
Identification of specific carbonic anhydrase inhibitors via in situ click chemistry, phage-display and synthetic peptide libraries: comparison of the methods and structural study†
Michael
Kugler‡
 a,
Martin
Hadzima
ab,
Rastislav
Dzijak
a,
Robert
Rampmaier
a,
Pavel
Srb
a,
Lukáš
Vrzal
a,
Zdeněk
Voburka
a,
Pavel
Majer
a,
Pavlína
Řezáčová
*a and
Milan
Vrabel
*a
a,
Martin
Hadzima
ab,
Rastislav
Dzijak
a,
Robert
Rampmaier
a,
Pavel
Srb
a,
Lukáš
Vrzal
a,
Zdeněk
Voburka
a,
Pavel
Majer
a,
Pavlína
Řezáčová
*a and
Milan
Vrabel
*a
aInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nám. 2, 16000, Prague, Czech Republic. E-mail: rezacova@uochb.cas.cz; vrabel@uochb.cas.cz
bDepartment of Organic Chemistry, Faculty of Science, Charles University, Albertov 6, 12800, Praha 2, Czech Republic
First published on 18th November 2022
Abstract
The development of highly active and selective enzyme inhibitors is one of the priorities of medicinal chemistry. Typically, various high-throughput screening methods are used to find lead compounds from a large pool of synthetic compounds, and these are further elaborated and structurally refined to achieve the desired properties. In an effort to streamline this complex and laborious process, new selection strategies based on different principles have recently emerged as an alternative. Herein, we compare three such selection strategies with the aim of identifying potent and selective inhibitors of human carbonic anhydrase II. All three approaches, in situ click chemistry, phage-display libraries and synthetic peptide libraries, led to the identification of more potent inhibitors when compared to the parent compounds. In addition, one of the inhibitor–peptide conjugates identified from the phage libraries showed greater than 100-fold selectivity for the enzyme isoform used for the compound selection. In an effort to rationalize the binding properties of the conjugates, we performed detailed crystallographic and NMR structural analysis, which revealed the structural basis of the compound affinity towards the enzyme and led to the identification of a novel exosite that could be utilized in the development of isoform specific inhibitors.
Introduction
Various high-throughput screening methods of large compound libraries composed of separately prepared derivatives have dominated drug discovery programs for decades.1–3 Besides these traditional medicinal chemistry approaches aiming to find an optimal ligand fitting to the active site of the target enzyme, new approaches employing the enzyme as a template ‘catalyzing’ the formation of new molecules from two or more fragments emerged recently as an alternative strategy.4–9 In contrast to the classical fragment-based drug discovery technique10,11 the so-called kinetic target-guided synthesis employs the biological target to chemically assemble its own inhibitors through the formation of covalent bonds between the fragments in a templated manner.12–14 The principle of this technique is based on the chelate effect, where two building blocks react with each other only when brought in close proximity upon simultaneous binding to the target enzyme.15 This strategy was employed by Sharpless and coworkers in 2002; they used the biocompatible 1,3-dipolar cycloaddition between alkyne and azide building blocks to identify a potent acetylcholinesterase inhibitor.16 The method was coined ‘in situ click chemistry’ and has since been successfully applied to other medicinally relevant targets.15,17–20A conceptually different selection method was more recently developed by Derda and co-workers. The authors employed phage-display libraries where each peptide sequence contained the same inhibitor attached to the N-terminus. After screening with the target enzyme, consensus sequences enriched in panning were identified and were shown to act synergistically with the inhibitor.21,22 Later, the same group applied the concept to select inhibitor–peptide conjugates for bovine carbonic anhydrase and concanavalin A.23
These strategies undoubtedly represent an attractive alternative to common medicinal chemistry approaches. However, based on the different principles, each approach can lead to diverse results and this makes it difficult to select the appropriate methodology for a given target. With the aim to compare various available techniques and to evaluate their advantages and limitations, we used three different selection methods on a single target enzyme. In particular, we used the in situ click chemistry approach, phage-display libraries, and synthetic peptide–inhibitor libraries to identify potent and selective sulfonamide inhibitors of the human carbonic anhydrase II (hCA II). hCA II has been associated with several cancers including melanoma, esophageal, renal and lung cancer, which makes it a potential target for therapy.24 Carbonic anhydrases play a crucial role in various physiological processes and are also associated with many pathological conditions (e.g. obesity, Alzheimer, glaucoma and cancer) that make them an important target in medicinal chemistry.25,26 There are 15 known isoforms of human carbonic anhydrases, and the identification of isoform selective inhibitors is of great importance to unveil their physiological role and to improve the therapeutic outcome of currently used inhibitors.27–31
Besides comparing three different selection methods, we also provide comprehensive structural biology data based on crystallographic and NMR studies to explain and rationalize the molecular details of the interaction between the enzyme and the selected inhibitors.
