
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOleophobic coated composite materials based on multi-layer graphitic scaffolding: applications within aircraft propellant tanks and oil-spill clean-up†
Rachel L.
McLaren
 a,
Rosenildo C.
da Costa
a,
Anna C.
Booth
a,
David J.
Morgan
bc,
Christian J.
Laycock
a,
Michael E. A.
Warwick
d and
Gareth R.
Owen
*a
a,
Rosenildo C.
da Costa
a,
Anna C.
Booth
a,
David J.
Morgan
bc,
Christian J.
Laycock
a,
Michael E. A.
Warwick
d and
Gareth R.
Owen
*a
aSchool of Applied Science, University of South Wales, Treforest, CF37 4AT, UK. E-mail: gareth.owen@southwales.ac.uk
bCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK
cHarwellXPS, EPSRC National Research Facility for X-Ray Photoelectron Spectroscopy, Didcot, Oxon OX11 0FA, UK
dEnergy Safety Research Institute, Swansea University Bay Campus, Fabian Way, Swansea SA1 8EN, UK
First published on 1st December 2022
Abstract
The preparation of oleophobic materials coated with a composite based on a multi-layer graphitic scaffolding is reported herein. A range of substrates were employed for this purpose including Kevlar, carbon fibre, glass fibre, nylon and stainless steel mesh. These were utilised, in comparison with free-standing film versions of the composite material, to investigate their enhanced ability to facilitate water penetration whilst simultaneously retaining the oleophobic behaviour. The materials demonstrated efficient oil/water separations and reusability. The free-standing films and coated substrates were characterised in detail via a range of spectroscopic and analytical techniques. Contact angle measurements for aviation Jet A-1 fuel on various coated substrates ranged from 96.9–107.0° whilst for hexadecane and silicone oil, contact angles of 90.6–120.3° and 74.5–103.3° were recorded, respectively. These values were slightly lower than the contact angles for the corresponding free standing film versions which were 111.9°, 126.4° and 105.9° for Jet A-1 fuel, hexadecane and silicone oil, respectively. BET surface area analysis of composite and films showed type IIb isotherms with H3-type hysteresis. T-Plot analysis was carried out to quantify external surface area of the composite and film in comparison to the base multilayered graphitic material scaffold. The morphology of the materials was analysed by SEM imaging to show the extent and degree of coating on the composite material upon the substrates. The application of these coated substrates as membranes within the context of aircraft propellant tanks and oil-spill removal was also explored, suggesting that coated carbon fibre and coated nylon serve as promising candidates for oil/water separation within these applications.
Design, System, ApplicationWithin this article we report the development of a composite material which has been coated on a range of grid-like substrates (including Kevlar, carbon fibre, glass fibre, nylon and stainless steel mesh) for the purposes of oil/water separations. The composite material is oleophobic and has been built on a multi-layer graphitic scaffolding. The substrates where utilised to provide enhanced water penetration whilst at the same time maintaining the oleophobic nature of the material. The newly formed materials have been analysed and characterised via a range of spectroscopic and analytical techniques. Our investigations have confirmed the oleophobic nature of the new materials. The coated substrates have been successfully demonstrated in two different examples of oil/water separation application, in the context of oil-spill removal and as membranes within aircraft propellant tanks. This has demonstrated the readily and commercially available multi-layer graphitic scaffolds as promising candidates for future oil/water separation applications. |
Introduction
The application of oleophobic/hydrophilic materials has shown promise for the purpose of oil-spill clean-up, oil/water separation and for the fabrication of anti-fogging, anti-fouling and detergent-free self-cleaning surfaces.1–8 In the context of oil-spills, the presence of oil within water systems has a detrimental effect to aquatic life.9 Large scale ocean oil-spills, of course, can have significant environmental impact. In addition, oil/water separation could also prove advantageous within aircraft fuel tanks where tanks often suffer from an ingress of water through openings. The accumulation of water within the propellant can cause a number of issues. For example, it leads to reduced fuel-storage capacity, an increased fuel tank mass, a risk of biocontamination and the possibility that the water may freeze, thereby obstructing normal operation. Although scavenger pumps and drainage valves are commonly utilised to remove this water, in practice, a more desirable means of separation is needed. The implementation of a membrane which only allows passage of water, whilst retaining the propellant within the tank, would be a solution on a practical scale.Unfortunately, it can be notoriously difficult to fabricate materials which possess higher contact angles to oil than water, since theoretically, a surface should always be more wettable to oil than water due to the higher surface tension associated with the latter. Consequently, for the majority of materials, the water contact angle (WCA) exceeds that of the oil contact angle (OCA).10,11 Despite this, much research has been carried out investigating materials which allow the wetting by water whilst simultaneously producing high OCAs.1,12–14 In recent examples, a combination of polyelectrolytes with fluorosurfactants have been employed for the fabrication of oleophobic/hydrophilic surfaces.1,2,7,15–23 Polyelectrolytes are believed to spontaneously undergo a one-to-one interaction with fluorosurfactants in aqueous solution to form polyelectrolyte–surfactant complexes.24–26 The fluorosurfactant component provides a combination of a sufficiently small dispersive component and a sufficiently large polar component associated with the surface tension values, thus providing surfaces with simultaneous oleophobic/hydrophilic behaviour.27–29 Fabrication of these surfaces can proceed by layer-by-layer deposition, covalent approaches, dip-coating, UV irradiation and copolymerisation.7 Surfaces comprising of SiO2 nanoparticles with poly(diallyldimethylammonium chloride) (PDDA) and a fluorosurfactant such as sodium perfluorooctanoate (PFO) are a known combination of reagents used.17,18 In these cases, large hexadecane contact angles exceeding 150° have been achieved.15,19,20 In these investigations, it has been shown that the addition of SiO2 nanoparticles provides the necessary roughness component required for the amplification of oleophobic/hydrophilic behaviour.30–33
As part of our ongoing investigations34,35 focusing on commercially derived plasma-synthesised multi-layer graphitic material (MLG),36 we became interested in utilising MLG as a scaffold for the development of composite materials.37 MLG refers to an exfoliated graphite-based material which has undergone consecutive argon and oxygen plasma treatments and therefore consists of covalently bound oxygen moieties and exfoliated stacks of layers.35 As such, this is referred to as a graphitic material rather than exfoliated graphite, due to the existence of covalently bound moieties.38 We recently reported a preliminary account on the synthesis of free-standing oleophobic/hydrophilic films comprising of MLG in conjunction with a PDDA/PFO complex.39 During this investigation, we found that the resulting MLG–PP film (i.e. the synthesised film containing the MLG, PDDA and PFO components) exhibited OCAs of up to 126.4° and allowed the ready passage of water through the material whilst also preventing oil passage for a period of at least three weeks. Water passage was slow however, taking up to 2.5 hours for complete passage through the film. Subsequent reduction in film thickness improved the rate of water passage however with limited practical application due to brittleness. As a result, we embarked on a further investigation aimed at supporting the MLG–PP composite onto various substrates to provide more robust platforms whilst at the same time providing larger channels to facilitate the passage of water. Thus, in this report the composite is deposited on a range of substrates including Kevlar, carbon fibre, glass fibre, nylon and stainless steel mesh. These substrates possess a range of mechanical strengths, flexibilities, spacings and physical properties that could influence binding of the MLG–PP as well as passage of liquids. Herein, we provide a detailed account of our investigations in addition to verifying their effectiveness in providing oil/water separation. We further demonstrate their potential within the context of oil-spill clean-up and aircraft propellant tank applications.
