
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and detonation performance of novel tetrazolyl–triazine nitrogen-rich energetic materials†
Paul
Richardson‡
 a,
Alexandros A.
Kitos‡
a,
Michael
Triglav
a,
Jeffrey S.
Ovens
a,
Isabelle
Laroche
b,
Stéphanie
Delisle
b,
Benoit
Jolicoeur
b,
Jaclyn L.
Brusso
*a and
Muralee
Murugesu
*a
a,
Alexandros A.
Kitos‡
a,
Michael
Triglav
a,
Jeffrey S.
Ovens
a,
Isabelle
Laroche
b,
Stéphanie
Delisle
b,
Benoit
Jolicoeur
b,
Jaclyn L.
Brusso
*a and
Muralee
Murugesu
*a
aDepartment of Chemistry and Biomolecular Sciences, University of Ottawa, 10 Marie Curie, Ottawa, Ontario K1N 6N5, Canada. E-mail: jbrusso@uottawa.ca; m.murugesu@uottawa.ca
bGeneral Dynamics Produits de défense et Systèmes tactiques-Canada, 5, Montée des Arsenaux, Repentigny, Québec, J5Z 2P4, Canada
First published on 26th October 2023
Abstract
The properties of energetic materials (EMs) are significantly influenced by specific features of their components and including multiple nitrogen-rich (N-rich) heterocycles within a single rigid framework is perhaps one of the most impactful and modern techniques employed in the design and development of novel high-performing explosives. In this regard, coupling tetrazole and s-triazine moieties is an attractive approach given their high nitrogen content and heats of formation resulting from the multiple N–N and/or C–N bonds in their frameworks. With this in mind, herein we report the synthesis of 2,4,6-tris(1H-tetrazol-5-yl)-1,3,5-triazine (H3TTT, 1), a new N-rich (73.7%) EM, along with a series of its salts (3–7). All compounds were physically characterized by IR, multinuclear (1H, 13C) NMR spectroscopy, gas pycnometry, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). The molecular structure of the TTT3− moiety was confirmed through single-crystal X-ray diffraction (SCXRD) analysis of the triethylammonium (TEA) salt (2). Key energetic parameters were assessed, revealing neutral H3TTT (1) exhibits excellent thermal stability over 247 °C as confirmed through DSC studies, while the decomposition temperatures of the energetic salts (3–7) were found to be lower than the parent material 1 (Tdec ranges from 144 °C (7) to 217 °C (3)). In terms of detonation performance, the highest values were observed for 4 (Pdet = 24.8 GPa, Vdet = 8061 m s−1) and 5 (Pdet = 24.6 GPa, Vdet = 7984 m s−1), as well as for 1 (Pdet = 22.4 GPa, Vdet = 7430 m s−1), where all three outperform TNT (Pdet = 19.5 GPa; Vdet = 6881 m s−1). The results of the thermochemical calculations indicate that H3TTT (1) and its salts 4 and 5 are characterized by ballistic parameters comparable to those of the commonly used JA-2 propellant. By using this simple and straightforward approach to EM development, we have generated a series of materials that may be employed as green alternatives to TNT secondary explosives and low-erosion, environmentally benign high-nitrogen ingredients for gun-propellants.
Introduction
Over the decades, the demand for energetic materials (EMs) has continuously grown as these materials are used in a wide range of applications such as mining, welding, fireworks, defence, aerospace, etc. Historically, materials such as 2,4,6-trinitrotoluene (TNT) have been employed due to their energetic performance, facile synthesis and relatively low cost. However, with an ever-growing demand for new, higher-performing materials with a specific application in mind, a “one trick pony” is not a viable option. Furthermore, stability and detonation properties must be appropriately balanced – it is common to find an energetic material that possesses large values of detonation velocity or detonation pressure (Vdet and Pdet, respectively) will also have low sensitivity to various external stimuli (e.g., impact, temperature, friction, etc.).1,2 As such, over the years, the synthesis of new EMs spurred a flurry of new materials by design with unique structural and physical properties for the targeted application.3 With that said, there is an urgent need for green smokeless energetic materials to curb their environmental impact upon application in various fields. To mitigate these issues, the field of EMs has shifted away from materials that contain heavy metals and/or produce significant amounts of greenhouse gases such as CO2. In this regard, nitrogen-rich (N-rich) energetic materials have become highly desirable, as the primary decomposition product of these compounds is the environmentally benign N2 gas.4–11When targeting energetic performance, molecules with high nitrogen-content are highly desirable and they may also overcome sensitivity problems. Firstly, N–N linkages take advantage of large bonding energies: whether single- (160 kJ mol−1), double- (418 kJ mol−1), or triple-bonded (954 kJ mol−1), compounds with a large number of N–N bonds possess high heats of formation, indicative of the chemical energy stored within that can be released upon decomposition.1,12 For example, the 3,6-bis(tetrazol-5-yl)-1,2,4,5-tetrazine (Chart 1), synthesised by Curtius et al.,13 shows an extremely positive energy of formation of 4976 kJ kg−1, which can be explained by its high nitrogen content of above 77%.14 Secondly, there are many examples of N-rich aromatic heterocycles in EM research.
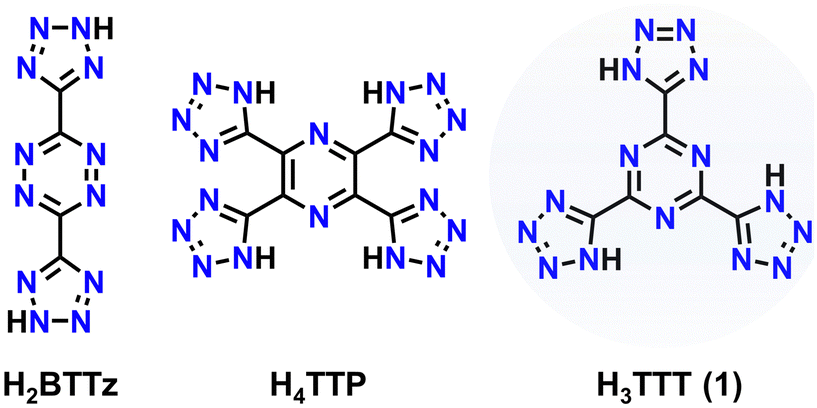 | ||
| Chart 1 Structures of 3,6-bis(tetrazol-5-yl)-1,2,4,5-tetrazine (H2BTTz), 2,3,5,6-tetra(tetrazol-5-yl)-pyrazine (H4TTP) and 2,4,6-tris(tetrazol-5-yl)-1,3,5-triazine (H3TTT). | ||
Functional groups such as triazoles, tetrazoles, tetrazines and more possess multiple covalent N–N linkages, while also imparting stability through the electron delocalization of the aromatic ring system. Such heterocycles also allow selective structural modification through judicious choice of substituents.15–21 Furthermore, many N-rich heterocycles such as imidazole, triazole, or tetrazole possess N atoms that have an acidic proton, which may be easily removed in the presence of a base, thereby yielding an anionic species. When combined with appropriate cationic species, energetic organic salts may be isolated. This approach enables the generation of many N-rich EMs that are also tunable through anion–cation interactions. Selecting specific organic and/or N-rich cations, the energetic properties of the initial material may be tailored to enhance particular physical properties of the system.