Results and discussion
Selection of hCA II inhibitors via in situ click chemistry
Inspired by the work on bovine carbonic anhydrase,20 we first applied the in situ click methodology to find improved inhibitors of the human enzyme homologue. The method relies on the formation of a triazole linkage as a result of a reaction between fragments that contain alkyne and azide in the presence of the target hCA II enzyme. For the selection process, we used 4-ethynylbenzenesulfonamide (Inh1) as the starting inhibitor and a series of azido amides as the second component. The pool of azido amides was generated by solid-phase synthesis from the corresponding amino acids using imidazole-1-sulfonyl azide diazotransfer reagent (for details see ESI†).32,33 For the selection, we incubated the mixture of azido amides, the alkyne-containing Inh1 and recombinant hCA II in TRIS buffer (pH 7.8) at 37 °C for 40 hours and then analyzed the reaction mixture in UPLC-MS. We used bovine serum albumin (BSA) in a control experiment to determine the unspecific formation of click products under these conditions (see ESI†). Analysis of the data led to the identification of leucine-containing hit compound (Hit1) that we resynthesized on a larger scale for further studies (Fig. 1A). | ||
| Fig. 1 Overview of the three different methods used for selection of carbonic anhydrase II inhibitors. A) In situ click chemistry, B) phage-display library, C) synthetic OBOC library, D) structural formulas of hits identified via the different selection methods. | ||
Selection of hCA II inhibitors from phage-display peptide library
Previously, it has been shown that it is possible to attach various functional molecules to the N-terminus of phage peptide libraries.21–23,34 The methodology is based on the construction of a phage library where each peptide sequence contains an N-terminal serine residue. Mild oxidative cleavage with NaIO4 generates an N-terminal aldehyde moiety that can be used for subsequent decoration of the peptides using hydroxylamine derivatives. Using this strategy, and the respective hydroxylamine-modified sulfonamide (Inh2), we constructed a phage library containing the desired sulfonamide ligand (see ESI†). The resulting library was incubated with bead-immobilized hCA II, and the phages that bound the enzyme were eluted and amplified (ESI†). Two additional rounds of panning were performed, and the eluted phages were finally sequenced to obtain two consensus peptide sequences (Hit2 and Hit3). These sequences were resynthesized on a larger scale for subsequent studies (Fig. 1B).Selection of hCA II inhibitors from synthetic peptide library
Recently, sulfonamide-modified peptide libraries were used to find selective ligands for carbonic anhydrases I and II.35–37 With the aim of comparing such an approach with the above selection methods, we used a modified version of the one-bead-one-compound (OBOC) technique, which can be used for the production of a relatively large library of peptide–inhibitor conjugates using the split-and-mix protocol.38–40 To introduce the inhibitor to the peptide sequences, we used an alkyne-containing lysine derivative in the last synthetic step and covalently attached the inhibitor via copper-catalyzed alkyne-azide cycloaddition (CuAAC) using an azide-containing sulfonamide (Inh3). In this way, we produced a peptide library where each bead contained a different peptide sequence but the same inhibitor. With the library in hand, we performed the screening experiment using fluorescently labeled hCA II to identify beads which bind the enzyme. Inspection of the library under a fluorescent stereomicroscope enabled the identification of fluorescent beads that were picked, and the peptide sequences were identified using standard sequencing by Edman degradation. Based on the analysis, two peptide sequences termed Hit4 and Hit5 were selected and resynthesized to evaluate the binding properties in more detail (Fig. 1C).Carbonic anhydrase inhibition
Having identified five different conjugates (Hit1–5) from three different selection methods, we determined the respective Ki values for hCA II using the stopped-flow carbon dioxide hydration assay.41,42 To evaluate if any of the selection procedures led to the identification of potentially isoform-selective inhibitors, we also determined the Ki values for the carbonic anhydrase IX (hCA IX) (Table 1).| Method | Inhibitor | K i (hCA II) [nM] | K i (hCA IX) [nM] | Selectivity indexa |
|---|---|---|---|---|
| a Selectivity index is the ratio between Ki (hCA IX) and Ki (hCA II). | ||||
| In situ click chemistry | Inh1 | 55 ± 9 | 150 ± 30 | 3 |
| Hit1 | 2.5 ± 1.4 | 11 ± 2 | 4 | |
| Phage-display peptide library | Inh2 | 471 ± 44 | 844 ± 83 | 2 |
| Hit2 | 16.0 ± 1.3 | 348 ± 26 | 22 | |
| Hit3 | 13.0 ± 1.0 | 1368 ± 82 | 105 | |
| OBOC synthetic peptide library | Inh3 | 485 ± 19 | 1200 ± 150 | 2.5 |
| Hit4 | 66.0 ± 4.2 | 69.0 ± 7 | 1.1 | |
| Hit5 | 36.0 ± 3 | 104 ± 6 | 3 | |
| Standard | AAZ 43 | 12.1 | 25.7 | 2 |
The first hit compound (Hit1), identified through the in situ click chemistry approach, has approximately a 20-fold lower Ki value when compared to the parent Inh1. This value is in good agreement with a previous study performed with bovine carbonic anhydrase.20Hit1 was the most potent hCA II inhibitor identified using the three different selection methods on both hCA II and hCA IX. Hit1 is approximately 4-times more selective for hCA II than it is for hCA IX. The two peptide–inhibitor conjugates identified from phage-display libraries showed Ki values that are 29-fold (Hit2) and 36-fold (Hit3) better when compared to the parent sulfonamide Inh2. For comparison, previous experiments performed with bovine carbonic anhydrase led to the identification of a tetrameric peptide with tryptophan as the first amino acid after the inhibitor.23 The two peptides identified from the inhibitor-modified synthetic OBOC peptide library showed a 7-fold (Hit4) and 13-fold (Hit5) improvement in potency relative to the starting sulfonamide Inh3.
Several interesting conclusions can be drawn from these results. First, all selection methods investigated here provide more potent inhibitors when compared to the starting molecules, with a ca. 20-fold increase in potency on average. Second, the structurally more complex peptide–inhibitor conjugates (Hit2–5) are less potent than the small molecule Hit1 identified via the in situ click approach. This can be explained, at least in part, by the higher potency of Inh1 that we used as the starting molecule for the selection when compared to Inh2 or Inh3. Third, all the identified hits are more potent towards hCA II when compared to hCA IX. This indicates that all selection methods yield better binders toward the enzyme which is used for the selection. Fourth, we obtained only moderate improvements in selectivity between the two enzyme isoforms for inhibitor conjugates identified via the in situ click approach and from synthetic peptide libraries (4-fold for Hit1, 3-fold for Hit5 and basically no selectivity for Hit4). In contrast, the peptide–inhibitor conjugates identified from phage libraries are 22-fold (Hit2) and even 105-fold (Hit3) more selective towards hCA II when compared to hCA IX. These results show that the phage-display peptide libraries yielded in this case not only more potent but also more selective inhibitors. Fifth, by comparing the structure of the peptide inhibitors (Hit4–5 containing the inhibitor attached via flexible, lysine linker while Hit2–3 contain the inhibitor warhead that is closer to the peptide backbone), one can speculate that at least for hCA II, it is more advantageous to have the peptidic part closer to the enzyme active site where the inhibitor binds, which leads to improved selectivity towards the hCA II isoform. This is best manifested by over a 100-fold improvement in selectivity of Hit3.