Results and discussion
Synthesis of substrates coated by MLG–PP composite
The synthesis of the MLG–PP composite was achieved by a two-step process comprised of adsorption of PDDA onto MLG followed by PDDA/PFO complexation upon addition of PFO as outlined within our previous work (Scheme 1).39 The non-covalently bound composite, MLG–PP, was produced as a thick black gummy material (Fig. S1, ESI†) which could be redispersed in methanol. Free-standing films such as that in Fig. S2 (top left; ESI†) were formed by allowing the methanol solvent to evaporate from these dispersions. The substrate materials of choice were consequently coated by pouring methanolic dispersions of MLG–PP over the materials and allowing the methanol solvent to evaporate overnight (Fig. S3†).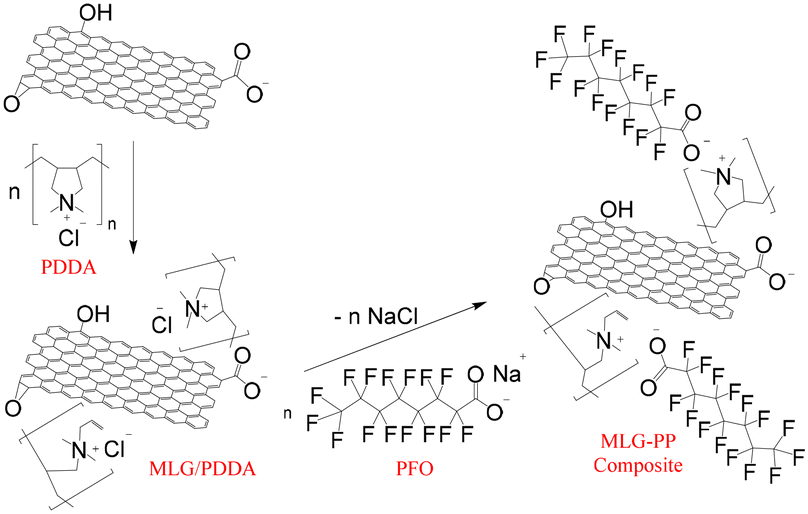 | ||
| Scheme 1 Synthesis of MLG–PP composite consisting of a two-step process involving adsorption of PDDA onto MLG followed by complexation of PFO with the PDDA/MLG mixture. | ||
The resulting coating on Kevlar, carbon fibre and glass fibre substrates appeared uniform, affording a black layer of MLG–PP composite across the entire surface of the substrate, from both above and below. The coating could not be peeled off as a film, suggesting that it had become well integrated onto the substrate through non-covalent interactions and physical integration between the fibres. In contrast, the coating upon nylon was much less uniform, forming dark regions where it was more concentrated, and lighter regions where it covered the substrate to a lesser extent. The coating upon stainless steel mesh also appeared uniform. In this case, however, the coating formed as a film only on the bottom surface of the mesh and was integrated across the apertures of the mesh. As a result, a thin layer of MLG–PP composite could be peeled off.
Oil/water passage studies on MLG–PP coated substrates
To test how well the MLG–PP coated substrates enabled water passage, whilst maintaining simultaneous oleophobic behaviour, a burette was filled with silicone oil dyed with oil red O and deionised (DI) water dyed with brilliant blue R, as described within our previous report.39 Both liquids were deposited upon the coated substrates via a drop-by-drop approach (approximately 1 drop per second). The water was added to the substrate first since it was at the bottom of the burette. The silicone oil was deposited once all the water had been added. The results of these tests are outlined in the following section. Photographs are shown for the coated carbon fibre test in Fig. 1 whilst photographs for the remaining substrates are included in the ESI† (Fig. S3–S14). | ||
| Fig. 1 Coated carbon fibre at various stages during oil/water passage tests (a) before liquid deposition, (b) water penetrated coated carbon fibre after approximately 5 seconds, (c) oil remained on top for at least 3 weeks and (d) silicone oil could easily be poured off the surface. | ||
In general, the coated substrates showed substantially improved water passage compared with that of MLG–PP films.39 This is as expected due to the holes formed by the mesh networks of the substrates. We were pleased to find that they also maintained the same oleophobic characteristics as observed for the MLG–PP films.
During tests on the coated Kevlar, it was observed that water passage occurred after approximately 10 seconds from initial deposition, as shown in Fig. S4.† This initial water passage through the material helped further passage of water. Thus, water penetration became almost instantaneous thereafter. A total of 5 mL of water passed through the coated substrate. Upon silicone oil deposition, it was found that no silicone oil passed through the coated substrate. In fact, the silicone oil remained on the surface of the coated substrate for at least 3 weeks. Furthermore, the silicone oil could be easily poured off the coated substrate by simply tilting the material.
The coated carbon fibre meanwhile was more effective than coated Kevlar in allowing water penetration. Complete passage of water took place approximately 5 seconds after initial deposition, as shown in Fig. 1(b). As with the previous example, initial water passage through the material helped the further passage of water. Again, upon silicone oil deposition, there was no penetration through the coated carbon fibre substrate, and it remained on the surface for at least 3 weeks (Fig. 1(c)). The oil could be poured off by tilting the material (Fig. 1(d)).
Similarly, the coated glass fibre also allowed water passage after approximately 5 seconds (Fig. S7†). In contrast to the coated Kevlar and the coated carbon fibre, however, the silicone oil passage was only prevented for up to 2 hours after its deposition on the surface (Fig. S7(c)†). Prior to this, silicone oil could be easily removed by pouring off the surface. This difference in behaviour was attributed to the larger mesh size. The pore sizes were visibly larger in the coated glass fibre substrate, as can be seen in the photograph of the coated glass fibre (Fig. S3† (bottom-left)).
The coated stainless steel mesh meanwhile was observed to exhibit slower water penetration than the aforementioned materials. Water passage required 5 minutes to reach completion (Fig. S11(b)†). This slow passage was attributed to the film-like morphology covering the apertures of the mesh, as outlined above, limiting the holes available for facile passage. Upon deposition of silicone oil, the oil droplets were not observed to penetrate the material, and instead, remained on the top of the coated substrate for at least 3 weeks.
Finally, the coated nylon was found to be the most effective of the coated substrates by allowing rapid water passage immediately after deposition. Again, following deposition, the silicone oil remained on the surface for at least a 3 week period and could easily be poured off (Fig. S9†). The efficiency of this oil/water separation was explored in further detail by dropwise addition of 2.08 g of water onto the coated membrane, followed by 2.01 g of silicone oil. Water passage occurred as before, with 1.88 g (90%) of the water recovered in a short period whilst the oil remained on the coated substrate. The unrecovered water was likely to have been retained by the coated membrane due to its hydrophilicity.
In all cases, the initial passage of water helped to assist the further passage of water. This can be understood as a preconditioning of the coated substrates which can be rationalised by the presence of hydrogen bonding between water molecules residing on the surface of the materials resulting in cohesive properties. As a control experiment, it was confirmed that all uncoated substrates allowed free passage of both silicone oil and water (Fig. S13†). As a result, it was concluded that the MLG–PP composite was effective in preventing the passage of silicone oil. In fact, all coated substrates, with the exception of coated glass fibre, prevented the passage of silicone oil through them for at least 3 weeks, where the silicone oil could be observed on the surface of the substrates with very little wetting. The results of these tests are summarised pictorially in Fig. 2.
 | ||
| Fig. 2 Schematic outlining the passage of water and oil through MLG–PP film and coated substrates. Water passage is represented by blue arrows, whilst red arrows represent silicone oil passage. *Note: on some occasions, the glass fibre coated substrate allowed oil penetration after 2 hours. | ||
Next, the reusability of the coated substrates was investigated, along with the durability of the MLG–PP coatings. To test the reusability of the coated substrates, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of water and red-dyed silicone oil (ca. 2 g) was pipetted onto the coated nylon substrate. On this occasion undyed water was utilised to assist in the observation of any traces of oil passing through due to the contrast between the dyed and undyed liquids. Once the water had passed through the membrane, the oil on the surface was carefully removed by pipette before another batch of the 1:1 mixture was applied. This process was repeated five times. No oil was observed to penetrate the membrane until the fifth repeat when a small quantity of the silicone oil passed through. This penetration may have been due to physical damage to the membrane during the procedure. The durability of the MLG–PP coating on the different substrates was also tested by washing used membranes repeatedly with water and acetone, to remove any residual silicone oil, before air drying for a minimum of 24 hours. Of the five coated substrates, no differences were observed in the appearance of the coated Kevlar, carbon fibre or glass fibre substrates. However, it was found that the coating was partially washed away from the nylon substrate and stainless steel mesh. Photographs of these two washed substrates are included in the ESI† (Fig. S10(a) and S12(a)) along with the other materials. Irrespective of this, all coated substrates were re-tested under the same silicone oil/water testing conditions, and it was clear that there was no decrease in their oil/water separation ability (see Fig. S5, S6, S8, S10 and S12†). As such, it is apparent that sufficient concentration of the MLG–PP composite remained on the materials to provide the necessary oleophobic behaviour. Interestingly, it was found that upon re-testing of coated glass fibre after washing, it became more effective at retaining the silicone oil on top of its surface. In this latter test, the oil remained on the surface for at least 3 weeks. This may be due to movement of the fibres during the washing stage, altering the size of the grid. Unlike many oleophobic/hydrophilic surfaces, PDDA/PFO reagents are present within both the surface regions, as well as the bulk of the MLG–PP composite. It is believed that they remain intercalated and held in place within slit pores of the MLG (vide infra).35 As a result, this may improve the wear resistance of these coated substrates.
