
Antioxidant mechanisms and products of four 4′,5,7-trihydroxyflavonoids with different structural types†
Ban
Chen
 ab,
Jiangtao
Su
ab,
Yuchen
Hu
ab,
Shuqin
Liu
c,
Xiaojian
Ouyang
d,
Rongxin
Cai
e,
Xiangyu
You
ab and
Xican
Li
*c
ab,
Jiangtao
Su
ab,
Yuchen
Hu
ab,
Shuqin
Liu
c,
Xiaojian
Ouyang
d,
Rongxin
Cai
e,
Xiangyu
You
ab and
Xican
Li
*c
aKey Laboratory of Fermentation Engineering (Ministry of Education), Hubei University of Technology, Wuhan, 430000, China
bCooperative Innovation Centre of Industrial Fermentation (Ministry of Education & Hubei Province), Hubei University of Technology, Wuhan, 430000, China
cSchool of Chinese Herbal Medicine, Guangzhou University of Chinese Medicine, Guangzhou, 510000, China. E-mail: lixican@126.com
dIncreasePharm Hengqin Inst. Co., Ltd., Zhuhai, 519000, China
eGuangdong Food Industry Institute Co., Ltd., Guangzhou, 510000, China
First published on 3rd November 2022
Abstract
4′,5,7-OHs are common substituents of natural flavonoids, a type of effective phenolic antioxidant. However, the antioxidant processes between 4′,5,7-trihydroxyflavonoids with different structural types have not been compared systematically, and the antioxidant products are challenging to determine. This study compared four 4′,5,7-trihydroxyflavonoids, including apigenin, genistein, kaempferol, and naringenin. In quantum chemical analyses, the four 4′,5,7-trihydroxyflavonoids showed different thermodynamic properties, and the C4′–OH (or C3–OH of kaempferol) possessed the strongest activity. Moreover, the reaction rate constants were larger when a hydrogen atom was transferred from C4′–OH (or C3–OH of kaempferol) than from C5–OH. When different atoms were linked to 2,2-diphenyl-1-picrylhydrazyl radical (DPPH˙), the C3′–DPPH adducts showed the smallest energy. In experimental assays, the scavenging ability for neutral free radicals, radical cations, and radical anions was negatively correlated with the corresponding theoretical parameters. Finally, mass spectroscopy detected the apigenin–DPPH˙, genistein–DPPH˙, and naringenin–DPPH˙ adduct peaks. In conclusion, the structural type of 4′,5,7-trihydroxyflavonoids can affect the antioxidant ability, site, and speed, but not the mechanism. After hydrogen abstraction at C4′–OH, 4′,5,7-trihydroxyflavones, 4′,5,7-trihydroxyisoflavones, and 4′,5,7-trihydroxyflavanones will produce antioxidant products via C3′–radical linking.
Introduction
Excessive free radicals can damage normal cells and cause oxidative stress in the body, which is related to many diseases, such as diabetes,1 arthritis,2 and atherosclerosis.3 A large number of studies have reported that natural flavonoids are effective antioxidants, most of which possess the ability to scavenge free radicals and prevent oxidative stress-related diseases.4–6As typical phenolic antioxidants, natural flavonoids include various structural types, such as flavones, isoflavones, flavonols, and flavanones. They usually have 4′,5,7-OHs as antioxidant sites and scavenge free radicals through hydrogen atom transfer (HAT), single electron transfer followed by proton transfer (SET-PT), and sequential proton loss electron transfer (SPLET) mechanisms.7 Under the three mechanisms, hydroxyl is oxidized by free radicals to an unstable phenoxy radical intermediate; the intermediate can form stable radical adducts to terminate the free-radical-chain reaction. The covalent linkage is essentially a radical adduct formation (RAF) or radical coupling reaction.8
Now RAF has become a hot issue in free radical chemistry, because it not only plays a crucial role in the antioxidant process of phytophenols but also provides a valuable theory for the biosynthesis and biodegradation of phenolic dimers (e.g., homodimers, chlorogenic acid dimers, and quercetin Diels–Alder anti-dimers).9–11 It can be affected by several structural factors, including the aromaticity of the phenyl ring, conjugated π bond, steric hindrance, and oligomerization degree. Possibly owing to the complication, there have been few studies regarding the RAF products of flavonoids despite having accumulated substantial antioxidant studies. In addition, it is unclear whether the structural type of 4′,5,7-trihydroxyflavonoids affects the antioxidant mechanism and RAF products.
To overcome the limitations of RAF product identification, we introduced ultrahigh-performance liquid chromatography quadrupole-Orbitrap mass spectrometers (UHPLC-Q-Orbitrap-MS/MS), and meanwhile the quantum chemical calculation was adopted as the aid of experimental evidence. The studied flavonoids, apigenin (AP), genistein (GE), kaempferol (KA), and naringenin (NA), have been selected as the representatives, because they belong to four common structural types of flavonoids (e.g., flavones, isoflavones, flavonols, and flavanones) and all possess 4′,5,7-OHs (Fig. 1). Their structural similarity offers the possibility for detailed structure–activity relationship analysis.
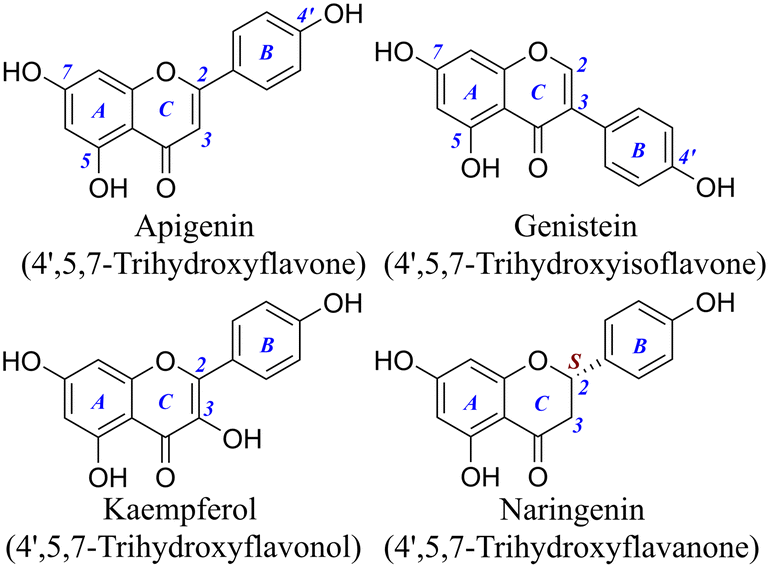 | ||
| Fig. 1 Structures of the four 4′,5,7-trihydroxyflavonoids (the C3–OH of kaempferol is not considered as a phenolic hydroxyl). | ||
Undoubtedly, this study provides an in-depth understanding of the antioxidant mechanisms of 4′,5,7-trihydroxyflavonoids with various structural types. More importantly, it also benefits RAF or radical coupling reaction research on phytophenols.
Results and discussion
As mentioned above, the hydrogen abstraction reaction usually occurs before RAF, mainly involving HAT, SET-PT, and SPLET mechanisms. In the HAT mechanism, a flavonoid directly detaches a phenolic hydrogen atom by homolytic cleavage of the O–H bond. The SET-PT and SPLET mechanisms are two-step reactions where electron transfer (ET) and proton transfer (PT) occur in separate steps. In the SET-PT mechanism, a flavonoid first undergoes ET to form a radical cation, and the radical cation consecutively loses a proton. In the SPLET mechanism, a proton is transferred from the O–H bond by heterolytic cleavage; then, an electron is donated from the anion created in the first step.12,13The three mechanisms are characterized by some important chemical thermodynamic parameters. Specifically, the HAT mechanism is governed by bond dissociation enthalpy (BDE), the two steps of the SET-PT mechanism are governed by ionization potential (IP) and proton dissociation enthalpy (PDE), and the two steps of the SPLET mechanism are governed by proton affinity (PA) and electron transfer enthalpy (ETE). The smaller these parameters are, the stronger the corresponding reactivity.14 This study calculated them based on quantum chemistry to clarify antioxidant activities, reaction sites, and preferred mechanisms of the four flavonoids.