Practical considerations on the use of the three selection methods
Having experience in using the three different selection approaches, we would like to briefly point out to some of the advantages and limitations of each methodology.While being straightforward, the in situ click chemistry approach requires a relatively large amount of the enzyme, which must retain its biological activity at an elevated temperature (37 °C) for an extended period of time that is needed for the templated reaction to take place. For this reason, we were unable to perform similar experiments with the much more labile recombinant hCA IX. The analysis of the hit click products and the interpretation of the UPLC-MS data can be complicated, and we recommend to perform carefully-designed control experiments, such as using ‘blank’ proteins (e.g. BSA) for the selection.
The preparation of synthetic peptide libraries containing the desired modification (sulfonamide inhibitor in this work) is easily achievable by a skilled synthetic organic chemist. The screening process is quite straightforward; however, special care should be taken to avoid identifying false positives. The introduction of rigorous blocking steps, the use of control experiments, and the rescreening of hit peptide beads are a generally good practice.44,45 The limited size of the synthetic peptide library constrains the diversity of sequences that can be practically achieved using this methodology. On the other hand, unnatural building blocks can easily be included in the synthetic steps.
The work with phage-display libraries requires a background in biology and access to biochemistry instrumentation. In contrast to synthetic libraries, the diversity (sequence coverage) of the biological libraries is usually much greater. An additional advantage of the method is in the identification of the hit sequences that is done by DNA sequencing, a method that can nowadays be performed on a large scale.
Structural analysis
With the focus on the more selective peptide–inhibitor conjugates, we next performed a structural analysis study with the aim of thoroughly inspecting and understanding the binding properties of the conjugates. We co-crystallized hCA II with the hit compounds Hit2–5 and determined the crystal structures of Hit2, Hit3, Hit4 at high resolution ranging from 0.98 Å to 1.03 Å resolution (Table S1†). Despite extensive efforts, we were unable to successfully co-crystallize hCA II in complex with Hit5.In the crystal structures, the inhibitor could be unambiguously modelled with full occupancy into a well-defined electron density map in the enzyme active site. However, the peptidic parts reaching beyond the active site cleft, were mostly disordered and only two, one, and none of amino acid residues, could be additionally modelled (Fig. S1†). The lack of a well-defined electron density can be attributed to the dynamic disorder caused by the inherent flexibility of the peptidic part or by the static disorder triggered by a presence of several different binding modes.
The crystal structures of partially modeled peptide–inhibitor conjugates revealed specific binding of the sulfonamide moiety into the active site of hCA II. The sulfonamide group made interactions with the zinc ion and residues located at the bottom of the active site cavity that are typical for organic sulfonamide-containing inhibitors.46 In addition to interactions with catalytic residues, hit compounds made additional interactions within the active site cavity through their peptidic part (Fig. 2). Hit2 made interatomic interactions with residues at the opening of the active site cavity Q92, V121, F131, V135, V143, L198, T199, T200, P201, L202, and W209, reaching 46 interatomic contacts in total (Fig. 2A). Hit4 made 28 interatomic interactions with Q92, V121, F131, V143, L198, T199–200, P202, and W209 (Fig. 2C). Five additional contacts were observed for Hit3; a total of 33 interatomic interactions with Q92, V121, F131, V135, V143, L198, T199, T200, P202, L204, and W209 were identified (Fig. 2B).
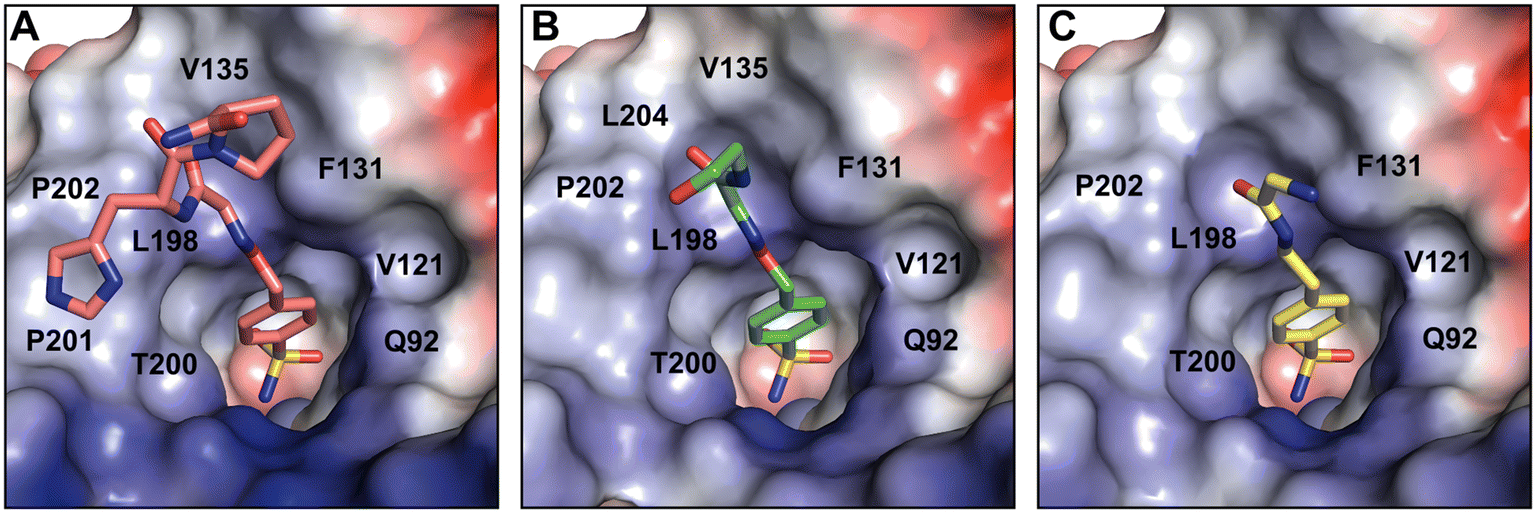 | ||
| Fig. 2 Peptide–inhibitor conjugates Hit2 (A), Hit3 (B), and Hit4 (C) bound to the hCA II active site. The hit compounds are depicted as sticks with differently colored carbon atoms: Hit2 (salmon), Hit3 (green), Hit4 (yellow). Oxygen, nitrogen, and sulfur atoms are shown in red, blue, and orange, respectively. The zinc ion is represented by the grey sphere and protein is represented by its solvent accessible surface colored according to the electrostatic potential (red for negative, blue for positive). The residues involved in interactions with the compounds are labelled with the exception of the residues H94, H96, H119, V143, T199, and W209 at the bottom of the cavity that are not labeled for the sake of clarity. | ||
