
Novel quinoline-piperazine hybrids: the design, synthesis and evaluation of antibacterial and antituberculosis properties†
Karunanidhi
Gnanavelu
ab,
Vinay Kumar
K. S.
a,
Sumesh
Eswaran
 *a and
Karthikeyan
Sivashanmugam
*b
*a and
Karthikeyan
Sivashanmugam
*b
aAnthem Biosciences Pvt. Ltd., #49, Bommasandra Industrial Area, Bommasandra, Bangalore 560099, Karnataka, India. E-mail: sumesh.e@anthembio.com
bSchool of Bio-Sciences and Technology, Vellore Institute of Technology, Vellore, Tamil Nadu 632014, India
First published on 1st November 2022
Abstract
A communicable disease such as tuberculosis (TB), which takes ∼10 million lives worldwide every year, is one of the major concerns for future generations. The intake of multiple antibiotics is increasing because of the emergence of multiple drug-resistant TB (MDR-TB) to pathogens which do not respond to the first-line TB drugs. Even though numerous drugs are available on the market, there is a huge need for MDR-TB drugs. Herein, our emphasis was to synthesise a series of 2,4,6-substituted quinoline conjugated piperazine coupled sulfonamides, as well as amides, and to study and evaluate their in vitro antibacterial activity against both susceptible and resistant pathogens of Gram-positive and Gram-negative bacteria. Furthermore, their antituberculosis activity was assessed against non-virulent, virulent and MDR pathogens. Few compounds displayed inhibitory activity against bacterial growth, but two compounds displayed significant inhibitory activity against all the TB strains (lowest MIC of 10g is 0.07 μM and 11e is 1.1 μM), which are more effective than other 1st line and 2nd line TB drugs. These two compounds are less cytotoxic, and could be developed as antibiotics or MDR-TB drugs by improving their hydrophilicity.
Introduction
Communicable diseases are the prime reason of concern for future generations. The infection rate is increasing as a result of new pathogens and threatening human health. Statistically, about 58% of deaths reported were due to infectious diseases in the USA during the timeframe of 1980 to 1992. Among these deaths, half were due to bacterial pathogens.1 This was also evident from the increased (up to 65%, 34.8 billion defined daily doses, DDDs) use of antibiotics worldwide during 2000 to 2015.2 In particular, low and middle income countries (LMICs) are leading an increase in the consumption of antibiotics used against antimicrobial resistance (AMR) or multi drug resistance (MDR). Increase in the rate of AMR/MDR in the developing countries is because of conditions such as lack of awareness, sanitation, and inadequate funding to prevent infections.3 In 2016, the UK reported that 0.7 million people lost their lives every year due to AMR, and estimated that this would increase to 10 million deaths by 2050.4 Among the LMICs, India is one of the major antibiotic consumers2 with major bacterial infections caused by Escherichia coli (8.4% to 92.9%), Klebsiella pneumoniae (4.1% to 79.4%) and Mycobacterium tuberculosis.5 A survey of antimicrobial susceptibility test (AST) results in India showed a 13% mortality burden due to MDR and this topic remains largely unstudied.6Tuberculosis (TB) caused by the bacillus M. tuberculosis remains the microbial disease of main concern which is still the leading killer in the world, ranking higher than HIV/AIDS until the COVID-19 (coronavirus) pandemic.5 From the last five years of surveys conducted by WHO, 30 high-mortality burden TB countries are listed which cover about 84% of the global incidence.3 In 2019, TB cases were estimated at about 9–10 million worldwide, and an average of 50% global TB cases were shared by India (27%), China (14%) and Russia (8%). As per the WHO tuberculosis assessment report, TB alone accounted for 1.5 million deaths and India alone accounted for ∼30% of these deaths.3 The concern of health ministries all over the world increases every year as the infection numbers keep proliferating to as high as 10 million people every year.7
The appearance of multiple drug-resistant TB (MDR-TB),8 which does not respond to the first-line TB drugs, has posed a serious threat to controlling and treating TB. Between 2005 and 2006, 53 out of 221 people (23%) died from MDR-TB due to the failure of the second line defence TB drugs and this outbreak was named as extensively drug-resistant (XDR-TB).8 The effect of MDR-TB on HIV infected people is a leading cause of death which takes about 0.2 million people's lives every year.7 Recently, the COVID-19 pandemic in 2020 has severely impacted the mortality of TB infected patients.9 This prompted WHO to declare TB an emergency in global health which was aimed to save ∼30 million people during 2018–2022.5,7
The alarming signals arising from the previous facts prompted an urgent need for the development of new drugs to combat the microbial mechanism of action, which would possibly be different from the existing ones.10 Based on currently available drugs, the quinolone nucleus has gained the majority of the attention.11 The wide range of drugs available with this heterocyclic core in antibiotics field such as nalidixic acid, clinafloxacin, gemifloxacin and many others.11,12 In addition to this, the majority of quinolone conjugated piperazines are marketed as antibiotics such as sparfloxacin, gatifloxacin, grepafloxacin, lomefloxacin and norfloxacin; and ciprofloxacin and levofloxacin are used as antibacterial agents against resistant strains and also used to treat TB (Fig. 1).11–13 However, quinoline derivatives are also used to treat TB, and, more significantly, quinoline containing bedaquiline is a drug used against MDR-TB and XDR-TB as an ATP synthase inhibitor.14 Similarly, quinoline derivatives are well known for use as antimalarial agents.15 Recently, quinoline-piperazine derivatives have been reported being used as anti-cancer agents16 and have enzyme metabolic inhibition activity.17 Furthermore, PF-622, a drug developed as a fatty acid amide hydrolase (FAAH) inhibitor, has a quinoline conjugated piperazine amide (Fig. 1) which acted as an important molecular framework18 and these moieties are least focused on for determining antimicrobial or antituberculosis activities.