:1 mixture of water and red-dyed silicone oil (ca. 2 g) was pipetted onto the coated nylon substrate. On this occasion undyed water was utilised to assist in the observation of any traces of oil passing through due to the contrast between the dyed and undyed liquids. Once the water had passed through the membrane, the oil on the surface was carefully removed by pipette before another batch of the 1:1 mixture was applied. This process was repeated five times. No oil was observed to penetrate the membrane until the fifth repeat when a small quantity of the silicone oil passed through. This penetration may have been due to physical damage to the membrane during the procedure. The durability of the MLG–PP coating on the different substrates was also tested by washing used membranes repeatedly with water and acetone, to remove any residual silicone oil, before air drying for a minimum of 24 hours. Of the five coated substrates, no differences were observed in the appearance of the coated Kevlar, carbon fibre or glass fibre substrates. However, it was found that the coating was partially washed away from the nylon substrate and stainless steel mesh. Photographs of these two washed substrates are included in the ESI† (Fig. S10(a) and S12(a)) along with the other materials. Irrespective of this, all coated substrates were re-tested under the same silicone oil/water testing conditions, and it was clear that there was no decrease in their oil/water separation ability (see Fig. S5, S6, S8, S10 and S12†). As such, it is apparent that sufficient concentration of the MLG–PP composite remained on the materials to provide the necessary oleophobic behaviour. Interestingly, it was found that upon re-testing of coated glass fibre after washing, it became more effective at retaining the silicone oil on top of its surface. In this latter test, the oil remained on the surface for at least 3 weeks. This may be due to movement of the fibres during the washing stage, altering the size of the grid. Unlike many oleophobic/hydrophilic surfaces, PDDA/PFO reagents are present within both the surface regions, as well as the bulk of the MLG–PP composite. It is believed that they remain intercalated and held in place within slit pores of the MLG (vide infra).35 As a result, this may improve the wear resistance of these coated substrates.
It is also worth mentioning that additional tests were conducted on the coated nylon. Firstly, 1.41 g of silicone oil was deposited on the material prior to the addition of 4.13 g of water. Water passage began within 30 seconds of first contact with the material and continued at a steady rate thereafter. In total, 4.01 g (97%) of the water was recovered, whilst the silicone oil remained on top of the coated substrate as before. This highlights the efficient oil/water separation by the material and confirms that prior wetting of the substrate with water is not required for oleophobic behaviour.
Finally, the separation of stabilised silicone oil-in-water emulsions was attempted. It was found that the coated nylon material was partially effective in that it separated the emulsion into emulsions of distinct droplet sizes. This has previously been documented as a “size-sieving” effect40 which may be more related to the size of the pores within the nylon material than the MLG–PP composite itself. This will be investigated as part of a follow up study exploring different emulsions, oil to water ratios and pores sizes within the MLG–PP coated materials.
Contact angle measurements
Once we had gained an appreciation of the extent of the oleophobic behaviour shown by MLG–PP films39 and coated substrates, contact angle measurements were conducted using silicone oil, hexadecane and Jet A-1 fuel. The results of the measurements are shown in Table 1. Corresponding contact angle images are depicted in Fig. S14.† Contact angles between 90–150° indicate oleophobic behaviour, thus, it can be said that all coated substrates display oleophobicity (apart from Kevlar and nylon for the case of silicone oil).6 In all substrates, the contact angles exceeded 90.6° for hexadecane, 96.9° for Jet A-1 fuel and 74.5° for silicone oil. In general, it was found that the contact angles decreased for oils in the order of hexadecane > Jet A-1 fuel > silicone oil. This order correlates well with the reported surface tension values of 27, 24 and 21 mN m−1 at 20 °C for hexadecane, Jet A-1 fuel and silicone oil, respectively.41,42 The higher the surface tension, the lesser extent the liquid will wet the surface. It is noticeable that the coated substrates exhibit slightly lower OCAs compared with the original MLG–PP film. In particular, relatively low contact angles for silicone oil are associated with coated Kevlar and coated nylon: 78.3° and 74.5°, respectively. This difference is likely to be due to the existence of larger spaces within the meshes which may become occupied by the oils, thereby reducing their contact angles. These holes can be clearly observed via Scanning Electron Microscopy (SEM) (vide infra).| Material | Average contact angle (°) | ||
|---|---|---|---|
| Silicone oil | Hexadecane | Jet A-1 fuel | |
| MLG–PP film | 105.9 (± 4.1) | 126.4 (± 0.5) | 111.9 (± 3.0) |
| Kevlar | 78.3 (± 3.9) | 116.1 (± 5.1) | 103.6 (± 1.8) |
| Carbon fibre | 103.3 (± 1.9) | 117.6 (± 0.9) | 107.0 (± 1.9) |
| Glass fibre | 90.3 (± 3.2) | 120.3 (± 1.6) | 106.0 (± 1.5) |
| Nylon | 74.5 (± 0.9) | 90.6 (± 2.7) | 97.2 (± 1.8) |
| Stainless steel mesh | 92.3 (± 0.7) | 109.3 (± 1.6) | 96.9 (± 1.5) |
In all cases, it was observed that water wets the materials almost instantaneously, thereby the corresponding WCAs were considered as 0°. This oleophobic/hydrophilic behaviour is due to the presence of the fluorocarbon chain within PFO and the surface roughness of the material. The fluorocarbon chains extend out towards the air/solid interface which provide a sufficiently low dispersive component of their surface tension, as detailed in other reported investigations.1,2,7,15–22
It is likely that the nanoscale roughness of the MLG scaffold in the MLG–PP film and coated substrates leads to an increased liquid repellence behaviour. It also plays a role in providing an increased oleophobic behaviour since the MLG provides a larger oleophobic surface area for liquid contact. This increase in surface area increases the interaction between polar moieties present in the MLG material with water droplets. As such, the oleophobic and hydrophilic behaviour becomes amplified.30
The oleophobic properties found in the original MLP–PP films are reproduced within the coated substrates. The substrates, in all examples, apart from the glass fibre, fully block the passage of the silicone oil. On the other hand, the substrates provide enhanced properties with respect to the water passage by providing larger channels in which water is able to pass through at rapid rates. The porous substrates therefore open up the possibility to utilise these materials for oil/water separations. These are further explored later in this article.