Previously, we reported the synthesis and characterization of a new planar N-rich EM 2,3,5,6-tetra(1H-tetrazol-5-yl)pyrazine, dubbed H4TTP (Chart 1).22 This molecule can be synthesized from readily available starting materials and was found to possess not only a large N-content (71.6%), but also high thermal stability (Tdec = 260 °C) and excellent detonation parameters (Vdet = 8.66 km s−1, Pdet = 28.0 GPa). Notably, the final step in the synthesis of H4TTP involves the reaction of tetracyanopyrazine (TCP) with sodium azide (NaN3) at 100 °C to induce a [2+3] dipolar azide–nitrile cycloaddition to form a tetrazole ring. It was through this research that we became interested in other nitrile containing precursors that would exhibit similar reactivity and, subsequently, yield new tetrazole-containing EMs. With this in mind, herein we report the synthesis of 2,4,6-tris(1H-tetrazol-5-yl)-1,3,5-triazine (H3TTT, 1), a new planar N-rich (73.7%) EM, along with a series of its energetic salts (3–7). All reported materials were physically characterized through IR and multinuclear (1H, 13C) NMR spectroscopy to determine the identity of the synthesized compounds. Gas pycnometry, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were used in conjunction with calculated heats of formation to determine the energetic performance. The molecular structure of the TTT3− moiety was confirmed through single-crystal X-ray diffraction (SCXRD) studies of the triethylammonium (TEA) organic salt. In terms of detonation performance, the highest values were observed for the hydrazinium, 4, (Pdet = 24.8 GPa, Vdet = 8061 m s−1) and hydroxylammonium, 5, (Pdet = 24.6 GPa, Vdet = 7984 m s−1) salts as well as for 1 (Pdet = 22.4 GPa, Vdet = 7430 m s−1), where all three outperform TNT (Pdet = 19.5 GPa; Vdet = 6881 m s−1).
Experimental section
Materials and physical measurements
Reagents were purchased from Sigma Aldrich or Alfa Aesar and used without further purification. The synthesis of tricyanotriazine (TCT) was performed using a previously reported synthesis.23 The synthesis of H3TTT (1) was performed under anaerobic conditions and utilized dry solvent obtained from a JC Meyer solvent system. The synthesis of all metal-free organic salts (2–7) was performed under aerobic/ambient conditions. FT-IR spectra (4000–650 cm−1) were recorded using an Agilent Technologies Cary 630 FT-IR spectrometer. 1H- and 13C-NMR spectra were recorded on a Bruker AVANCE II 400 or Bruker AVANCE III HD 600 spectrometer. The 15N-NMR spectroscopic data was recorded with a Bruker AVANCE III HD 600 spectrometer at a frequency of 60.8433718 MHz and a relaxation delay (d1) of 5 s. Chemical shifts in 1H- and 13C-NMR spectra are reported relative to Me4Si and 15N-NMR to MeNO2 at room temperature. TGA performed at a heating rate of 10 °C min−1 in flowing high-purity nitrogen on a Q5000 Thermogravimetric Analyzer. DSC experiments were performed with a TA Instruments Q10 instrument at a heating rate of 5 °C min−1. Densities were measured at room temperature using a Micromeritics AccuPyc II 1345 gas pycnometer.Safety precautions
The starting materials and compounds reported herein are high-energy density materials. Their sensitivity to various external stimuli, e.g. towards impact, friction and electrical discharge are not known. Although no safety issues were encountered throughout this research, these N-rich energetic materials should be handled with extreme care, following all the standard safety precautions along with the use of equipment such as leather gloves, leather coat, face shields and ear plugs.Synthetic details
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture (5 mL) of dichloromethane (DCM)/tetrahydrofuran (THF), triethylamine (TEA, 0.091 g, 0.726 g mL−1, 91 μL, 0.9 mmol) was added dropwise. The initially cloudy tan suspension changed to a clear yellow solution upon the complete addition of the TEA. The reaction was stirred for 5 minutes, filtered, and the filtrate was left to crystalize through slow diffusion of Et2O, yielding large yellow rods suitable for SCXRD studies. Yield: 0.056 g (32%). 1H-NMR (600 MHz, DMSO-d6): 1.20 ppm (triplet, –CH3), 3.32 ppm (quartet, –CH2–), 10.59 ppm (broad singlet, –NH). 13C-NMR (600 MHz, DMSO-d6): 8.52 ppm (–CH3), 45.43 ppm (–CH2–), 159.97 ppm (–CN4), 166.83 ppm (NCN). IR: v = 2990 (w), 2630 (w), 1528 (m), 1457 (m), 1376 (s), 1357 (s), 1299 (m), 1258 (m), 1161 (m), 1034 (m), 1032 (m), 810 (s) cm−1.
:1 mixture (5 mL) of dichloromethane (DCM)/tetrahydrofuran (THF), triethylamine (TEA, 0.091 g, 0.726 g mL−1, 91 μL, 0.9 mmol) was added dropwise. The initially cloudy tan suspension changed to a clear yellow solution upon the complete addition of the TEA. The reaction was stirred for 5 minutes, filtered, and the filtrate was left to crystalize through slow diffusion of Et2O, yielding large yellow rods suitable for SCXRD studies. Yield: 0.056 g (32%). 1H-NMR (600 MHz, DMSO-d6): 1.20 ppm (triplet, –CH3), 3.32 ppm (quartet, –CH2–), 10.59 ppm (broad singlet, –NH). 13C-NMR (600 MHz, DMSO-d6): 8.52 ppm (–CH3), 45.43 ppm (–CH2–), 159.97 ppm (–CN4), 166.83 ppm (NCN). IR: v = 2990 (w), 2630 (w), 1528 (m), 1457 (m), 1376 (s), 1357 (s), 1299 (m), 1258 (m), 1161 (m), 1034 (m), 1032 (m), 810 (s) cm−1.
General synthetic method of salts 3–7
To a suspension of 1 (86 mg, 0.3 mmol) in H2O (10 mL) was added 0.9 mmol of organic cation source and the reaction was stirred for 15 minutes. Upon complete solubilization of the reactants, the solvent was removed under reduced pressure yielding the targeted salt as a tan solid.Single-crystal X-ray crystallography
Single-crystal X-ray diffraction (SCXRD) data (ω- and ϕ-scans) were collected for compound 2 on a Bruker AXS SMART single crystal diffractometer at 203 K using Mo Kα radiation (λ = 0.71073 Å). Data reduction and absorption corrections (multi-scan) were performed using APEX2 and SADABS, respectively.24 The structure was solved using direct methods with SHELXT25 and refined by the full-matrix least-squares methods on F2 with SHELXL-2018/3.26 All non-hydrogen atoms were refined anisotropically. Carbon-bound hydrogen atoms were included and refined in calculated positions (riding model) and refined isotropically. The crystallographic data and structure refinement summary for compound 2 are listed in Table S1 (ESI†).Results and discussion
