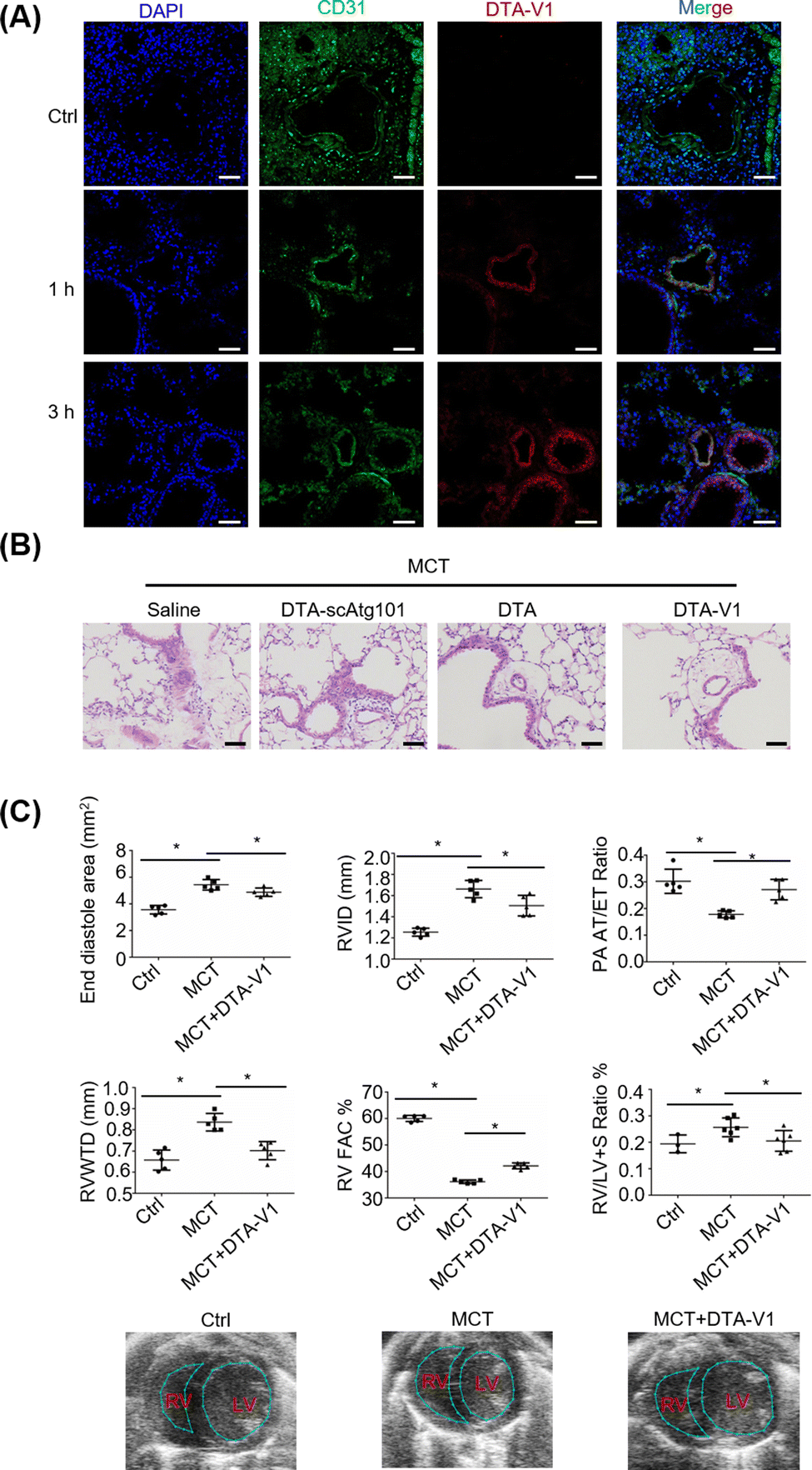
To validate the therapeutic effects of DTA-V1 in vivo, we further built a monocrotaline-induced PVR mouse model (MCT mice). DTA-V1 was injected into MCT mice through the tail vein. Cy3-tagged DTA-V1 was employed to examine the accumulation of DNA nanostructures in pulmonary vessels. Immunofluorescence results indicated that the red fluorescence increased in a time-dependent manner around the pulmonary vascular walls (Fig. 5A). The accumulation of DTA-V1 in the pulmonary vascular wall region became pronounced at the 3 h time point. Meanwhile, DTA-V1 colocalized with the vascular endothelium (CD31), indicating that DTA-V1 is indeed bound to endothelial cells. Next, we evaluated the therapeutic effects of DTA-V1 by intravenous administration of DTV-V1 at a dosage of 5 mg kg−1 three times. Hematoxylin–eosin staining showed that the vascular wall of the MCT mice was severely thickened (Fig. 5B). In contrast, DTA-V1 significantly attenuated PVR compared to the DTA-scAtg101 group and DTA (DNA triangles without aptamers) group. We further investigated the phenotype by echocardiographic analysis. As shown in Fig. 5C, the PVR model was successfully established by week 6 post-MCT exposure. MCT mice showed significant increases in the end diastole area, right ventricular interior dimension at diastole (RVID) and RV wall thickness (RVWTD), as well as significant reductions in RV fraction area change (FAC) % and pulmonary artery (PA) acceleration time/ejection time (AT/ET) ratio. These parameters suggest that MCT mice developed adverse RV remodeling, impaired RV contractility and PA dysfunction. However, DTA-V1 treatment led to significant protection against RV and PA dysfunction in MCT mice. Notably, the DTA-V1 treatment completely suppressed the increase of the end diastole area, RVID and RVWTD observed in MCT mice, and greatly restored the RV FAC % and PA AT/ET ratio. In agreement with these echocardiographic results, the calculated RV versus LV + S (septum) weight ratio showed a significant increase in MCT mice, indicative of RV hypertrophy, and this trend was completely eliminated by DTA-V1 treatment. These data suggested that silencing Atg101 indeed attenuated PVR in mice. The underlying mechanism might be attributed to the knocked down of Atg101, which subsequently downregulated Beclin-1 and upregulated VE-cadherin. The current study represents a new nanotechnology that uses nanomaterials to manipulate endothelial barrier functions for potential therapy of vascular disease.