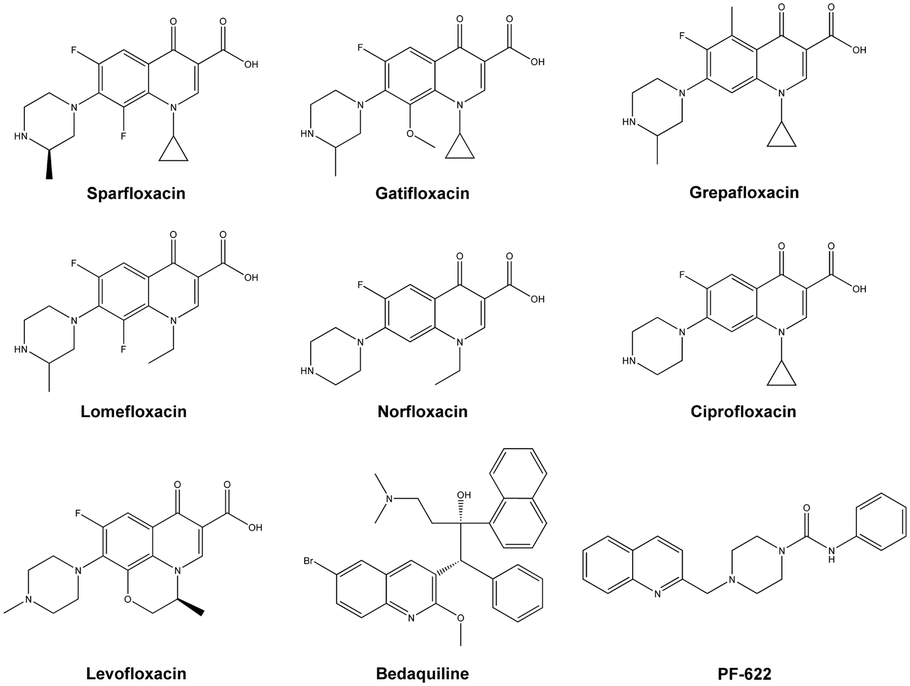 | ||
| Fig. 1 Quinolone and quinoline bearing antibiotic, anti-tuberculosis and FAAH inhibitor drugs. | ||
In general, the mode of action of quinolone drugs is via bacterial DNA destruction and they are becoming unable to function properly against MDR-TB. The overall effect of structural modification on quinolones helps researchers to understand how microbes become resistant to the mode of action of the drugs which is described comprehensively.1 In recent papers in the literature, the structure activity relationship of quinolones suggested that the 3rd and 4th positions should be unaltered to avoid a significant loss of biological activity.1 Because most of the pathogens developed resistance against the available quinolone TB drugs, we recommended modifying the main core quinolone to quinoline by not altering the 3rd position and then synthesised 27 novel quinoline conjugated piperazine coupled sulfonamide and benzamide hybrids to screen against AMR and MDR-TB pathogens.
Results and discussion
On the basis of these observations and as part of our industrial drug discovery programme to develop new antibacterial19,20 and antitubercular agents,21–23 we designed novel quinoline conjugated piperazine derivatives, wherein active pharmacophores, namely, piperazine coupled sulfonamides and amides, have been introduced at the 2nd position of the quinoline ring containing active fluoro and methoxy groups at the 6th position and improved biological activity was anticipated.Chemical synthesis
The reaction sequence employed for synthesis of the title compounds (7a and 7b) is shown in Scheme 1. The starting material, a 4-substituted aniline (1a–1b), was conveniently converted to 3-(4-substituted-phenylamino)-but-2-enoic acid ethyl ester (2a–2b) by heating with ethyl acetoacetate in the presence of a catalytic amount of p-toluene sulfonic acid (p-TsOH) in toluene at 110 °C for 8 h and a good yield was obtained.24 The compound 2a–2b was cyclised using