
Synthesis of bis-furyl-pyrrolo[3,4-b]pyridin-5-ones via Ugi–Zhu reaction and in vitro activity assays against human SARS-CoV-2 and in silico studies on its main proteins†
Ivette
Morales-Salazar
a,
Flora P.
Montes-Enríquez
a,
Carlos E.
Garduño-Albino
a,
M. A.
García-Sánchez
a,
Ilich A.
Ibarra
 b,
Yareli
Rojas-Aguirre
c,
Montserrat Elemi
García-Hernández
d,
Rosa Elena
Sarmiento-Silva
e,
Sofía Lizeth
Alcaraz-Estrada
*f,
Erik
Díaz-Cervantes
*g,
Eduardo
González-Zamora
*a and
Alejandro
Islas-Jácome
*a
b,
Yareli
Rojas-Aguirre
c,
Montserrat Elemi
García-Hernández
d,
Rosa Elena
Sarmiento-Silva
e,
Sofía Lizeth
Alcaraz-Estrada
*f,
Erik
Díaz-Cervantes
*g,
Eduardo
González-Zamora
*a and
Alejandro
Islas-Jácome
*a
aDepartamento de Química, Universidad Autónoma Metropolitana-Iztapalapa, Av. Ferrocarril San Rafael Atlixco 186, Col. Leyes de Reforma 1A Sección, Iztapalapa, Ciudad de México C.P. 09310, Mexico
bLaboratorio de Fisicoquímica y Reactividad de Superficies, Instituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior S/N, Ciudad Universitaria, Coyoacán, Ciudad de México C.P. 04510, Mexico
cDepartamento de Polímeros, Instituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior S/N, Ciudad Universitaria, Coyoacán, Ciudad de México C.P. 04510, Mexico
dDepartamento de Microbiología e Inmunología, Facultad de Medicina, Veterinaria y Zootecnia, Universidad Nacional Autónoma de México, Av. Universidad 3000, Ciudad Universitaria, Coyoacán, Ciudad de México C.P. 04510, Mexico
eLaboratorio de Virología y Laboratorio Mixto Internacional ELDORADO, Facultad de Medicina, Veterinaria y Zootecnia, Universidad Nacional Autónoma de México, Av. Universidad 3000, Ciudad Universitaria, Coyoacán, Ciudad de México C.P. 04510, Mexico
fDivisión de Medicina Genómica, Centro Médico Nacional 20 de Noviembre, ISSSTE, Félix Cuevas 540, Col. Del Valle Sur, Benito Juárez, Ciudad de México C.P. 03100, Mexico
gDepartamento de Alimentos, Centro Interdisciplinario del Noreste, Universidad de Guanajuato, Tierra Blanca, Guanajuato C.P. 37975, Mexico
First published on 18th November 2022
Abstract
An Ugi–Zhu three-component reaction (UZ-3CR) coupled in one pot manner to a cascade process (N-acylation/aza Diels–Alder cycloaddition/decarboxylation/dehydration) was performed to synthesize a series of bis-furyl-pyrrolo[3,4-b]pyridin-5-ones in 45 to 82% overall yields using ytterbium triflate as a catalyst, toluene as a solvent, and microwaves as a heat source. The synthesized molecules were evaluated in vitro against human SARS-CoV-2 through a time-of-addition approach, finding that compound 1e, at a concentration of 10.0 μM, exhibited a significant reduction at the initial infection stages, thus showing prophylactic potential. On the other hand, it was found that compound 1d, at the same concentration, was significantly active when applied post-infection, thus exhibiting a therapeutic profile. Moreover, compound 1f showed both, prophylactic and therapeutic activity. Then, to understand interactions between synthesized compounds and the main proteins related to the virus, docking studies were performed on spike-glycoprotein, main-protease, and Nsp3 protein, finding moderate to strong binding energies, matching accurately with the in vitro results. Additionally, a pharmacophore model was computed behind further rational drug design.
Introduction
The first confirmed case of infection by SARS-CoV-2 was recorded in Wuhan, P. R. C., in December 2019.1 The COVID-19 pandemic was declared in March 2020 by the World Health Organization's (WHO).2 Another respiratory syndrome of the coronavirus family to be reported were severe acute respiratory syndrome coronavirus (SARS-CoV-1)3 and Middle Eastern respiratory syndrome coronavirus (MERS-CoV).4 These two syndromes set the tone for a thorough understanding of SARS-CoV-2.5 A name analogous to SARS-CoV-2 is coronavirus due to its morphology since the homo-trimers of its spike-glycoprotein transmembrane form a crown and protrude from the viral surface.6 This virus belongs to the Coronaviridae family and has the feature of having a very large RNA genome.7 This virus shows a tendency to be transmitted from animals to humans, so it has been reported around 60 coronaviruses isolated from bats.8 The acute respiratory syndrome has caused a high mortality rate around the world. According to WHO portal, around 6.5m people have passed, from 2020 to September 2022.9To understand mechanistic of this virus, two very important proteins have been isolated: first, the spike-glycoprotein (PDB: 6VSB).10 The structure of this protein is divided into two subunits: the S1 that contains the receptor binding domain (RBD) and the S2 that is closely related to the junction between the virus and the host cell.11 The angiotensin-converting-enzyme 2 (ACE2) is responsible for binding SARS-CoV-2 with the host cell.12 Second, the main protease MPro, also called 3CLPro, (PDB: 6LU7),13 is a three-domain cysteine protease, which is crucial in the transcription and replication because it processes polyproteins that are translated into viral RNA,14 as well as the Nsp3 protein (PDB: 7KXB).15 Also, 3CLPro has 11 cleavages.16 During the pandemic period, several compounds have been probed in vitro and in vivo in desire to counter SARS-CoV-2, e.g. antivirals and antiretrovirals (Fig. 1A), among other drugs, some of them exhibiting moderate to good potential activity, but eventually with undesirable side effects.17a,b Indeed, there are three compounds currently approved to treat the disease caused by SARS-CoV-2. These are paxlovid, remdesivir and molnupiravir; and of them, only paxlovid has been known to interact with the SARS-CoV-2 main viral proteases. Paxlovid is an FDA-approved oral treatment, composed of nirmatrelvir, an inhibitor that blocks the SARS-CoV-2 Mpro enzyme activity, combined with ritonavir, an antiretroviral protease inhibitor that does not act against SARS-CoV-2, but rather slows down the metabolism of nirmatrelvir (Fig. 1B). Several reports have been published demonstrating their antiviral activity through different strategies, and it is worth mentioning that none of them were plaque assays. For instance, Hoffman17c through a protease FRET assay reported an inhibition constant Ki = 0.271 nM. Besides, Owen17d reported an EC50 of 74.5 nM in Vero E6 cells, determined through cell viability assays. Moreover, remdesivir and molnupiravir are viral polymerase inhibitors. Then, Rosales et al.,17e using Vero-TMPRSS2 or HeLa-ACE2 expressing cells, determined through cell viability assays the antiviral activity of their compounds including nirmatrelvir on different SARS-CoV-2 variants. They reported that these compounds maintained their activity across the different variants and cell types. Therefore, it is difficult to make precise and or accurate comparisons between the efficacy in inhibiting the infection from different compounds assayed.
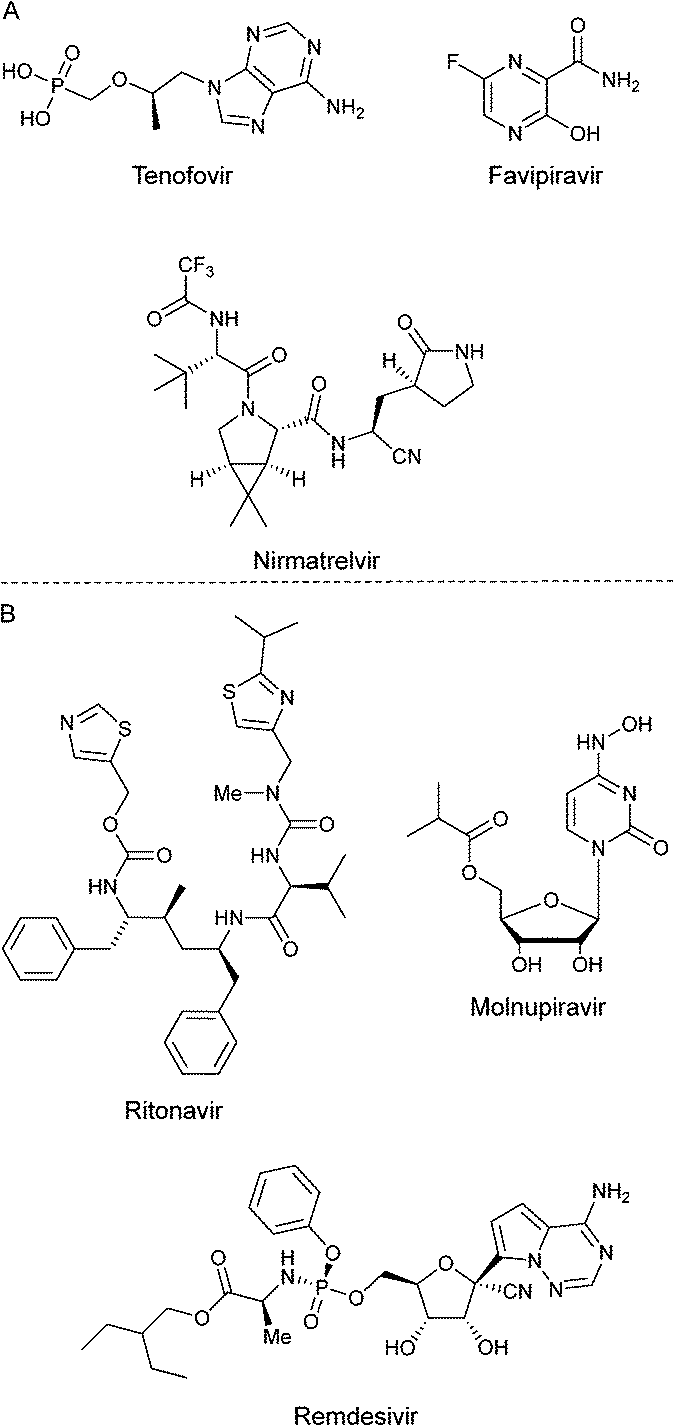 | ||
| Fig. 1 A) Some drugs tested against SARS-CoV-2 during pandemic of COVID-19. B) Compounds approved to treat COVID 19 disease. | ||
Besides, multicomponent reactions (MCRs) are powerful synthetic tools to assemble complex polyheterocyclic cores in a one pot manner.18 MCRs meet most green chemistry principles, becoming them in one of the most sustainable alternatives to synthesize compounds a la carte with pharmaceutical interest because they decrease both the number of reaction steps, and the consumption of resources like time, energy, and consumables for chromatography, all of them commonly overused in multistep synthetic methodologies.19 In this context, a recent variant of the Ugi-three component reaction (U-3CR), in which construction of the 5-aminooxazole core takes place efficiently, has been named as the Ugi–Zhu three-component reaction (UZ-3CR). Thus, aldehydes, amines and amino acid-derived α-isocyanoacetamides are sequentially combined in the same reactor to synthesize 5-aminooxazoles, which can be used as heterocyclic scaffolds for further post-Ugi–Zhu reactions, including aza-Diels–Alder cycloadditions toward pyrrolo[3,4-b]pyridine-5-ones,20 functionalizations of amine moiety, and cascade one pot processes taking advantage of both functional groups, oxazole and amine.21 Indeed, we recently published a comprehensive review about the Ugi-3CR and its variants, in which the UZ-3CR was recognized for its elegance, robustness, and utility towards not only 5-aminooxazoles, but also a wide variety of polyheterocyclic systems.22 In a previous proceedings-type communication,23 along with their syntheses, we reported preliminary results from docking studies between five bis-furanyl-pyrrolo[3,4-b]pyridin-5-ones and the main protease MPro, which revealed interesting ligand–target interactions that led us to extend and improve our research performing in vitro assays using human SARS-CoV-2, together with more robust in silico studies, for instance, a more accurate docking between six bis-furyl-pyrrolo[3,4-b]pyridin-5-ones and MPro again, but also to spike-glycoprotein, and the non-structural protein 3 of the virus (Nsp3), even including a proposal of a pharmacophore model, which may be useful for further investigations, just like anti-SARS-CoV-2 rational drug design. It is noteworthy that vaccines should not be considered as the unique defences against coronavirus.