A comparison of the binding modes of the hit compounds revealed that the distal parts of peptide–inhibitor conjugates were oriented towards the hydrophobic part of the active site, specifically residues V135, L198, P201, P202, and L204. We may assume that the interaction of these distal parts of the compounds with the amino acid residues at the outer rim of the active site and on the adjacent enzyme surface is dynamic or transient, and it could not be captured in the static crystal structure. We thus performed NMR binding studies that were able to identify the ligand-induced perturbations of each residue by following the HN spectra of 15N hCA II.47
We first performed an NMR study of Inh2 that was used as a reference for a subsequent NMR study of peptide–inhibitor conjugates. The minimum changes in the positions of HN peaks for the free and inhibitor-bound hCA II were calculated and plotted against the protein sequence (Fig. S2C†) to identify residues that are significantly affected by inhibitor binding. These were subsequently highlighted in the crystal structure of hCA II to visualize the most affected regions of the protein surface (Fig. S2A and B†). As expected, the most affected were residues in the enzyme active site cavity directly interacting with Inh2. Additionally, the analysis also identified other residues whose position was affected indirectly: residues belonging to the central anti-parallel β-sheet supporting the enzyme active site (N67, F70, I91, Q92, V121 and V143), residues in helical region between β-strands 5 and 6 (F131, Q136 and Q137), and loops located on the opposite side of the active site cleft (N62, K170 and G171).
The HN spectrum for Inh2-bound hCA II was used as a reference for subsequent analysis of the interactions of the peptide–inhibitor conjugates Hit2, Hit3, Hit4, and Hit5 (Fig. 3 and S3 and S4†). The binding interfaces of inhibitor conjugates partially overlap with those induced by binding of the Inh2 (represented in grey in Fig. 3). Additional changes in chemical shifts could be attributed to enzyme areas involved in a transient or disordered interaction of the peptide part that could not be captured by X-ray crystallography (represented in yellow, orange and red in Fig. 3).
 | ||
| Fig. 3 NMR chemical shift perturbations of 15N hCA II upon binding of peptide–inhibitor conjugates Hit2 (A), Hit3 (B), Hit4 (C), and Hit5 (D). Residues perturbed by ligand binding are mapped on the surface of 15N hCA II. Residues perturbed in the same way as by binding of Inh2 (the reference spectra) are highlighted in grey. Normalized combined chemical shifts of residues perturbed upon peptide–inhibitor conjugate binding (relative to the Inh2 binding) are colored yellow, orange, and red for RMSD values over 1, 2 or 3, respectively. For visualization of binding for each hit conjugate, the crystal structure was used, with the exception of Hit5 where the crystal structure of Hit4 was used instead. Views are rotated by 180° along the vertical axis exposing the side opposite to the enzyme active site are shown in the Fig. S3.† | ||
Residues perturbed by the interactions with peptide conjugates generally clustered into two regions. The first one are residues 124–140 that line the opening of the active site and belong to a short helical region connecting strands 5 and 6. The second cluster is formed by residues on the opposite side of the active site cleft and are also affected by compound bindings: this cluster includes residues N62, T169, K170, and G171. These residues belong to flexible loops connecting individual strands of the central β sheet loop (N62 in the loop connecting strands 1 and 2, the other residues in the loop preceding strand 8). Compounds Hit2 and Hit3 selected through the phage-display library approach bind more to the first regions. Hit4 and Hit5 from the synthetic peptide library affect more the second region. The extent of interacting residues on the protein surface correlates with the compound Ki values: spectra for Hit2 and Hit3 identified numerous residues with higher chemical shifts, while Hit4 binding affected the least number of residues (Fig. 3).
Our data prove that Inh2 serves as an anchoring element and its presence is indispensable for the interaction of peptide conjugates with the enzyme active site. When we used the peptide sequences present in Hit2–4 without the attached sulfonamide warhead, no significant chemical shifts were observed (Fig. S5†). Therefore, we may conclude that the sulfonamide warhead dominates the interaction with the enzyme and it is only strengthened by the amino acid residues when placed in the proper spatial orientation and proximity to the enzyme active site. The contribution of the individual parts of the peptide conjugates to the binding affinity was investigated further.