polyphosphoric acid and a catalytic amount POCl3, as per a previously reported protocol, to give the 4-hydroxy quinoline derivatives 3a–3b at 75 °C.25 In addition, compounds 3a–3b were alkylated with dimethyl sulfate in toluene under the reflux temperature to give substituted 2-methylquinoline (4a–4b) with a good yield.26 The compounds obtained, 4a–4b, were brominated using NBS and AIBN in acetonitrile to give 5a–5bvia a modified protocol with an improved yield of about 20% as against the reported 52% yield.27 Intermediates 5a–5b were coupled with N-Boc piperazine in DMF in the presence of potassium carbonate (K2CO3) at 60 °C and produced Boc-protected 6a–6b. The Boc group of 6a–6b was cleaved with trifluoroacetic acid (TFA) in dichloromethane (DCM) at 0 °C to give a key intermediate, 6-substituted-4-methoxy-2-[(piperazin-1-yl)methyl]-quinolines (7a–7b), as a TFA salt (Scheme 2).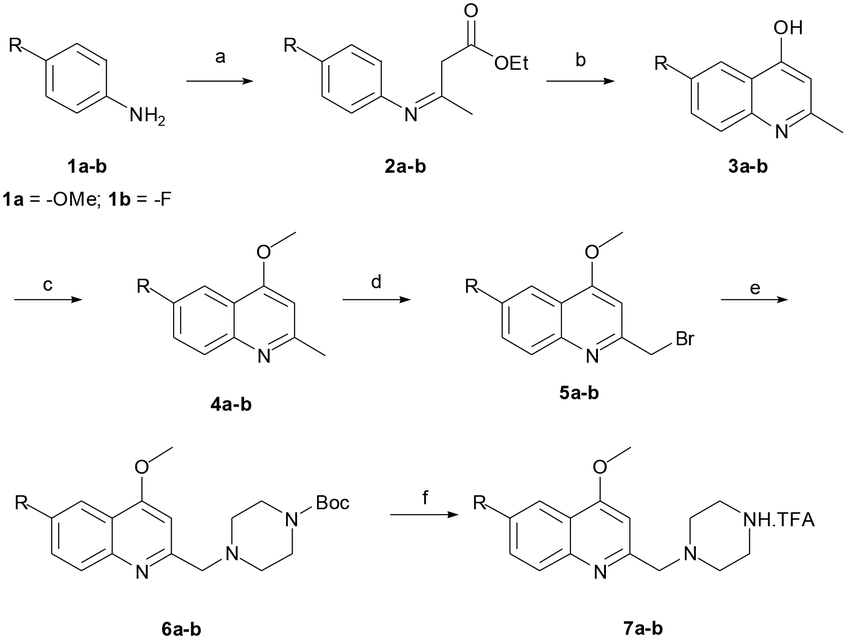 | ||
| Scheme 1 Synthesis of 6-substituted-4-methoxy-2-[(piperazin-1-yl)methyl]quinoline trifluoroacetic acid salt (7a–7b). Reagents and conditions: (a) ethyl acetoacetate, p-TsOH, toluene, 110 °C, 8 h, (b) PPA, POCl3, 75 °C, 4 h, (c) Me2SO4, toluene, 110 °C, 8 h, (d) NBS, AIBN, MeCN, 65 °C, 12 h, (e) Boc-piperazine, K2CO3, DMF, 60 °C, 6 h, and (f) TFA, DCM, 0 °C to 25 °C, 2 h. | ||
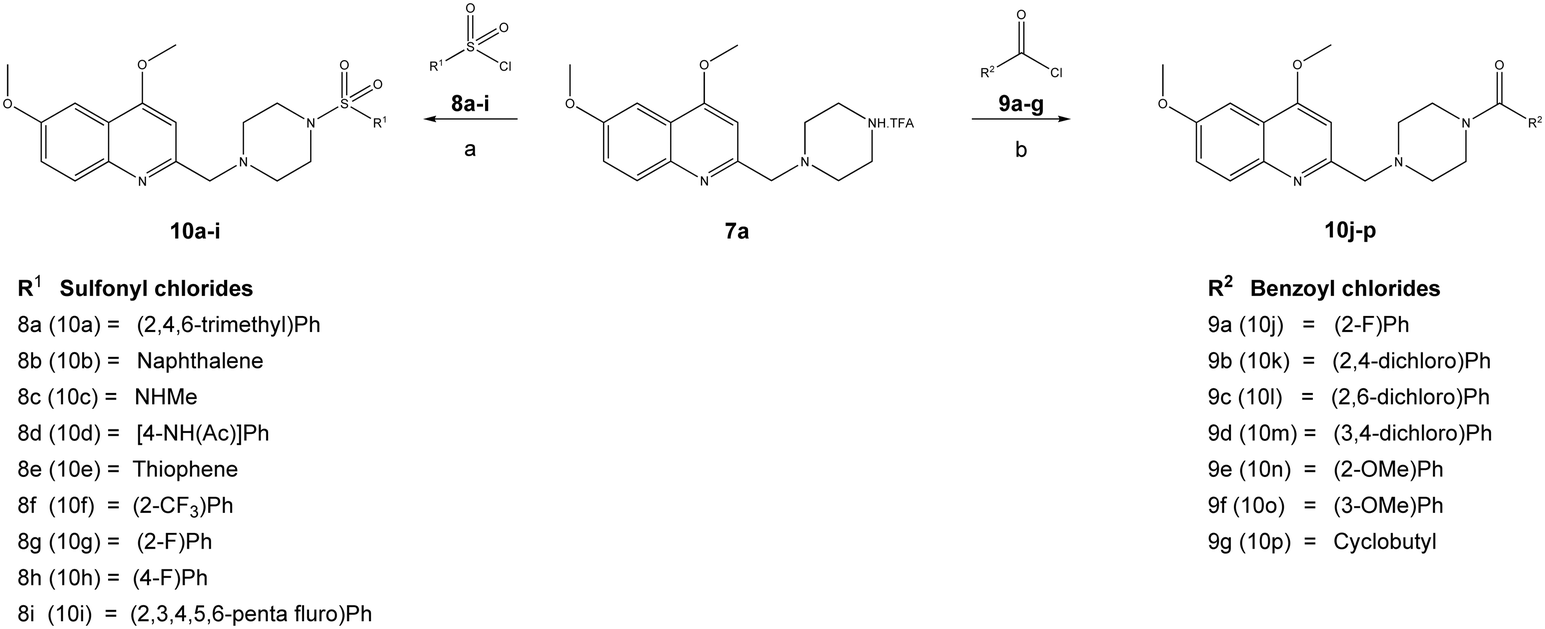 | ||
| Scheme 2 Synthesis of 4-(4,6-dimethoxy-quinolin-2-ylmethyl)-piperazine-1-sulfonamide (10a–10i) and 4-(4,6-dimethoxy-quinolin-2-ylmethyl)-piperazine-1-amide (10j–10p) derivatives. Reagents and conditions: (a) DIPEA, DCM, various sulfonyl chlorides (8a–8i), 0–10 °C, 8 h, and (b) Et3N, DCM, various acid chlorides (9a–9g), 0–10 °C, 4 h. | ||
Furthermore, the TFA salt of quinoline conjugated piperazine derivatives (7a–7b) were reacted with sulfonyl chlorides and acid chlorides in the presence of Hünig's base to yield the corresponding quinoline-piperazine hybrids of sulfonamides 10a–10i and 11a–11f and then amides 10j–10p and 11g–11k (Scheme 3).