Results and discussion
Synthesis
With the aim to synthesize the in vitro assayed bis-furyl-pyrrolo[3,4-b]pyridin-5-ones 1a–f, an UZ-3CR was coupled in one pot fashion to a cascade process: aza-Diels–Alder/N-acylation/aromatization (decarboxylation/dehydration). In some of our previous works,24 it was found that dichloromethane (DCM), scandium(III) triflate (ScOTf3), and microwaves are the best solvent, catalyst, and heat source, respectively, to assemble the polyheterocyclic core pyrrolo[3,4-b]pyridin-5-one. However, for the present work, it was necessary to perform a new screening towards optimal reaction conditions due to special stereoelectronic nature of the reactants for the UZ-3CR, particularly due to the presence of furyl or tetrahydrofuryl motifs. Thus, the model reaction between furan-2-carbaldehyde (2a), furan-2-ylmethanamine (3a), 2-isocyano-1-morpholino-3-phenylpropan-1-one (6a) and maleic anhydride (9) to synthesize the bis-furyl-pyrrolo[3,4-b]pyridin-5-one 1a, a series of Lewis acids (Sc(OTf)3, Yb(OTf)3 and InCl3), and a Brønsted acid (NH4Cl) were examined. Also, the reaction was probed varying the solvent: MeOH, EtOH, CH3CN and PhMe at 65 to 80 °C using both conventional and dielectric MW heating conditions. It was found that toluene was the best solvent, being the unique among all that perfectly dissolved all reactants. Finally, ytterbium(III) triflate in catalytic amounts gave the highest yield (Table 1, entry 6).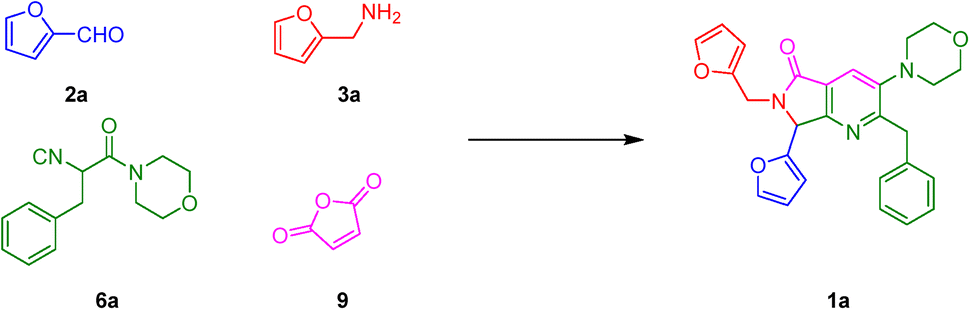
First, it was necessary to synthesize the α-isocyanoacetamides 6a–b in three reaction steps according to the Zhu and Bienaymé protocol starting from their corresponding amino acids phenylalanine and tyrosine, respectively.25 Once achieved them, according to the UZ-3CR mechanism, a condensation occurs between aldehydes 2a–b and amines 3a–b to access to imines 4a–f, which become in Lewis' iminium-like cations 5a–f after reacting with catalytic amounts of ytterbium(III) triflate. Then, an α-nucleophilic attack by the isocyanides 6a–b to 5a–f afforded the 5-aminooxazoles 8a–fvia a non-prototropic chain-ring tautomerization of 7a–f. Then, after addition of maleic anhydride (9), an aza-Diels–Alder cycloaddition followed by a N-acylation/decarboxylation/dehydration26 took place under a cascade-type one pot process towards the corresponding bis-furyl-pyrrolo[3,4-b]pyridin-5-ones 1a–f in 45 to 82% overall yields (Scheme 1). It is worth highlighting the structural complexity of the products, as well as the formation of several new C–C and C–N bonds, while leaving only a couple of water molecules and one carbon dioxide, denoting the high atomic economy and green approach coming from this synthetic methodology. Also, the use of furan and THF-substituted starting materials for the structural decoration of the pyrrolo[3,4-b]pyridin-5-one core has not been reported in the literature, as far as we know.
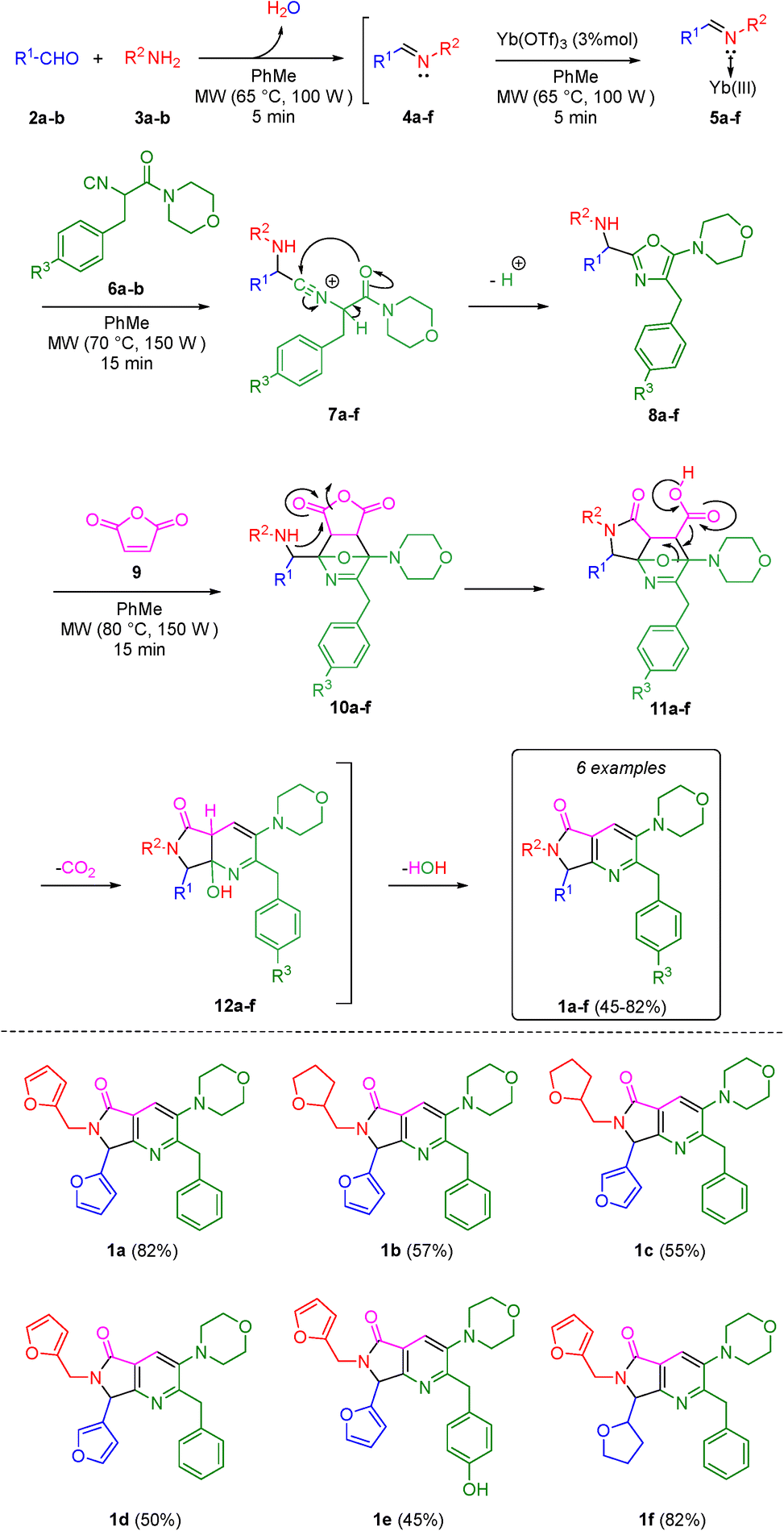 | ||
| Scheme 1 Synthesis of pyrrolo[3,4-b]pyridin-5-ones via a one-pot process (Ugi–Zhu/aza-Diels–Alder/N-acylation/aromatization). | ||
In vitro assays
As described in the Experimental section, cell viability was explored in the initial concentration range of 0.001 to 100 μM by the crystal violet assay, after exposing the cells 48 h to the compounds. Fig. 2 shows that up to 10 μM, there is an average cell viability ≥100% when compared to the vehicle control (V.C.) for all the compounds. However, at 100 μM, the highest concentration tested, five out of the six compounds of the series herein reported (1a–e) significantly decreased the Vero E6 cells viability. The IC50 value for each compound is as follows: 1a – 276.09 μM, 1b – 76.9 μM, 1c – 289.81 μM, 1d – 85.06 μM, 1e – 153.01 μM and 1f – 612.26 μM.