Elemental surface analysis of MLG–PP
To gain an understanding of the elemental composition and nature of the components within the MLG–PP composite, X-ray photoelectron spectroscopy (XPS) was carried out. XPS quantitative data for this material and its component precursors are presented in Table 2. The corresponding spectra are shown in Fig. 3–5. Further survey spectra and additional XPS spectra are provided for each material within Fig. S15–S19.†| Orbital | Atomic concentration (%) | ||||
|---|---|---|---|---|---|
| MLG | PDDA | MLG/PDDA | PFO | MLG–PP composite | |
| C 1s | 93.4 | 69.3 | 73.5 | 28.5 | 55.1 |
| N 1s | 0.3 | 7.4 | 8.2 | — | 3.4 |
| O 1s | 6.3 | 11.2 | 6.0 | 4.7 | 4.7 |
| Cl 2p | — | 7.6 | 9.1 | — | 1.1 |
| Na 1s | — | 2.5 | 2.3 | 4.7 | 0.3 |
| S 2p | — | 2.0 | — | — | — |
| F 1s | — | 1.0 | 62.1 | 35.4 | |
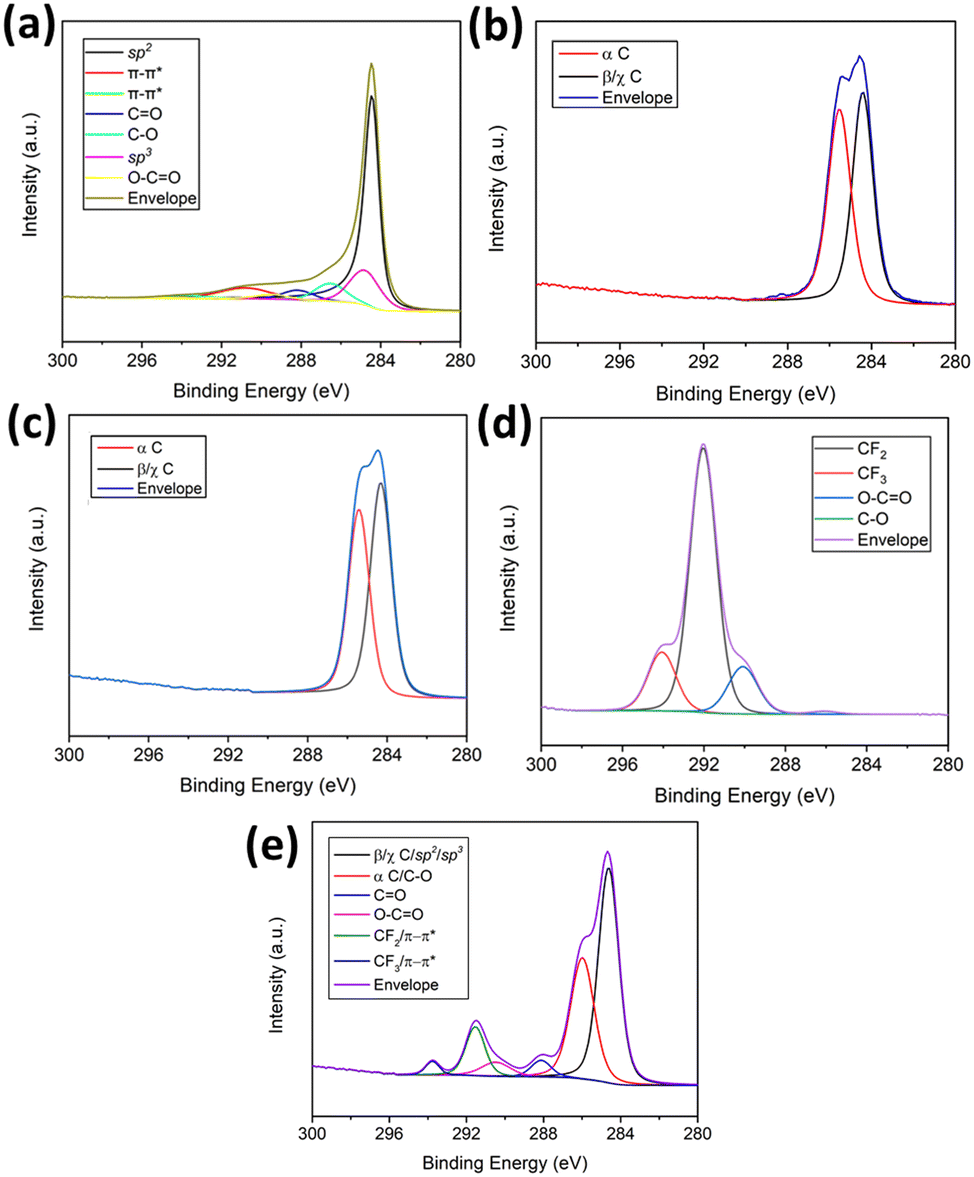 | ||
| Fig. 3 High-resolution deconvoluted spectra of C 1s orbital for (a) MLG (b) PDDA (c) MLG/PDDA (d) PFO and (e) MLG–PP composite. | ||
 | ||
| Fig. 4 High-resolution deconvoluted spectrum of O 1s orbital for MLG. | ||
 | ||
| Fig. 5 High-resolution deconvoluted spectra for MLG–PP composite showing (a) Cl 2p orbital (b) Na 1s orbital (c) N 1s (d) O 1s (e) F 1s. | ||
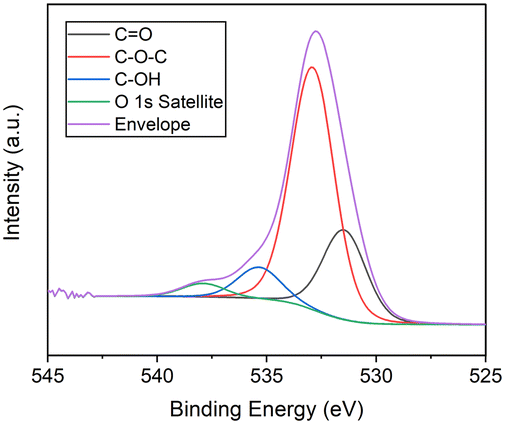
Initially, the surface elemental composition was examined for the MLG precursor. The data confirmed the presence of carbon, nitrogen and oxygen corresponding to atomic percentage compositions (at%) of 93.4, 0.3 and 6.3, respectively. Deconvolution of the high-resolution C 1s spectrum (Fig. 3(a)) indicated the presence of seven carbon environments. These possess binding energies (eV) of 284.4, 284.8, 286.2, 287.7, 289.3, 290.8 and 293.9, which correspond to sp2, sp3, C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, O–CO and two π–π* satellite components, respectively. π–π* satellite structures arise due to π → π* transitions providing evidence of the π bonding network.43 This evidence, in conjunction with a large proportion of sp2 character, originates from extensive π bonding throughout the honeycomb lattice, as expected within graphitic materials.44 Moreover, the presence of oxygen arises due to functionality in the form of C–OH, C–O–C and O–CO, CO, typical of partially oxidised graphene-based materials containing hydroxy, epoxide, carboxyl, and carbonyl functional groups.45 These are also observed within the O 1s spectrum (Fig. 4) which displays four oxygen environments at 531.5, 532.9, 535.4 and 538.0 eV, corresponding to the CO, C–O–C, C–OH and the O 1s satellite structure functionalities. The oxygen functionalities are introduced during the plasma treatment and processing of the MLG precursor. A trace amount of nitrogen is also present as evidenced by the N 1s state at 400.4 eV (Fig. S15†). This is related to a nitrogen containing contaminant likely to be introduced during plasma treatment.
O, O–CO and two π–π* satellite components, respectively. π–π* satellite structures arise due to π → π* transitions providing evidence of the π bonding network.43 This evidence, in conjunction with a large proportion of sp2 character, originates from extensive π bonding throughout the honeycomb lattice, as expected within graphitic materials.44 Moreover, the presence of oxygen arises due to functionality in the form of C–OH, C–O–C and O–CO, CO, typical of partially oxidised graphene-based materials containing hydroxy, epoxide, carboxyl, and carbonyl functional groups.45 These are also observed within the O 1s spectrum (Fig. 4) which displays four oxygen environments at 531.5, 532.9, 535.4 and 538.0 eV, corresponding to the CO, C–O–C, C–OH and the O 1s satellite structure functionalities. The oxygen functionalities are introduced during the plasma treatment and processing of the MLG precursor. A trace amount of nitrogen is also present as evidenced by the N 1s state at 400.4 eV (Fig. S15†). This is related to a nitrogen containing contaminant likely to be introduced during plasma treatment.
XPS analysis was carried out on PDDA to enable comparisons to be made between the MLG–PP composite and MLG and to understand any surface elemental changes which take place between the various materials. Several investigations have discussed the adsorption of PDDA upon graphitic surfaces.46–52 It has been suggested that an electrostatic interaction exists between the negatively charged structures of graphene oxide or reduced graphene oxide and the positive charge of the PDDA cation.50–53 It is well-established that the presence of –COOH and –OH functional groups on the surface of such materials are capable of ionising in aqueous solution, thereby, providing the surface with a negative charge.54–56 Other researchers have suggested that the unsaturated pendant double bonds, which are the minor component in the PDDA mixtures, undergo π–π interactions with graphitic networks. This is typically evidenced by the presence of a shake-up peak at 290.9 eV associated with the π → π* transitions within the XPS spectrum.49,57 Under the conditions of our XPS analysis of PDDA, the π–π* shake-up peak was not observed within the XPS C 1s spectrum, (Fig. 3(b)). Instead, two peaks at 284.4 and 285.6 eV were observed, which we assigned as the carbons β and γ to the quaternary ammonium group (284.4 eV) and the carbon environments α to the quaternary ammonium group (285.6 eV) within the polyelectrolyte.58 It should be noted, however, that the 13C{1H} NMR spectrum provides evidence of some sp2 carbons, which is consistent with the presence of the unsaturated species (see Fig. S20†). A more comprehensive analysis and implication of the impact of the XPS technique on the structure of PDDA has been reported by ourselves elsewhere.58
Through analysis of PDDA, it was possible to compare how the binding energies associated with the elements became altered when the polyelectrolyte PDDA is mixed with MLG. The main features of the resulting C 1s spectrum resembled those of PDDA (cf.Fig. 3(c) versus (b)). None of the peaks associated with the π–π* structure or the C–O, CO and O–CO functional groups on MLG were apparent. Of course, any sp2 and sp3 associated with MLG would be expected to overlap with that of the carbon environments from PDDA. As such, this suggests a large proportion of PDDA within the surface regions of the MLG/PDDA mixture. The XPS data also indicates a substantial change in the overall oxygen content reducing from 11.2 at% in PDDA to 6.0 at% in MLG/PDDA (see Table 2, and Fig. S17† for further details). PDDA is known to retain water and thus be apparent in the XPS data. This content is likely to be reduced in the MLG/PDDA mixture. It may also be possible that the O 1s state became attenuated due to the lack of oxygen within the newly formed surface regions. Also, within MLG/PDDA, an N 1s peak was also present at 401.5 eV providing evidence of the quaternary ammonium groups within PDDA (see Fig. S17†). This is very similar to the binding energy associated with free PDDA, where N+ electrostatically interacts with Cl−/OH− ions. Because of this, the N 1s peak cannot be used as clear evidence of complexation occurring between carboxylate functionality present on MLG's surface.