Contribution of individual amino acids in peptide conjugate Hit3 to the binding affinity
We set to deconvolute the contribution of individual amino acids on the binding and selectivity of Hit3 identified from phage libraries. Toward this goal, we prepared four truncated (t) versions of the Hit3 conjugate Hit3-t1–4 (Fig. 4). | ||
| Fig. 4 Structures of the truncated inhibitors originating from Hit3. | ||
First, we evaluated the Ki values of the truncated versions of Hit3 against hCA II and hCA IX (Table 2). Interestingly, by extending the inhibitory sulfonamide warhead with a single amino acid (Ser, Hit3-t1), the potency of the inhibitor improved by 20-fold (from 471 nM to 23 nM). The Ki value further dropped upon the addition of the leucine residue (Hit3-t2), slightly increased upon proline addition (Hit3-t3), and dropped down again to 8.6 nM for the conjugate Hit3-t4 containing four amino acid residues. We concluded that the peptidic part of the conjugate contributes to the improved potency, and the effect is most pronounced after the addition of the first amino acid. On the other hand, the addition of the second and third amino acid residue had a great effect on compound selectivity. Two truncated versions Hit3-t2 and Hit3-t3, were more selective towards hCA II than the parent full-length conjugate Hit3 (Ki (hCA IX)/Ki (hCA II) ca. 165-fold vs. 105-fold).
| Inhibitor | K i (hCA II) [nM] | K i (hCA IX) [nM] | K i (hCA I) [nM] | K i (hCA XII) [μM] |
|---|---|---|---|---|
| a Values from Table 1 are shown for comparison. | ||||
| AAZ 48 | 12 | 25.2 | 250 | 0.0057 |
| Inh2 | 471 ± 44a | 844 ± 83a | 601 ± 48 | 5.4 ± 1.2 |
| Hit3-t1 | 23 ± 1.6 | 1800 ± 150 | 346 ± 22 | 5.1 ± 1.0 |
| Hit3-t2 | 14.6 ± 0.9 | 2500 ± 200 | 161.8 ± 35.3 | 14.0 ± 3.2 |
| Hit3-t3 | 17.4 ± 1.0 | 2800 ± 160 | 243.5 ± 31.4 | 9.6 ± 1.1 |
| Hit3-t4 | 8.6 ± 0.9 | 936 ± 63 | 75.5 ± 6.0 | 6.1 ± 2.8 |
| Hit3 | 13.0 ± 1.0a | 1368 ± 82a | 104.7 ± 28.9 | 25.7 ± 11.1 |
To further assess compound selectivity, we tested inhibition of two additional hCA isoforms. Isoform hCA I is ubiquitous cytosolic enzyme expressed in red blood cells and gastrointestinal tract49 while hCA XII is a membrane-associated isoform that, similarly to hCA IX, controls the extracellular pH of cancer cells.50 Comparison of Ki values (Table 2) reveals Hit3 as well as all truncated variants are selective towards hCA II over other isoforms. Inhibition constants for hCA I are in the high nanomolar range and selectivity index (Ki (hCA I)/Ki (hCA II)) ranges from 8 to 15. Inhibition of hCA XII by all tested compounds is in the micromolar range and consequently the selectivity toward hCA II is three orders of magnitude higher.
The crystal structures of Hit3-t1–4 bound to hCA II were determined at atomic resolution (Table S2†). Similar to the crystal structures of the parent compound Hit3, the sulfonamide inhibitor parts could be modelled with full occupancy into a well-defined electron density map. The quality of the electron density map for the peptide part was lower, and the residues at the second and third positions could not be modeled unambiguously (Fig. S6†). Serine at position 1 could be modelled in all compounds; thus, the interactions of Hit3-t1 with the hCA II active site are fully resolved in the crystal structure. Serine made extensive interactions with residues Q92, V121, F131, G132, V135, V143, L198, T199–200, P202, and W209 (Fig. 5A): this explains why the addition of this residue had a large positive effect on inhibitory potency.
 | ||
| Fig. 5 Truncated versions of conjugates Hit3-t1 (A), Hit3-t2 (B), and Hit3-t4 (C) bound to the hCA II active site. The hit compounds are depicted in sticks with differently colored carbon atoms: Hit3-t1 (salmon), Hit3-t2 (green), Hit3-t4 (yellow). Oxygen, nitrogen, and sulfur atoms are shown in red, blue, and orange, respectively. The zinc ion is represented by the grey sphere. The protein is represented by its solvent accessible surface and colored according to electrostatic potential (red for negative, blue for positive). The residues involved in interactions with hit compounds are highlighted. For clarity, residues H94, H96, H119, V143, T199, and W209 at the bottom of the cavity are not labeled. | ||
In Hit3-t2 and Hit3-t3, residues at position 2 and 3 could not be modelled because, the electron density map was low or missing (Fig. S6†). The binding pose of Hit3-t3 was virtually identical to Hit3-t2; thus, the structure of hCA II in complex with Hit3-t3 was not refined and not deposited to PDB. The binding poses of serine in Hit3-t2 and Hit3-t3 differ slightly from that of Hit3-t1 (Fig. 5A and B) suggesting that the residues at position 2 and 3, although disordered in our crystal structure, impose a steric strain on the first residue and divert it from its original pose within the enzyme active cavity.
The addition of phenylalanine at position 4 in Hit3-t4 residues stabilized the binding pose of the peptidic part and three out of four amino acid residues could be modelled for this compound (Fig. S6†). Hit3-t4 interacted with virtually the same residues as Hit3-t1 (with the exception of G132, Fig. 5C). Leucine at position 2 and proline at position 3 of Hit3-t4 did not form any additional contacts with the hCA II residues and are oriented toward the solvent. Based on the position of the carboxyl group of this terminal proline, the interaction of terminal phenylalanine, although not captured in the crystal structure, can be expected in the hydrophobic patch formed by I91, V121 and F131 (Fig. 5C).