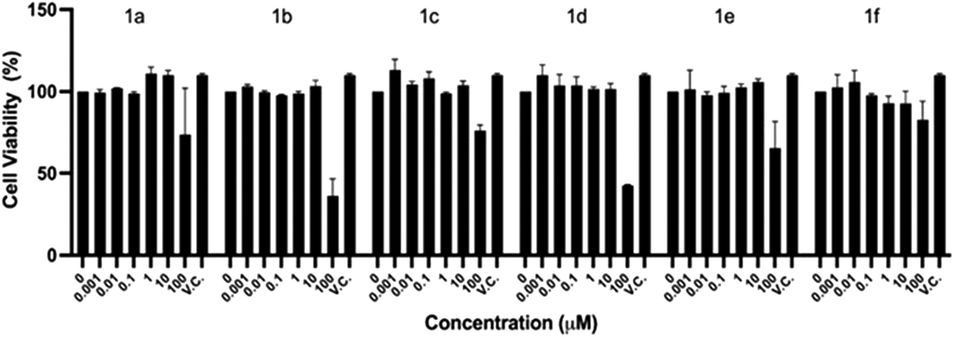 | ||
| Fig. 2 Vero E6 cell viability assays. The vehicle control (V.C.) refers to cells that were cultured in DMEM medium plus 0.5% DMSO, which was the DMSO volume used in the diluent to solubilize the compounds. The experiment was performed two independent times in triplicate and the bars depict the standard deviation of the mean (n = 6). | ||
Fig. 3A shows that under EC1, compound 1e significantly inhibited the infection by up to 77%. This striking result reflects the prophylactic potential of the polyheterocycle 1e. With these time-of-addition experiments, it can be inferred that 1e is preventing the viral infection, by interfering in one or more of the pathways involved in the initial steps of the viral replication cycle: a) cell entry stage, when the attachment and concentration of the virus to the cell surface is occurring; b) cell receptor interaction; and/or c) receptor-mediated endocytosis. Compounds 1a, 1c, and 1f also showed biological activity under this experimental condition, but lower than 1e. Where, compounds 1a, 1c and 1f displayed 19.4%, 9.4% and 31.94% of inhibition of infection respectively, indicating a moderate prophylactic potential. The biological activity of 1a, 1c and 1f was comparable, which suggests, as described above, that they may be interacting with proteins in the initial stages of the replication cycle, one possibility being the spike glycoprotein, thus reducing the overall viral load, but not as strongly as 1e. These findings lead us to hypothesize that the biological behaviour displayed by 1e is due to the –OH group in the phenyl ring, coming from the tyrosine amino acid.
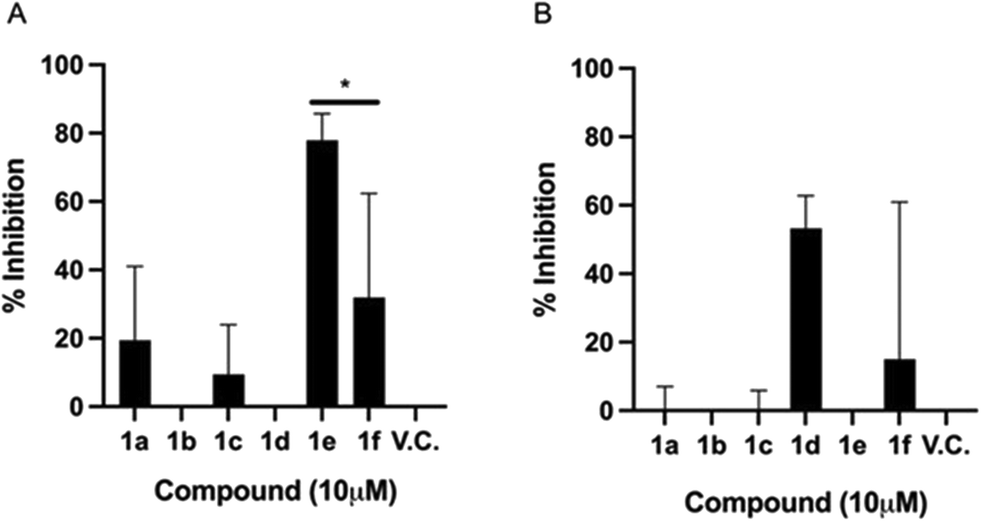 | ||
| Fig. 3 % inhibition of SARS-CoV-2 infection on Vero E6 cells. A) EC1: the compounds were added to the culture medium immediately after viral exposure of the cells. B) EC2: the compounds were added 1 h post-infection. The experiments were performed two independent times in triplicate (n = 6)*. p-Value = 0.66 using Mann–Whitney tests. The bars depict the standard deviation of the mean. | ||
Fig. 3B shows that the inhibitory effect of 1e is lost if added when the cells have already been infected. Similarly, the antiviral activity of 1a and 1c is deprived in post-infection conditions. Interestingly, compound 1d showed a 53% infection inhibition, thus indicating a potential therapeutic profile, as it has an effect once the infection has been established. The furan-3-yl moiety seems to be crucial to exert the observed therapeutic activity.
To note, 1f derivative displays both a mild prophylactic and therapeutic effects. It has been postulated that this kind of double effect arises when cell mediated immune responses are stimulated, thus suggesting that the compound could display immunomodulatory effects.29
The time-of-addition approach allowed identifying three compounds that can act as prophylactics, which may prevent the viral infection: 1a, 1c, and 1e; a compound acting at the onset of the viral infection with therapeutic potential: 1d; and a compound that is presumably generating a robust prophylactic and therapeutic antiviral response: 1f. Further animal studies are required to substantiate this potential. From a chemical structure point of view, compound 1a and 1e differ only in the hydroxyl group found in the phenyl ring. However, the synthesis of compound 1e is considerably more difficult than that of compound 1a because compound 1e precursor comes from tyrosine amino acid. Thus, it is well known that hydrogen-bond donors like OH group of phenols are labile (pKa ∼ 10), interfering in Ugi dehydration harsh reaction conditions. Moreover, compound 1f is synthesized using tetrahydro-2-furan carboxaldehyde, raising its production costs. However, the synthetic procedure to obtain 1e and 1f is worthy when observing this encouraging antiviral behaviour.
Several compounds bearing pyrimidine, isoxazole, as well as pyranyl–pyrimidinone moieties have been synthesized and evaluated through in silico models against SARS-CoV-2, showing promising results.30 These previous works motivated us to synthesize the pyrrolo[3,4-b]pyridin-5-ones that are herein reported. Striking results were obtained after the in vitro evaluation against SARS-CoV-2 as we find that five out of the six compounds demonstrated potential to inhibit the SARS-CoV-2 infection. As far as we know, this is the first time that this type of heterocyclic compounds is studied in biological in vitro models using the whole human coronavirus SARS-CoV-2. The three compounds (paxlovid, remdesivir and molnupiravir) that are currently commercially available are of a different chemical nature, so their comparison with the pyrrolo[3,4-b]pyridin-5-ones that we are reporting, is not decisive at this moment.
In silico studies
The results show the whole studied molecules into preferred cavities of the SARS-CoV-2-MPro, spike-glycoprotein surface, and Nsp3, respectively (Fig. 4A–C). For the case of the SARS-CoV-2-MPro, the six synthesized molecules present more negative values for the LE than the co-crystal molecule, which means a better ligand–target interaction (Table 2).