As shown in Table 1, for each of the four flavonoids, the order of BDEs between different sites in water or methanol was C4′–OH < C7–OH < C5–OH. That was to say, the HAT activity of C4′–OH and C5–OH was the strongest and the weakest, respectively.
| Methanol | Water | |||||||
|---|---|---|---|---|---|---|---|---|
| C3–OH | C5–OH | C7–OH | C4′–OH | C3–OH | C5–OH | C7–OH | C4′–OH | |
| AP, apigenin; GE, genistein; KA, kaempferol; NA, naringenin. | ||||||||
| AP | — | 408.33 | 401.43 | 370.87 | — | 397.39 | 390.45 | 369.29 |
| GE | — | 407.24 | 403.73 | 364.72 | — | 396.03 | 392.67 | 358.89 |
| KA | 346.16 | 398.45 | 397.57 | 362.86 | 334.81 | 387.46 | 386.92 | 359.64 |
| NA | — | 459.29 | 406.36 | 369.25 | — | 447.98 | 395.18 | 364.00 |
The weak activity of C5–OH might be related to the formation of an intramolecular hydrogen bond (IHB) with the adjacent carbonyl. Such weak interactions could be described using the interaction region indicator (IRI) in Fig. 2;15 the isosurface maps clearly showed an IHB between C5–OH and C4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. Thus, the C5–OH required more energy for homolysis.
O. Thus, the C5–OH required more energy for homolysis.
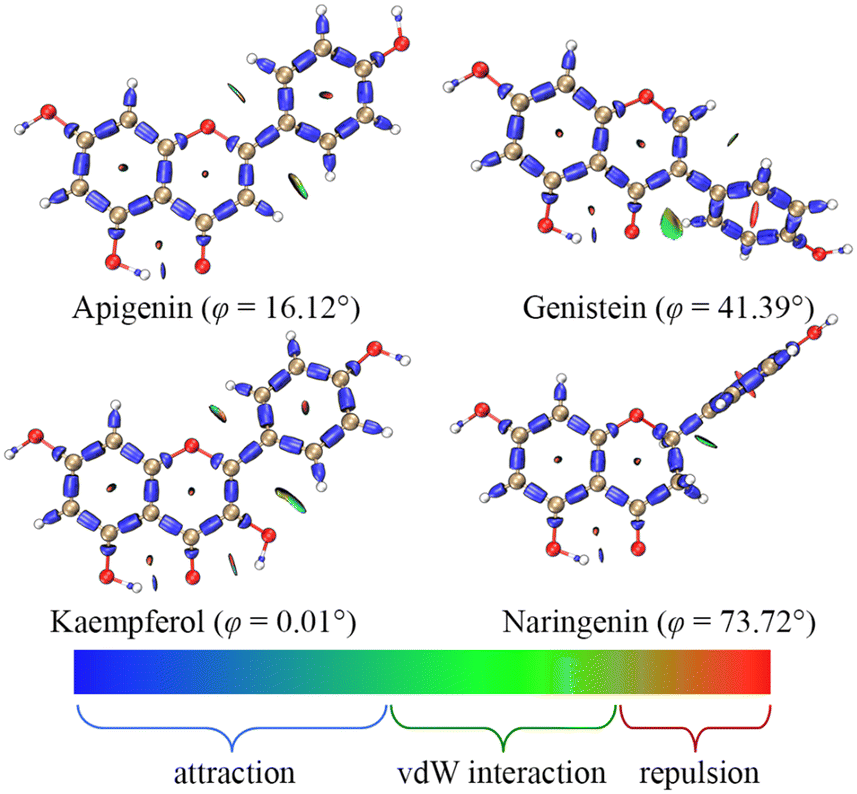 | ||
| Fig. 2 The dominant conformations and interaction region indicators of the four flavonoids; φ is the dihedral angle between the A ring and the B ring. | ||
The strong activity of C4′–OH was probably due to the more uniform distribution of electrons after HAT. The electron delocalization was related to the planarity of an aromatic system, which was reflected by the dihedral angle between the A ring and the B ring. The dihedral angle shown in Fig. 2 indicated a high level of electron delocalization for KA. The distribution of lone electrons of free radical intermediates was further described using spin density and spin population.16,17 Compared with HAT at C5–OH, HAT at C4′–OH led to a situation where the distribution of lone electrons was wider, and the standard deviation (SD) of the spin population and the enthalpy (H) of the free radical intermediate were smaller (Fig. 3). In particular, when HAT occurred at C3–OH of KA, the lone electron might be bidirectionally delocalized to the A ring and the B ring through the C2C3 double bond. Hence, the spin population SD, free radical intermediate enthalpy, and BDEs of C3–OH were further smaller than those of C4′–OH. In contrast, when the C2C3 double bond of KA was saturated to form aromadendrin, the BDE of C3–OH increased significantly to 448.20 kJ mol−1 in water, which was larger than that of C4′–OH (370.17 kJ mol−1).
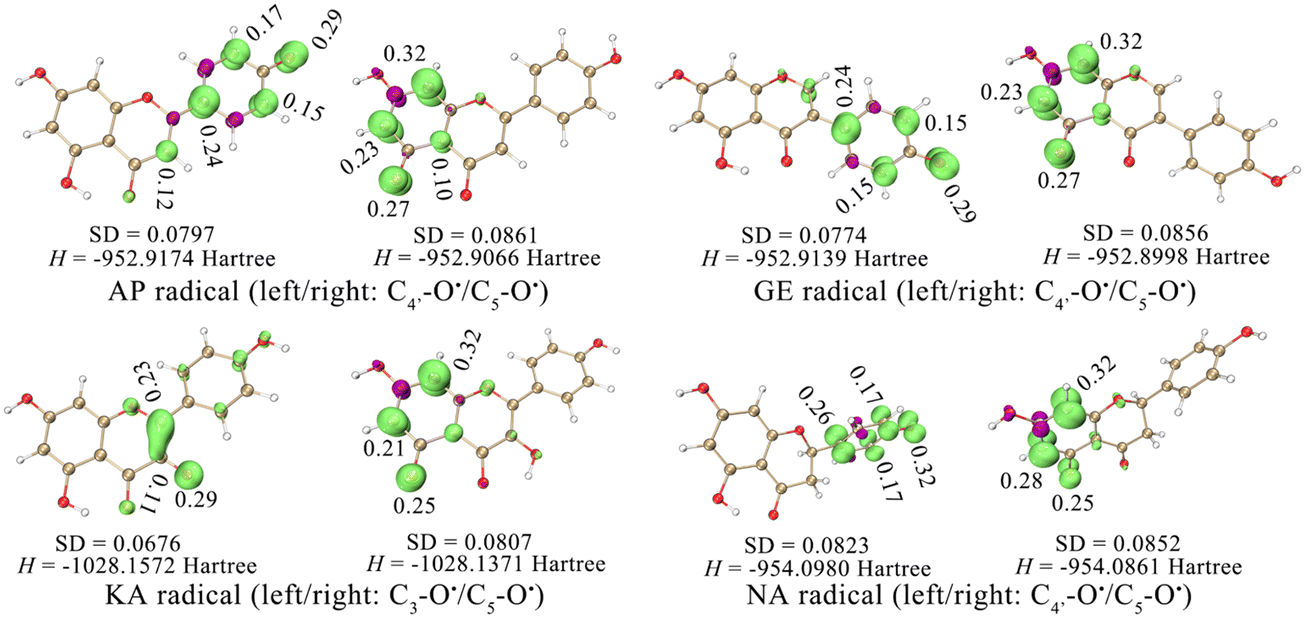 | ||
| Fig. 3 Standard deviation (SD) of the spin population and the enthalpy (H) of free radical intermediates. In the isosurface map, lime and purple colours correspond to the regions where the spin population is positive and negative, respectively (isovalue = 0.008). | ||
Therefore, spin density and spin population measurement could be used as a concise and efficient method to analyse the HAT site in a flavonoid. From the perspective of the minimum BDE of different molecules, the HAT activity order of the four flavonoids was KA > GE > NA > AP, indicating that the structural type of 4′,5,7-trihydroxyflavonoids affected the antioxidant activity and site mainly through the spin population, which was reflected by molecular planarity and conjugation.