To follow the interactions of individual amino acids of the Hit3 compound in solution, we performed NMR binding studies similar to what was described for the full-length peptide–inhibitor conjugates (Fig. 6 and S7 and S8†).
 | ||
| Fig. 6 NMR chemical shift perturbations of 15N hCA II upon binding of truncated versions of conjugates Hit3-t1 (A), Hit3-t2 (B), Hit3-t3 (C), and Hit3-t4 (D). Residues perturbed upon peptide–inhibitor conjugate binding are mapped on the surface of 15N hCA II. Normalized combined chemical shifts with values over 1, 2 or 3 are colored yellow, orange, and red, respectively. For visualization of binding for each truncated hit conjugate, the determined crystal structure was used, with the exception of Hit3-t3 where the crystal structure of Hit3-t2 was used instead due to a missing crystal structure. Views rotated by 180° along the vertical axis exposing the side opposite to the enzyme active site are shown in Fig. S3.† | ||
We found that the binding patches for all four truncated conjugates are similar and correspond to the ones observed with the addition of Hit3-t1 (Fig. 6 and S2†). This confirms the major contribution of the first amino acid (Ser) to the specific binding. Enzyme residues that were the most affected by the peptide binding belong to region 125–140 (loop with two short helixes connecting β-strands 5 and 6). The highest contribution came from residues F131, G132, K133, V135, Q136 and extends further away from the active site cleft towards residues G129 and D130. It is clear that this surface patch is interacting with the peptidic part although it could not be captured in the X-ray structure. Unlike most of the residues in the enzyme active site cleft, this region is not conserved between hCA II and hCA IX. This region in hCA II is formed by residues TKYGDFGK![[F with combining low line]](https://www.rsc.org/images/entities/char_0046_0332.gif)
![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[K with combining low line]](https://www.rsc.org/images/entities/char_004b_0332.gif) AVQ
AVQ![[V with combining low line]](https://www.rsc.org/images/entities/char_0056_0332.gif)
![[Q with combining low line]](https://www.rsc.org/images/entities/char_0051_0332.gif) QPDG, while hCA IX has the sequence TAFARVDEALGRPGG. There are only 3 conserved residues (shown in bold) and none of them is involved in the interaction with Hit3 compounds (interacting residues are underlined in the hCA II sequence). Interaction in this variable region thus ensures binding to hCA II but not hCA IX; thus it represents a suitable enzyme exosite for targeting with isoform specific inhibitors.
QPDG, while hCA IX has the sequence TAFARVDEALGRPGG. There are only 3 conserved residues (shown in bold) and none of them is involved in the interaction with Hit3 compounds (interacting residues are underlined in the hCA II sequence). Interaction in this variable region thus ensures binding to hCA II but not hCA IX; thus it represents a suitable enzyme exosite for targeting with isoform specific inhibitors.
To provide peptide–inhibitor conjugates with a more defined mode of binding into this exosite, rigidification of the peptide backbone by e.g. stapling51,52 or cyclization53,54 could be employed to improve the potency and selectivity.
Conclusion
In this work, we investigated three different selection methods with the aim of finding ways to improve the potency and selectivity of sulfonamide inhibitors that target human carbonic anhydrase isoenzymes. Among the three methods used, the in situ click chemistry approach yielded the most potent inhibitor, with a Ki value in the low nanomolar range (Table 1) but with a low selectivity towards hCA II over hCA IX. The synthetic peptide libraries provided peptide–inhibitor conjugates with improved potency but relatively poor selectivity (Table 1). The selection from the phage-display library led to the identification of potent and selective inhibitor conjugates (Table 1). The most potent peptide–inhibitor conjugates that were identified have single digit nanomolar inhibitory activity against hCA II and are more selective for this enzyme isoform when compared to hCA I, hCA IX and hCA XII. Our results point to the possibility of using sulfonamides in combination with peptide backbones to construct a new chemotype of carbonic anhydrase inhibitors that utilize simultaneous binding to both the active site and the exosite.Our detailed crystallographic and NMR structural analysis study revealed that the sulfonamide warhead is the main anchoring part that is important for the binding and inhibition activity of the conjugates. The peptide backbone serves as a supplementary structural element that provides additional interactions in close proximity to the active site and positively affects compound affinity and specificity towards the hCA II isoform. Although the binding mode of the peptide backbone could not be unambiguously resolved by X-ray crystallography, the NMR study provided evidence of transient or dynamic interactions that are responsible for the observed improvement in potency and selectivity. We identified a region of amino acid residues located in the vicinity of the enzyme active site that are the key determinant for selective binding of peptide conjugates originating from the phage-display library selection campaign. This enzyme exosite, formed by residues 125–140, could serve as a suitable area to be targeted by isoform specific inhibitors.
In conclusion, all three methods investigated in this work represent an appealing alternative to commonly used approaches based on high-throughput screening methods of small molecule libraries. In line with our results, we find the phage-display libraries have great potential to improve otherwise poor and unspecific inhibitors. Especially in combination with various cyclization strategies, the phage or mRNA displayed peptide libraries55–61 could serve as a valuable source of novel peptide–inhibitor conjugates with advanced properties.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
Diffraction data were collected on BL14.1 and BL14.2 at the BESSY II electron storage ring operated by the Helmholtz-Zentrum Berlin. The authors would like to acknowledge Dr. Václav Veverka from IOCB CAS Prague, for his help with NMR analysis and Dr. Jiří Brynda and Anna Maliukova from IOCB CAS Prague, for their help with enzyme inhibition assays. This work was supported by Gilead Sciences and IOCB Research Centre, by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grants agreement No. 677465) and by European Regional Development Fund (Project No. CZ.02.1.01/0.0/0.0/16_019/0000729). The PhD scholarship for M. H. from the Department of Organic Chemistry Faculty of Science, Charles University, Prague is also acknowledged.References
- A. Carnero, Clin. Transl. Oncol., 2006, 8, 482–490 CrossRef CAS PubMed.