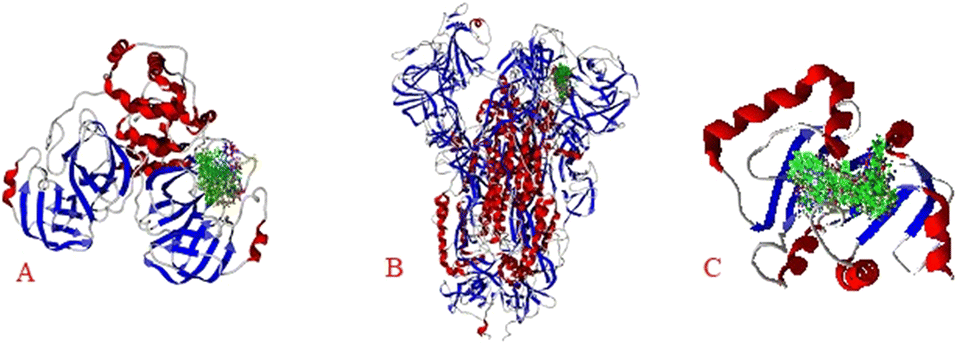 | ||
| Fig. 4 Docking of the eight studied molecules with: A) SARS-CoV-2-MPro, B) spike-glycoprotein surface, and C) Nsp3. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Protein | Molecule | LE | E | H-bond | Electro | vdW |
| LE means the ligand efficiency, which is the energy per atom provided for the ligand (LE = E/#heavy atoms); E are the whole ligand–target energies; H-bond are the hydrogen bond interactions; Electro are the electrostatic interactions; vdW are the van der Waals interactions. | ||||||
| MPro | 1a | −5.32 | −180.95 | −4.00 | 1.81 | −16.58 |
| 1b | −5.41 | −183.80 | −1.04 | 0.01 | 76.82 | |
| 1c | −5.67 | −192.83 | −1.58 | −0.27 | −52.00 | |
| 1d | −5.35 | −181.97 | −2.40 | 1.53 | −10.75 | |
| 1e | −5.68 | −198.88 | −3.65 | 0.09 | −21.23 | |
| 1f | −5.71 | −194.06 | −1.31 | 0.32 | −45.63 | |
| Co-crystal | −5.04 | −211.79 | −8.38 | −0.61 | −54.52 | |
| P8 (ref. 33) | −5.44 | −255.79 | −7.99 | −0.11 | −60.73 | |
| Spike-glycoprotein | 1a | −4.26 | −144.90 | −3.91 | −0.24 | −42.27 |
| 1b | −4.66 | −158.39 | −3.14 | 1.39 | −39.43 | |
| 1c | −4.70 | −159.95 | −3.57 | −0.25 | −28.46 | |
| 1d | −4.42 | −150.34 | −0.91 | −1.52 | −8.61 | |
| 1e | −5.50 | −192.34 | −4.19 | −0.07 | 2.50 | |
| 1f | −4.49 | −152.53 | −2.00 | 1.24 | 25.78 | |
| Co-crystal | −3.54 | −173.29 | −5.51 | −5.74 | 0.56 | |
| P8 (ref. 33) | −5.04 | −209.43 | −4.90 | −1.21 | −48.36 | |
| Nsp3 | 1a | −4.63 | −157.25 | −4.12 | 0.07 | −39.84 |
| 1b | −4.72 | −160.61 | −5.67 | 0.02 | −47.19 | |
| 1c | −4.86 | −165.38 | −7.97 | −0.09 | −44.07 | |
| 1d | −4.94 | −167.85 | −2.07 | 0.09 | −49.48 | |
| 1e | −4.57 | −160.09 | −4.82 | 0.10 | −34.26 | |
| 1f | −4.72 | −160.43 | −4.05 | −0.19 | 197.02 | |
| Co-crystal | −4.63 | −134.32 | −3.58 | 0.00 | −34.25 | |
| P8 (ref. 33) | −5.01 | −235.65 | −13.19 | 0.12 | −19.70 | |
In this order, the molecules 1f, 1e, and 1c are the better interacting molecules, with LE values more negatives, −5.71 kcal mol−1, −5.68 kcal mol−1, and −5.67 kcal mol−1, respectively. Note that these three molecules also are better than the P8 molecule, considering the LE values. Despite the molecules 1f, 1e, and 1c presenting different values, they only vary by 0.04 kcal mol−1 and can interact in similar forms. Considering the principal energies which promote a better interaction between the studied ligands and this protein, it is worth mentioning that hydrogen bond and van der Waals interactions are determinant. However, the size of the molecule is also a key factor in the more negative value of LE for 1f. In this case, electrostatic interaction is insignificant in determining the better interacting ligand. On the other hand, the interaction of the studied ligands with the spike-glycoprotein surface shows significant results correlated theoretically with the experimental results from in vitro assays. As the spike-glycoprotein is the first interacting protein for SARS-CoV-2, this interaction is crucial for the effectiveness of possible drugs. In this way, 1e is the ligand with the more negative value of LE (−5.50 kcal mol−1), being the next better P8 (−5.04 kcal mol−1), and 1c (−4.70 kcal mol−1). Note that, comparing the synthesized molecules, the difference between 1e and 1c is 0.7 kcal mol−1, which is a significant difference. On the other hand, analysing the principal energies, hydrogen bonds are the more significant interactions between the studied ligands and the spike-glycoprotein of the SARS-CoV-2, with a value of −4.19 kcal mol−1. With these results, it can be preliminary concluded that this difference may explain the experimental results, which demonstrate that 1e is the molecule with putative prophylactic antiviral activity. Finally, to explain the 1d effect once the infection has been established (1 h post-infection), as well as the 1f prophylactic and therapeutic effect, Table 2 shows the higher affinity of both compounds with the other key protein of the SARS-CoV-2, the Nsp3.
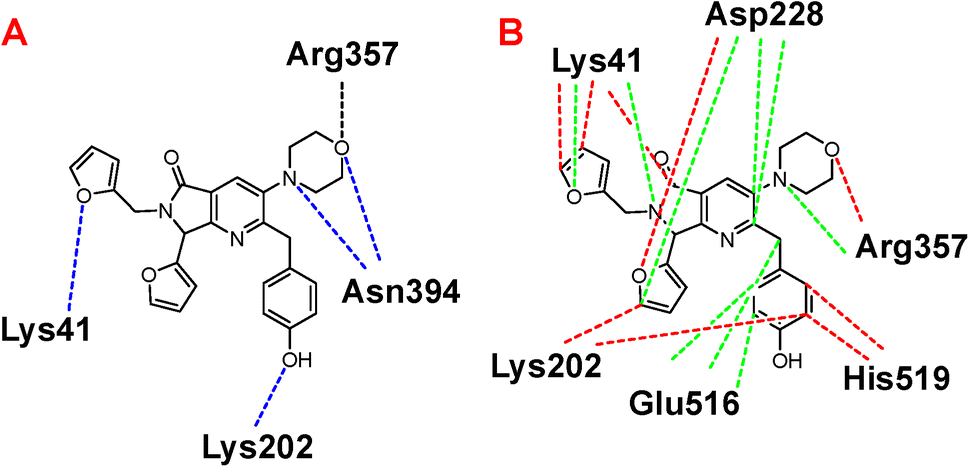
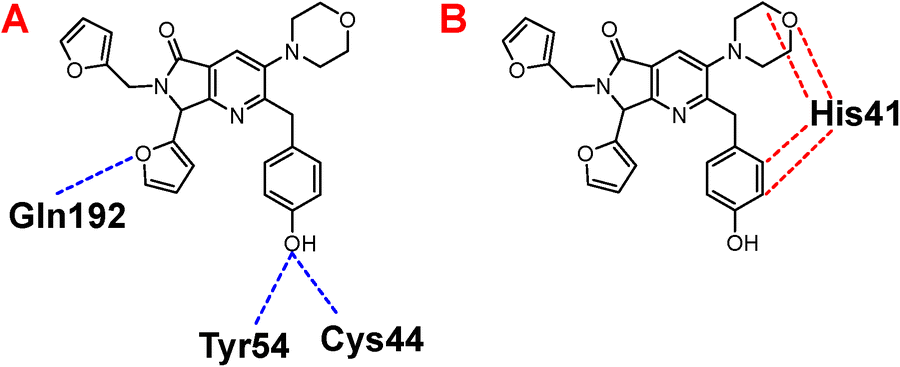
Once highlighted the importance of 1e, it has decided to evaluate the principal interactions of this one with the studied proteins. Fig. 5A shows the hydrogen bond interactions between 1e and SARS-CoV-2-MPro, in which Cys44, Tyr54, and Gln192 are the primary interacting residues, specifically the hydroxyl group of the ligand. In the case of the electrostatic interactions, these are mainly repulsive, with His41, see Fig. 5B. Regarding the spike-glycoprotein from SARS-CoV-2, Fig. 6A shows the hydrogen bond interactions from this one with 1e, in which the primary residues are Lys41, Lys202, Arg357, and Asn394, which interact with the oxygens into the ligand. Fig. 6B depicts the electrostatic interactions between 1e and the spike-glycoprotein active site, which also Lys41, Lys202, and Arg357 are the central interacting residues, moreover Asp228 and His519. Note that there are both attractive and repulsive interactions.
 | ||
| Fig. 5 A) Hydrogen bond and B) electrostatic interactions between 1e and SARS-CoV-2-MPro. Blue lines represent the hydrogen bond interactions, and green lines represent the electrostatic repulsion interactions. | ||
 | ||
| Fig. 6 A) Hydrogen bond and B) electrostatic interactions between 1e and the Nsp3 from SARS-CoV-2. Blue lines represent the hydrogen bond interactions; green lines depict the attraction, and red lines the electrostatic repulsion interactions. | ||
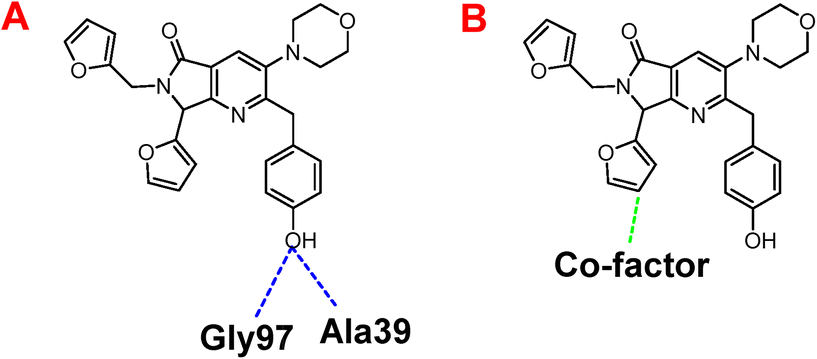
Besides, Fig. 7A shows the hydrogen interactions between the Nsp3 and 1e, which shows similar interactions in the hydroxyl group, interacting the Gly97 and Ala30. In the case of electrostatic interactions, only interacts 1e with the cofactor of the protein (Fig. 7B).
 | ||
| Fig. 7 A) Hydrogen bond, and B) electrostatic interactions between 1e and the spike-glycoprotein from SARS-CoV-2. Blue lines represent the hydrogen bond interactions, and red lines the electrostatic repulsion interactions. | ||
The hydrophobic interactions between 1e and SARS-CoV-2 proteins, MPro, spike-glycoprotein, and Nsp3 were evaluated to conclude this section. Fig. 8A shows the hydrophobic interactions between 1e and the SARS-CoV-2-MPro, highlighting the arrangement of the two-ring fragment of 1e with the studied protein's hydrophobic surface the main interactions to promote better possesses in this kind of protein.31
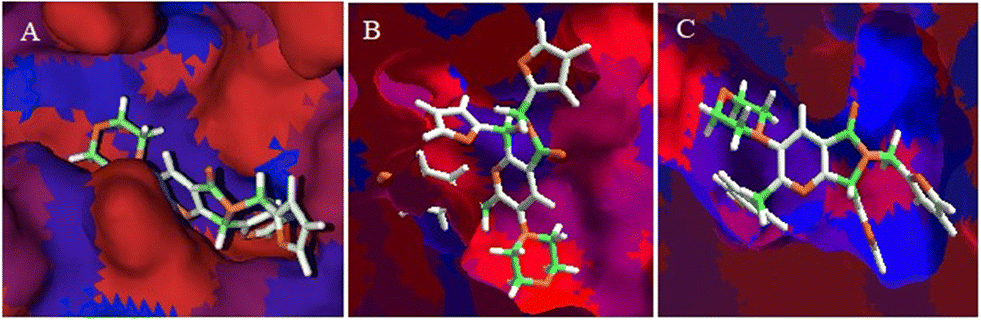 | ||
| Fig. 8 Hydrophobic interactions between 1e with A) MPro, B) Spike-glycoprotein, and C) Nsp3 proteins from SARS-CoV-2. Blue surfaces represent hydrophobic sites, red surfaces hydrophilic sites, green sticks depict hydrophobic fragments of the ligand, and orange sticks are hydrophilic fragments. | ||
On the other hand, the interaction between 1e and the spike-glycoprotein is more important than the first mentioned protein, due to this is the first contact protein from SARS-CoV-2 with a possible inhibitory molecule. In this order, Fig. 8B shows how the hydrophilic surface on the downside plays a crucial role in ligand–target interaction, which is the six-member ring of the 1e arrangement in this π-zone. Moreover, the hydrophobic site in the central zone of the cavity is essential to the bioactive structure of ligands, in which the two-ring member fragment of 1e is placed. Finally, Fig. 8C shows the higher hydrophobic surface of Nsp3 respect to the other proteins, which promotes a higher interaction of the aromatic–hydrophobic rings of 1e.