Since the SET-PT or SPLET mechanism contains two steps, IP plus PDE or PA plus ETE should be considered as the final characterization parameters. By comparing BDE, IP plus PDE, and PA plus ETE (Tables 2 and 3), the following conclusions could be drawn: (a) the ET activity of the four flavonoids was different from the PT activity both in the SET-PT and SPLET mechanisms; (b) hydrogen abstraction reaction of the four flavonoids in water or methanol occurred preferentially via the HAT rather than the SET-PT or SPLET mechanism. (c) Under the three mechanisms, the activity order (e.g., KA > GE > NA > AP) and the active site (e.g., C3–OH or C4–OH) remained the same.
| Methanol | Water | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IP | C3–OH | C5–OH | C7–OH | C4′–OH | IP | C3–OH | C5–OH | C7–OH | C4′–OH | |
| The values in brackets are the sum of IP (ionization potential) and PDE (proton dissociation enthalpy). AP, apigenin; GE, genistein; KA, kaempferol; NA, naringenin. | ||||||||||
| AP | 521.38 | — | 45.55 (566.92) | 38.65 (560.02) | 17.42 (538.80) | 499.43 | — | 56.96 (556.39) | 50.01 (549.44) | 28.85 (528.28) |
| GE | 494.97 | — | 70.86 (565.83) | 67.35 (562.32) | 33.33 (528.31) | 472.98 | — | 82.04 (555.02) | 78.68 (551.67) | 44.90 (517.88) |
| KA | 480.52 | 24.23 (504.76) | 76.52 (557.05) | 75.65 (556.17) | 48.01 (528.53) | 459.06 | 34.74 (493.80) | 87.39 (546.45) | 86.85 (545.91) | 59.57 (518.63) |
| NA | 520.54 | — | 44.41 (564.95) | 97.34 (617.88) | 12.67 (533.21) | 498.30 | — | 55.87 (554.17) | 108.67 (606.97) | 24.69 (522.99) |
| Methanol | Water | |||||||
|---|---|---|---|---|---|---|---|---|
| C3–OH | C5–OH | C7–OH | C4′–OH | C3–OH | C5–OH | C7–OH | C4′–OH | |
| The values outside brackets are the PA (proton affinity), and the values in brackets are the ETE (electron transfer enthalpy). AP, apigenin; GE, genistein; KA, kaempferol; NA, naringenin. | ||||||||
| AP | — | 155.50 (411.46) | 128.99 (431.07) | 134.45 (404.39) | — | 159.79 (396.53) | 134.00 (415.38) | 140.11 (388.11) |
| GE | — | 154.15 (411.72) | 127.97 (434.39) | 151.79 (376.55) | — | 158.69 (396.27) | 133.26 (418.34) | 156.18 (361.64) |
| KA | 143.96 (360.83) | 146.67 (410.41) | 127.54 (428.66) | 139.01 (389.55) | 147.93 (345.80) | 151.40 (394.99) | 132.66 (413.19) | 144.44 (374.12) |
| NA | — | 151.36 (413.63) | 126.09 (491.83) | 151.33 (381.92) | — | 155.92 (398.19) | 131.15 (475.76) | 155.68 (367.24) |
The ET sites were confirmed by the electrostatic potential (ESP) and electron density difference (EDD).18,19 In the ESP results, the minima were mainly distributed around the O atoms, indicating that these regions were rich in electrons (Fig. 4 and S1†). The EDD in Fig. 5 describes the electron density change before and after ET,20,21 implying that in the ET process of SET-PT and SPLET mechanisms for the four flavonoids, electrons were transferred mainly from the C4′–OH (or C3–OH of KA) and their conjugated sites. Combining ESP and EDD illustrated that the ET sites of the four flavonoids were mainly the C4′–OH (or C3–OH of KA). The PT sites could be verified by the bond dipole moment, which was calculated based on the localized molecular orbitals (LMOs).22,23 Specifically, for each LMO identified as a two-centre one, the LMO dipole moment exhibits a deviation of the centroid of the LMO electron distribution from the centre of the bonding region, which can be used to reveal the bond polarity and is thus considered a bond dipole moment. The bond dipole moment (Table S1†) showed a positive correlation with PA for C7–OH and C4′–OH, indicating that their heterolysis ability was governed by the polarity. However, C5–OH possessed the largest bond dipole moment and PA due to the IHBs, which was consistent with organic chemistry theory.
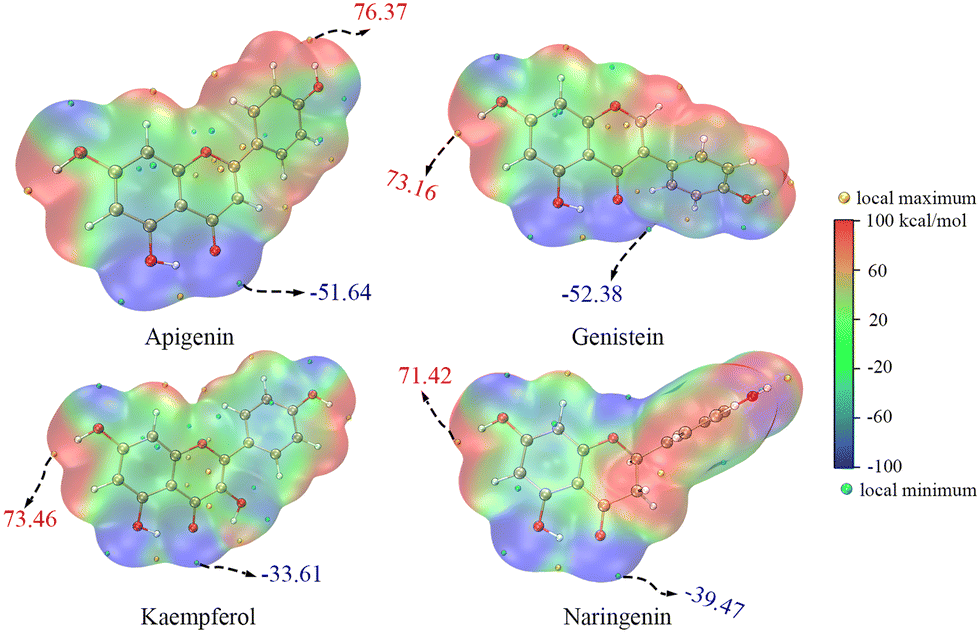 | ||
| Fig. 4 The electrostatic potential of neutral molecules of the four flavonoids (the values in the figure are the global extrema; the electrostatic potential of anions of the four flavonoids is shown in Fig. S1†). | ||
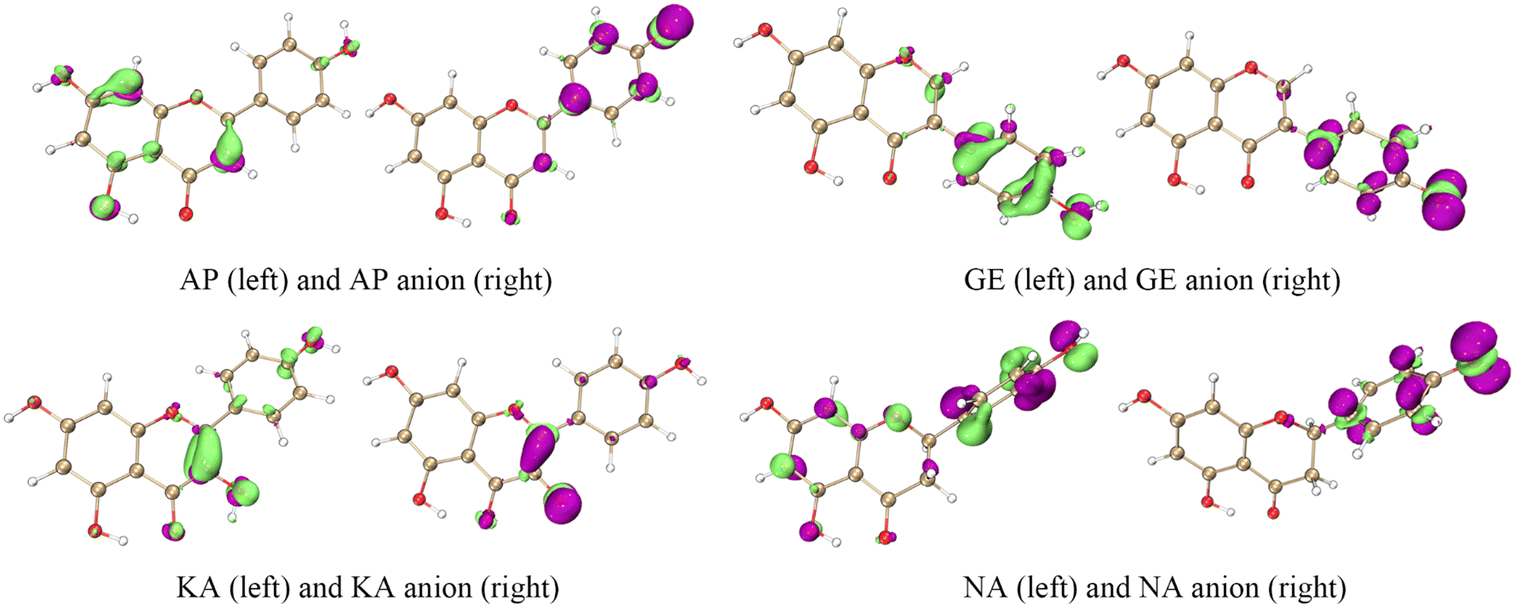 | ||
| Fig. 5 The isosurface maps of electron density difference for the four flavonoids; left, the difference between neutral molecules and radical cations; right, the difference between anions and free radical intermediates. In the isosurface map, lime and purple colours correspond to the regions where the electron density is increased and decreased, respectively (isovalue = 0.005). AP, apigenin; GE, genistein; KA, kaempferol; NA, naringenin. | ||