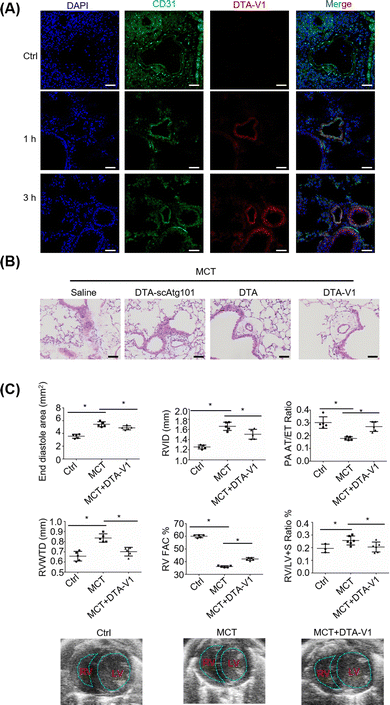 |
| | Fig. 5 DTA-V1 attenuated PVR in vivo. (A) CLMS analysis of the targeting capability of DTA-V1 in vivo. DTA-V1 was modified with Cy5, and the vascular endothelium was labeled with CD31. Scale bar: 50 μM. (B) H & E staining showed the attenuation of PVR. Scale bar: 50 μm. (C) Echocardiography analysis of the therapeutic effects on PVR mice. All the data of the figures represent the mean value ± SD (n = 5). p-Values were calculated using one-way ANOVA, *p < 0.05 indicates connected groups. | |
Conclusions
In summary, we proposed and demonstrated a new strategy that uses DNA nanostructures to specifically target and restore the endothelial integrity, and subsequently attenuate the PVR. The targeting effect was highly associated with the nanomaterial structural design. By targeting endothelial cells, DNA nanostructures carrying siAtg101 drug exhibited superior performance in protecting endothelial integrity, as demonstrated by CLSM imaging and transwell assay. In vivo study demonstrated that the functionalized DNA nanostructures significantly attenuated the PVR in a monocrotaline-induced mouse model by protecting the endothelial barrier. The underlying protection mechanism was attributed to the regulation of cell autophagy and subsequent VE-cadherin expression. The current proof-of-concept study provided a new strategy and nanomaterial platform for the therapy of endothelial dysfunction for vascular endothelium-related diseases. Future work exploiting self-assembled nanoarchitectures for noncancerous disease treatment is highly promising and desirable.37–39
Author contributions
Qian Liu: investigation, methodology, writing-original draft, funding acquisition. Di Wu: investigation, formal analysis, writing-original draft. Binfeng He: methodology, investigation, formal analysis. Xiaotong Ding: investigation. Yu Xu: investigation. Ying Wang: investigation, writing-original draft. Mingzhou Zhang: investigation. Hang Qian: conceptualization, writing-review & editing, supervision, funding acquisition. David Tai Leong: conceptualization, writing-review & editing, supervision. Guansong Wang: conceptualization, writing-review & editing, supervision, funding acquisition.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work is funded by the National Natural Science Foundation of China (81670047 and 32071379) and the Natural Science Foundation of Chongqing, China (No. cstc2020jcyj-msxmX0622, cstc2021jcyj-msxmX1179). Qian Liu and Di Wu contributed equally to this work.