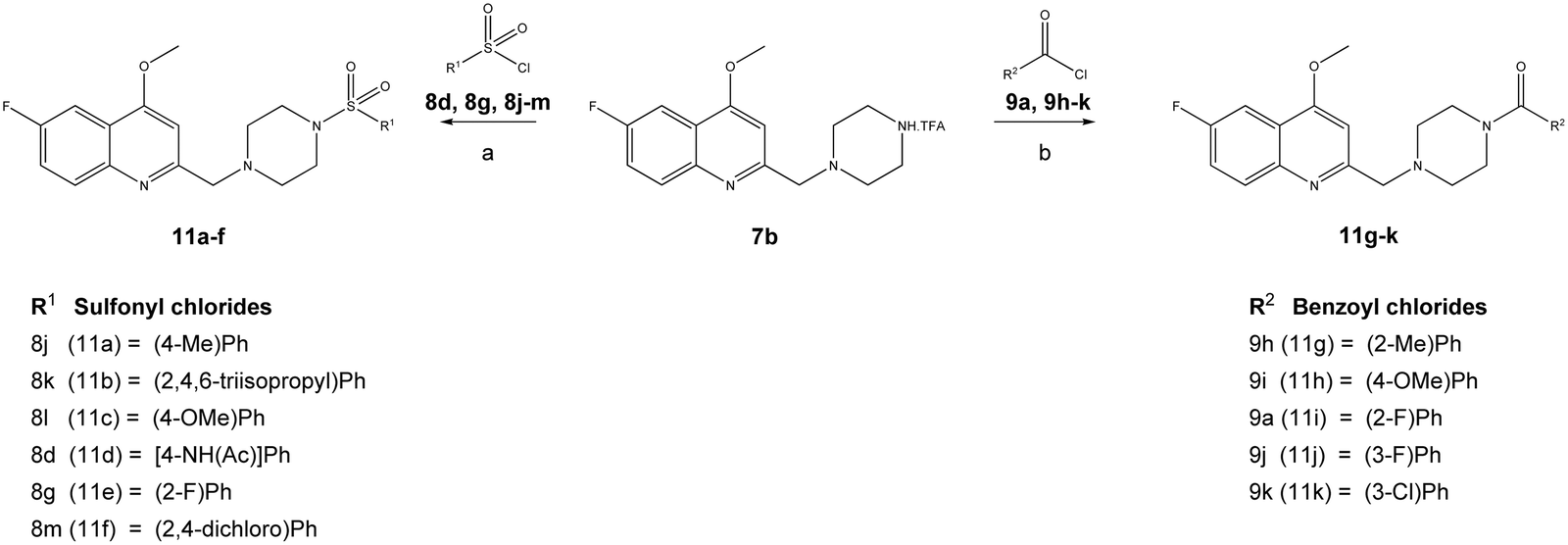 | ||
| Scheme 3 Synthesis of [4-(6-fluoro-4-methoxy-quinolin-2-ylmethyl)-piperazine-1-sulfonyl]-amide (11a–11f) and 4-(6-fluoro-4-methoxy-quinolin-2-ylmethyl)-piperazine-1-amide (11g–11k) derivatives. Reagents and conditions: (a) DIPEA, DCM, various sulfonyl chlorides (8d, 8g, 8j–8m), 0–10 °C, 8 h, and (b) DIPEA, DCM, various benzoyl chlorides (9a, 9h–9k), 0–10 °C, 4 h. | ||
Comprehensive antibacterial profile of the novel compounds
The newly synthesised compounds 10a–10p and 11a–11k were screened for their in vitro antibacterial activity against Gram-positive pathogens such as methicillin-susceptible Staphylococcus aureus (MSSA) (ATCC 25923), methicillin-resistant Staphylococcus aureus (MRSA) (ATCC 33591), methicillin-resistant Staphylococcus epidermidis (clinical MRSE) (AB 735), vancomycin-resistant Enterococci faecalis (VREF) (ATCC 700802) and Enterococci faecium (ATCC 49624); and Gram-negative pathogens such as susceptible E. coli (AB 117), resistant clinical strains of K. pneumoniae (AB 356), Pseudomonas aeruginosa (AB 645), Acinetobacter baumannii (BAA 747) and Moraxella catarrhalis (ATCC 25238). Because M. tuberculosis has both Gram-positive and Gram-negative bacterial characteristics,28 we preferred to use both Gram-positive and Gram-negative pathogens to test against the compounds.Antimicrobial susceptibility testing (AST) was done as reported previously29 by determining the minimum inhibitory concentration (MIC) of antimicrobial agents which was done using sub-cultured soybean casein digest agar medium (SCDA). The culture turbidity was adjusted as per McFarland's standard (∼108 CFU mL−1) which was further diluted to ∼105 CFU mL−1. The concentration used for screening the title compounds, with serial dilution, was from 64 μg mL−1 to 0.03 μg mL−1. The drugs linezolid, trimethoprim and ciprofloxacin were used as control standards with concentrations ranging from 64 μg mL−1 to 0.03 μg mL−1. The triplicate assessment results (Table 1) were read after 22–24 h of incubation at 37 °C. The MIC data suggested that the 4,6-dimethoxy quinoline piperazine coupled sulfonamide having a fluorine at the 2nd position on the benzene ring (10g) showed excellent activity against almost all the bacteria, a lower MIC of 0.03 μg mL−1 against S. aureus and an MIC of 0.06 μg mL−1 against M. catarrhalis were observed which were comparable with the standard drugs used as controls. Similarly, trifluorocarbon substitution at the 2nd position (10f) on the benzene ring showed moderately good results against Gram-positive bacteria but it significantly inhibited the growth of the Gram-negative pathogen M. catarrhalis (MIC, 1 μg mL−1). However, 4-methoxy-6-fluro-quinoline piperazine coupled sulfonamide having a fluorine at the 2nd position on the benzene ring (11e) showed excellent activity against almost all the bacteria and a lower MIC of 0.03 μg mL−1 against M. catarrhalis was observed. Likewise, methyl substitution at the 4th position on the benzene ring (11a) and N-acetyl substitution at the 4th position on the benzene ring (11d) showed average bacterial inhibition. The rest of the compounds tested failed to restrict the growth of pathogens and none of our compounds failed to stop the evolution of K. pneumonia, P. aeruginosa and A. baumannii. It was strange that the quinoline conjugated piperazine amides/benzamides (10j–10p and 11g–11k) were inactive against all the pathogens even at a higher concentration (64 μg mL−1) and it was evident that sulfonamide derivatives were more biologically active than amides. Overall, we observed that good activity was attributed to the presence of fluoro (10g and 11e) or trifluorocarbon (11f) substitution on the aryl group at the 2nd position. These encouraging results from the antibacterial studies impelled us to go on to the preliminary screening of the title compounds for in vitro anti-TB activity.