XPS analysis of PFO provided evidence for the presence of several elements including carbon, oxygen, sodium, and fluorine. Initially, the C 1s spectrum was examined. This revealed four peaks at 286.0, 290.0, 292.0 and 294.0 eV (Fig. 3(d)). These relate to carbons within various environments including C–O, O–CO, CF2 and CF3.15 The presence of fluorine was also indicated by deconvolution of the peaks at 689.2 eV and 690.2 eV. These correspond to CF3 and CF2 environments, respectively (see Fig. S18(b)†).59 Furthermore, peaks at 532.8 and 1072.4 eV corresponded to O 1s and Na 1s environments, relating to oxygen and sodium moieties within the fluorosurfactant (Fig. S18(c) and (d),† respectively).
Finally, XPS analysis was conducted on the MLG–PP composite, providing evidence of carbon, nitrogen, oxygen, chlorine, sodium, and fluorine. These elements relate to a contribution of MLG, PDDA and PFO within the surface regions. The C 1s spectrum (Fig. 3(e)) shows the presence of six peaks located at 284.6, 286.0, 288.1, 290.5, 291.5 and 293.8 eV. The latter three binding energies correspond to the O–CO, CF2 and CF3 environments of PFO, whilst the former two correspond to the β/γ and α environments of PDDA. The remaining peak at 288.1 eV and a slight broadening of the peak at 286.0 eV indicate the presence of C 1s environments originating from MLG. The peak at 288.1 eV corresponds to CO within MLG, whilst the broadening of PDDA's peak is indicative of C–O within MLG (Fig. 3(a)). Furthermore, it is likely that the remaining environments expected for MLG overlap with the C 1s environments of PDDA and PFO.
There is a substantial reduction in the atomic concentration of chlorine between PDDA and MLG–PP composite. This is attributed to the complex formation between polyelectrolyte and fluorosurfactant, resulting in an exchange of counterions, and the formation of NaCl [as evidenced by peaks at 199.8 and 1070.8 eV corresponding to Cl 2p 3/2 and Na 1s orbitals, respectively (see Fig. 5(a) and (b))]. The washing procedure for MLG–PP composite consisted of several dispersion–centrifugation–decantation cycles to remove NaCl, hence this is only present in trace quantities (0.3 at% for sodium atomic concentration as shown in Table 2). Furthermore, it is observed that a small quantity of Cl− bound to quaternary ammonium groups still exists as indicated by the Cl 2p 3/2 peak at 196.8 eV, suggesting that PFO did not complex fully with every available quaternary ammonium group present on PDDA. As such, the complex is not formed in a 1:1 stoichiometry contrarily to previous reports.24–26 This is likely due to inaccessibility of PFO to all PDDA quaternary ammonium species across the surface of the MLG. Whilst we did not directly quantify the stoichiometry, we found that upon washing of MLG–PP composite, both PDDA and PFO were present in the filtrate, suggesting they were in excess upon addition with respect to the quantity that could be adsorbed on to the MLG.
Four nitrogen species were also present within the composite, one as the major component, consistent with the N 1s spectrum for PDDA,58 as shown in the N 1s spectrum for MLG–PP (Fig. 5(c)). A shift in the binding energy for the quaternary ammonium species was also observed from 401.7 eV in PDDA to 402.1 eV in MLG–PP. This increase in binding energy by 0.5 eV is attributed to the complexation between the quaternary ammonium groups of PDDA with the carboxylate functionality of PFO. This shift may be attributed to the ability of oxygen to form strong electrostatic interactions with the positively charged nitrogen species.
Analysis of the O 1s spectrum for MLG–PP composite also provides evidence for the presence of oxygen, displaying a peak at 530.8 eV (Fig. 5(d)). This was attributed to the contribution of oxygen functionalities from MLG and PFO. The presence of fluorine within the F 1s spectrum resembled that of the CF3 and CF2 environments present within PFO, corresponding to 687.9 and 688.9 eV, respectively (Fig. 5(e)). If a 1:1 stoichiometry existed between PDDA and PFO, there should be 15 fluorine atoms for every 1 nitrogen atom. It was found that the ratio between nitrogen to fluorine was approximately 13:1, giving further evidence that at least a small portion of the quaternary ammonium groups were not complexed with a PFO moiety within the composite.
Morphology of membranes
Our investigation subsequently focused on the morphology, surface area, porosity and structure of the materials. SEM analysis was conducted to gain an insight to the structure and morphology of MLG, MLG–PP composite and the coated substrates. Initially SEM images were acquired for MLG, as shown in Fig. S21.† These showed that the material consisted of aggregates which ranged in size between several hundred microns to several hundred nanometres. The aggregates contained a powdery and clumped appearance, consistent with stacks of partially oxidised graphitic sheets held together via extensive van der Waals forces.The morphology became significantly altered upon formation of the MLG–PP composite (Fig. 6). Large aggregates were formed, which possessed lengths of several hundred microns to several millimetres. The powdery texture was lost, and the new aggregates possessed smoother surfaces. Representative SEM images were also acquired for a mixture of PDDA and PFO for comparative purposes. These images, which are depicted within Fig. S22,† show that the resulting material is comprised of large aggregates approximately 2 mm in length. The PDDA/PFO complexes also possess a smooth morphology which appear scaly with a rippling effect at higher magnifications. This scaly rippling effect was also observed within the MLG–PP composite at higher magnifications supporting the presence of the PDDA/PFO mixture upon the MLG material (Fig. 6(d)). This rippling effect was also observed to some degree within aggregates of MLG–PP film (Fig. S23†).39 Interestingly, the physical texture of MLG–PP composite varied across the sample. When physically handling the material, it was found that some areas were brittle and could be snapped easily, whilst others were more flexible and could be stretched. The PDDA/PFO mixture had a more flexible texture, typical of many polymer-containing materials. This suggests that within the MLG–PP composite there was a contribution of both rigidity, originating from MLG, and flexibility, due to the presence of the polymeric components. Additionally, the MLG–PP film was also analysed by AFM, the results of which are presented elsewhere.39 The AFM analysis revealed that the MLG–PP film exhibited a textured and rough surface.
 | ||
| Fig. 6 SEM image of MLG–PP composite at magnifications of (a) 105× (b) 415× (c) 4.15k× and (d) 20.76k×. | ||
SEM imaging was subsequently conducted on the coated substrates. A range of representative images are shown in Fig. 7. These demonstrated that the coating on the substrates were not always homogenous and that large pores within the substrates were not always fully coated with the composite. Some regions of each coated substrate appeared homogenous whilst others did not. Each of the coated substrates were consequently inspected at higher magnifications to further understand the nature of the coating with the MLG–PP composite material.
 | ||
| Fig. 7 SEM images obtained in wide field mode of coated (a) Kevlar (b) carbon fibre (c) glass fibre (d) nylon and (e) stainless steel mesh at magnifications of 20×. | ||
The Kevlar, carbon fibre and glass fibre substrates, prior to coating, contained individual strands with diameters of 15, 7.5 and 15.1 μm respectively, as shown in Fig. 8(a), 9(a) and 10(a). These materials formed a crosshatched arrangement with many gaps between strands that run perpendicular to one another, as well as between the grid type structure. These gaps thus lead to channels and pores through the materials.
 | ||
| Fig. 8 SEM images of (a) uncoated Kevlar at a magnification of 1.73k× and coated Kevlar at magnifications of (b) 56× (c) 589× (d) 519× (e) 10.38k× and (f) 10.38k×. | ||
 | ||
| Fig. 9 SEM images of (a) uncoated carbon fibre at a magnification of 1.38k× and coated carbon fibre at magnifications of (b) 461× (c) 2.08k× (d) 830k× (e) 4.15k× and (f) 1.04k×. | ||
 | ||
| Fig. 10 SEM images of (a) uncoated glass fibre at a magnification of 1.38k×; and coated glass fibre at magnifications of (b) 208× (c) 1.38× (d) 1.38k× (e) 4.15k× and (f) 10.38k×. | ||
Upon coating, it is observed that some of the pores in these three materials remain uncoated. This can be seen in the images in Fig. 7(a)–(c). Representative high magnification SEM images reveals the presence of gaps within the three coated substrates corresponding to widths of 64 and 90 μm in coated Kevlar (Fig. 8(b) and (c)), 52, 15 and 14 μm in coated carbon fibre (Fig. 9(b)–(d)) and 101 μm in coated glass fibre (Fig. 10(b)). In the case of Kevlar and glass fibre, these appear to be resulting from spacings between the fibres in the materials. On the other hand, for the carbon fibre material, the smaller holes appear to be the result of tears in the MLG–PP coating. Furthermore, additional pores were also observed at the junctions between coated regions of the substrate and the fibres themselves. For example, two areas which were measured in the coated glass fibre corresponded to 114 and 232 μm2 (Fig. 10(c)).