Notes and references
- M. Howard, B. J. Zern, A. C. Anselmo, V. V. Ahuvaev, S. Mitragotri and V. Muzykantov, ACS Nano, 2014, 8, 4100–4132 CrossRef CAS PubMed.
- B. L. Chung, M. J. Toth, N. Kamaly, Y. J. Sei, J. Becraft, W. J. M. Mulder, Z. A. Fayad, O. C. Farokhzad, Y. Kim and R. Langer, Nano Today, 2015, 10, 759–776 CrossRef PubMed.
- S. Sindhwani, A. M. Syed, J. Ngai, B. R. Kingston, L. Maiorino, J. Rothschild, P. MacMillan, Y. Zhang, N. U. Rajesh, T. Hoang, J. L. Y. Wu, S. Wilhelm, A. Zilman, S. Gadde, A. Sulaiman, B. Ouyang, Z. Lin, L. Wang, M. Egeblad and W. C. W. Chan, Nat. Mater., 2020, 19, 566–575 CrossRef CAS PubMed.
- S. Li, Y. Zhang, J. Wang, Y. Zhao, T. Ji, X. Zhao, Y. Ding, X. Zhao, R. Zhao, F. Li, X. Yang, S. Liu, Z. Liu, J. Lai, A. K. Whittaker, G. J. Anderson, J. Wei and G. Nie, Nat. Biomed. Eng., 2017, 1, 667–679 CrossRef CAS PubMed.
- J. K. Tee, M. I. Setyawati, F. Peng, D. T. Leong and H. K. Ho, Nanotoxicology, 2019, 13, 682–700 CrossRef CAS PubMed.
- F. Peng, J. K. Tee, M. I. Setyawati, X. Ding, H. L. A. Yeo, Y. L. Tan, D. T. Leong and H. K. Ho, ACS Appl. Mater. Interfaces, 2018, 10, 31938–31946 CrossRef CAS PubMed.
- W. Jinping, Z. Luyao, P. Fei, S. Xinghua and D. T. Leong, Chem. Mater., 2018, 30, 3759–3767 CrossRef.
- M. I. Setyawati, C. Y. Tay, B. H. Bay and D. T. Leong, ACS Nano, 2017, 11, 5020–5030 CrossRef CAS PubMed.
- C. Y. Tay, M. I. Setyawati and D. T. Leong, ACS Nano, 2017, 11, 2764–2772 CrossRef CAS PubMed.
- M. I. Setyawati, V. N. Mochalin and D. T. Leong, ACS Nano, 2016, 10, 1170–1181 CrossRef CAS PubMed.
- M. I. Setyawati, C. Y. Tay, D. Docter, R. H. Stauber and D. T. Leong, Chem. Soc. Rev., 2015, 44, 8174–8199 RSC.
- N. Ni, W. Wang, Y. Sun, X. Sun and D. T. Leong, Biomaterials, 2022, 287, 121640 CrossRef CAS PubMed.
- F. Peng, M. I. Setyawati, J. K. Tee, X. Ding, J. Wang, M. E. Nga, H. K. Ho and D. T. Leong, Nat. Nanotechnol., 2019, 14, 279–286 CrossRef CAS PubMed.
- M. I. Setyawati, C. Y. Tay, S. L. Chia, S. L. Goh, W. Fang, M. J. Neo, H. C. Chong, S. M. Tan, S. C. Loo, K. W. Ng, J. P. Xie, C. N. Ong, N. S. Tan and D. T. Leong, Nat. Commun., 2013, 4, 1673 CrossRef CAS PubMed.
- J. K. Tee, L. X. Yip, E. S. Tan, S. Santitewagun, A. Prasath, P. C. Ke, H. K. Ho and D. T. Leong, Chem. Soc. Rev., 2019, 48, 5381–5407 RSC.
- J. A. Leopold and B. A. Maron, Int. J. Mol. Sci., 2016, 17, 761 CrossRef PubMed.
- C. Guignabert, L. Tu, B. Girerd, N. Ricard, A. Huertas, D. Montani and M. Humbert, Chest, 2015, 147, 529–537 CrossRef PubMed.
- Z. Y. Dai, M. M. Zhu, Y. Peng, H. Jin, N. Machireddy, Z. J. Qian, X. M. Zhang and Y. Y. Zhao, Am. J. Respir. Crit. Care Med., 2018, 198, 788–802 CrossRef CAS PubMed.
- N. Hosokawa, T. Sasaki, S. I. Iemura, T. Natsume, T. Hara and N. Mizushima, Autophagy, 2009, 5, 973–979 CrossRef CAS PubMed.
- J. Du, Z. Xu, Q. Liu, Y. Yang, H. Qian, M. Hu, Y. Fan, Q. Li, W. Yao, H. Li, G. Qian, B. F. He, D. Zhou, C. Mao and G. S. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 42544–42555 CrossRef CAS PubMed.