Synthesis and structural analysis
The synthetic pathway towards H3TTT (1) involves using inexpensive, readily available starting materials in a two-step reaction affording an overall yield of 64% (Scheme 1). Cyanuric chloride reacted with potassium cyanide (KCN) to yield tricyanotriazine (TCT), which subsequently reacted with NaN3 to isolate H3TTT (1). The initial step to isolate the TCT was performed as previously reported in the literature,23 while the second step to form the three tetrazole functional groups was performed analogously to the synthesis of H4TTP.22 Furthermore, the inclusion of an organic base readily deprotonates the H3TTT starting material that leads to the isolation of a TTT3− anion counterbalanced by three organic cations upon removal of solvent H2O under reduced pressure (Scheme 1).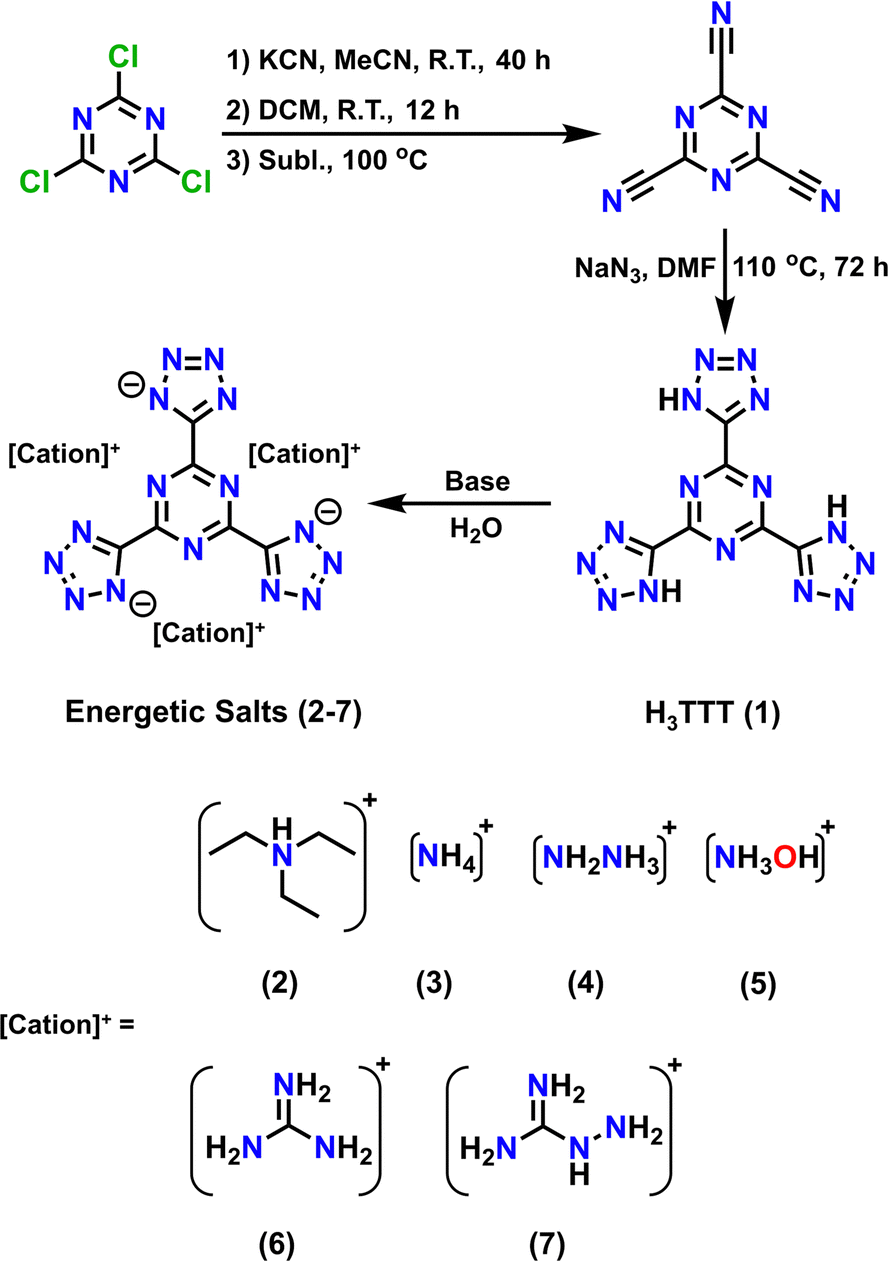 | ||
| Scheme 1 Synthetic pathway for the synthesis of 2,4,6-tris(1H-tetrazol-5-yl)-1,3,5-triazine (H3TTT, 1) and its energetic salts (2–7). Reagents and conditions are indicated in the scheme. | ||
While 1 is readily isolatable, obtaining a crystal structure of the neutral species proved challenging. While 13C-NMR and IR spectra (Fig. S1 and S2, ESI†) are consistent with the formation of H3TTT, the poor solubility of 1 in various solvents renders its crystallization suitable for single-crystal X-ray diffraction difficult. More specifically, 1 was found to only exhibit low solubility in polar aprotic solvents, such as dimethyl formamide (DMF), dimethyl sulfoxide (DMSO), and N-methyl-2-pyrrolidone (NMP), limiting its crystallization. Furthermore, once dissolved, 1 remains in solution, even after the addition of excess antisolvent, and only precipitates as microcrystalline solid (Fig. S3, ESI†) upon solvent reduction through heating and vacuum. However, upon adding various organic bases, 1 readily dissolves in water and various organic polar solvents, suggesting the formation of soluble organic salts. Subsequent removal of the solvent under vacuum affords the triethylammonium ([TEAH]3[TTT], 2), ammonium ([NH4]3[TTT], 3), hydrazinium ([N2H5]3[TTT], 4), hydroxylammonium ([NH3OH]3[TTT], 5), guanidinium ([GuH]3[TTT], 6) and aminoguanidinium ([AGuH]3[TTT], 7) salts of TTT3− (Scheme 1).
While recrystallization attempts were performed on all materials isolated, single crystals suitable for SCXRD analysis were only obtained for 2 through slow diffusion of a DCM/THF solution with Et2O. Compound 2 crystallizes as pale-yellow rods in the trigonal P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group, with two molecules per unit cell and a crystallographic density of 1.11 g cm−3 (Fig. 1(A)). Additional crystallographic details and metrical parameters can be found in Tables S1–S3 (ESI†). Upon close inspection, some deviations in the bond distances within both the central triazine ring (1.331(6) Å to 1.334(6) Å) and in the pendant anionic tetrazole rings (1.314 (10) Å to 1.346(7) Å) are observed, along with deviations from the ideal 120° angles for a six-membered aromatic ring. Moreover, 2 exhibits a center of symmetry located directly in the centroid of the central triazine ring. Furthermore, the central TTT3− anion is quasi-planar, with a twist angle of 1.98° between the mean calculated plane of the central triazine ring and the mean calculated plane of the outer tetrazole moieties. Within the unit cell, three triethylammonium (TEAH+) cations balance the overall charge of the three negatively charged tetrazole functional groups of the TTT3− anion (Fig. 1(B)). When viewed along the crystallographic a-axis, the formation of a bilayer of repeating TTT3− anions and TEAH+ cations can be observed (Fig. S4, ESI† left). An ethyl group of the TEAH+ extends down into close contact with the tetrazole group, while the other two groups do not come into close contact with the TTT3− anion – rather one extends into the center of the bilayer, while the other extends to the outer edge of the bilayer. These molecular features hold significant importance in the overall packing of the crystal structure (vide infra). Along the crystallographic c-axis (Fig. S4, ESI† right), the molecules organize into a set of repeating pairs of three TEAH+ cations and one TTT3− anion, which subsequently organizes the aforementioned bilayer into hexagonal columns with a central void running through the middle of the column.
space group, with two molecules per unit cell and a crystallographic density of 1.11 g cm−3 (Fig. 1(A)). Additional crystallographic details and metrical parameters can be found in Tables S1–S3 (ESI†). Upon close inspection, some deviations in the bond distances within both the central triazine ring (1.331(6) Å to 1.334(6) Å) and in the pendant anionic tetrazole rings (1.314 (10) Å to 1.346(7) Å) are observed, along with deviations from the ideal 120° angles for a six-membered aromatic ring. Moreover, 2 exhibits a center of symmetry located directly in the centroid of the central triazine ring. Furthermore, the central TTT3− anion is quasi-planar, with a twist angle of 1.98° between the mean calculated plane of the central triazine ring and the mean calculated plane of the outer tetrazole moieties. Within the unit cell, three triethylammonium (TEAH+) cations balance the overall charge of the three negatively charged tetrazole functional groups of the TTT3− anion (Fig. 1(B)). When viewed along the crystallographic a-axis, the formation of a bilayer of repeating TTT3− anions and TEAH+ cations can be observed (Fig. S4, ESI† left). An ethyl group of the TEAH+ extends down into close contact with the tetrazole group, while the other two groups do not come into close contact with the TTT3− anion – rather one extends into the center of the bilayer, while the other extends to the outer edge of the bilayer. These molecular features hold significant importance in the overall packing of the crystal structure (vide infra). Along the crystallographic c-axis (Fig. S4, ESI† right), the molecules organize into a set of repeating pairs of three TEAH+ cations and one TTT3− anion, which subsequently organizes the aforementioned bilayer into hexagonal columns with a central void running through the middle of the column.