In general, the coating process resulted in MLG–PP film formation across much of the substrate. Many regions acquired film morphology over the strands, whilst other regions consisted of film suspended between strands, as observed in Fig. 8(d), 9(e), (f), 10(d) and (e). Furthermore, some regions do not acquire MLG–PP film morphology at all, as evidenced by the large pores present. In regions where coating consists of film formation suspended between strands, it is observed that the morphology differs significantly. Some regions comprise a smooth film formation, as shown in Fig. 8(e), 9(e), 10(c) and (e), likely to be due to the presence of large concentrations of PDDA and PFO composite polymer. These regions possess no pores observable by SEM imaging. Other regions, such as those in Fig. 8(f), 9(e), (f) and 10(f), possess a similar aggregated morphology to that of MLG–PP films and thus consist of a more cluster-like appearance. In this case, pores are also present within the film only areas. For example, it is observed that coated Kevlar possesses pores with approximate areas of 1.0, 4.0 and 6.0 μm2 (Fig. 8(f)). In regions where film formation occurs over the strands directly, a smooth morphology is observed. These can be contrasted with strands which do not acquire film formation. In the latter case these appear much darker in the images. This is clearly observed within images associated with coated carbon fibre and coated glass fibre (Fig. 9(f) and 10(d)). It is evident that pores are formed both within the film itself and in regions where film formation is not present. In the latter case, these regions may not have become coated during the process or may have become coated, but the flexible nature of the substrate may have led to tearing of the film. For the coated carbon fibre, this latter situation is evident in Fig. 9(d). The images also show that there are a large variety of pores present within the coated substrates that could potentially allow the passage of fluids through them.
Nylon and stainless steel mesh also consist of a crosshatched arrangement. However, they do not consist of the parallel fibrous arrangements present in the other substrates. Nylon contains an aperture and width of 0.1 mm, whilst stainless steel mesh consists of square-shaped apertures of 0.28 mm, as shown in images of these uncoated substrates (Fig. 11(a) and 12(a)). Upon coating these substrates, it was evident that some of these apertures acquired a suspended film across them, whilst others were either partially coated or remained uncoated. As such, large pores were created in the latter case, as can be seen in Fig. 11(b) and 12(b). The images show that the coated stainless steel mesh possesses large pores with areas of 0.02 and 0.04 mm2. It was also observed that coated regions displayed varying morphologies, similar to those of the previous coated substrates. Some regions displayed a smooth morphology consisting of film formation containing primarily PDDA/PFO, as evidenced by Fig. 11(c) and 12(c). Other regions comprised of a coating that shows similarity to that of MLG–PP films with a cluster-like appearance, as shown in Fig. 11(d) and 12(d). Pores were also apparent within the coated areas themselves. Representative high magnification images of coated nylon reveal the presence of pores with areas corresponding to 3 and 5 μm2 in Fig. 11(e). Smaller pores of around 0.2 μm2 were also apparent in the stainless steel mesh (Fig. 12(e)). Furthermore, pores also formed between the spacing between suspended film and the substrate itself as evidenced by the 13 μm width pore within coated nylon (Fig. 11(f)); this is likely due to the flexibility of the substrate. Moreover, a large split can be observed within a coated region of the stainless steel mesh, possessing a width of 31 μm (Fig. 12(f)). This is most likely to be formed due to tearing as a result of movements of the substrate. It is therefore shown that coated nylon and coated stainless steel mesh also contain large pores where the coatings are non-uniform. Additional SEM images for the coated substrates are provided within Fig. S24–S28.†
 | ||
| Fig. 11 SEM images of (a) uncoated nylon at a magnification of 125×; and coated nylon at magnifications of (b) 415× (c) 10.38k× (d) 10.39k× (e) 20.75k× and (f) 4.15k×. | ||
 | ||
| Fig. 12 SEM images of (a) uncoated stainless steel mesh at a magnification of 106×; and coated stainless steel mesh at magnifications of (b) 208× (c) 10.38k× (d) 10.38k× (e) 9.96k× and (f) 692×. | ||
The somehow lower OCAs for coated Kevlar and coated nylon are consistent with the SEM images for these two materials, which show the presence of larger pores (Fig. 8(c) and 11(b)). Here, it is likely that the silicone oil can occupy space within the pore thereby generating these lower contact angles.
The largest pores present in the images of the coated nylon and coated stainless steel mesh are characterised by widths of 100 and 200 μm, respectively. On the other hand, the largest observed pores of coated Kevlar, coated carbon fibre and coated glass fibre are 90, 52 and 232 μm wide. The pores in the coated substrates were therefore found to reduce in size in the order: glass fibre > stainless steel mesh > nylon > Kevlar > carbon fibre. The presence of the largest pores in the glass fibre explains why it was the only coated substrate which sometimes allowed oil to pass through. Additionally, the flexible, fibrous nature of the coated Kevlar, carbon fibre and glass fibre, may assist the passage of water due to physical movement of fibres as the substrate becomes saturated with water. Whilst many of the pores observed within the substrates are sufficiently large to accommodate the passage of oil through them, the MLG–PP coating acts as a barrier. It is evident that the fluorocarbon chains of the PFO moieties are effective in “repelling” oil droplets away from these pores.7
Further analysis and characterisation of MLG, MLG–PP composite and MLG–PP film
| Material | BET surface area (m2 g−1) | BJH adsorption volume of pores between 1.7–300 nm (cm3 g−1) | BJH average pore width (adsorption) (4 V Å−1) (nm) |
|---|---|---|---|
| MLG | 670.9 | 1.16 | 6.3 |
| MLG–PP composite | 4.3 | 0.03 | 16.7 |
| MLG–PP film | 1.8 | 0.01 | 23.1 |
To further explore the characteristics of these materials BJH and t-plot analysis were carried out. The BJH pore size distributions (PSDs) are displayed in Fig. S30.† The corresponding BJH data and t-plot data are provided in Table S1.† It was found that the BJH pore volume associated with the three materials deviates quite substantially, with values of 1.16, 0.03 and 0.01 cm3 g−1 corresponding to MLG, MLG–PP composite and MLG–PP film, respectively. T-Plot data also revealed a drastic decrease in the external surface area (comprising of pores >2 nm in size) corresponding to composite and film formation, where MLG, MLG–PP composite and MLG–PP film possessed values of 489.3, 4.3 and 1.8 m2 g−1, respectively. As such, the composite formation results in a severe loss of pore volume and pore area, which coincides with a large decrease in the BET surface area. It is likely that mesopores and macropores within the MLG–PP composite are reduced in number due to the presence of PDDA and PFO moieties occupying and blocking slit pores. A further decrease in pore volume and area is observed upon MLG–PP film formation, which is also consistent with the material possessing the smallest BET surface area. Again, it is likely that the film formation process further reduces the number pores as a result of the more ordered alignment of MLG stacks and PDDA/PFO moieties.
The BJH pore size distributions vary between the three materials, as shown in Fig. S30.† MLG possesses a PSD between 1.7–84.5 nm, suggesting the presence of micropores (pores < 2 nm), mesopores (pores between 2 and 50 nm) and macropores (pores exceeding 50 nm) within the material. The MLG–PP composite and MLG–PP film materials meanwhile possess much larger PSDs corresponding to 4.1–123.8 and 5.9–130.3 nm respectively, indicating the presence of mesopores and macropores only. This finding is confirmed by a lack of t-plot micropore area and volume for these materials. This absence of micropores is also consistent with the absence of a sharp nitrogen uptake within the BET curves at P/P0 < 1 (inset in Fig. S29†). Moreover, the loss of small mesopores with widths between 1.7–4.1 nm for MLG–PP composite and 1.7–5.9 nm for MLG–PP film, suggests it is the small pores which were most impacted by the presence of PDDA and PFO. It is, however, observed that the PSD widens towards the macroporous region for MLG–PP composite and MLG–PP film, suggesting that larger pores were introduced into the materials, albeit in very low amounts. The presence of PDDA and PFO moieties could be responsible for a widening of some slit pores in some instances.