BDE, IP, PDE, and PA are usually used to describe the activity of different antioxidant mechanisms from the perspective of chemical thermodynamics. However, chemical kinetics illustrate the speed of an antioxidant reaction, which is difficult to observe experimentally due to the products' instability and the reaction's rapidity. In computational chemistry, there are many methods to explore the kinetics of a reaction, and transition state (TS) theory is one of them.24 Accordingly, the TS, activation free energy (ΔG≠), and intrinsic reaction coordinate of the four flavonoids to scavenge ˙OH were calculated (Fig. S2–S5†). Based on these results, the reaction rate constants (k) in Table 4 were obtained using the KiSThelP 2021 program;25 they suggested that when HAT occurred at the strong activity site (e.g., C4′–OH or C3–OH of KA), the order of the k values was somewhat similar to that of HAT activity. In addition, when HAT occurred at the weak activity site (e.g., C5–OH), the k values were smaller than those at the strong activity site (e.g., C4′–OH or C3–OH of KA), and the k values were somewhat negatively correlated with the activation free energy (ΔG≠) in each group. In summary, although it was difficult to observe experimentally, the structural type also affected the antioxidant speed of 4′,5,7-trihydroxyflavonoids (i.e., the stronger the HAT activity, the faster the reaction speed). This might be because the change in the molecular skeleton affected the activation energy of hydroxy groups.
| Group | Reaction sites | ΔG≠ | k |
|---|---|---|---|
| AP, apigenin; GE, genistein; KA, kaempferol; NA, naringenin. | |||
| Strong | AP–C4′–OH | 101.02 | 7.70 × 10−12 |
| GE–C4′–OH | 48.54 | 1.87 × 10−10 | |
| KA–C3–OH | 68.50 | 6.71 × 10−10 | |
| NA–C4′–OH | 135.29 | 2.03 × 10−11 | |
| Weak | AP–C5–OH | 92.78 | 2.72 × 10−14 |
| GE–C5–OH | 98.71 | 6.89 × 10−14 | |
| KA–C5–OH | 45.63 | 1.47 × 10−10 | |
| NA–C5–OH | 88.03 | 3.59 × 10−14 | |
The quantum chemical calculation results were partly verified by relevant experiments, including 2,2-diphenyl-1-picrylhydrazyl radical (DPPH˙)-scavenging and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonate) radical (ABTS˙+)-scavenging assays. DPPH˙ and ABTS˙+ are two artificial free radicals that can exist stably in vitro, and they are often used to evaluate the antioxidant capacity of molecules in methanol and water, respectively.26 The activity orders of the four flavonoids in the two assays were KA > GE > NA > AP and KA > GE > AP > NA (Table 5), which were negatively correlated with the calculated BDE and IP, respectively. Hence, DPPH˙ and ABTS˙+ were scavenged via HAT and ET mechanisms, which was consistent with the conclusions reported in the literature.27,28
| DPPH˙-scavenging | ABTS˙+-scavenging | ˙O2−-scavenging | |
|---|---|---|---|
| The IC50 value (μM) was defined as the concentration of 50% radical scavenging or relative reducing power, calculated by linear regression analysis, and expressed as the mean ± SD (n = 3). The IC50 values with different superscripts (a, b, c, d, or e) in the same column have statistically significant differences (P < 0.05). Trolox ((±)-6-hydroxyl-2,5,7,8-tetramethylchromane-2-carboxylic acid) is the positive control for the antioxidant assays. The dose–response curves are shown in Fig. S6.† AP, apigenin; GE, genistein; KA, kaempferol; NA, naringenin. | |||
| AP | 2760.69 ± 108.63e | 18.20 ± 0.69d | 68.85 ± 0.94b |
| GE | 1230.73 ± 75.44c | 10.16 ± 0.61b | 88.77 ± 1.09c |
| KA | 15.51 ± 0.30a | 4.73 ± 0.08a | 81.57 ± 2.62c |
| NA | 1730.30 ± 62.51d | 13.01 ± 0.04c | 24.17 ± 2.92a |
| Trolox | 19.79 ± 0.40b | 10.32 ± 1.28b | 962.78 ± 16.27d |
To obtain biological evidence, we conducted an ˙OH-scavenging assay. As shown in Fig. 6, the four flavonoids efficiently scavenged ˙OH, a typical reactive oxygen species occurring in cells.29 However, in the salicylic acid-based chromogenic method, the ˙OH-scavenging activity decreased in the order KA > GE > NA > AP, while in the cell counting kit-8 (CCK-8) assay, the activity decreased in the order NA > AP > KA > GE. This finding indicated that the four flavonoids mainly underwent the HAT mechanism to scavenge ˙OH in the salicylic acid-based chromogenic method and multiple mechanisms to scavenge ˙OH in the CCK-8 assay.