| Compounds | Gram-positive panel MIC (μg mL−1) | Gram-negative panel MIC (μg mL−1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| S. aureus (MSSA) ATCC 29213 | S. aureus (MRSA) ATCC 33591 | S. epidermidis (clinical – MRSE) AB 735 | E. faecalis (VREF) ATCC 700802 | E. faecium ATCC 49624 | E. coli (clinical – ESBL) AB 117 | K. pneumoniae (clinical) AB 356 | P. aeruginosa (clinical) AB 645 | A. baumannii (clinical) BAA 747 | M. catarrhalis ATCC 25238 | |
| a Susceptible bacterial strains. b Resistant bacterial strains. c MIC values >64 μg mL−1 refers to no inhibition of bacterial growth. | ||||||||||
| 10a | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 10b | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 10c | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 10d | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 10e | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 10f | 4 | 32 | 16 | 64 | 8 | 16 | >64 | >64 | >64 | 1 |
| 10g | 0.03 | 8 | 32 | 1 | 2 | 8 | >64 | >64 | >64 | 0.06 |
| 10h | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 10i | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 10j | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 10k | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 10l | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 10m | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | 64 |
| 10n | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 10o | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 10p | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | 2 |
| 11a | 64 | >64 | 32 | >64 | >64 | >64 | >64 | >64 | >64 | 16 |
| 11b | >64 | >64 | >64 | >64 | >64 | >64 | >64 | 64 | 64 | 64 |
| 11c | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 11d | >64 | >64 | 64 | >64 | >64 | >64 | >64 | >64 | >64 | 32 |
| 11e | 0.5 | 0.06 | 16 | 2 | 0.5 | 16 | >64 | >64 | >64 | 0.03 |
| 11f | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 11g | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 11h | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 11i | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 11j | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 11k | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | 64 |
| Linezolid | 2 | 4 | 8 | 4 | 4 | 8 | >64 | >64 | >64 | 4 |
| Trimethoprim | 4 | 8 | >8 | 0.25 | 0.03 | 16 | 2 | >8 | >8 | 4 |
| Ciprofloxacin | 0.5 | 1 | 4 | 0.5 | 2 | 8 | 0.125 | >8 | 0.5 | 0.03 |
Anti-tuberculosis studies
The title compounds were evaluated against non-virulent Micobacterium smegmatis (ATCC 607) using a broth micro dilution method as per the Clinical and Laboratory Standards Institute (CLSI) standards and adopting the protocol previously reported for anti-TB screening.29–31The compounds were dissolved in DMSO at a concentration of 4.0 mg mL−1 (1 mL DMSO was used for dissolution). Middlebrook 7H9 broth with 10% albumin, dextrose, and catalase (ADC) and the test solutions were prepared together at a higher concentration of 64 μg mL−1 followed by serial dilutions up to 0.015 μg mL−1 (147 μM to 0.04 μM). The plates were inoculated with M. smegmatis (∼105 CFU mL−1) and this was verified by 10-fold diluted inoculum on Middlebrook 7H11 agar plates (four weeks incubation at 37 °C). The first line TB drug, rifampicin, and the second line TB drug, levofloxacin, served as positive controls and a drug free broth containing DMSO served as a negative control, respectively. The bacterial growth inhibition was recorded after 2 d of incubation at 37 °C because it had a fast growing ability and the observed MICs are shown in Table 2. Likewise, the title compounds were screened against virulent M. tuberculosis H37Rv (ATCC 25618) and multi-drug resistant M. tuberculosis H37Rv (MDR-TB) (ATCC 35822) as per the previous protocol, except that the MIC reading was taken after 21 d (Table 2) due to their delayed growth.