The SEM images outlined in the previous section provided evidence of large macropores within the MLG–PP film, possessing widths of approximately 0.5–2 μm in size.39 It is noteworthy that although the area associated with these pores contributes to the BET and t-plot external surface area data, it does not contribute to BJH data, which only analyses pores between 1.7–300 nm.62,63
Application of coated substrates as membranes
With the oleophobic properties of the coated substrates demonstrated, along with the expedient passage of water, the next steps were to explore their applications as membranes. Our aims were to test the MLG–PP coated substrates as membranes in “real world” applications, such as for oil-spill clean-ups and within aircraft propellant tanks. The results of these investigations are outlined in the following sections. | ||
| Fig. 13 Photographs outlining how coated carbon fibre supported by stainless steel mesh was utilised to remove silicone oil from the surface of water. | ||
 | ||
| Fig. 14 Images of the water separation tests using coated nylon membrane which allows water to be removed whilst simultaneously retaining Jet A-1 fuel. In the case with no membrane present the fuel freely passes through. | ||
Experimental
All solvents and reagents were purchased from commercial suppliers and used with no further purification. The MLG material was synthesised and provided by Perpetuus Carbon Technologies.36 MLG–PP composite was made in accordance with our previous report.39 MLG–PP films and coated substrates were synthesised within regular medium gauge non-stick oven trays with exterior dimensions of 24.5 cm by 17.5 cm and interior dimensions of 23.5 cm by 16 cm. The nylon material was purchased from Sefar Ltd. A5 size meshes were purchased from The Mesh Company and consist of industry standard woven wire mesh stainless steel grade (#50).Synthesis of coated substrates
MLG–PP composite (2.5 g) was dispersed in 150 mL of MeOH via bath sonication for 15 minutes. This dispersion was poured into a baking tray containing the Kevlar, carbon fibre and glass fibre. All substrates were covered by the dispersion. The methanol was allowed to slowly evaporate overnight, affording coated substrates. Coated nylon was synthesised using the same procedure, however, a dispersion containing 0.26 g of MLG–PP composite in 150 mL MeOH was used.Conclusions
Within this work, the further development of a unique composite based on multi-layer graphitic (MLG) material with the polyelectrolyte/surfactant complex PDDA and PFO (MLG–PP) has been achieved. The composite was used to synthesise freestanding MLG films and to coat various substrates including Kevlar, carbon fibre, glass fibre and nylon. The films and substrates all showed oleophobic behaviour to silicone oil, hexadecane and Jet A-1 fuel. The coated carbon fibre showed promise as a membrane for the clean-up of oil on the surface of water with some degree of reusability demonstrated. It was also shown that coated nylon can be used within the context of aircraft propellant tanks to provide a membrane which retains Jet A-1 fuel and allows the release of accumulated water. OCAs for the composite film exhibited high values up to 126.4° using silicone oil, 105.9° for hexadecane and 111.9° for Jet A-1 fuel. Coating of the composite on a variety of substrates provided a strategy to improve the mechanical stability of the materials and also led to high OCAs for silicone oil on coated carbon fibre (103.3°), hexadecane on coated carbon fibre (120.3°) and Jet A-1 fuel on coated carbon fibre (107.0°).The MLG–PP composite, MLG–PP films and MLG–PP coated substrates were investigated in detail using XPS, XRD, BET/BJH/t-plot, FT-IR, Raman spectroscopy and SEM techniques. SEM analysis provided evidence of a unique porous network within the MLG–PP films. Furthermore, by utilising the MLG–PP on grids and meshes, additional pores and channels were incorporated into the materials. These served to decrease the time for water passage through these materials with respect to the MLG–PP films and enabled the demonstration of their application in oil/water separation.
The successful coating of the MLG–PP composite onto four grid-like substrates highlights the general effectiveness of the MLG–PP as a coating for a range of porous materials for oil/water separation applications. The flexibility and strength of the resulting materials can, of course, be defined by the properties of the parent substrate. The demonstration of the oil/water separations on the laboratory scale highlights the potential for such materials to be applied within “real world” applications, such as for oil-spill clean-ups and within aircraft propellant tanks. The results of this investigation are currently being optimised and developed with a focus on reusability and durability. This offers potential for future utilisation on larger scales.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the European Social Fund (ESF) via the Welsh Government through a KESS2 PhD studentship (R. M.). It was co-sponsored by Perpetuus Carbon Technologies. The company is also thanked for providing the raw MLG material. We are also grateful to the HarwellXPS EPSRC National Facility for carrying out the XPS data collections. We are additionally grateful to Dr. Greg Shaw at Cardiff University for carrying out XRD and Raman analysis.References
- S. J. Hutton, J. M. Crowther and J. P. S. Badyal, Chem. Mater., 2000, 12, 2282–2286 CrossRef CAS.
- R. A. Lampitt, J. M. Crowther and J. P. S. Badyal, J. Phys. Chem. B, 2000, 104, 10329–10331 CrossRef CAS.
- J. A. Howarter and J. P. Youngblood, Adv. Mater., 2007, 19, 3838–3843 CrossRef CAS.
- J. A. Howarter and J. P. Youngblood, Macromol. Rapid Commun., 2008, 29, 455–466 CrossRef CAS.
- X. Zhu, H.-E. Loo and R. Bai, J. Membr. Sci., 2013, 436, 47–56 CrossRef CAS.
- J. Yong, F. Chen, Q. Yang, J. Huo and X. Hou, Chem. Soc. Rev., 2017, 46, 4168–4217 RSC.
- Y. Wang and X. Gong, J. Mater. Chem. A, 2017, 5, 3759–3773 RSC.
- H. Yu, M. Wu, G. Duan and X. Gong, Nanoscale, 2022, 14, 1296–1309 RSC.
- H. Wake, Estuarine, Coastal Shelf Sci., 2005, 62, 131–140 CrossRef CAS.
- J. Drelich, J. L. Wilbur, J. D. Miller and G. M. Whitesides, Langmuir, 1996, 12, 1913–1922 CrossRef CAS.
- C. W. Extrand and Y. Kumagai, J. Colloid Interface Sci., 1997, 191, 378–383 CrossRef CAS PubMed.
- A. Nakajima, S. Koizumi, T. Watanabe and K. Hashimoto, Langmuir, 2000, 16, 7048–7050 CrossRef CAS.
- X. Han and X. Gong, ACS Appl. Mater. Interfaces, 2021, 13, 31298–31309 CrossRef CAS PubMed.
- Z. Xiong, H. Yu and X. Gong, Langmuir, 2022, 38, 8708–8718 CrossRef CAS PubMed.
- J. Yang, Z. Zhang, X. Xu, X. Zhu, X. Men and X. Zhou, J. Mater. Chem., 2012, 22, 2834 RSC.
- P. S. Brown, O. D. L. A. Atkinson and J. P. S. Badyal, ACS Appl. Mater. Interfaces, 2014, 6, 7504–7511 CrossRef CAS PubMed.
- P. S. Brown and B. Bhushan, Sci. Rep., 2015, 5, 14030 CrossRef PubMed.
- P. S. Brown and B. Bhushan, Sci. Rep., 2015, 5, 8701 CrossRef PubMed.
- H. Yoon, S.-H. Na, J.-Y. Choi, S. S. Latthe, M. T. Swihart, S. S. Al-Deyab and S. S. Yoon, Langmuir, 2014, 30, 11761–11769 CrossRef CAS PubMed.
- J. Yang, L. Yin, H. Tang, H. Song, X. Gao, K. Liang and C. Li, Chem. Eng. J., 2015, 268, 245–250 CrossRef CAS.
- J. Li, L. Yang, H. Liu, G. Li, R. Li, Y. Cao and H. Zeng, ACS Appl. Mater. Interfaces, 2020, 12, 45266–45273 CrossRef CAS PubMed.
- F. Li, Z. Wang, S. Huang, Y. Pan and X. Zhao, Adv. Funct. Mater., 2018, 28, 1706867 CrossRef.
- F. R. Sultanov, C. Daulbayev, B. Bakbolat and Z. A. Mansurov, Eurasian Chem.-Technol. J., 2018, 20, 195 CrossRef CAS.