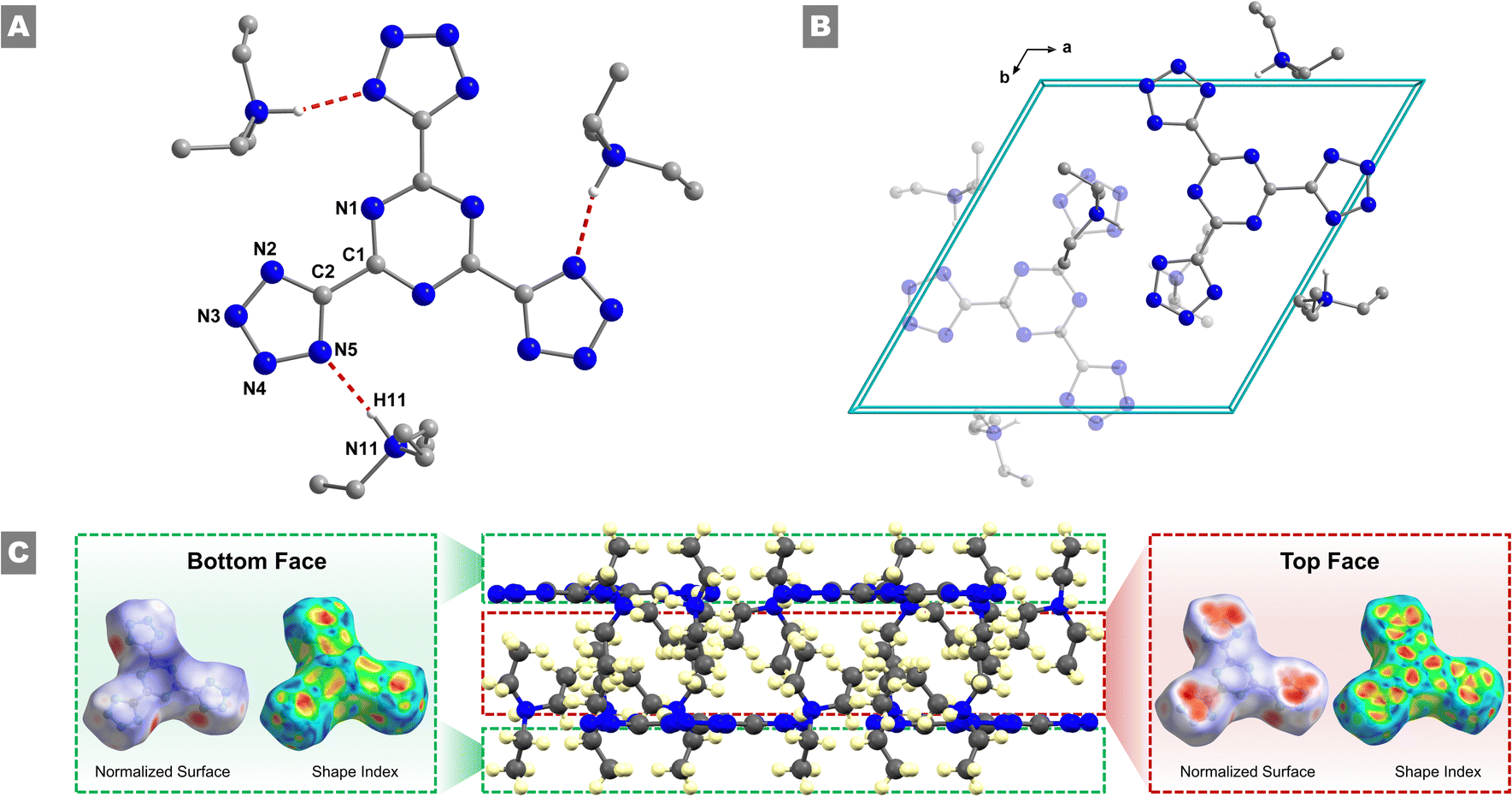 | ||
| Fig. 1 (A) Partially labelled representation of the molecular structure of 2 highlighting the intramolecular N–H⋯N hydrogen bonds. (B) View of the packing arrangement of 2 along the crystallographic c-axis. (C) Supramolecular packing interactions of the TTT3− anion, comparing the bottom and top faces of the TTT3− anion of 2. The “bottom” face shows little interaction with neighboring molecules, while the “top” face takes part in a number of intermolecular interactions, highlighting the bilayer packing arrangement of 2. Disordered atoms associated with the triethylammonium cations have been removed for clarity. Color code: N, blue; C, grey. | ||
The packing arrangement of molecules in the crystal lattice plays a critical role on the density of a material and consequently its energetic performances. Therefore, to gain subtle details of the packing arrangements and intermolecular interactions Hirshfeld surface analysis along with 2D fingerprint plots were investigated. The Hirshfeld surfaces of the TTT3− anion of 2 were generated utilizing the Crystal Explorer 17.5 software (Fig. 1(A) and Fig. S5, ESI†).27 The normalized Hirshfeld surface highlights the nature of contacts that interact with the surface – blue regions indicate the absence of intermolecular interactions, while red regions indicate their presence. Along the plane of the TTT3− anion, regions of red can be seen in relation to the tetrazole function groups. These are correlated with the intermolecular N⋯H interactions between the tetrazole groups and the ammonium nitrogen of the TEAH+ cation. Further analysis shows that this interaction occurs with a distance of 1.94 Å, indicating a strong hydrogen bonding interaction. A matching set of red regions can be seen on the opposing side of the pocket from the N⋯H interaction, which can be attributed to a C⋯H interaction between a carbon atom of an ethyl group of TEAH+ and the tetrazole ring. Additional interactions can be observed through the top and bottom of the TTT3− anion. To avoid confusion, consider the previously discussed bilayer nature of the crystal structure – the “top” of the TTT3− anion was chosen as the face, which is directed toward the interior of the bilayer, and the “bottom” of the TTT3− was chosen as the face that is directed to the exterior of the bilayer (Fig. 1(C)). The bottom of the TTT3− anion was found to not take part in intermolecular interactions, as confirmed by the lack of any red regions in the Hirshfeld surface. This ensures the bilayer nature of the crystal packing, where the exterior of the bilayer is occupied solely by alkyl groups of the TEAH+ cations. However, when viewing the top of the TTT3− anion, three red regions immediately reveal themselves on top of the tetrazole functional groups. These interactions occur due to the previously discussed ethyl group of the TEAH+, which extends down toward the face of the aromatic tetrazole ring. In further detail, the large red region can be subdivided into four different sub-regions, utilizing the shape index of the Hirshfeld surface (Fig. S6, ESI†). The outer three regions are associated with the three protons of the methyl group, with distances ranging from 1.86 Å to 2.40 Å, while the central region is associated with the carbon atom of the methyl group, with a distance of 2.41 Å at its closest point to the ring. Overall, the Hirshfeld analysis confirms the bilayer packing arrangement of the crystal structure, as well as the importance of the ethyl groups of the TEAH+ cations to stabilizing the TTT3− anions in the lattice.
Spectroscopy
Compounds 1–7 were analyzed utilizing 13C-NMR to determine the presence of the TTT3− anion, as well as the cations in 6 and 7. Compound 2 was analyzed via both 1H and 13C-NMR due to the presence of alkyl functionalities in the TEAH+ cations. The 13C-NMR spectrum of 1 in DMSO-d6 reveals the presence of two peaks at 155.80 ppm and 164.71 ppm (Fig. S1, ESI†). These peaks are associated with the C atom of the tetrazole functional group (–CN4) and the C atom on the central triazine ring (NCN), respectively. NMR predictor software was employed to provide information on the region of potential signals.28,29 Through these simulations, 1 was shown to have predicted signals at 152.9 ppm (–CN4) and 167.5 ppm (NCN), which are in good agreement with the experimental values and support the presence of the H3TTT moiety in 1. The 15N-NMR spectrum of H3TTT (1) was recorded in an acetone-d6/DMSO-d6 (3:1 volume ratio) mixture of solvents, and chemical shifts are given with respect to CH3NO2 as external standard. The 15N-NMR spectrum (Fig. S7, ESI†) of 1, shows signals at δ = −375.77 ppm assigned to the s-triazine and at δ = −119.07 and 9.28 ppm assigned to the tetrazole groups. The chemical shifts are in agreement with other tetrazole- and/or triazine-based compounds in the literature.30–34
Compound 2 was initially analyzed through 1H-NMR spectroscopy (Fig. S8, ESI†) in DMSO-d6. The primary features found in the spectrum are the presence of a triplet centred at 1.20 ppm and a quartet centred at 3.32 ppm (Fig. S8, ESI† inset). These signals are associated with the –CH3 and –CH2– groups of the TEAH+ cation. Indeed, upon integration of these signals, the triplet integrates to a value of 3.01, and the quartet integrates to 2.00, confirming these signals are associated with the proton environments of an ethyl group. The complete 1H spectrum further reveals the presence of a broad singlet at 10.59 ppm. This signal is associated with the NH proton of the TEAH+ cation. Finally, some minor solvent impurities from the synthetic process were also observed: 1.75 ppm (THF), 1.91 ppm (acetone), 2.50 ppm (DMSO, multiplet), 2.77 ppm (DMF), 3.52 ppm (H2O), 5.76 ppm (DCM).35 Further confirmation of the structure was revealed through 13C-NMR (Fig. S9, ESI†). The primary peaks are all singlets, and are found at 8.52 ppm, 45.43 ppm, 159.97 ppm, and 166.83 ppm. The peaks present at 8.52 ppm (–CH3) and 45.43 ppm (–CH2–) signals are associated with the carbon environments of the ethyl groups of the TEAH+ cation. The singlets at 159.97 ppm (–CN4) and 166.83 ppm (NCN) are associated with the TTT3− anion. Of particular note is the shift in the signal associated with the carbon on the tetrazole functional group. In neutral 1, the tetrazole singlet appears at 155.80 ppm, whereas in the anion, the signal shifts downfield to 159.97 ppm. This is attributed to the deprotonation of the tetrazole moiety, which deshields the carbon atom and subsequently shifts the signal.