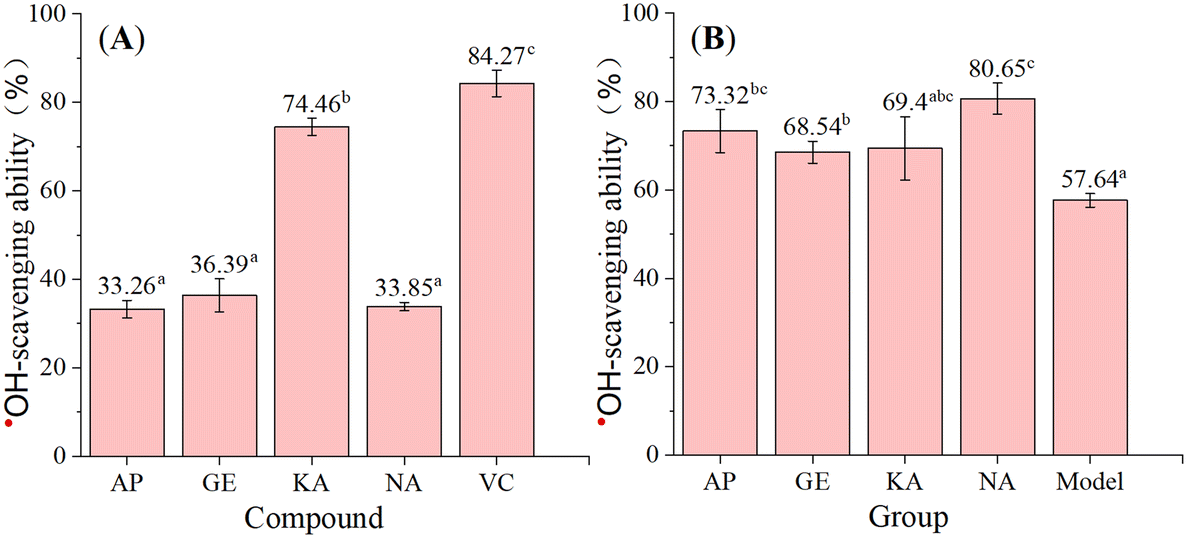 | ||
| Fig. 6 The ˙OH-scavenging ability of the four flavonoids based on the acid-based chromogenic method (A) and CCK-8 assay (B). VC means vitamin C, and the activity values with different superscripts (a, b, or c) have statistically significant differences (P < 0.05). AP, apigenin; GE, genistein; KA, kaempferol; NA, naringenin. | ||
According to previous studies, after prolonged incubation, the Fenton reaction can produce O2 (H2O2 + 2Fe3+ → 2Fe2+ + O2 + 2H+) and ˙O2− (O2 + Fe2+ → Fe3+ + ˙O2−) in addition to ˙OH (Fe2+ + H2O2 → OH− + ˙OH + Fe3+), and the scavenging of radical anions such as ˙O2− involves PT through the SET-PT and SPLET mechanisms.30 In the CCK-8 assay, HT22 cells were incubated with samples for more than 12 hours; thus, the assay might imply the ˙O2−-scavenging activity of the four flavonoids, which was negatively correlated with the calculated PDEs. The interaction of each flavonoid with ˙O2− was explored using the pyrogallol autoxidation assay improved by our team.31 Interestingly, the order of ˙O2−-scavenging activity (Table 5) was the same as that of the CCK-8 assay, confirming the PT-based mechanism in radical anion-scavenging.
The above experimental results validated the quantum chemical calculations and suggested that although radicals with different electrical properties or sources in different solvents were scavenged by different mechanisms, HAT, ET, and PT are the three crucial pathways. More importantly, they provided evidence for HAT as the primary thermodynamic mechanism and C4′–OH (or C3–OH of KA) as the primary active site.
Therefore, RAF product identification should be based on C4′–OH (or C3–OH of KA) reacting with a neutral free radical via the HAT mechanism. As seen in Fig. 3, the lone electron was mainly distributed on C4′–O (or C3–O of KA) and its conjugated C atoms (i.e., spin population >0.1); these atoms could be considered potential RAF linking sites. Accordingly, the possible RAF products with the heat of formation (ΔE) are shown in Table 6. The data showed that after C4′–OH lost an H atom, its ortho C-radical adduct was always preferentially generated for AP, GE, and NA. However, the ΔE values of all possible RAF products of KA were positive, suggesting that KA could not form RAF products, although its HAT activity was the strongest. Such results could be explained by steric hindrance, which was described using the IRI in Fig. 7. After DPPH˙ was linked to C3′, the A, B, and C rings of a flavonoid maintained a certain coplanarity, and the steric hindrance of the RAF products was weak (e.g., the IRI isosurface had a small distribution range); meanwhile, after DPPH˙ was linked to C2–OH, the C ring was flipped to the other side of the plane, leading to apparent steric hindrance.
| Potential linking sites | Potential RAF products | ΔE (kJ mol−1) | |
|---|---|---|---|
| AP, apigenin; GE, genistein; KA, kaempferol; NA, naringenin. | |||
| AP | C3, C1′, C3′, C4′–O | (1) C3–DPPH (2) C1′–DPPH (3) C3′–DPPH (4) C4′–O–DPPH | (1) −101.17 (2) 55.18 (3) −111.25 (4) 52.06 |
| GE | C1′, C3′, C4′–O | (1) C1′–DPPH (2) C3′–DPPH (3) C4′–O–DPPH | (1) 89.65 (2) −112.72 (3) 58.17 |
| KA | C2, C3, C3–O | (1) C2–DPPH (2) C3–DPPH (3) C3–O–DPPH | (1) 27.55 (2) 34.33 (3) 71.10 |
| NA | C1′, C3′, C4′–O | (1) C1′–DPPH (2) C3′–DPPH (3) C4′–O–DPPH | (1) 38.37 (2) −112.96 (3) 107.58 |
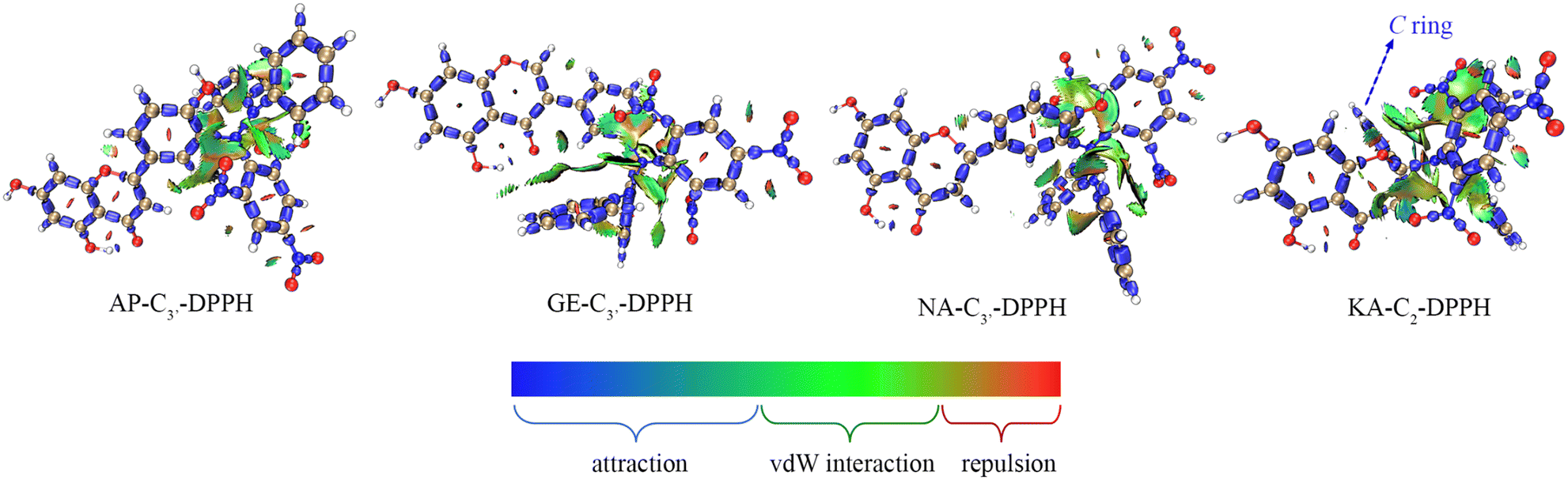 | ||
| Fig. 7 The interaction region indicator isosurface maps of the four flavonoid–DPPH adducts. | ||
In this study, UHPLC-Q-Orbitrap-MS/MS analysis was employed for the experimental evidence of RAF product identification. DPPH˙ was mixed with the four 4′,5,7-trihydroxyflavonoids for UHPLC-Q-Orbitrap-MS/MS analysis. As seen in Fig. 8, AP, GE, and NA mixed DPPH˙ gave evident chromatographic peaks (RT 10.10 min, 9.81 min, and 10.07 min, respectively) with molecular ion peaks at m/z values of 662.1165, 662.1116, and 664.1323 in the MS/MS field, and KA did not show an adduct signal in the MS/MS spectra. Because the m/z values were equal to the sum of the molecular masses of each flavonoid radical intermediate and DPPH˙, it was preliminarily proposed that there were flavonoid–DPPH adducts in the reaction products.