- M. Antonietti, C. Burger, J. Conrad and A. Kaul, Macromol. Symp., 1996, 106, 1–8 CrossRef CAS.
- P. M. Macdonald and A. Tang, Langmuir, 1997, 13, 2259–2265 CrossRef CAS.
- M. Antonietti, J. Conrad and A. Thuenemann, Macromolecules, 1994, 27, 6007–6011 CrossRef CAS.
- F. M. Fowkes, Ind. Eng. Chem., 1964, 56, 40–52 CrossRef CAS.
- F. M. Fowkes, Y. C. Huang, B. A. Shah, M. J. Kulp and T. B. Lloyd, Colloids Surf., 1988, 29, 243–261 CrossRef CAS.
- D. K. Owens and R. C. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741–1747 CrossRef CAS.
- C.-T. Hsieh, J.-M. Chen, R.-R. Kuo, T.-S. Lin and C.-F. Wu, Appl. Surf. Sci., 2005, 240, 318–326 CrossRef CAS.
- T. “Leo” Liu and C.-J. “CJ” Kim, Science, 2014, 346, 1096–1100 CrossRef CAS PubMed.
- H. C. Kang and A. M. Jacobi, Langmuir, 2011, 27, 14910–14918 CrossRef CAS PubMed.
- X. Du, X. Li and J. He, ACS Appl. Mater. Interfaces, 2010, 2, 2365–2372 CrossRef CAS PubMed.
- R. L. McLaren, C. J. Laycock, D. J. Morgan and G. R. Owen, New J. Chem., 2020, 44, 19144–19154 RSC.
- R. L. McLaren, C. J. Laycock, E. Brousseau and G. R. Owen, New J. Chem., 2021, 45, 12071–12080 RSC.
- I. Walters and D. Walters, Perpetuus Research & Development Limited, EU Pat., EP3129323B1, 2019 Search PubMed.
- S. Malik, F. M. Ruddock, A. H. Dowling, K. Byrne, W. Schmitt, I. Khalakhan, Y. Nemoto, H. Guo, L. Kumar Shrestha, K. Ariga and J. P. Hill, Beilstein J. Nanotechnol., 2018, 9, 801–808 CrossRef CAS PubMed.
- A. Bianco, H.-M. Cheng, T. Enoki, Y. Gogotsi, R. H. Hurt, N. Koratkar, T. Kyotani, M. Monthioux, C. R. Park, J. M. D. Tascon and J. Zhang, Carbon, 2013, 65, 1–6 CrossRef CAS.
- R. L. McLaren, R. C. da Costa, C. J. Laycock, D. J. Morgan, M. E. A. Warwick and G. R. Owen, New J. Chem., 2021, 45, 19210–19214 RSC.
- Z. Chu, Y. Feng and S. Seeger, Angew. Chem., Int. Ed., 2015, 54, 2328–2338 CrossRef CAS PubMed.
- J. B. Boreyko, G. Polizos, P. G. Datskos, S. A. Sarles and C. P. Collier, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7588–7593 CrossRef CAS PubMed.
- J. T. Edwards, in 55th AIAA Aerospace Sciences Meeting, American Institute of Aeronautics and Astronautics, Reston, Virginia, 2017, pp. 1–58 Search PubMed.
- A. Theodosiou, B. F. Spencer, J. Counsell and A. N. Jones, Appl. Surf. Sci., 2020, 508, 144764 CrossRef CAS.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS PubMed.
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- B. Kim, H. Park and W. M. Sigmund, Langmuir, 2003, 19, 2525–2527 CrossRef CAS.
- B. Kim and W. M. Sigmund, Langmuir, 2003, 19, 4848–4851 CrossRef CAS.
- J. H. Rouse and P. T. Lillehei, Nano Lett., 2003, 3, 59–62 CrossRef CAS.
- D. Yang, J. Rochette and E. Sacher, J. Phys. Chem. B, 2005, 109, 4481–4484 CrossRef CAS PubMed.
- S. Wang, D. Yu, L. Dai, D. W. Chang and J.-B. Baek, ACS Nano, 2011, 5, 6202–6209 CrossRef CAS PubMed.
- S. N. Lim, W. Ahn, S.-H. Yeon and S. Bin Park, Chem. - Asian J., 2014, 9, 2946–2952 CrossRef CAS PubMed.
- C. Zhu, P. Wang, L. Wang, L. Han and S. Dong, Nanoscale, 2011, 3, 4376 RSC.
- Y. Xiang, M. K. Banks, R. Wu, W. Xu and S. Chen, Mater. Chem. Phys., 2018, 220, 58–65 CrossRef CAS.
- S. Yang, X. Feng, S. Ivanovici and K. Müllen, Angew. Chem., 2010, 122, 8586–8589 CrossRef.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- T. Szabó, O. Berkesi, P. Forgó, K. Josepovits, Y. Sanakis, D. Petridis and I. Dékány, Chem. Mater., 2006, 18, 2740–2749 CrossRef.
- H. Dautzenberg, E. Görnitz and W. Jaeger, Macromol. Chem. Phys., 1998, 199, 1561–1571 CrossRef CAS.
- R. L. McLaren, G. R. Owen and D. J. Morgan, Results Surf. Interfaces, 2022, 6, 100032 CrossRef.
- D. Briggs and G. Beamson, High Resolution XPS of Organic Polymers: The Scienta ESCA300 Database, Wiley & Sons, Chichester, 1992 Search PubMed.
- K. S. W. Sing, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- S. Lowell and J. E. Shields, Powder Surface Area and Porosity, Springer Netherlands, Dordrecht, 3rd edn, 1991 Search PubMed.
- W. Sun, W. Liu and Y. Hu, J. Cent. South Univ. Technol., 2008, 15, 373–377 CrossRef CAS.
- W. Chen, X. Zhang, M. Mamadiev and Z. Wang, RSC Adv., 2017, 7, 927–938 RSC.
- M.-L. Lin, T. Chen, W. Lu, Q.-H. Tan, P. Zhao, H.-T. Wang, Y. Xu and P.-H. Tan, J. Raman Spectrosc., 2018, 49, 46–53 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- S. Claramunt, A. Varea, D. López-Díaz, M. M. Velázquez, A. Cornet and A. Cirera, J. Phys. Chem. C, 2015, 119, 10123–10129 CrossRef CAS.
- R. P. Vidano, D. B. Fischbach, L. J. Willis and T. M. Loehr, Solid State Commun., 1981, 39, 341–344 CrossRef CAS.
- D. C. Elias, R. R. Nair, T. M. G. Mohiuddin, S. V. Morozov, P. Blake, M. P. Halsall, A. C. Ferrari, D. W. Boukhvalov, M. I. Katsnelson, A. K. Geim and K. S. Novoselov, Science, 2009, 323, 610–613 CrossRef CAS PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- I. K. Moon, J. Lee, R. S. Ruoff and H. Lee, Nat. Commun., 2010, 1, 73 CrossRef PubMed.
- C. Hontoria-Lucas, A. J. López-Peinado, J. D. D. López-González, M. L. Rojas-Cervantes and R. M. Martín-Aranda, Carbon, 1995, 33, 1585–1592 CrossRef CAS.
- J. M. Walter, Raman Spectroscopy of Graphite, https://jumwalter.de/plaintext/geo-und-mehr/raman-spectroscopy-of-graphite.html, 2013.
- S. Reich and C. Thomsen, Philos. Trans. R. Soc., A, 2004, 362(18), 2271–2288 CrossRef CAS PubMed.
- A. Inam, R. Brydson and D. V. Edmonds, Mater. Charact., 2020, 163, 110264 CrossRef CAS.
- M. S. Seehra, U. K. Geddam, D. Schwegler-Berry and A. B. Stefaniak, Carbon, 2015, 95, 818–823 CrossRef CAS PubMed.
- S. Baena-Zambrana, S. L. Repetto, C. P. Lawson and J. K.-W. Lam, Prog. Aerosp. Sci., 2013, 60, 35–44 CrossRef.
- Aircraft Accident Report AAR 1/2010 – Boeing 777-236ER, G-YMMM, 17 January 2008 – (https://www.gov.uk), Report on the accident to Boeing 777-236ER, G-YMMM, at London Heathrow Airport on 17 January 2008, Technical Report, Air Accident Investigation Branch, Department of Transport (UK), 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Containing details coated substrate preparation, further characterisation and application instrumentation. See DOI: https://doi.org/10.1039/d2me00197g |
| This journal is © The Royal Society of Chemistry 2023 |