Compounds 3–7 were solubilized in DMSO-d6 and analyzed utilizing 1H- and 13C-NMR spectroscopy (Fig. 2 and Fig. S10, ESI†). All compounds show the presence of the TTT3− anion through signals that are within close agreement with the 159.97 ppm and 166.83 ppm signals of 2 (ranges of 160.16–160.81 ppm for –CN4 tetrazole functional groups and ranges of 166.37–167.08 ppm for NCN triazine carbons). Furthermore, for compounds 6 and 7, the guanidine carbon of the guanidinium (6) and aminoguanidinium (7) cations are also found in the 13C-NMR spectra as a singlet with a value of 158.67 ppm for 6 and 159.43 ppm for 7. These values are similar to previously reported guanidinium and aminoguanidinium cations in other energetic salts.36,37
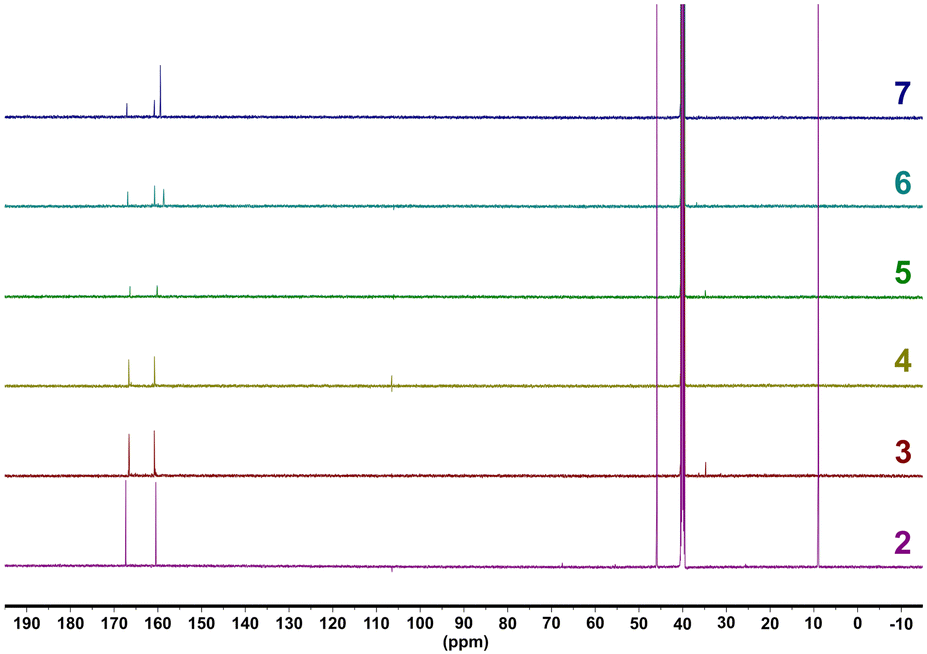 | ||
| Fig. 2 13C-NMR spectra for compounds 3–7. Compound 2 is included for comparative purposes. Measurements were performed at 600 MHz in D2O over 5 minutes. | ||
Physicochemical properties
Determination of the decomposition temperature of an EM is essential for determining its potential for industrial applications. In general, compounds that exhibit thermal stability above 160 °C are considered to be thermally stable materials and thus safe to handle, barring any other sensitivities.1 Therefore, the decomposition temperatures of compounds 1–7 were determined through thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements (Fig. 3 and Fig. S11, S12, ESI†). The physicochemical properties of all energetic compounds are listed in Table 1. As the temperature increases, 1 maintains a constant mass percent (Fig. 3), and begins a slight decrease upon reaching 200 °C. Upon reaching 247 °C, a sharp increase in the mass percent is observed to a maximum of ∼400%, followed by an immediate drop to 30%. This is indicative of a detonation of the material, as the pressure generated by explosive decomposition results in a false mass percentage increase. This behaviour was further confirmed through DSC measurements, which show a single, exothermic process beginning at ∼220 °C and reaching a maximum at 255 °C (Fig. 3, inset). This range of processes perfectly encompasses the initial mass loss and subsequent explosive decomposition temperature measured by TGA. Therefore, the decomposition temperature of 1 was found to be 247 °C.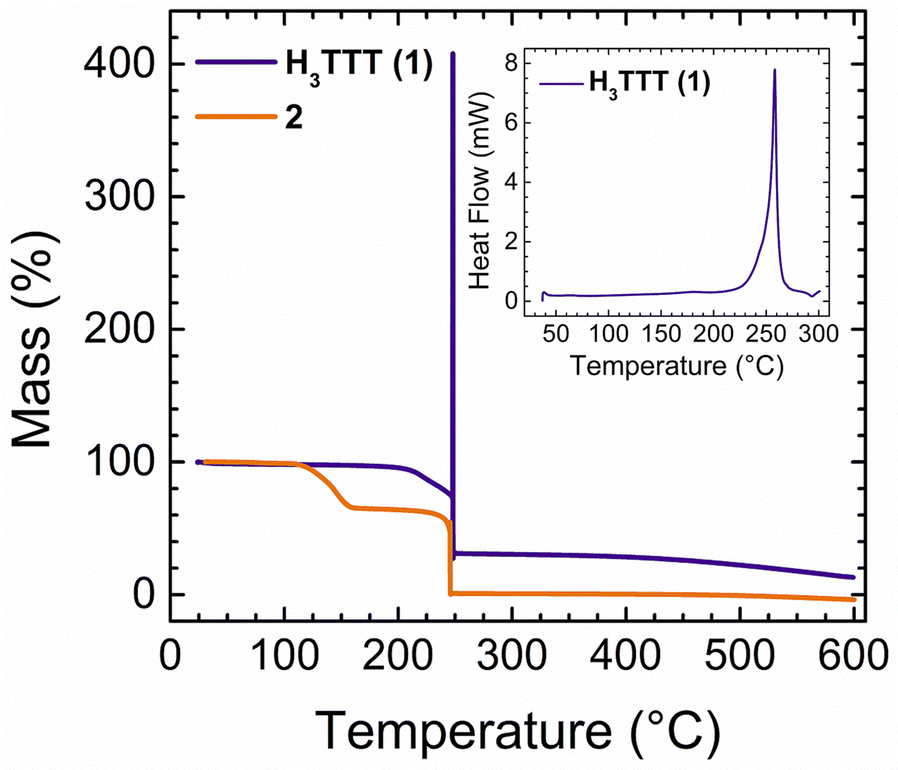 | ||
| Fig. 3 Thermogravimetric analysis (TGA) of compounds 1 and 2, performed using a temperature ramp of 10 °C min−1. The significant increase in mass percent observed at 247 °C in 1 is associated with the detonation of the material. (inset) Differential scanning calorimetry (DSC) of 1, also performed at 10 °C min−1. | ||
| Compound | N + Oa (%) | Ωb (%) | T dec (°C) | P (g cm−3) |
|
V det (m s−1) | P det (GPa) |
|---|---|---|---|---|---|---|---|
|
a Combined nitrogen and oxygen content.
b Oxygen balance (%) based on CO2 for CaHbNcOd: OB (%) = 1600 × (d − 2a − b/2)/MW, MW = molecular weight.