- D. G. Brown and J. Boström, J. Med. Chem., 2018, 61, 9442–9468 CrossRef CAS PubMed.
- D. Weigelt and I. Dorange, in Lead Generation, ed. J. Holenz, 2016, pp. 93–132, DOI:10.1002/9783527677047.ch05.
- D. Rideout, Science, 1986, 233, 561–563 CrossRef CAS PubMed.
- J. Inglese and S. J. Benkovic, Tetrahedron, 1991, 47, 2351–2364 CrossRef CAS.
- I. Huc and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2106–2110 CrossRef CAS PubMed.
- R. Nguyen and I. Huc, Angew. Chem., Int. Ed., 2001, 40, 1774–1776 CrossRef CAS.
- M. Jaegle, E. L. Wong, C. Tauber, E. Nawrotzky, C. Arkona and J. Rademann, Angew. Chem., Int. Ed., 2017, 56, 7358–7378 CrossRef CAS PubMed.
- M. Wichert, N. Krall, W. Decurtins, R. M. Franzini, F. Pretto, P. Schneider, D. Neri and J. Scheuermann, Nat. Chem., 2015, 7, 241–249 CrossRef CAS PubMed.
- D. A. Erlanson, R. S. McDowell and T. O'Brien, J. Med. Chem., 2004, 47, 3463–3482 CrossRef CAS.
- D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke and H. Jhoti, Nat. Rev. Drug Discovery, 2016, 15, 605–619 CrossRef CAS.
- M. Y. Unver, R. M. Gierse, H. Ritchie and A. K. H. Hirsch, J. Med. Chem., 2018, 61, 9395–9409 CrossRef CAS PubMed.
- D. Bosc, V. Camberlein, R. Gealageas, O. Castillo-Aguilera, B. Deprez and R. Deprez-Poulain, J. Med. Chem., 2020, 63, 3817–3833 CrossRef CAS PubMed.
- A. Lossouarn, P. Y. Renard and C. Sabot, Bioconjugate Chem., 2021, 32, 63–72 CrossRef CAS.
- T. Hirose, N. Maita, H. Gouda, J. Koseki, T. Yamamoto, A. Sugawara, H. Nakano, S. Hirono, K. Shiomi, T. Watanabe, H. Taniguchi, K. B. Sharpless, S. Ōmura and T. Sunazuka, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15892–15897 CrossRef CAS.
- W. G. Lewis, L. G. Green, F. Grynszpan, Z. Radić, P. R. Carlier, P. Taylor, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 1053–1057 CrossRef CAS.
- S. K. Mamidyala and M. G. Finn, Chem. Soc. Rev., 2010, 39, 1252–1261 RSC.
- N. Willand, M. Desroses, P. Toto, B. Dirié, Z. Lens, V. Villeret, P. Rucktooa, C. Locht, A. Baulard and B. Deprez, ACS Chem. Biol., 2010, 5, 1007–1013 CrossRef CAS PubMed.
- A. Bhardwaj, J. Kaur, M. Wuest and F. Wuest, Nat. Commun., 2017, 8, 1 CrossRef.
- V. P. Mocharla, B. Colasson, L. V. Lee, S. Röper, K. B. Sharpless, C.-H. Wong and H. C. Kolb, Angew. Chem., Int. Ed., 2005, 44, 116–120 CrossRef CAS PubMed.
- S. Ng, M. R. Jafari, W. L. Matochko and R. Derda, ACS Chem. Biol., 2012, 7, 1482–1487 CrossRef CAS PubMed.
- S. Ng, E. Lin, P. I. Kitov, K. F. Tjhung, O. O. Gerlits, L. Deng, B. Kasper, A. Sood, B. M. Paschal, P. Zhang, C.-C. Ling, J. S. Klassen, C. J. Noren, L. K. Mahal, R. J. Woods, L. Coates and R. Derda, J. Am. Chem. Soc., 2015, 137, 5248–5251 CrossRef CAS.
- K. F. Tjhung, P. I. Kitov, S. Ng, E. N. Kitova, L. Deng, J. S. Klassen and R. Derda, J. Am. Chem. Soc., 2016, 138, 32–35 CrossRef CAS.
- J. Haapasalo, K. Nordfors, S. Jarvela, H. Bragge, I. Rantala, A. K. Parkkila, H. Haapasalo and S. Parkkila, Neuro. Oncol., 2007, 9, 308–313 CrossRef CAS.
- C. T. Supuran and A. Scozzafava, Bioorg. Med. Chem., 2007, 15, 4336–4350 CrossRef CAS.
- C. T. Supuran, Curr. Top. Med. Chem., 2007, 7, 825–833 CrossRef CAS PubMed.
- C. Capasso and C. T. Supuran, in Targeting Carbonic Anhydrases, 2014, pp. 6–16, DOI:10.4155/fseb2013.13.43.
- C. B. Mishra, M. Tiwari and C. T. Supuran, Med. Res. Rev., 2020, 40, 2485–2565 CrossRef CAS.
- B.-H. Jonsson and A. Liljas, Biophys. J., 2020, 119, 1275–1280 CrossRef CAS PubMed.
- C. T. Supuran, Expert Opin. Drug Discovery, 2017, 12, 61–88 CrossRef CAS.
- C. T. Supuran, Expert Opin. Drug Discovery, 2020, 15, 671–686 CrossRef CAS.
- E. D. Goddard-Borger and R. V. Stick, Org. Lett., 2007, 9, 3797–3800 CrossRef CAS PubMed.
- G. T. Potter, G. C. Jayson, G. J. Miller and J. M. Gardiner, J. Org. Chem., 2016, 81, 3443–3446 CrossRef CAS.