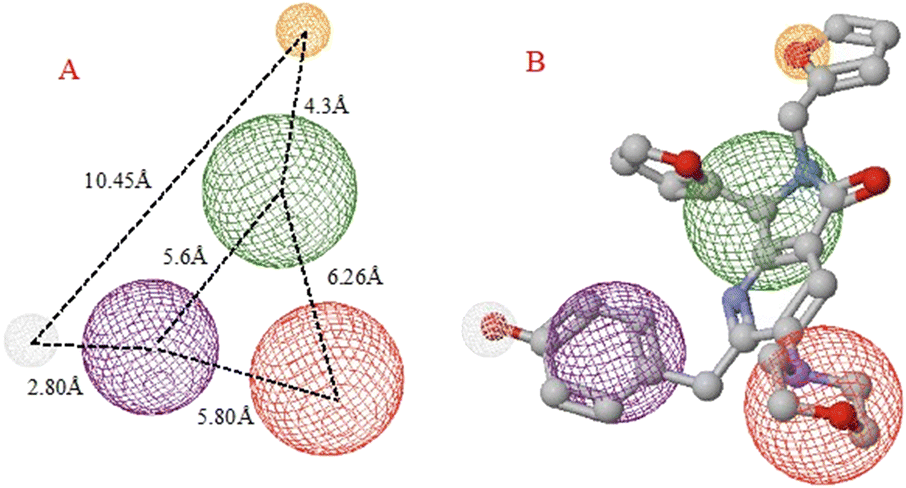 | ||
| Fig. 9 A) SARS-CoV-2 spike-glycoprotein pharmacophoric model. B) Orange spheres depict hydrogen acceptor fragments, green spheres are the hydrophobic–aromatic fragments, red spheres represent hydrophilic fragments, purple spheres are aromatic fragments, and gray spheres depict the hydrogen donor fragment. | ||
Regarding the prophylactic and therapeutic effects, docking studies reveal the importance of the hydrophobicity, the lengths of some rings, and the center of the molecule, which varies in each one depending on its moieties. For example, comparing 1c with 1e and 1f, 1e presents three aromatic rings at the edges of the molecule, in contrast with 1c and 1f, which present only two. The difference between 1c and 1f is the distance between the molecule's center and the five-ring-non-aromatic ring.
 | ||
| Fig. 10 Designed molecules A) P1, B) P2, and C) P3. | ||
Considering that the best target is the SARS-CoV-2-MPro, a molecular docking was carried on this target, obtaining the results exposed in Table 3.
| Molecule | LE | E | H-bond | Electro | vdW |
|---|---|---|---|---|---|
| P1 | −5.47 | −196.91 | −8.42 | −0.57 | −46.03 |
| P2 | −5.46 | −191.00 | −4.30 | 0.85 | −52.18 |
| P3 | −5.69 | −199.21 | −4.72 | 0.10 | 29.59 |
Table 3 depicts that the P3 molecule presents a better interaction with SARS-CoV-2-MPro than another molecule studied above. This derivative (P3) is like 1e but modifies the non-aromatic ring of the molecule, conferring more hydrophilic behaviour to this site and therefore obtaining a better candidate to the drug, which is clear in the hydrophobicity analysis depicted in Fig. 11.
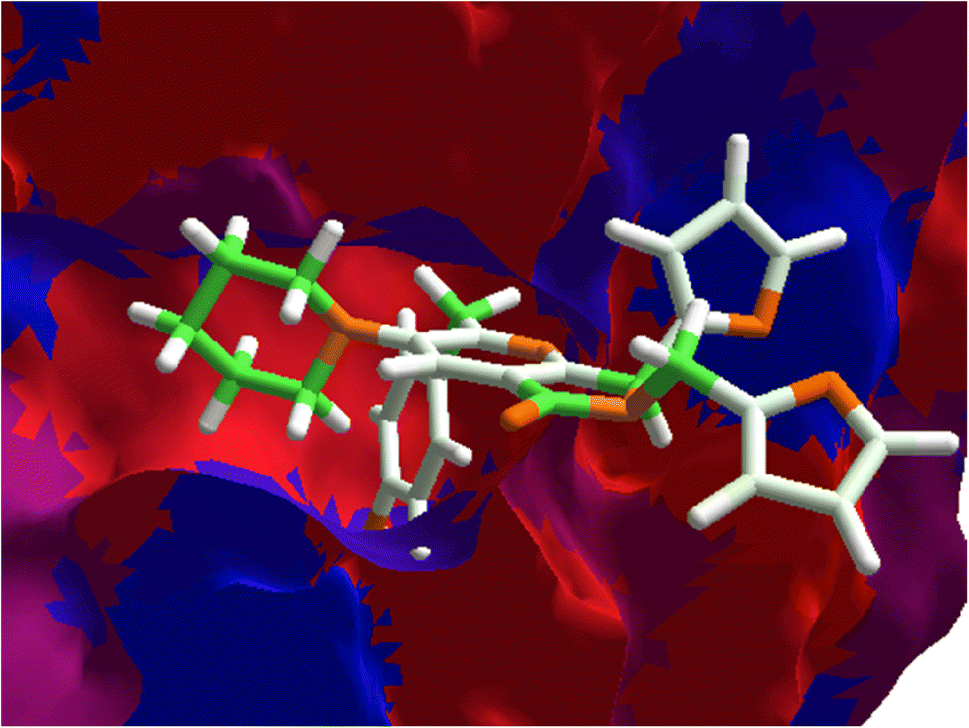 | ||
| Fig. 11 Hydrophobic interactions between P3 with SARS-CoV-2-MPro. Blue surfaces represent hydrophobic sites, red surfaces hydrophilic sites, green sticks depict hydrophobic fragments of the ligand, and orange sticks are hydrophilic fragments. | ||
Conclusions
A series of six bis-furyl-pyrrolo[3,4-b]pyridin-5-ones was synthesized via the Ugi–Zhu reaction and evaluated in vitro against human SARS-CoV-2. Using a time-of-addition approach, it was identified the compound 1e, with a promising prophylactic profile, another one (1d), with therapeutic potential, and a third one (1f), showing both, prophylactic and therapeutic activity. However, further observations will be needed to evaluate activity individually or in combination in animal models. Also, further studies to evaluate the antiviral activity against another coronavirus will be helpful for the identification of effective and safe pan-coronavirus inhibitors. Regarding the in silico results, it can be concluded that 1e is not the better ligand interacting with the SARS-CoV-2-MPro. However, the difference between this one and 1f and 1c is only 0.04 kcal mol−1, which is an insignificant highlight difference, and can be considered that the three ligands interact similarly with the SARS-CoV-2-MPro. However, in the case of the SARS-CoV-2 spike-glycoprotein, 1e is the best ligand, which can determine the prophylactic effect corroborated with the in vitro assays. As well as interaction with the Nsp3 protein can explain the prophylactic and therapeutic effects of 1d and 1f. Also, the pharmacophoric model shows the importance of the hydrophilic, hydrophobic, and hydrophobic–aromatic sites in the cavities into the SARS-CoV-2 spike-glycoprotein, as well as the favourable coupling of 1e on the cavity of the SARS-CoV-2 spike-glycoprotein, which was demonstrated designing a second generation of compounds which present higher ligand–target interactions, highlights the hydrophilic interactions. Further in-depth studies will be also needed to determine the mechanism of action at a molecular level and to evaluate their in vivo activity. Despite the synthetic cost, the encouraging antiviral activity exhibited by this series makes it worthy of continuing researching the bis-furyl-pyrrolo[3,4-b]pyridin-5-ones as promising bioactive molecules. As far as we know, this is the first time that bis-furyl-pyrrolo[3,4-b]pyridin-5-ones were synthesized and evaluated in vitro against the whole human SARS-CoV-2. Finally, through this work, we hope to provide valuable knowledge to the scientific community devoted to antiviral drug discovery and to contribute to the fight against the COVID-19 pandemic.Experimental section
Synthesis
1H and 13C nuclear magnetic resonance (NMR) spectra were acquired on a Bruker AMX Advance III spectrometer (500 MHz). The solvent used for NMR experiments was deuterated chloroform (CDCl3). Chemical shifts are reported in parts per million (δ/ppm). Coupling constants are reported in Hertz (J/Hz). Internal reference for NMR spectra was tetramethylsilane (TMS) at 0.00 ppm. Multiplicities of the signals are reported using the standard abbreviations: singlet (s), doublet (d), triplet (t), quartet (q) and multiplet (m). NMR spectra were analyzed using the MestReNova software (Ver. 12.0.0-20080). Infrared (IR) spectra were acquired on a Perkin Elmer 1600 spectrometer using the attenuated total reflectance (ATR) method. The maximum absorbance peaks are reported in reciprocal centimetres (νmax/cm−1). IR spectra were analysed using the Report Builder software (Ver. 2.01). High-resolution mass spectroscopy (HRMS) spectra were acquired by Electrospray ionization (ESI) on a Micro-TOF II spectrometer Bruker Daltonics. HRMS samples were injected directly (Apollo source) and analysed by time-of-flight method (TOF). HRMS spectra were analysed using the Compass 1.5 analysis software. Microwave-assisted reactions were performed in closed-vessel mode on a CEM Discover SP MW-reactor. Reaction progress was monitored by thin-layer chromatography (TLC) and the spots were visualized under ultraviolet (UV) light (254 or 365 nm). Flash columns packed with silica-gel 60, 230–400 mesh particle size and glass preparative plates (20 × 20 cm) coated with silica-gel 60 doped with UV indicator (F254) were used to purify the products. Mixtures of hexanes (Hex) and ethyl acetate (EtOAc) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 1:2 (v/v) proportion were used to run TLC, silica-gel columns, preparative plates, and to measure the retention factor (Rf) values (using the same mobile phase for all the experiments). All starting reagents and solvents were used as received (without further purification, distillation, nor dehydration). Chemical structures were drawn using the ChemDraw Professional software (Ver. 15.0.0.106). The purity for all synthesized products (>98%) was assessed by NMR.