c Thermal decomposition temperature (onset) under nitrogen (determined by the DSC exothermal peak, 5°Cmin−1).
d Density measured by gas pycnometer at 25 °C.
e Crystal density.
f Calculated molar enthalpy of formation in solid state.
g Detonation velocity.
h Detonation pressure.
|
|||||||
| H3TTT (1) | 73.67 | −75.74 | 247 | 1.78 | 1190 | 7430 | 22.4 |
| 2 | 42.82 | −195.67 | 245 | 1.11e | 460 | 5072 | 6.5 |
| 3 | 74.97 | −85.64 | 214 | 1.77 | 365 | 6888 | 16.8 |
| 4 | 77.14 | −81.82 | 165 | 1.83 | 854 | 8061 | 24.8 |
| 5 | 78.10 | −62.45 | 217 | 1.75 | 634 | 7984 | 24.6 |
| 6 | 72.70 | −93.43 | 163 | 1.68 | 457 | 6557 | 14.6 |
| 7 | 74.53 | −89.86 | 144 | 1.64 | 838 | 6973 | 16.8 |
| H4TTP 22 | 71.58 | −81.76 | 260 | 1.95 | 1383 | 8655 | 28.0 |
| TNT 38 | 60.77 | −73.97 | 295 | 1.65 | −67 | 6881 | 19.5 |
| RDX 39 | 81.06 | −21.61 | 204 | 1.80 | 70 | 8795 | 34.9 |
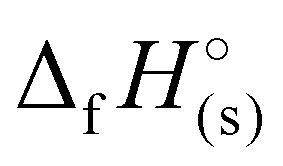
For 2, no mass loss is observed until 125 °C, upon which a decrease of 40% is observed, which ends at 150 °C. This loss is correlated with the decomposition of the TEAH+ cations present in the salt. A subsequent and sharp decomposition down to 0% is observed at 245 °C, which is related to the explosive decomposition of the TTT3− anion. This decomposition temperature falls in line with the aforementioned decomposition temperature of 247 °C for 1. For compounds 3–7, TGA curves did not yield the distinct sharp behavior seen previously for 1 and 2 (Fig. S11, ESI†). However, the mass percentage decreases not attributed to solvent can be observed in 3–7, yielding a range of decomposition temperatures from 144 °C (7) to 217 °C (3). All decomposition temperatures of the salts were further confirmed through DSC measurements (Fig. S12, ESI†) and found to be lower than the parent material 1. In most cases the decomposition temperatures surpass 160 °C, indicating that these compounds can be identified as thermally stable EMs.
One of the most important physical properties of an EM in the solid-state is its density, which directly relates to its energetic properties, where increasing the density of an explosive leads to an increase in its detonation performance.1 The experimental densities were measured using a gas pycnometer at 25 °C in a helium atmosphere. The densities of all compounds (except 2) range from 1.64 to 1.83 g cm−3 as given in Table 1 and are similar to those previously reported for the metal-free organic salts of H4TTP.40 It is noteworthy that all compounds herein have higher densities than TNT (1.65 g cm−3) and 4 possesses a density of 1.83 g cm−3, which is higher than that of the benchmark explosive RDX (1.80 g cm−3).
Furthermore, all compounds (except 2) show excellent nitrogen and oxygen contents (Table 1) in their molecular backbone, ranging from 72.70 to 78.10%, significantly higher than TNT (60.77%). Among all, 4 and 5 have the highest nitrogen and oxygen content of 77.14 and 78.10%, which are higher than H4TTP (71.58%) and slightly lower than RDX (81.06%). The oxygen balance of all compounds ranges from −62.45 to −89.86%, falling in the same range as H4TTT (−81.76%) and TNT (−73.97%) but significantly lower than RDX (−21.61%).
Molecular electrostatic potentials (ESP) provide insight into the sensitivity towards impact of a given energetic material.41–43 Typically, molecules containing regions of large positive charge (i.e., electron-deficient regions) over the molecular framework tend to indicate increased sensitivity to impact.1 The ESP of H3TTT (1) in the region between V(r) ≤ −0.1194 Hartree and V(r) ≥ +0.1949 Hartree computed at the 0.001 electron bohr−3 hypersurface is shown in Fig. S13 (ESI†). The color gradient reflects the electrostatic potential distribution from red (electron-rich regions) to green (zero potential) to blue (electron-deficient regions). H3TTT exhibits large positive electrostatic potential regions along the tetrazole rings while a slightly electron-deficient region over the triazine ring suggests a potentially increased sensitivity to impact. We are noting here that this requires further experimental validation. Computational details are provided in the ESI.†
Energetic properties
To assess the viability of 1–7 as potential candidates for energetic applications, the heat of formation calculations of all N-rich compounds were performed. Utilizing the modified complete basis set method (CBS-4M) and the Gaussian 09 (Revision E.01) software,44,45 the gas-phase enthalpies of formation for 1–7 were calculated into arbitrary units. These values were subsequently converted into units of kJ mol−1 utilizing the atomization energy method (eqn (1)):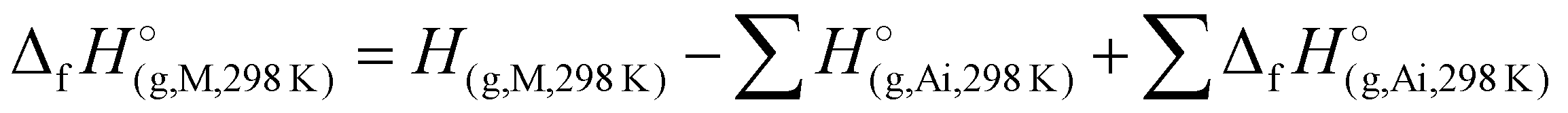 | (1) |
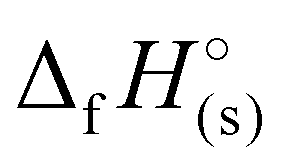 ) were determined for all compounds. For 1, the standard molar enthalpy of formation was calculated using the aforementioned gas-phase enthalpy of formation in conjunction with the standard molar enthalpy of sublimation, estimated using Trouton's rule (eqn (2)):48
) were determined for all compounds. For 1, the standard molar enthalpy of formation was calculated using the aforementioned gas-phase enthalpy of formation in conjunction with the standard molar enthalpy of sublimation, estimated using Trouton's rule (eqn (2)):48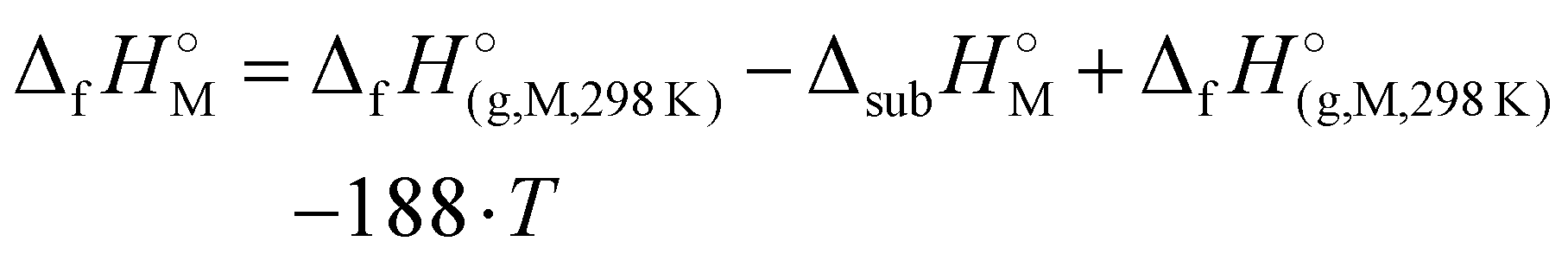 | (2) |
For 2–7, the solid-state heats of formation were calculated through the employment of the gas-phase heats of formation along with the heat of phase transition (lattice energy) utilizing Hess’ law (the Born–Haber energy cycle). The lattice energy and subsequently the lattice enthalpy (ΔUL and ΔHL, respectively) were calculated employing the experimentally determined densities of all salts following the methodology developed by Jenkins, Tudela, and Glasser.49 Utilizing all previously assembled calculations, the standard molar enthalpy of formation (solid-state heat of formation, 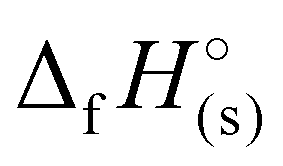 ) for each compound was calculated (Table 1). All the newly synthesised compounds show positive
) for each compound was calculated (Table 1). All the newly synthesised compounds show positive 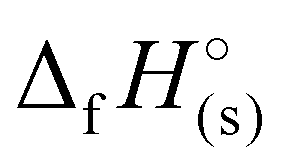 (365 to 1190 kJ mol−1), significantly higher than that of TNT (−67 kJ mol−1) and RDX (70 kJ mol−1) as shown in Table 1. Among all, H3TTT (1) possesses the highest
(365 to 1190 kJ mol−1), significantly higher than that of TNT (−67 kJ mol−1) and RDX (70 kJ mol−1) as shown in Table 1. Among all, H3TTT (1) possesses the highest 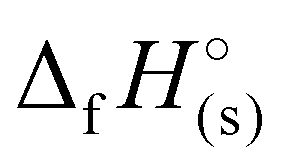 of 1190 kJ mol−1, slightly lower than H4TTP (1383 kJ mol−1). However, the overall lower values of
of 1190 kJ mol−1, slightly lower than H4TTP (1383 kJ mol−1). However, the overall lower values of 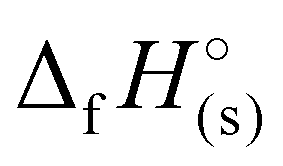 calculated for the various TTT3− salts (3–7) will lead to reduced energetic performance compared to the family of metal-free H4TTP salts.