- K. F. Geoghegan and J. G. Stroh, Bioconjugate Chem., 1992, 3, 138–146 CrossRef CAS.
- J. E. Jee, J. Lim, Y. S. Ong, J. Oon, L. Gao, H. S. Choi and S. S. Lee, Org. Biomol. Chem., 2016, 14, 6833–6839 RSC.
- J. Li, K. Shi, Z. F. Sabet, W. Fu, H. Zhou, S. Xu, T. Liu, M. You, M. Cao, M. Xu, X. Cui, B. Hu, Y. Liu and C. Chen, Sci. Adv., 2019, 5, eaax0937 CrossRef CAS PubMed.
- N. Kanfar, M. Tanc, P. Dumy, C. T. Supuran, S. Ulrich and J. Y. Winum, Chem. – Eur. J., 2017, 23, 6788–6794 CrossRef CAS.
- A. Furka, F. Sebestyen, M. Asgedom and G. Dibo, Int. J. Pept. Protein Res., 1991, 37, 487–493 CrossRef CAS.
- K. S. Lam, S. E. Salmon, E. M. Hersh, V. J. Hruby, W. M. Kazmierski and R. J. Knapp, Nature, 1991, 354, 82–84 CrossRef CAS.
- R. A. Houghten, C. Pinilla, S. E. Blondelle, J. R. Appel, C. T. Dooley and J. H. Cuervo, Nature, 1991, 354, 84–86 CrossRef CAS.
- R. G. Khalifah, J. Biol. Chem., 1971, 246, 2561–2573 CrossRef CAS.
- M. Kugler, J. Holub, J. Brynda, K. Pospíšilová, S. E. Anwar, D. Bavol, M. Havránek, V. Král, M. Fábry, B. Grüner and P. Řezáčová, J. Enzyme Inhib. Med. Chem., 2020, 35, 1800–1810 CrossRef CAS.
- M. A. A. Najm, W. R. Mahmoud, A. T. Taher, S. E. S. Abbas, F. M. Awadallah, H. A. Allam, D. Vullo and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2022, 37, 2702–2709 CrossRef CAS PubMed.
- V. V. Komnatnyy, T. E. Nielsen and K. Qvortrup, Chem. Commun., 2018, 54, 6759–6771 RSC.
- D. Pei and G. Appiah Kubi, Expert Opin. Drug Discovery, 2019, 14, 1097–1102 CrossRef CAS PubMed.
- V. M. Krishnamurthy, G. K. Kaufman, A. R. Urbach, I. Gitlin, K. L. Gudiksen, D. B. Weibel and G. M. Whitesides, Chem. Rev., 2008, 108, 946–1051 CrossRef CAS.
- X. Cheng, V. Veverka, A. Radhakrishnan, L. C. Waters, F. W. Muskett, S. H. Morgan, J. Huo, C. Yu, E. J. Evans, A. J. Leslie, M. Griffiths, C. Stubberfield, R. Griffin, A. J. Henry, A. Jansson, J. E. Ladbury, S. Ikemizu, M. D. Carr and S. J. Davis, J. Biol. Chem., 2013, 288, 11771–11785 CrossRef CAS.
- S. Sersen, K. Traven, J. Kljun, I. Turel and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2019, 34, 388–393 CrossRef CAS PubMed.
- S. Lindskog, Pharmacol. Ther., 1997, 74, 1–20 CrossRef CAS PubMed.
- A. Waheed and W. S. Sly, Gene, 2017, 623, 33–40 CrossRef CAS.
- Y. H. Lau, P. de Andrade, Y. Wu and D. R. Spring, Chem. Soc. Rev., 2015, 44, 91–102 RSC.
- X. Li, S. Chen, W.-D. Zhang and H.-G. Hu, Chem. Rev., 2020, 120, 10079–10144 CrossRef CAS PubMed.
- K. Shinbara, W. Liu, R. H. P. van Neer, T. Katoh and H. Suga, Front. Chem., 2020, 8 Search PubMed.
- J. S. R. Samuel and Q. Nir, Curr. Top. Med. Chem., 2018, 18, 526–555 CrossRef PubMed.
- S. S. Kale, C. Villequey, X.-D. Kong, A. Zorzi, K. Deyle and C. Heinis, Nat. Chem., 2018, 10, 715–723 CrossRef CAS PubMed.
- S. Ng and R. Derda, Org. Biomol. Chem., 2016, 14, 5539–5545 RSC.
- G. K. Mothukuri, S. S. Kale, C. L. Stenbratt, A. Zorzi, J. Vesin, J. Bortoli Chapalay, K. Deyle, G. Turcatti, L. Cendron, A. Angelini and C. Heinis, Chem. Sci., 2020, 11, 7858–7863 RSC.
- K. Maola, J. Wilbs, J. Touati, M. Sabisz, X.-D. Kong, A. Baumann, K. Deyle and C. Heinis, Angew. Chem., Int. Ed., 2019, 58, 11801–11805 CrossRef CAS.
- A. I. Ekanayake, L. Sobze, P. Kelich, J. Youk, N. J. Bennett, R. Mukherjee, A. Bhardwaj, F. Wuest, L. Vukovic and R. Derda, J. Am. Chem. Soc., 2021, 143, 5497–5507 CrossRef CAS PubMed.
- T. Passioura and H. Suga, Chem. Commun., 2017, 53, 1931–1940 RSC.
- Y. Huang, M. M. Wiedmann and H. Suga, Chem. Rev., 2019, 119, 10360–10391 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00330a |
| ‡ Current address: Gene Center and Department of Biochemistry, Ludwig-Maximilians-Universität München, Feodor-Lynen-Straße 25, 81377, Munich, Germany. |
| This journal is © The Royal Society of Chemistry 2023 |