:1 or 1:2 (v/v) proportion were used to run TLC, silica-gel columns, preparative plates, and to measure the retention factor (Rf) values (using the same mobile phase for all the experiments). All starting reagents and solvents were used as received (without further purification, distillation, nor dehydration). Chemical structures were drawn using the ChemDraw Professional software (Ver. 15.0.0.106). The purity for all synthesized products (>98%) was assessed by NMR.
Synthesis and characterization of the bis-furyl-pyrrolo[3,4-b]pyridin-5-ones 1a–f
:1 or 1:2 (v/v) proportions as mobile phase to isolate the corresponding bis-furyl-pyrrolo[3,4-b]pyridin-5-ones 1a–f.
:1, v/v); FT-IR (ATR) νmax/cm−1 1713 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1H NMR (500 MHz, CDCl3) δ: 7.91 (s, 1H), 7.37 (dd, J = 1.9, 0.9 Hz, 1H), 7.33 (dd, J = 1.9, 0.9 Hz, 1H), 7.21–7.10 (m, 5H), 6.44 (dd, J = 3.3, 0.9 Hz, 1H), 6.39 (dd, J = 3.5, 1.9 Hz, 1H), 6.30 (dd, J = 3.2, 1.9 Hz, 1H), 6.25 (dd, J = 3.3, 0.6 Hz, 1H), 5.56 (s, 1H), 5.22 (d, J = 15.6 Hz, 1H), 4.30 (d, J = 13.9 2H), 4.06 (d, J = 15.6 Hz, 1H), 3.81–3.78 (m, 4H), 2.86–2.78 (m, 4H). 13C NMR (125 MHz, CDCl3) δ: 166.3, 162.1, 157.7, 150.2, 148.3, 148.0, 143.6, 142.6, 139.4, 128.8, 128.3, 126.3, 124.2, 111.2, 110.6, 110.5, 108.7, 67.2, 58.7, 53.1, 40.2, 37.0. HRMS: m/z calcd for: C27H26N3O4+ 456.1920, found 456.1918.
:2, v/v); FT-IR (ATR) νmax/cm−1 1685 (CO); 1H NMR (500 MHz, CDCl3) δ: 7.90 (s, 1H), 7.36 (dd, J = 1.9, 0.8 Hz, 1H), 7.21–7.19 (m, 5H), 6.45 (ddd, J = 3.3, 0.8, 0.4 Hz, 1H), 6.38 (dd, J = 3.3, 1.9 Hz, 1H), 5.81 (s, 1H), 4.35–4.28 (m, 2H), 4.08–4.03 (m, 2H), 3.88–3.83 (m, 1H), 3.81–3.78 (m, 4H), 3.75–3.69 (m, 1H), 3.15 (d, J = 10.5 Hz, 1H), 2.86–2.77 (m, 4H), 1.90–1.84 (m, 2H), 1.69–1.64 (m, 3H). 13C NMR (125 MHz, CDCl3) δ: 167.1, 161.9, 158.0, 148.2, 143.4, 139.3, 130.8, 128.7, 128.2, 126.2, 124.1, 111.2, 110.5, 77.4, 68.0, 67.1, 59.9, 53.0, 43.0, 40.1, 28.6, 25.8. HRMS: m/z calcd for C27H30N3O4+ 460.2226, found 460.2231.
:1, v/v); FT-IR (ATR) νmax/cm−1 1688 (CO); 1H NMR (500 MHz, CDCl3) δ: 7.90 (s, 1H), 7.62 (ddd, J = 9.5, 1.6, 0.4 Hz, 1H), 7.38 (dd, J = 2.0, 1.5 Hz, 1H), 7.23–7.11 (m, 5H), 5.97 (dd, J = 1.9, 0.9 Hz, 1H), 5.72 (s, 1H), 4.37–4.26 (m, 2H), 4.17–4.06 (m, 1H), 3.89–3.84 (m, 1H), 3.82–3.79 (m, 4H), 3.77–3.71 (m, 1H), 2.84–2.80 (m, 4H), 2.08–1.94 (m, 1H), 1.91–1.82 (m, 1H), 1.66–1.51 (m, 1H). 13C NMR (125 MHz, CDCl3) δ: 167.2, 162.0, 160.0, 148.1, 143.9, 142.8, 139.4, 128.9, 128.3, 126.3, 124.5, 124.3, 120.2, 108.7, 68.0, 67.3, 58.2, 53.2, 44.3, 42.4, 40.2, 29.5 ppm; HRMS: m/z calcd for C27H30N3O4+ 460.2231, found 460.2231.
:1, v/v); FT-IR (ATR) νmax/cm−1 1680 (CO); 1H NMR (500 MHz, CDCl3) δ: 7.91 (s, 1H), 7.61 (ddd, J = 1.4, 0.9, 0.4 Hz, 1H), 7.41 (ddd, J = 2.0, 1.5, 0.5 Hz, 1H), 7.35 (dd, J = 1.9, 0.9 Hz, 1H), 7.22–7.11 (m, 5H), 6.31 (dd, J = 3.2, 1.9 Hz, 1H), 6.25 (d, J = 3.3, 0.7 Hz, 1H), 6.03 (ddd, J = 1.9, 0.9, 0.4 Hz, 1H), 5.45 (s, 1H), 5.26 (d, J = 15.6 Hz, 1H), 4.30 (d, J = 13.9 Hz, 2H), 4.03 (d, J = 15.7 Hz, 1H), 3.82–3.79 (m, 4H), 2.85–2.79 (m, 4H). 13C NMR (125 MHz, CDCl3): d 166.2, 162.2, 159.5, 150.3, 148.1, 144.1, 142.6, 139.3, 128.8, 128.3, 126.3, 124.3, 120.0, 110.5, 108.7, 67.2, 56.9, 53.1, 40.1, 36.5. HRMS: m/z calcd for C27H26N3O4+ 456.1900, found 456.1918.
:2, v/v); FT-IR (ATR) νmax/cm−1 3432 (OH), 1648 (CO); 1H NMR (500 MHz, CDCl3) δ: 7.90 (s, 1H), 7.37 (ddd, J = 1.8, 0.9, 0.2 Hz, 1H), 7.34 (dd, J = 1.9, 0.9 Hz, 1H), 7.22 (d, J = 4.2 Hz, 1H), 7.22 (d, J = 8.8 Hz, 1H), 7.01 (d, J = 8.8 Hz, 1H), 6.43 (ddd, J = 3.3, 0.9, 0.4 Hz, 1H), 6.39 (ddd, J = 3.3, 1.9, 0.3 Hz, 1H), 6.30 (dd, J = 3.2, 1.9 Hz, 1H), 6.25 (dd, J = 3.2, 0.7 Hz, 1H), 5.55 (s, 1H), 5.24 (d, J = 15.7 Hz, 1H), 4.28 (q, J = 7.1 Hz, 1H), 4.06 (d, J = 15.7 Hz, 1H), 3.84–3.80 (m, 4H), 2.87–2.80 (m, 4H). 13C NMR (125 MHz, CDCl3): d 166.3, 161.8, 157.9, 153.8, 150.3, 149.7, 148.3, 148.0, 143.7, 142.7, 137.1, 129.9, 124.4, 120.9, 111.3, 110.7, 110.6, 108.8, 67.3, 58.7, 53.2, 39.5, 37.1. HRMS: m/z calcd for C27H26N3O5+ 472.1974, found 472.1978.
:2, v/v); FT-IR (ATR) νmax/cm−1 1682 (CO); 1H NMR (500 MHz, CDCl3) δ: 7.84 (s, 1H), 7.31 (dd, J = 1.8, 0.9 Hz, 1H), 7.25–7.14 (m, 5H), 6.31 (dd, J = 3.3, 0.8 Hz, 1H), 6.29 (dd, J = 3.1, 1.9 Hz, 1H), 5.32 (dd, J = 15.4, 0.7 Hz, 1H), 4.77 (d, J = 2.7 Hz, 1H), 4.69–4.64 (m, 1H), 4.48 (d, J = 15.5 Hz, 1H), 4.37 (d, J = 14.0 Hz, 1H), 4.23 (d, J = 14.0 Hz, 1H), 3.86–3.79 (m, 4H), 3.73–3.59 (m, 2H), 2.91–2.78 (m, 5H), 1.85–1.67 (m, 2H). 13C NMR (125 MHz, CDCl3): d 167.5, 161.1, 157.9, 150.6, 147.6, 142.3, 139.4, 128.9, 128.2, 126.2, 123.7, 110.3, 108.5, 78.9, 68.5, 67.1, 61.6, 53.0, 39.8, 38.0, 25.6, 24.7. HRMS: m/z calcd for C27H30N3O4+ 460.2225, found 460.2231.
O); 1H NMR (500 MHz, CDCl3) δ: 7.91 (s, 1H), 7.37 (dd, J = 1.9, 0.9 Hz, 1H), 7.33 (dd, J = 1.9, 0.9 Hz, 1H), 7.21–7.10 (m, 5H), 6.44 (dd, J = 3.3, 0.9 Hz, 1H), 6.39 (dd, J = 3.5, 1.9 Hz, 1H), 6.30 (dd, J = 3.2, 1.9 Hz, 1H), 6.25 (dd, J = 3.3, 0.6 Hz, 1H), 5.56 (s, 1H), 5.22 (d, J = 15.6 Hz, 1H), 4.30 (d, J = 13.9 2H), 4.06 (d, J = 15.6 Hz, 1H), 3.81–3.78 (m, 4H), 2.86–2.78 (m, 4H). 13C NMR (125 MHz, CDCl3) δ: 166.3, 162.1, 157.7, 150.2, 148.3, 148.0, 143.6, 142.6, 139.4, 128.8, 128.3, 126.3, 124.2, 111.2, 110.6, 110.5, 108.7, 67.2, 58.7, 53.1, 40.2, 37.0. HRMS: m/z calcd for: C27H26N3O4+ 456.1920, found 456.1918.