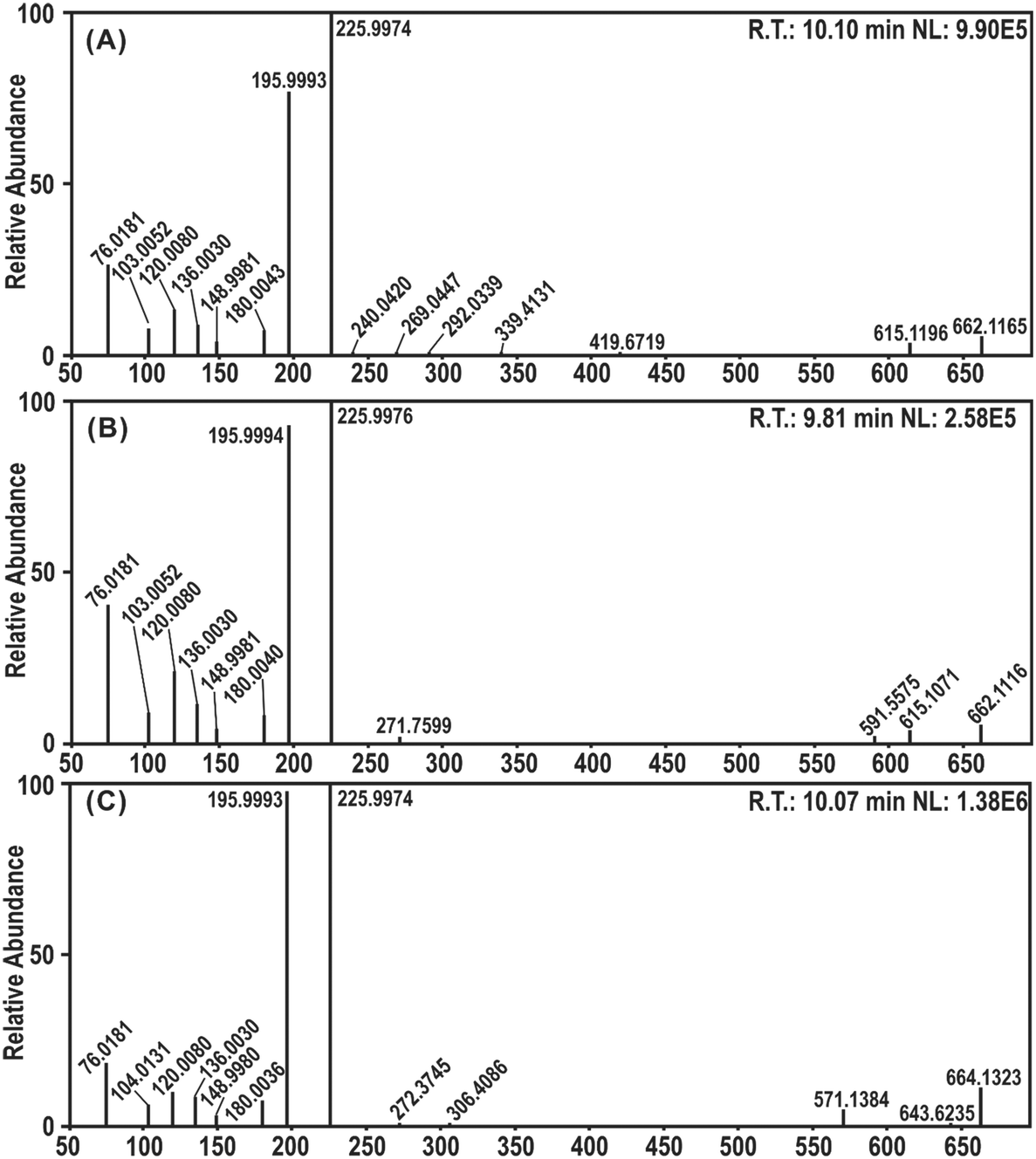 | ||
| Fig. 8 UHPLC-Q-Orbitrap-MS/MS analysis of the DPPH˙ reaction products with flavonoids: (A) MS/MS spectra of the apigenin–DPPH adduct; (B) MS/MS spectra of the genistein–DPPH adduct; (C) MS/MS spectra of the naringenin–DPPH adduct. R.T., retention time; NL, normalized level. | ||
Experimental
Chemicals
Mouse hippocampal neuronal HT22 cells were obtained from American Type Culture Collection (Rockville, MD, USA); Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), and trypsin were obtained from Gibco (Waltham, MA, USA); CCK-8 was from ApexBio Technology (Boston, MA, USA). Apigenin, genistein, kaempferol, naringenin, and naringenin chalcone were purchased from Alfa Biotechnology (Chengdu, China); salicylic acid was purchased from J&K Scientific (Beijing, China); H2O2 (30%), pyrogallol, Trolox ((±)-6-hydroxyl-2,5,7,8-tetramethylchromane-2-carboxylic acid), DPPH radical, TPTZ (tripyridyltriazine), and neocuproine were purchased from Sigma-Aldrich (Shanghai China); (NH4)2ABTS was purchased from Amresco (OH, USA). Other important chemical reagents included CuSO4, FeCl3·6H2O, HCl, K2S2O8, C4H11NO3 (Tris), NaH2PO4, Na2HPO4, CH3COONH4, and CH3COOH, and they were purchased from Zhiyuan Chemical Reagent Co., Ltd. (Tianjin, China); some HPLC grade reagents, such as methanol, ethanol, and formic acid, were purchased from Merck KGaA (Darmstadt, Germany).Computational details
All quantum chemical calculations were performed in the aqueous and methanol solvation model based on density (SMD) using the Gaussian 16 C.01 program.32,33 The geometry optimization and vibration frequency were calculated using the B3LYP-D3(BJ)/6-311+G(d,p) level.34–36 The optimized structures at the local minimum were ensured by the absence of an imaginary frequency, and the TS was ensured by only one imaginary frequency. Single-point energy calculations were further performed using M06-2X-D3 combined with a larger basis set def2-TZVPD with optimized geometries.37,38 All output files were visualized using GaussView 6.0 and VMD 1.9.3 programs.39,40 The calculation formulas for the antioxidant parameters are eqn (1)–(5):| BDE = H(ArO•) + H(H•) − H(ArOH) | (1) |
| IP = H(ArOH•+) + H(e−) − H(ArOH) | (2) |
| PDE = H(ArO•) + H(H+) − H(ArOH•+) | (3) |
| PA = H(ArO−) + H(H+) − H(ArOH) | (4) |
| ETE = H(ArO•) + H(e−) − H(ArO−) | (5) |
The wave function-based results, such as spin density, spin population, IRI, ESP, EDD, and LMO dipole moment, were obtained from the Multiwfn 3.8 program based on the corresponding output files.43 The reaction rate constants were calculated based on TS theory using the KiSThelP 2021 program,25 and the results considered the one-dimensional quantum mechanical tunneling effect through the Wigner correction.44
Antioxidant colorimetric assays
In all antioxidant colorimetric assays involved in this paper, the flavonoid and control solutions were prepared with methanol and fully dissolved by ultrasonic vibration at 40 °C.45The antioxidant colorimetric assays included DPPH˙-scavenging, ABTS˙+-scavenging, ˙OH-scavenging, and ˙O2− scavenging. The main chemical reagents in these assays were as follows: 0.1 M DPPH˙ methanol solution, 7.4 mM (NH4)2ABTS aqueous solution, 2.6 mM K2S2O8 aqueous solution, Fenton's reagent (40 mM H2O2 + 10 mM FeCl2·4H2O), 10 mM salicylic acid methanol solution, 0.05 M Tris-HCl buffer (pH 7.4), and 60 mM pyrogallol (7.5 mg in 1 mM HCl).