calculated for the various TTT3− salts (3–7) will lead to reduced energetic performance compared to the family of metal-free H4TTP salts.
Using the calculated values of the heats of formation and experimental densities, the detonation pressures (Pdet) and velocities (Vdet) were calculated using the CHEETAH Version 4.0 thermochemical computer code with BKWS product library.50 As can be seen in Table 1, the calculated detonation velocities lie between Vdet = 6557 and 8061 m s−1. The highest values in terms of detonation performance were observed for 4 (8061 m s−1) and 5 (7984 m s−1), both of which outperform TNT (6881 m s−1). In comparison with H4TTP (8655 m s−1) and RDX (8795 m s−1), a slight decrease in the performance is observed due to the lower densities of the TTT3−-based energetic salts. In terms of detonation pressures, 4 (24.8 GPa) and 5 (24.6 GPa) show remarkable results, which are significantly greater than that of TNT (19.5 GPa). Both of these, as well as the parent compound 1 (Pdet = 22.4 GPa, Vdet = 7430 m s−1), outperform TNT (Pdet = 19.5 GPa; Vdet = 6881 m s−1) and therefore have the potential to act as EMs, specifically as green alternatives to TNT secondary explosives.
Ballistic properties
To evaluate the suitability of 1–7 as additives in high-energy gun propellants, their ballistic parameters were calculated using the CHEETAH Version 4.0 code with the BLAKE product library for a loading density of 0.2 g cm−3.50 It should be noted that the heats of formation (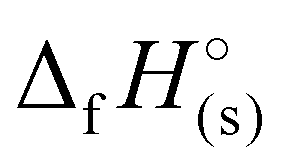 ) and densities of the compounds in Table 1 were used to calculate the ballistic properties. All calculated thermochemical parameters for gun propellants, including the adiabatic flame temperature (Tf), the maximum pressure (Pmax) at a given loading density, the impetus (fp), the covolume (η) and the ratio of heat capacities (γ), are presented in Table 2 and compared to H4TTP and JA-2. The JA-2 propellant is commonly used for tank ammunition applications and consists of nitrocellulose, nitroglycerine, and diethylene glycol dinitrate in relative amounts of roughly 60%, 15%, and 25%, respectively.51 Neutral H3TTT (1) was found to have the highest flame temperature of 3205 K compared to its energetic salts 2–7 (ranging from 1379–2298 K; Table 2), and was found to be higher compared to H4TTP (3041 K) while slightly lower (8% decrease) than that of JA-2 (3486 K). Furthermore, the flame temperature of 1 (3205 K) is considered mildly hot, while the flame temperatures of the energetic salts 4 and 5 (2028 K and 2298 K, respectively) are considered relatively cold temperatures. Interest in flame temperatures is due in part to barrel erosion concerns where a lower flame temperature limits the reaction occurring between gun barrel steel and the hot combustion gases, while also limiting thermal expansion and contraction during repeated firings.52,53 Regarding the maximum pressure (Table 2), the hydrazinium salt 5 was found to have the highest pressure of 227.1 MPa, followed by the neutral H3TTT (210.9 MPa) and the energetic salt 4 (196.6 MPa). Consequently, the impetus follows a similar trend where the hydrazinium salt 5 posses the highest impetus of 859 J g−1 followed by the neutral H3TTT (840 J g−1) and the energetic salt 4 (743 J g−1). A similar trend was also observed for the calculated covolume (η) and the ratio of heat capacities (γ) as evident in Table 2. Noteably, these parameters are comparable to or slightly lower than those of the commonly used JA-2 propellant, suggesting that H3TTT (1) and its salts 4 and 5 may be suitable as low-erosion and environmentally benign high-nitrogen ingredients for gun-propellants.54 In addition, favoring the formation of nitrogen gas over CO during the propellant combustion will positively influence the life span of the gun barrel, since CO can react with the steel of the gun barrel to form iron carbide resulting in higher chemical erosion of the barrel.52,53
) and densities of the compounds in Table 1 were used to calculate the ballistic properties. All calculated thermochemical parameters for gun propellants, including the adiabatic flame temperature (Tf), the maximum pressure (Pmax) at a given loading density, the impetus (fp), the covolume (η) and the ratio of heat capacities (γ), are presented in Table 2 and compared to H4TTP and JA-2. The JA-2 propellant is commonly used for tank ammunition applications and consists of nitrocellulose, nitroglycerine, and diethylene glycol dinitrate in relative amounts of roughly 60%, 15%, and 25%, respectively.51 Neutral H3TTT (1) was found to have the highest flame temperature of 3205 K compared to its energetic salts 2–7 (ranging from 1379–2298 K; Table 2), and was found to be higher compared to H4TTP (3041 K) while slightly lower (8% decrease) than that of JA-2 (3486 K). Furthermore, the flame temperature of 1 (3205 K) is considered mildly hot, while the flame temperatures of the energetic salts 4 and 5 (2028 K and 2298 K, respectively) are considered relatively cold temperatures. Interest in flame temperatures is due in part to barrel erosion concerns where a lower flame temperature limits the reaction occurring between gun barrel steel and the hot combustion gases, while also limiting thermal expansion and contraction during repeated firings.52,53 Regarding the maximum pressure (Table 2), the hydrazinium salt 5 was found to have the highest pressure of 227.1 MPa, followed by the neutral H3TTT (210.9 MPa) and the energetic salt 4 (196.6 MPa). Consequently, the impetus follows a similar trend where the hydrazinium salt 5 posses the highest impetus of 859 J g−1 followed by the neutral H3TTT (840 J g−1) and the energetic salt 4 (743 J g−1). A similar trend was also observed for the calculated covolume (η) and the ratio of heat capacities (γ) as evident in Table 2. Noteably, these parameters are comparable to or slightly lower than those of the commonly used JA-2 propellant, suggesting that H3TTT (1) and its salts 4 and 5 may be suitable as low-erosion and environmentally benign high-nitrogen ingredients for gun-propellants.54 In addition, favoring the formation of nitrogen gas over CO during the propellant combustion will positively influence the life span of the gun barrel, since CO can react with the steel of the gun barrel to form iron carbide resulting in higher chemical erosion of the barrel.52,53
| Compound | T f (K) | P max (MPa) | f p (J g−1) | η (cm3 g−1) | γ |
|---|---|---|---|---|---|
| a Adiabatic flame temperature. b Maximum pressure. c Impetus. d Covolume. e Ratio of heat capacities. | |||||