| Compounds | MIC (μg mL−1) M. smegmatis (ATCC 607) | MIC (μg mL−1) M. tuberculosis H37Rv (ATCC 25618) | MIC (μg mL−1) M. tuberculosis H37Rv (MDR-TB) (ATCC 35822) |
|---|---|---|---|
| MIC >64 μg mL−1 refers to no inhibition of bacterial growth. | |||
| 10a | >64 | >64 | >64 |
| 10b | >64 | >64 | >64 |
| 10c | >64 | >64 | >64 |
| 10d | >64 | >64 | >64 |
| 10e | 32 | >64 | >64 |
| 10f | >64 | >64 | >64 |
| 10g | 0.031 | 0.063 | 0.5 |
| 10h | >64 | >64 | >64 |
| 10i | >64 | >64 | >64 |
| 10j | >64 | >64 | >64 |
| 10k | >64 | >64 | >64 |
| 10l | >64 | >64 | >64 |
| 10m | >64 | >64 | >64 |
| 10n | >64 | >64 | >64 |
| 10o | >64 | >64 | >64 |
| 10p | >64 | >64 | >64 |
| 11a | >64 | >64 | >64 |
| 11b | >64 | >64 | >64 |
| 11c | >64 | >64 | >64 |
| 11d | >64 | >64 | >64 |
| 11e | 0.5 | 1 | 4 |
| 11f | >64 | >64 | >64 |
| 11g | >64 | >64 | >64 |
| 11h | >64 | >64 | >64 |
| 11i | >64 | >64 | >64 |
| 11j | >64 | >64 | >64 |
| 11k | >64 | >64 | >64 |
| Levofloxacin | 0.125–0.25 | 2 | 4 |
| Rifampicin | 0.5 | 4 | >64 |
Compound 10g emerged as a potent inhibitory agent against all the bacterial strains. The lowest MIC values of 0.06 μg mL−1 (0.14 μM) and 0.5 μg mL−1 (1.1 μM) were observed against M. tuberculosis and MDR-TB, and 0.031 μg mL−1 (0.07 μM) for M. smegmatis which performed better than the 2nd line TB drug (levofloxacin, lowest MIC observed 0.35 μM). Compound 11e showed an activity equivalent to those of the control samples against all three strains (MIC of 0.5 μg mL−1 or 1.1 μM against M. smegmatis; 1 μg mL−1 (2.7 μM) against M. tuberculosis; and 4 μg mL−1 or 11 μM against MDR-TB) (Fig. 2). Interestingly, compound 10e had shown moderate activity against M. smegmatis but it failed to inhibit the growth of M. tuberculosis and MDR-TB which was confirmed by trials in triplicate. Compound 10e has a thiophene heterocycle which is connected to the piperazine of the quinoline hybrid, and the heterocyclic ring interaction with M. smegmatis might be the reason for the good activity. The rest of the title compounds were inactive against all the strains. Based on the data obtained, the 10g and 11e molecules could be developed as antibiotics and anti-TB drugs because they significantly inhibited the growth of non-virulent-TB, virulent-TB and MDR-TB and both Gram-positive/Gram-negative bacteria.