- Z. Li, G. Luo, W. P. Hu, J. L. Hua, S. Geng, P. K. Chu, J. Zhang, H. Wang and X. F. Yu, Angew. Chem., Int. Ed., 2020, 59, 20568–20576 CrossRef CAS PubMed.
- C. Y. Tay, M. I. Setyawati and D. T. Leong, ACS Nano, 2017, 11, 2764–2772 CrossRef CAS PubMed.
- Q. Hu, H. Li, L. Wang, H. Gu and C. Fan, Chem. Rev., 2019, 119, 6459–6506 CrossRef CAS PubMed.
- H. Zhao, L. Li, F. Li, C. Liu, M. Huang, J. Li, F. Gao, X. Ruan and D. Y. Yang, Adv. Mater., 2022, 21, e2109920 CrossRef PubMed.
- J. Tang, J. Qu, C. Zhu, C. Yao and D. Y. Yang, Adv. Funct. Mater., 2021, 32, 2107267 CrossRef.
- Z. You, H. Qian, C. Wang, B. He, J. Yan, C. Mao and G. Wang, Biomaterials, 2015, 67, 137–150 CrossRef CAS PubMed.
- H. Li, B. He, X. Liu, J. Li, Q. Liu, W. Dong, Z. Xu, G. Qian, H. Zuo, C. Hu, H. Qian, C. Mao and G. Wang, Theranostics, 2017, 7, 2537–2554 CrossRef CAS PubMed.
- T. Roy, B. D. James and J. B. Allen, Macromol. Biosci., 2021, 21, e2000337 CrossRef PubMed.
- L. Y. Wan, W. F. Yuan, W. B. Ai, Y. W. Ai, J. J. Wang, L. Y. Chu, Y. Q. Zhang and J. F. Wu, Expert Opin. Drug Delivery, 2019, 16, 207–218 CrossRef CAS PubMed.
- S. Ko, H. Liu, Y. Chen and C. Mao, Biomacromolecules, 2008, 9, 3039–3043 CrossRef CAS PubMed.
- H. Lee, A. K. R. Lytton-Jean, Y. Chen, K. T. Love, A. I. Park, E. D. Karagiannis, A. Sehgal, W. Querbes, C. S. Zurenko, M. Jayaraman, C. G. Peng, K. Charisse, A. Borodovsky, M. Manoharan, J. S. Donahoe, J. Truelove, M. Nahrendorf, R. Langer and D. G. Anderson, Nat. Nanotechnol., 2012, 7, 389–393 CrossRef CAS PubMed.
- K. E. Bujold, A. Lacroix and H. F. Sleiman, Chemistry, 2018, 4, 495–521 CrossRef CAS.
- S. J. Lee, A. Smith, L. Guo, T. P. Alastalo, M. Li, H. Sawada, X. Liu, Z. H. Chen, E. Ifedigbo, Y. Jin, C. Feghali-Bostwick, S. W. Ryter, H. P. Kim, M. Rabinovitch and A. M. K. Choi, Am. J. Respir. Crit. Care Med., 2011, 183, 649–658 CrossRef CAS PubMed.
- H. Popelka and D. J. Klionsky, Autophagy, 2015, 11, 1953–1955 CrossRef CAS PubMed.
- M. Zachari and I. G. Ganley, Essays Biochem., 2017, 61, 585–596 CrossRef PubMed.
- A. Leonard, M. W. Millar, S. A. Slavin, K. M. Bijli, D. A. D. Santos, D. A. Dean, F. Fazal and A. Rahman, Cell Signalling, 2019, 61, 120–129 CrossRef CAS PubMed.
- K. Ariga, X. F. Jia, J. Song, J. P. Hill, D. T. Leong, Y. Jia and J. Li, Angew. Chem., Int. Ed., 2020, 59, 15424–15446 CrossRef CAS PubMed.
- Y. Wu, W. Jiang, S. Huo, S. Li, Y. Xu, S. Ding, J. Zhou, H. Liu, W. Lv and Y. Wang, Biomaterials, 2021, 279, 121237 CrossRef CAS PubMed.
- C. Xuan, L. Hao, X. Liu, Y. Zhu, H. Yang, Y. Ren, L. Wang, T. Fujie, H. Wu, Y. Chen, X. Shi and C. Mao, Biomaterials, 2020, 252, 120018 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Di
Wu‡
a,
Binfeng
He
a,
Xiaotong
Ding
a,
Yu
Xu
a,
Ying
Wang
c,
Mingzhou
Zhang
a,
Hang
Qian
ab,
Di
Wu‡
a,
Binfeng
He
a,
Xiaotong
Ding
a,
Yu
Xu
a,
Ying
Wang
c,
Mingzhou
Zhang
a,
Hang
Qian