:2, v/v); FT-IR (ATR) νmax/cm−1 1685 (CO); 1H NMR (500 MHz, CDCl3) δ: 7.90 (s, 1H), 7.36 (dd, J = 1.9, 0.8 Hz, 1H), 7.21–7.19 (m, 5H), 6.45 (ddd, J = 3.3, 0.8, 0.4 Hz, 1H), 6.38 (dd, J = 3.3, 1.9 Hz, 1H), 5.81 (s, 1H), 4.35–4.28 (m, 2H), 4.08–4.03 (m, 2H), 3.88–3.83 (m, 1H), 3.81–3.78 (m, 4H), 3.75–3.69 (m, 1H), 3.15 (d, J = 10.5 Hz, 1H), 2.86–2.77 (m, 4H), 1.90–1.84 (m, 2H), 1.69–1.64 (m, 3H). 13C NMR (125 MHz, CDCl3) δ: 167.1, 161.9, 158.0, 148.2, 143.4, 139.3, 130.8, 128.7, 128.2, 126.2, 124.1, 111.2, 110.5, 77.4, 68.0, 67.1, 59.9, 53.0, 43.0, 40.1, 28.6, 25.8. HRMS: m/z calcd for C27H30N3O4+ 460.2226, found 460.2231.
:1, v/v); FT-IR (ATR) νmax/cm−1 1688 (CO); 1H NMR (500 MHz, CDCl3) δ: 7.90 (s, 1H), 7.62 (ddd, J = 9.5, 1.6, 0.4 Hz, 1H), 7.38 (dd, J = 2.0, 1.5 Hz, 1H), 7.23–7.11 (m, 5H), 5.97 (dd, J = 1.9, 0.9 Hz, 1H), 5.72 (s, 1H), 4.37–4.26 (m, 2H), 4.17–4.06 (m, 1H), 3.89–3.84 (m, 1H), 3.82–3.79 (m, 4H), 3.77–3.71 (m, 1H), 2.84–2.80 (m, 4H), 2.08–1.94 (m, 1H), 1.91–1.82 (m, 1H), 1.66–1.51 (m, 1H). 13C NMR (125 MHz, CDCl3) δ: 167.2, 162.0, 160.0, 148.1, 143.9, 142.8, 139.4, 128.9, 128.3, 126.3, 124.5, 124.3, 120.2, 108.7, 68.0, 67.3, 58.2, 53.2, 44.3, 42.4, 40.2, 29.5 ppm; HRMS: m/z calcd for C27H30N3O4+ 460.2231, found 460.2231.
:1, v/v); FT-IR (ATR) νmax/cm−1 1680 (CO); 1H NMR (500 MHz, CDCl3) δ: 7.91 (s, 1H), 7.61 (ddd, J = 1.4, 0.9, 0.4 Hz, 1H), 7.41 (ddd, J = 2.0, 1.5, 0.5 Hz, 1H), 7.35 (dd, J = 1.9, 0.9 Hz, 1H), 7.22–7.11 (m, 5H), 6.31 (dd, J = 3.2, 1.9 Hz, 1H), 6.25 (d, J = 3.3, 0.7 Hz, 1H), 6.03 (ddd, J = 1.9, 0.9, 0.4 Hz, 1H), 5.45 (s, 1H), 5.26 (d, J = 15.6 Hz, 1H), 4.30 (d, J = 13.9 Hz, 2H), 4.03 (d, J = 15.7 Hz, 1H), 3.82–3.79 (m, 4H), 2.85–2.79 (m, 4H). 13C NMR (125 MHz, CDCl3): d 166.2, 162.2, 159.5, 150.3, 148.1, 144.1, 142.6, 139.3, 128.8, 128.3, 126.3, 124.3, 120.0, 110.5, 108.7, 67.2, 56.9, 53.1, 40.1, 36.5. HRMS: m/z calcd for C27H26N3O4+ 456.1900, found 456.1918.
:2, v/v); FT-IR (ATR) νmax/cm−1 3432 (OH), 1648 (CO); 1H NMR (500 MHz, CDCl3) δ: 7.90 (s, 1H), 7.37 (ddd, J = 1.8, 0.9, 0.2 Hz, 1H), 7.34 (dd, J = 1.9, 0.9 Hz, 1H), 7.22 (d, J = 4.2 Hz, 1H), 7.22 (d, J = 8.8 Hz, 1H), 7.01 (d, J = 8.8 Hz, 1H), 6.43 (ddd, J = 3.3, 0.9, 0.4 Hz, 1H), 6.39 (ddd, J = 3.3, 1.9, 0.3 Hz, 1H), 6.30 (dd, J = 3.2, 1.9 Hz, 1H), 6.25 (dd, J = 3.2, 0.7 Hz, 1H), 5.55 (s, 1H), 5.24 (d, J = 15.7 Hz, 1H), 4.28 (q, J = 7.1 Hz, 1H), 4.06 (d, J = 15.7 Hz, 1H), 3.84–3.80 (m, 4H), 2.87–2.80 (m, 4H). 13C NMR (125 MHz, CDCl3): d 166.3, 161.8, 157.9, 153.8, 150.3, 149.7, 148.3, 148.0, 143.7, 142.7, 137.1, 129.9, 124.4, 120.9, 111.3, 110.7, 110.6, 108.8, 67.3, 58.7, 53.2, 39.5, 37.1. HRMS: m/z calcd for C27H26N3O5+ 472.1974, found 472.1978.
:2, v/v); FT-IR (ATR) νmax/cm−1 1682 (CO); 1H NMR (500 MHz, CDCl3) δ: 7.84 (s, 1H), 7.31 (dd, J = 1.8, 0.9 Hz, 1H), 7.25–7.14 (m, 5H), 6.31 (dd, J = 3.3, 0.8 Hz, 1H), 6.29 (dd, J = 3.1, 1.9 Hz, 1H), 5.32 (dd, J = 15.4, 0.7 Hz, 1H), 4.77 (d, J = 2.7 Hz, 1H), 4.69–4.64 (m, 1H), 4.48 (d, J = 15.5 Hz, 1H), 4.37 (d, J = 14.0 Hz, 1H), 4.23 (d, J = 14.0 Hz, 1H), 3.86–3.79 (m, 4H), 3.73–3.59 (m, 2H), 2.91–2.78 (m, 5H), 1.85–1.67 (m, 2H). 13C NMR (125 MHz, CDCl3): d 167.5, 161.1, 157.9, 150.6, 147.6, 142.3, 139.4, 128.9, 128.2, 126.2, 123.7, 110.3, 108.5, 78.9, 68.5, 67.1, 61.6, 53.0, 39.8, 38.0, 25.6, 24.7. HRMS: m/z calcd for C27H30N3O4+ 460.2225, found 460.2231.
In vitro assays
000 cells were plated on each well of a 96 well plate in DMEM with 8% of FBS at 37 °C and 5% CO2. The day of the experiment new media containing the compounds at different concentrations at a final volume of 100 μl per well was added and incubated 48 h at 37 °C and 5% CO2. Media without the compounds was used as a negative control and media without the compounds but containing the DMSO volume used to dissolve the compounds was included as the vehicle control. After 48 h, media was removed, cells were washed with PBS 1x, 100 μL of 10% formaldehyde was added in each well and incubated 1 h at room temperature. After incubation, formaldehyde was removed, 100 μL of crystal violet (0.1% crystal violet, 3.7% formaldehyde) was added and plates were incubated 1 h at room temperature. Subsequently crystal violet was removed, cells were washed with 100 μL of water two times, 100 μL of 10% of acetic acid were added and cells were incubated 1 h at room temperature. Finally, crystal violet release of viable cells was measured at 590 wavelengths in a plate absorbance reader (Sunrise, TECAN). Cell viability was calculated with respect to the negative control. All treatments were carried out three times in duplicates. IC50 values were calculated by polynomial regression using Curve Expert program version 1.4.
000 cells per well of Vero E6 were plated in a 96 well plate in DMEM with 8% of FBS at 37 °C and 5% CO2. The experiments were carried out using DMEM with 0.015% of trypsin (Sigma cat no # T4799). The antiviral activity was assessed through a time-of-addition approach in which two conditions were tested. In the first case, the compounds were added at the same time the cells were infected (experimental condition 1, EC1). The infection was allowed to take place for 1 h at 37 °C and 5% CO2. After incubation, the cells were washed with PBS 1x twice, DMEM medium with trypsin was added, and 24 h post-infection (pi), aliquots of the collected supernatant were made and stored at −80 °C until used for titration by plaque assay. The EC2 consisted of first performing the infection as described above. After 1 h cells were washed with 1x PBS and then DMEM with 0.015% trypsin containing the compounds at a concentration of 10 μM was added. 24 h pi, aliquots of the collected supernatant were made and stored at −80 °C until used for titration by plaque assay. All infections were carried out using a virus concentration of a MOI of 0.1.
000 cells were seeded in 24-well plates and grown to form a confluent monolayer. Cells were infected with serial dilutions of virus stocks in DMEM with 0.015% trypsin and incubated 1 h at 37 °C and 5% CO2. After the incubation period cells were washed 2 times with PBS 1x and then overlaid with DMEM + 0.015% trypsin containing 2% of agarose (ThermoFisher, Cat. No. R0492). The plates were incubated at 37 °C and 5% CO2 for 72 h. The cells were fixed with 10% formaldehyde in PBS 1x for 24 h at 4 °C, washed with water to remove agarose and strained with crystal violet (0.1%) prepared in 3.7% formaldehyde. After 30 min, the wells were washed with PBS, and plaques were counted to determine virus titers.