In the DPPH˙-scavenging assays, the prepared radical solution was treated with different samples and incubated in the dark at room temperature for 30 min, and the absorbance value was measured using a microplate reader (Multiskan FC, Thermo Fisher Scientific, Waltham, MA, USA) at 519 nm.45 The DPPH˙-scavenging percentage was calculated using eqn (6):
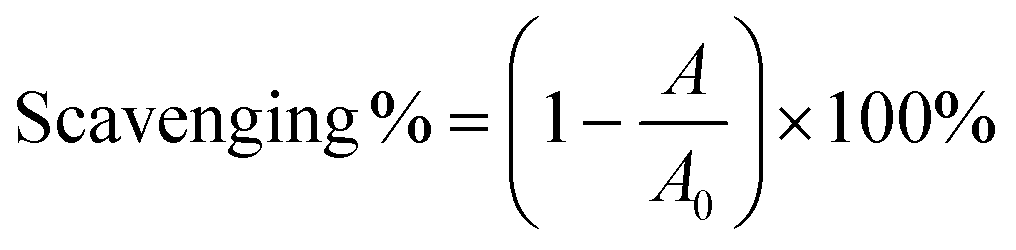 | (6) |
In the ABTS˙+-scavenging assays, the radical was produced by mixing 0.2 mL of (NH4)2ABTS solution with 0.2 mL of K2S2O8 solution. The mixture was incubated in the dark at 27 °C for 12 h to complete the radical generation process before dilution with distilled water (at a ratio of approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20). The ABTS˙+ solution was treated with different samples and incubated in the dark at 27 °C for 10 min, and the absorbance value was measured using a microplate reader at 734 nm.46 The ABTS˙+-scavenging percentage was calculated using eqn (6).
:20). The ABTS˙+ solution was treated with different samples and incubated in the dark at 27 °C for 10 min, and the absorbance value was measured using a microplate reader at 734 nm.46 The ABTS˙+-scavenging percentage was calculated using eqn (6).
The ˙OH-scavenging based on the salicylic acid chromogenic method was performed according to the method in Fig. 9, and the ˙OH-scavenging percentage was calculated using eqn (7):
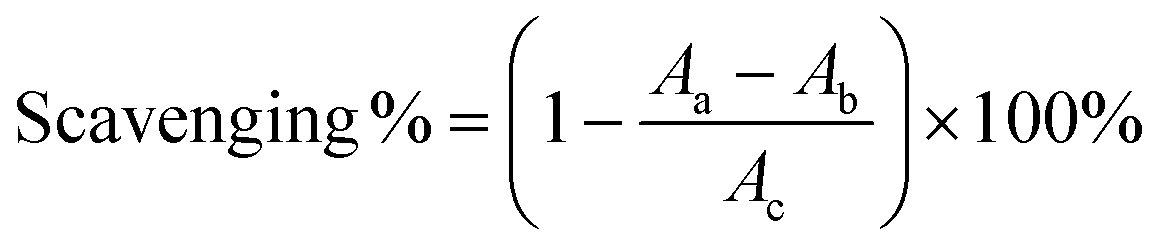 | (7) |
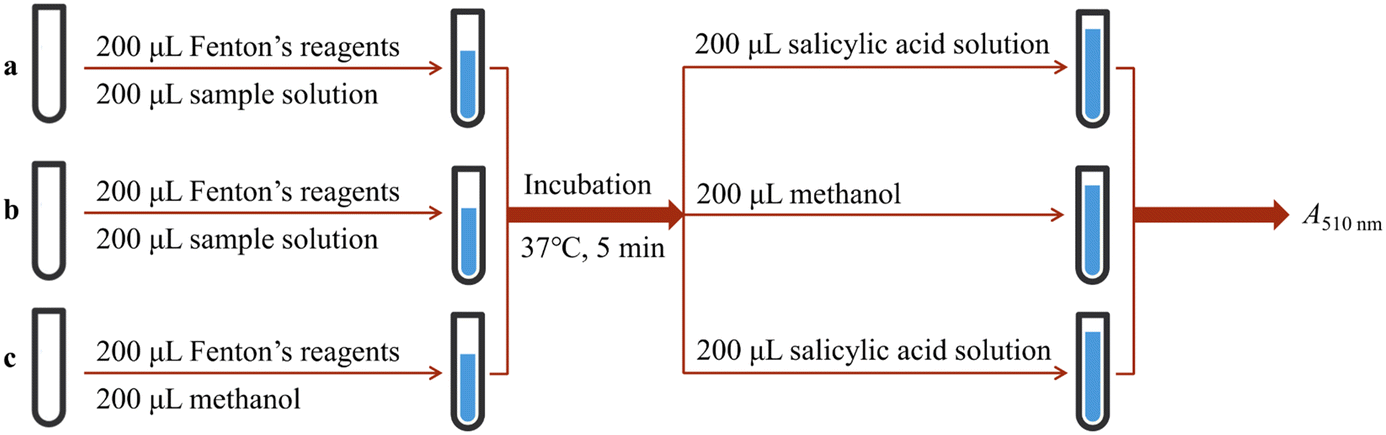 | ||
| Fig. 9 Experimental procedures for ˙OH-scavenging based on salicylic acid. The a–c represent different experimental groups. | ||
In the ˙O2−-scavenging assay, each sample was treated with pyrogallol in Tris-HCl buffer. The absorbance of the mixture at 325 nm was measured using a UV-vis spectrophotometer (Unico 2100, Unico, Shanghai, China) every 30 s for 5 min.31 The increased value (ΔA) was used to calculate the ˙O2− scavenging ability as seen in eqn (8):
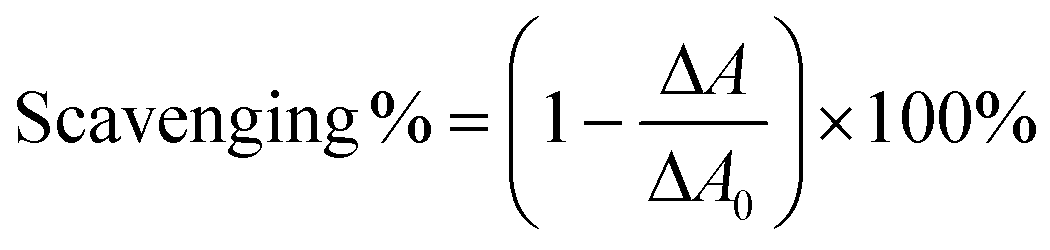 | (8) |
CCK-8 assay
The HT22 cells were cultured in DMEM supplemented with 10% FBS at 37 °C in 5% CO2 and were dissociated at a 1:3 ratio by 2.5% trypsin when the growth reached 90–95% confluence. The cells were seeded into 96-well plates at a density of 1 × 104 cells per well and were adhered for 24 h. Then, the cells were incubated with 5 μL of sample (for the sample group) or medium (for the model group) to a final concentration of 50 μM. After incubation for another 24 h, 100 μL of Fenton reagent (200 μM H2O2 + 200 μM FeCl2) was added to each well and incubated for 24 h. After the culture medium was discarded, 100 μL of CCK-8 was added to each well and incubated for 2 h. The survival was calculated according to the A450 nm values. Each sample test was repeated in five independent wells.
UHPLC-Q-Orbitrap-MS/MS analysis of DPPH˙ reaction products with flavonoids
The methanol solutions of each flavonoid and DPPH˙ were mixed with a molar ratio of 1:4, and the resulting mixture was incubated for 12 h in the dark at room temperature. Subsequently, the mixture was passed through a 0.22 μm filter for UHPLC-Q-Orbitrap-MS/MS analysis. The chromatography was performed on a C18 column (2.1 mm i.d. × 100 mm, 1.6 μm, Phenomenex Inc., Torrance, CA, USA) with a gradient elution of mobile phase A (methanol) and phase B (0.1% formic acid water) at a flow rate of 0.50 mL min−1, and the column temperature was maintained at 45 °C. The injection volume was 5 μL, and the gradient elution program was as follows: 0–2 min, maintained at 30% B; 2–10 min, 30 → 0% B; 10–12 min, 0 → 30% B.
The chromatography was combined with Quadrupole/Orbitrap tandem mass spectrometry (Thermo Fisher Scientific, Waltham, MA, USA). The MS detection was performed in negative electrospray ionization mode. The MS/MS spectra were obtained in the m/z range of 60–900. The final optimized mass parameters were set as follows: ion spray voltage, −4500 V; ion source heater temperature, 450 °C; curtain gas pressure, 30 psi; nebulizing gas pressure, 50 psi; Tis gas pressure, 50 psi. The declustering potential was set at −100 V, whereas the collision energy was set at −45 V with a collision energy spread of 15 V.