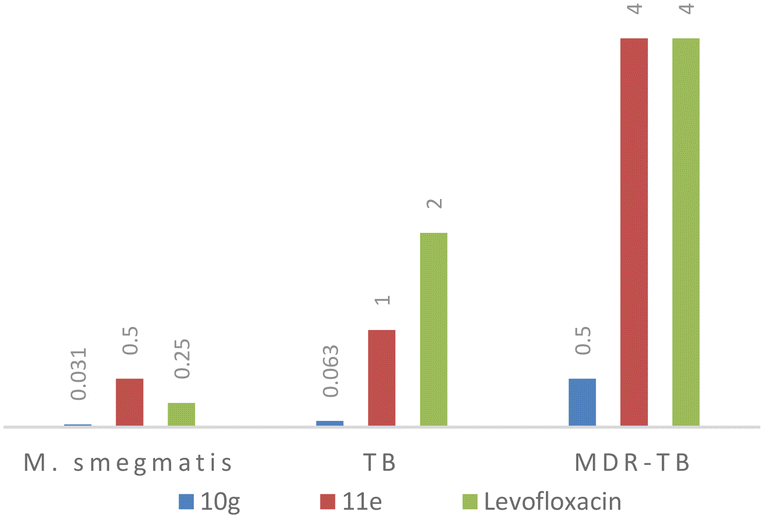 | ||
| Fig. 2 Comparison of the MIC data of active compounds (10g and 11e) with second line TB drug, levofloxacin. | ||
An in vitro cytotoxic assay was performed with potent compounds (10g and 11e) with concentrations ranging from 143 to 4.6 μM (64 to 1 μg mL−1, and DMSO was maintained as vehicle control) against HEK293 (normal human embryonic kidney cell line), HepG2 (human liver cancer cell line) and A549 (human lung carcinoma cell line). The active compounds showed zero inhibition up to 64 μg mL−1 against the HEK293 cell line. Compounds 10g and 11e did not inhibit the cell growth of HepG2 and A549 up to 32 μg mL−1 concentration, whereas minimal cytotoxic activity (<12%) was shown at a concentration of 64 μg mL−1 (Fig. 3).
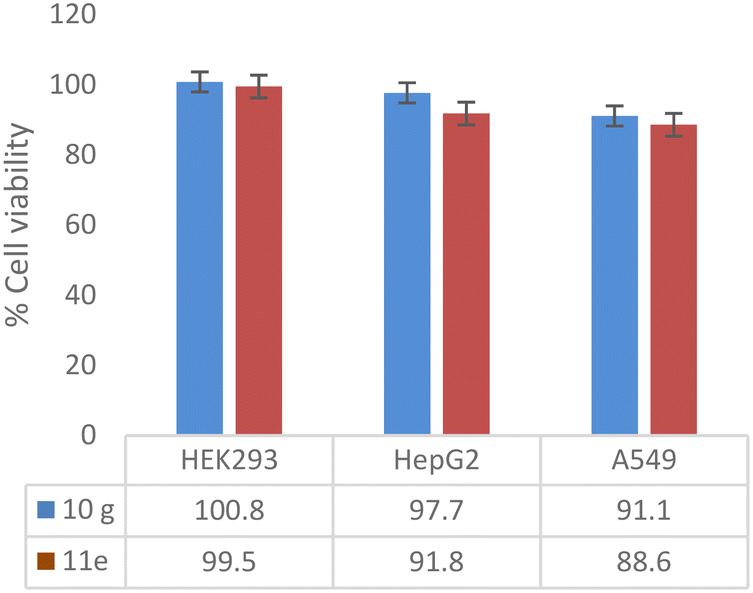 | ||
| Fig. 3 Cytotoxic assay of potent compounds (10g and 11e) at 143 μM concentration expressed as percentage cell viability of human cell lines. | ||
We had faced a solubility issue at higher concentrations (>64 μg mL−1) which was not acceptable for in vivo models and active pharmaceutical ingredient (API) development. Hence, we are currently working to enhance the solubility of active molecules by increasing their polarity/hydrophilicity (10e, 10g and 11e) by coupling glucose/polyethylene glycol/amino acids with the hydroxy group present on quinoline at the 4th position (3a–3b) and evaluating their biological activity. In addition, work is in progress to establish supplementary molecules with higher or efficient activity in multiple therapeutic areas.
Conclusion
The present research study covers the successful synthesis of novel 2,4,6-substituted quinoline conjugated piperazine coupled sulfonamides and amides. The sulfonamide-quinoline-piperazine hybrids were active in in vitro studies, and, in particular, the fluoro group at the 2nd position on the aryl group of sulfonamide emerged as potent inhibitors (10g and 11e) against M. tuberculosis, multi-drug resistant M. tuberculosis and M. smegmatis. The same compounds showed good activity against most of the Gram-positive and Gram-negative bacteria. These hybrids are less cytotoxic to human cells, and so they can be developed as antibiotics and MDR-TB drugs. Surprisingly, none of the amide-quinoline-piperazine hybrids were active against any of the pathogens studied.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to the management of Anthem Biosciences Pvt. Ltd., Bangalore, India for their invaluable support and allocation of resources for this work. The authors also wish to thank the analytical and molecular biology team of Anthem Biosciences for administering the biological activities.References
- L. R. Peterson, Clin. Infect. Dis., 2001, 33, S180–S186 CrossRef CAS.
- Y. Klein Eili, P. Van Boeckel Thomas, M. Martinez Elena, S. Pant, S. Gandra, A. Levin Simon, H. Goossens and R. Laxminarayan, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E3463–E3470 CAS.
- World Health Organization, Global tuberculosis report 2019, 2019 Search PubMed.
- O. N. Jim, Tackling drug-resistant infections globally: final report and recommendations, Government of the United Kingdom, 2016 Search PubMed.
- World Health Organization, Global antimicrobial resistance and use surveillance system (GLASS) report: 2021, Report 9240027335, 2021 Search PubMed.