In silico studies
To begin was modelled using the Avogadro package,32 the six studied molecules (1a–f), as well as the co-crystallized molecule and the P8 (ref. 33) compound (proposed in previous works of some of us). These molecules were optimized at the universal force field (UFF) level using the Gaussian 09 (G09)34 software. On the other hand, the main protease of the SARS-CoV-2 (SARS-CoV-2-MPro), the surface spike-protein and the Nps3 enzyme protein of the same virus were downloaded from the protein data bank, with the PDB code 6LU7,356VSB,10 and 7KXB15 respectively. Once downloaded, the selected targets have corrected the residues, removed the solvent, and adjusted the protonation using the chimera36 software. To perform the docking assay was used the scoring function Moldock37 through the software Molegro Virtual Docker.38 Finally, a pharmacophoric analysis was made using the ZincPharmer39 web server.Author contributions
Synthesis and characterization, I. M.-S., F. P. M.-E. and C. E. G.-A.; methodology, M. A. G.-S. and I. A. I.; validation, M. E. G.-H. and R. E. S.-S.; investigation, Y. R.-A and S. L. A.-E.; writing – original draft preparation, E. D.-C. and E. G.-Z.; funding acquisition and writing – review & editing, A. I.-J. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
I. M.-S. thanks CONACyT – México for her PhD scholarship (947606). All authors are gratefully acknowledged with Citlali Adelaida Arroyo-Gómez, Atilano Gutierrez-Carrillo, and Mónica A. Rincón-Guevara for IR, NMR and HRMS acquisitions, respectively. E. D.-C. thanks the computational resource provided by the Laboratorio Nacional de Caracterización de Propiedades Fisicoquímicas y Estructura Molecular (UG-UAA-CONACYT, Project: 123732). A. I.-J. acknowledges “Proyecto Apoyado por el Fondo Sectorial de Investigación para la Educación CONACyT-SEP CB-2017-2018 (A1-S-32582)” for financial support.Notes and references
- C. Huang, Y. Wang, X. Li, L. Ren, J. Zhao, Y. Hu, L. Zhang, G. Fan, J. Xu, X. Gu, Z. Cheng, T. Yu, J. Xia, Y. Wei, W. Wu, X. Xie, W. Yin, H. Li, M. Liu, Y. Xiao, H. Gao, L. Guo, J. Xie, G. Wang, R. Jiang, Z. Gao, Q. Jin, J. Wang and B. Cao, Lancet, 2020, 395, 497 CrossRef CAS.
- D. Cucinotta and M. Vanelli, Acta Biomed., 2020, 91, 157 Search PubMed.
- A. E. Gorbalenya, S. C. Baker, R. S. Baric, R. J. de Groot, C. Drosten, A. A. Gulyaeva, B. L. Haagmans, C. Lauber, A. M. Leontovich, B. W. Neuman, D. Penzar, S. Perlman, L. L. M. Poon, D. Samborskiy, I. A. Sidorov, I. Sola and J. Ziebuhr, Nat. Microbiol., 2020, 55, 536 Search PubMed.
- J. Cui, F. Li and Z. L. Shi, Nat. Rev. Microbiol., 2019, 17, 181 CrossRef CAS PubMed.
- Y. Yan, L. Chang and L. Wang, Rev. Med. Virol., 2020, 30, e2106 CrossRef CAS.
- B. Hu, H. Guo, P. Zhou and Z. Shi, Nat. Rev. Microbiol., 2021, 19, 141 CrossRef CAS PubMed.
- D. K. Rajendran, V. Rajagopal, S. Alagumanian, T. S. Kumar, S. P. S. Prabhakaran and D. Kasilingam, VirusDisease, 2020, 31, 161 CrossRef CAS.
- A. Banerjee, K. Kulcsar, V. Misra, M. Frieman and K. Mossman, Viruses, 2019, 11, 41 CrossRef PubMed.
- World Health Organization, Available online: https://www.who.int, (accessed on 24 July 2022).
- D. Wrapp, N. Wang, K. S. Corbett, J. A. Goldsmith, C.-L. Hsieh, O. Abiona, B. S. Graham and J. S. McLellan, Science, 2020, 367, 1260 CrossRef CAS PubMed.
- Y. Tian, C. Lian, Y. Chen, D. Wei, X. Zhang, Y. Ling, Y. Wang and L.-S. Yeap, Cell Discovery, 2020, 6, 75 CrossRef CAS PubMed.
- W. Liu, L. Liu, G. Kou, Y. Zheng, Y. Ding, W. Ni, Q. Wang, L. Tan, W. Wu, S. Tang, Z. Xiong and S. Zheng, J. Clin. Microbiol., 2020, 58, e00461-20 CrossRef PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289 CrossRef CAS PubMed.
- M. A. Jairajpuri and A. Ansari, Clin. Sci., 2020, 134, 2235 CrossRef CAS.
- C. A. Brosey, J. H. Houl, P. Katsonis, L. P. F. Balapiti-Modarage, S. Bommagani, A. Arvai, D. Moiani, A. Bacolla, T. Link, L. S. Warden, O. Lichtarge, D. E. Jones, Z. Ahmed and J. A. Tainer, Prog. Biophys. Mol. Biol., 2021, 163, 171 CrossRef CAS.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409 CrossRef CAS PubMed.
- (a) V. Soriano, C. de-Mendoza, B. Edagwa, A. Treviño, P. Barreiro, J. V. Fernandez-Montero and H. E. Gendelman, AIDS Rev., 2022, 24, 41 Search PubMed; (b) I. Zanella, D. Zizioli, F. Castelli and E. Quiros-Roldan, Pharmaceuticals, 2021, 14, 454 CrossRef CAS PubMed; (c) R. L. Hoffman, R. S. Kania, M. A. Brothers, J. F. Davies, R. A. Ferre, K. S. Gajiwala, M. He, R. J. Hogan, K. Kozminski, L. Y. Li, J. W. Lockner, J. Lou, M. T. Marra, L. J. Mitchell Jr., B. W. Murray, J. A. Nieman, S. Noell, S. P. Planken, T. Rowe, K. Ryan, G. J. Smith III, J. E. Solowiej, C. M. Steppan and B. Taggart, J. Med. Chem., 2020, 63, 12725 CrossRef CAS PubMed; (d) D. R. Owen, C. M. N. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, R. Ferre, K. S. Gajiwala, S. A. Gibson, S. E. Greasley, B. L. Hurst, E. P. Kadar, A. S. Kalgutkar, J. C. Lee, J. Lee, W. Liu, S. W. Mason, S. Noell, J. J. Novak, R. S. Obach, K. Ogilvie, N. C. Patel, M. Pettersson, D. K. Rai, M. R. Reese, M. F. Sammons, J. G. Sathish, R. S. P. Singh, C. M. Steppan, A. E. Stewart, J. B. Tuttle, L. Updyke, P. R. Verhoest, L. Wei, Q. Yang and Y. Zhu, Science, 2021, 374, 1586 CrossRef CAS; (e) R. Rosales, B. L. McGovern, M. L. Rodriguez, D. K. Rai, R. D. Cardin, A. S. Anderson, PSP study group, E. M. Sordillo, H. van Bakel, V. Simon, A. García-Sastre and K. M. White, bioRxiv, 2022, preprint, DOI:10.1101/2022.01.17.476685.
- Key reference: I. A. Ibarra, A. Islas-Jácome and E. González-Zamora, Org. Biomol. Chem., 2018, 16, 1402 RSC.
- R. C. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16, 2958 RSC.
- X. Sun, P. Janvier, G. Zhao, H. Bienaymé and J. Zhu, Org. Lett., 2001, 3, 877 CrossRef CAS PubMed.
- P. Janvier, X. Sun, H. Bienaymé and J. Zhu, J. Am. Chem. Soc., 2002, 124, 2560 CrossRef PubMed.
- J. C. Flores-Reyes, A. Islas-Jácome and E. González-Zamora, Org. Chem. Front., 2021, 8, 5460 RSC.
- I. Morales-Salazar, S. L. Castañón-Alonso, D. Canseco-González, E. Díaz-Cervantes, E. González-Zamora and A. Islas-Jácome, Chem. Proc., 2022, 8, 84 Search PubMed.
- (a) A. Islas-Jácome, L. E. Cárdenas-Galindo, A. V. Jerezano, J. Tamariz, E. González-Zamora and R. Gámez-Montaño, Synlett, 2012, 23, 2951 CrossRef; (b) A. Islas-Jácome, A. Gutiérrez-Carrillo, M. A. García-Garibay and E. González-Zamora, Synlett, 2014, 25, 0403 Search PubMed.
- A. Fayol, C. Housseman, X. Sun, P. Janvier, H. Bienaymé and J. Zhu, Synthesis, 2005, 1, 161 Search PubMed.
- A. Islas-Jácome, A. Rentería-Gómez, M. A. Rentería-Gómez, E. González-Zamora, J. O. C. Jiménez-Halla and R. Gámez-Montaño, Tetrahedron Lett., 2016, 57, 3496 CrossRef.
- N. S. Ogando, T. J. Dalebout, J. C. Zevenhoven-Dobbe, R. W. A. L. Limpens, Y. van der Meer, L. Caly, J. Druce, J. J. C. de Vries, M. Kikkert, M. Bárcena, I. Sidorov and E. J. Snijder, J. Gen. Virol., 2020, 101, 925 CrossRef CAS PubMed.
- S. T. Jones, V. Cagno, M. Janeček, D. Ortiz, N. Gasilova, J. Piret, M. Gasbarri, D. A. Constant, Y. Han, L. Vuković, P. Král, L. Kaiser, S. Huang, S. Constant, K. Kirkegaard, G. Boivin, F. Stellacci and C. Tapparel, Sci. Adv., 2020, 6, eaax9318 CrossRef CAS PubMed.
- P. Czuppon, F. Débarre, A. Gonçalves, O. Tenaillon, A. S. Perelson, J. Guedj and F. Blanquart, PLoS Comput. Biol., 2021, 17, e1008752 CrossRef CAS PubMed.
- F. K. Algethami, M. Cherif, S. Jlizi, N. Ben Hamadi, A. Romdhane, M. R. Elamin, M. A. Alghamdi and H. Ben Jannet, Molecules, 2021, 26, 6103–6121 CrossRef CAS.
- M. A. Almaraz-Girón, E. Calderón-Jaimes, A. Sánchez Carrillo, E. Díaz-Cervantes, E. Castañón Alonso, A. Islas-Jácome, A. Domínguez-Ortiz and S. L. Castañón-Alonso, Molecules, 2021, 26, 3817 CrossRef.
- M. Hanwell, D. Curtis, D. Lonie, T. Vandermeersch, E. Zurek and G. Hutchison, Aust. J. Chem., 2012, 4, 17 CAS.
- C. J. Cortés-García, L. Chacón-García, J. E. Mejía-Benavides and E. Díaz-Cervantes, PeerJ, 2020, 2, e10 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09, 2009 Search PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605 CrossRef CAS.
- R. Thomsen and M. H. Christensen, J. Med. Chem., 2006, 49, 3315 CrossRef CAS.
- J. Yang and C. Chen, Proteins, 2004, 55, 288 CrossRef CAS PubMed.
- D. R. Koes and C. J. Camacho, Nucleic Acids Res., 2012, 40, W409 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00350c |
| This journal is © The Royal Society of Chemistry 2023 |