Statistical analysis
The IC50 value was defined as the final concentration of 50% radical scavenging and calculated via the dose–response curves which were plotted using Origin 2017 (OriginLab, Northampton, MA, USA). The Shapiro–Wilk test was used to verify whether the data conformed to the normal distribution, and the measurement data conforming to a normal distribution were described as the mean ± SD. The significance of data differences was analyzed by one-way analysis of variance using SPSS 26.0 software (SPSS Inc., Chicago, IL, USA), with the statistical significance set at P < 0.05.Conclusions
The 4′,5,7-trihydroxyflavonoids with different structural types exert their antioxidant ability through the same mechanisms. Specifically, in water and methanol, they can undergo HAT, ET-based, and PT-based mechanisms to scavenge neutral radicals, radical cations, and radical anions, respectively. However, when C3 is substituted by hydroxy to form a 4′,5,7-trihydroxyflavonol, the C3–OH will be the active site and promote the antioxidant activity and speed; when the molecular skeleton transferred to other structural types of 4′,5,7-trihydroxyflavonoids, the antioxidant activity and speed will also change. Such a structure–activity relationship is mainly related to the structural parameters, conjugation level, and activation energy. After the C4′–OH 4′,5,7-trihydroxyflavonoid loses an H atom, the unsubstituted C3′ of flavones, isoflavones, and flavanones can link free radicals to produce adducts, while flavonol-radical adducts cannot exist stably because of steric hindrance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Natural Science Foundation of Guangdong Province (2017A030312009) and the Scientific Research Project of Department of Education of Hubei Province (B2017047).References
- J. C. Wu, Y. Zhao, K. Li, S. Muhammad, M. Z. Ju, L. H. Liu, Y. B. Huang, B. B. Wang, W. J. Ding, B. X. Shen and H. Huang, TrAC, Trends Anal. Chem., 2022, 157, 116734 CrossRef CAS.
- M. W. Chen, Q. J. Lu, Y. J. Chen, Y. K. Hou, Y. M. Zou, Q. Zhou, W. H. Zhang, L. X. Yuan and J. X. Chen, ACS Biomater. Sci. Eng., 2022, 8, 3361–3376 CrossRef CAS.
- S. Wang, J. W. Zhang, W. Li, D. L. Chen, J. S. Tu, C. M. Sun and Y. A. Du, Carbohydr. Polym., 2022, 296, 119940 CrossRef CAS.
- R. Q. Alkhatib, A. B. Almasarweh, N. M. Abdo, A. S. Mayyas, M. A. Al-Qudah and S. T. Abu-Orabi, Int. J. Food Prop., 2022, 25, 1920–1933 CrossRef CAS.
- H. F. Al-Sayyed, R. A. Al-Kurd, I. F. Mahmoud, S. M. AbdelQader, D. H. Sweidan, L. T. Rizeq, T. A. Arafat and M. M. Mwalla, Int. J. Food Prop., 2022, 25, 1290–1301 CrossRef CAS.
- M. Y. Cao, J. Wu, C. Q. Xie, L. Wu, Z. Gu, J. W. Hu and W. Xiong, Food Agric. Immunol., 2022, 33, 511–529 CrossRef CAS.
- C. J. Shang, Y. J. Zhang, C. F. Sun and L. L. Wang, J. Mol. Liq., 2022, 362, 119748 CrossRef CAS.
- H. Boulebd, Phytochemistry, 2021, 184, 112670 CrossRef CAS PubMed.
- M. S. El-Shall, Y. M. Ibrahim, E. H. Alsharaeh, M. Meot-Ner and S. P. Watson, J. Am. Chem. Soc., 2009, 131, 10066–10076 CrossRef CAS PubMed.
- J. F. Li, Y. Qin, X. L. Yu, Z. X. Xiong, L. F. Zheng, Y. Sun, J. R. Shen, N. S. Guo, L. Tao, Z. Y. Deng and X. R. Liu, J. Food Biochem., 2019, 43, e12654 CrossRef PubMed.
- X. C. Li, J. Y. Zeng, Y. P. Liu, M. S. Liang, Q. R. Liu, Z. Li, X. J. Zhao and D. F. Chen, Antioxidants, 2020, 9, 205 CrossRef CAS PubMed.
- A. Benayahoum, S. Bouakkaz, T. Bordjiba and M. Abdaoui, J. Mol. Liq., 2019, 275, 221–231 CrossRef CAS.
- Y. Xue, Y. Zheng, L. An, Y. Dou and Y. Liu, Food Chem., 2014, 151, 198–206 CrossRef CAS PubMed.
- A. Maria Mendoza-Wilson, S. Ivan Castro-Arredondo and R. Renato Balandran-Quintana, Food Chem., 2014, 161, 155–161 CrossRef.
- T. Lu and Q. Chen, Chemistry-Methods, 2021, 1, 231–239 CrossRef CAS.
- J. Luzon, J. Campo, F. Palacio, G. J. McIntyre, J. M. Rawson, R. J. Less, C. M. Pask, A. Alberola, R. D. Farley, D. M. Murphy and A. E. Goeta, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 144429 CrossRef.
- H. Nakata, D. G. Fedorov, K. Kitaura and S. Nakamura, Chem. Phys. Lett., 2015, 635, 86–92 CrossRef CAS.
- J. Zhang and T. Lu, Phys. Chem. Chem. Phys., 2021, 23, 20323–20328 RSC.
- T. Lu and F. Chen, J. Mol. Graphics Modell., 2012, 38, 314–323 CrossRef CAS.
- E. Hosseinzadeh, N. L. Hadipour and G. Parsafar, J. Photochem. Photobiol., A, 2017, 333, 70–78 CrossRef CAS.
- J. S. Cao, M. J. Wei and F. W. Chen, Acta Phys.-Chim. Sin., 2016, 32, 1639–1648 CAS.
- J. Pipek and P. G. Mezey, J. Chem. Phys., 1989, 90, 4916–4926 CrossRef CAS.
- J. W. Boughton and P. Pulay, J. Comput. Chem., 1993, 14, 736–740 CrossRef CAS.
- L. P. Viegas, Chem. Phys. Lett., 2022, 803, 139829 CrossRef CAS.
- S. Canneaux, F. Bohr and E. Henon, J. Comput. Chem., 2014, 35, 82–93 CrossRef CAS PubMed.
- A. Floegel, D. O. Kim, S. J. Chung and O. K. Chun, FASEB J., 2010, 24, 535.9-535.9 CrossRef.
- H. Dinh Quy, B. Mai Van and N. Pham Cam, J. Mol. Liq., 2021, 340, 117149 CrossRef.
- A. A. Shanty and P. V. Mohanan, Spectrochim. Acta, Part A, 2018, 192, 181–187 CrossRef CAS PubMed.
- J. Treml and K. Smejkal, Compr. Rev. Food Sci. Food Saf., 2016, 15, 720–738 CrossRef CAS.
- Q. Yi, J. Ji, B. Shen, C. Dong, J. Liu, J. Zhang and M. Xing, Environ. Sci. Technol., 2019, 53, 9725–9733 CrossRef CAS PubMed.
- X. Li, J. Agric. Food Chem., 2012, 60, 6418–6424 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. Nakatsuji, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. F. Peintinger, D. V. Oliveira and T. Bredow, J. Comput. Chem., 2013, 34, 451–459 CrossRef CAS PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView, Version 6, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- Z. Markovic, J. Tosovic, D. Milenkovic and S. Markovic, Comput. Theor. Chem., 2016, 1077, 11–17 CrossRef CAS.
- J. Rimarčík, V. Lukeš, E. Klein and M. Ilčin, J. Mol. Struct.: THEOCHEM, 2010, 952, 25–30 CrossRef.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- E. Wigner, Z. Phys. Chem. B, 1932, 19, 203–216 Search PubMed.
- X. Li, ChemistrySelect, 2018, 3, 13081–13086 CrossRef CAS.
- G. Wang, X. Li and H. Zeng, Acta Chim. Sin., 2009, 67, 974–982 CAS.
- J. Yao, Y. Cheng, M. Zhou, S. Zhao, S. Lin, X. Wang, J. Wu, S. Li and H. Wei, Chem. Sci., 2018, 9, 2927–2933 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1, Fig. S1–S6. See DOI: https://doi.org/10.1039/d2md00333c |
| This journal is © The Royal Society of Chemistry 2023 |