- S. Gandra, K. K. Tseng, A. Arora, B. Bhowmik, M. L. Robinson, B. Panigrahi, R. Laxminarayan and E. Y. Klein, Clin. Infect. Dis., 2019, 69, 563–570 CrossRef PubMed.
- World Health Organization, Global tuberculosis report 2020, Report 9240016090, 2020 Search PubMed.
- L. Phillips, Nature, 2013, 493, 14–17 CrossRef PubMed.
- R. Fukunaga, P. Glaziou, J. B. Harris, A. Date, K. Floyd and T. Kasaeva, Morb. Mortal. Wkly. Rep., 2021, 70, 427–430 CrossRef CAS PubMed.
- S. K. Verma, R. Verma, S. Verma, Y. Vaishnav, S. P. Tiwari and K. P. Rakesh, Eur. J. Med. Chem., 2021, 209, 112886 CrossRef CAS.
- T. D. M. Pham, Z. M. Ziora and M. A. T. Blaskovich, MedChemComm, 2019, 10, 1719–1739 RSC.
- H. B. Rode, D. M. Lade, R. Grée, P. S. Mainkar and S. Chandrasekhar, Org. Biomol. Chem., 2019, 17, 5428–5459 RSC.
- S. Behera, P. Mohanty, R. Behura, B. Nath, A. K. Barick and B. R. Jali, Biointerface Res. Appl. Chem., 2021, 12, 6078–6092 Search PubMed.
- A. S. Pym, A. H. Diacon, S.-J. Tang, F. Conradie, M. Danilovits, C. Chuchottaworn, I. Vasilyeva, K. Andries, N. Bakare, T. De Marez, M. Haxaire-Theeuwes, N. Lounis, P. Meyvisch, B. Van Baelen, R. P. G. van Heeswijk and B. Dannemann, Eur. Respir. J., 2016, 47, 564–574 CrossRef CAS PubMed.
- M. Foley and L. Tilley, Pharmacol. Ther., 1998, 79, 55–87 CrossRef CAS.
- R. S. Viswas, S. Pundir and H. Lee, J. Enzyme Inhib. Med. Chem., 2019, 34, 620–630 CrossRef CAS.
- O. Çakmak, S. Ökten, D. Alımlı, C. C. Ersanlı, P. Taslimi and Ü. M. Koçyiğit, J. Mol. Struct., 2020, 1220, 128666 CrossRef.
- A. Richard, G. J. Breitenbucher, P. Kanaka, S. Mark and X. Wei, US Pat., WO2006/074025A1, 2006 Search PubMed.
- S. Eswaran, A. V. Adhikari, N. K. Pal and I. H. Chowdhury, Bioorg. Med. Chem. Lett., 2010, 20, 1040–1044 CrossRef CAS PubMed.
- S. Eswaran, A. V. Adhikari and N. S. Shetty, Eur. J. Med. Chem., 2009, 44, 4637–4647 CrossRef CAS.
- S. Eswaran, A. V. Adhikari and R. Ajay Kumar, Eur. J. Med. Chem., 2010, 45, 957–966 CrossRef CAS.
- S. Eswaran, A. V. Adhikari, I. H. Chowdhury, N. K. Pal and K. D. Thomas, Eur. J. Med. Chem., 2010, 45, 3374–3383 CrossRef CAS.
- K. D. Thomas, A. V. Adhikari, I. H. Chowdhury, T. Sandeep, R. Mahmood, B. Bhattacharya and E. Sumesh, Eur. J. Med. Chem., 2011, 46, 4834–4845 CrossRef CAS PubMed.
- D. Szamosvári, M. Prothiwa, C. L. Dieterich and T. Böttcher, Chem. Commun., 2020, 56, 6328–6331 RSC.
- B. Medapi, J. Renuka, S. Saxena, J. P. Sridevi, R. Medishetti, P. Kulkarni, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem., 2015, 23, 2062–2078 CrossRef CAS PubMed.
- G. Anukumari, M. A. Rao and P. Dubey, Asian J. Chem., 2015, 27, 2947–2950 CrossRef CAS.
- J. Li, H. Jin, H. Zhou, J. Rothfuss and Z. Tu, MedChemComm, 2013, 4, 443–449 RSC.
- L. M. Fu and C. S. Fu-Liu, Tuberculosis, 2002, 82, 85–90 CrossRef CAS PubMed.
- A. Shah, M. A. Rather, Q. P. Hassan, M. A. Aga, S. Mushtaq, A. M. Shah, A. Hussain, S. A. Baba and Z. Ahmad, J. Appl. Microbiol., 2017, 122, 1168–1176 CrossRef CAS.
- M. A. Rather, A. M. Lone, B. Teli, Z. S. Bhat, P. Singh, M. Maqbool, B. A. Shairgojray, M. J. Dar, S. Amin, S. K. Yousuf, B. A. Bhat and Z. Ahmad, MedChemComm, 2017, 8, 2133–2141 RSC.
- Z. S. Bhat, H. Ul Lah, M. A. Rather, M. Maqbool, T. Ara, Z. Ahmad and S. K. Yousuf, MedChemComm, 2018, 9, 165–172 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General synthetic procedure, compound characterisation and NMR data on all tested compounds are available. See DOI: https://doi.org/10.1039/d2md00260d |
| This journal is © The Royal Society of Chemistry 2023 |