| H3TTT (1) | 3205 | 210.9 | 840 | 0.946 | 1.176 |
| 2 | 1379 | 122.2 | 465 | 1.094 | 1.153 |
| 3 | 1481 | 126.8 | 484 | 1.132 | 1.214 |
| 4 | 2028 | 196.6 | 743 | 1.175 | 1.218 |
| 5 | 2298 | 227.1 | 859 | 1.193 | 1.248 |
| 6 | 1434 | 122.4 | 468 | 1.128 | 1.209 |
| 7 | 1744 | 163.1 | 617 | 1.165 | 1.212 |
| H4TTP 22 | 3041 | 197.6 | 789 | 0.932 | 1.171 |
| JA-2 51 | 3486 | 289.0 | 1161 | 0.983 | 1.224 |
Conclusions
In conclusion, the reaction of tricyanotriazine with NaN3 through a [2+3] dipolar azide–nitrile cycloaddition led to the successful synthesis and characterization of 2,4,6-tris(1H-tetrazol-5-yl)-1,3,5-triazine (H3TTT, 1) and a series of its nitrogen-rich salts (3–7). The molecular structure of the TTT3− moiety was confirmed through single-crystal X-ray diffraction studies of the triethylammonium organic salt (2). The Hirshfeld surfaces and 2D fingerprint plots of 2 were studied, and the results indicated that the molecular packing and arrangement in the crystals are stabilized by N⋯H and H⋯H intermolecular interactions. Compound 1 (H3TTT) showed excellent thermal stability over 247 °C as confirmed through DSC studies, while the decomposition temperatures of the energetic salts (3–7) were found to be lower than the parent material 1 (temperature ranges from 144 °C (7) to 217 °C (3)). Due to the multiple N–N and C–N bonds in their backbones, all compounds possess positive heat of formation values (from 365 to 1190 kJ mol−1), while the experimental densities fall in the range of 1.64 to 1.83 g cm−3. The highest values in terms of detonation performance were observed for compounds 4 (Pdet = 24.8 GPa, Vdet = 8061 m s−1) and 5 (Pdet = 24.6 GPa, Vdet = 7984 m s−1), as well as for the parent compound 1 (Pdet = 22.4 GPa, Vdet = 7430 m s−1), where all three outperform TNT (Pdet = 19.5 GPa; Vdet = 6881 m s−1) and therefore have potential to act as green alternatives to TNT secondary explosives. The results of the thermochemical calculations indicate that H3TTT (1) and its salts 4 and 5 are characterized by ballistic parameters comparable to those of the commonly used JA-2 propellant. As highlighted here, introducing multiple tetrazolyl heterocycles in a single rigid framework proves to be a successful and straightforward synthetic approach towards novel energetic materials with high N-content, high thermal stability, and high heats of formation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science and Engineering Council of Canada and General Dynamics Ordnance and Tactical Systems. P. R. would like to thank the Government of Ontario for an Ontario Graduate Scholarship (OGS). A. A. K. is grateful to Mitacs postdoctoral fellow scholarship through the Mitacs Accelerate program.Notes and references
- T. M. Klapötke, Chemistry of High-Energy Materials, De Gruyter, 2019 Search PubMed.
- H. Gao and J. M. Shreeve, Azole-Based Energetic Salts, Chem. Rev., 2011, 111, 7377–7436 CrossRef CAS PubMed.
- O. T. O’Sullivan and M. J. Zdilla, Properties and Promise of Catenated Nitrogen Systems As High-Energy-Density Materials, Chem. Rev., 2020, 120, 5682–5744 CrossRef PubMed.
- J. Tang, H. Yang, Y. Cui and G. Cheng, Nitrogen-rich tricyclic-based energetic materials, Mater. Chem. Front., 2021, 5, 7108–7118 RSC.
- D. Herweyer, J. L. Brusso and M. Murugesu, Modern trends in “Green” primary energetic materials, New J. Chem., 2021, 45, 10150–10159 RSC.
- Y.-H. Joo, B. Twamley, S. Garg and J. M. Shreeve, Energetic Nitrogen-Rich Derivatives of 1,5-Diaminotetrazole, Angew. Chem., Int. Ed., 2008, 47, 6236–6239 CrossRef CAS PubMed.
- T. M. Klapötke and D. G. Piercey, 1,1′-Azobis(tetrazole): A Highly Energetic Nitrogen-Rich Compound with a N10 Chain, Inorg. Chem., 2011, 50, 2732–2734 CrossRef PubMed.
- Q. Wang, F. Pang, G. Wang, J. Huang, F. Nie and F.-X. Chen, Pentazadiene: a high-nitrogen linkage in energetic materials, Chem. Commun., 2017, 53, 2327–2330 RSC.
- D. Chen, H. Yang, Z. Yi, H. Xiong, L. Zhang, S. Zhu and G. Cheng, C8N26H4: An Environmentally Friendly Primary Explosive with High Heat of Formation, Angew. Chem., Int. Ed., 2018, 57, 2081–2084 CrossRef CAS.
- M. Xiao, X. Jin, J. Zhou and B. Hu, 1,2,5-Oxadiazole-1,2,3,4-tetrazole-based high-energy materials: molecular design and screening, Struct. Chem., 2021, 32, 1619–1628 CrossRef CAS.
- M. S. Manna, C. K. Das and S. Ghanta, Design of C–H–N–O based new hetero-cyclic high energy density molecules: a theoretical survey, Struct. Chem., 2021, 32, 1095–1104 CrossRef CAS.
- N. Saracoglu, Recent advances and applications in 1,2,4,5-tetrazine chemistry, Tetrahedron, 2007, 63, 4199–4236 CrossRef CAS.
- T. Curtius, A. Darapsky and E. Müller, Die sogenannten Pentazol-Verbindungen von J. Lifschitz, Ber. Dtsch. Chem. Ges., 1915, 48, 1614–1634 CrossRef CAS.
- T. M. Klapötke, D. G. Piercey, F. Rohrbacher and J. Stierstorfer, Synthesis and Characterization of Energetic Salts of the (C4N122−) Dianion, Z. Anorg. Allg. Chem., 2012, 638, 2235–2242 CrossRef.
- D. E. Chavez, M. A. Hiskey and R. D. Gilardi, 3,3′-Azobis(6-amino-1,2,4,5-tetrazine): A Novel High-Nitrogen Energetic Material, Angew. Chem., Int. Ed., 2000, 39, 1791–1793 CrossRef CAS.
- G.-H. Tao, B. Twamley and J. M. Shreeve, A thermally stable nitrogen-rich energetic material—3,4,5-triamino-1-tetrazolyl-1,2,4-triazole (TATT), J. Mater. Chem., 2009, 19, 5850–5854 RSC.
- W. Zhang, J. Zhang, M. Deng, X. Qi, F. Nie and Q. Zhang, A promising high-energy-density material, Nat. Commun., 2017, 8, 181 CrossRef PubMed.
- A. A. Larin, N. V. Muravyev, A. N. Pivkina, K. Yu Suponitsky, I. V. Ananyev, D. V. Khakimov, L. L. Fershtat and N. N. Makhova, Assembly of Tetrazolylfuroxan Organic Salts: Multipurpose Green Energetic Materials with High Enthalpies of Formation and Excellent Detonation Performance, Chem. – Eur. J., 2019, 25, 4225–4233 CrossRef CAS PubMed.
- M. Benz, M. S. Gruhne, T. M. Klapötke, N. Krüger, T. Lenz, M. Lommel and J. Stierstorfer, Evolving the Scope of 5,5′-Azobistetrazoles in the Search for High Performing Green Energetic Materials, Eur. J. Org. Chem., 2021, 4388–4392 CrossRef CAS.
- A. K. Yadav, V. D. Ghule and S. Dharavath, Dianionic nitrogen-rich triazole and tetrazole-based energetic salts: synthesis and detonation performance, Mater. Chem. Front., 2021, 5, 8352–8360 RSC.
- J. Singh, S. Lal, R. J. Staples and J. M. Shreeve, Functionalized planar aromatic rings as precursors to energetic N,N′-(4,6-dinitro-1,3-phenylene)dinitramide and its salts, Mater. Chem. Front., 2022, 6, 933–938 RSC.
- T. G. Witkowski, E. Sebastiao, B. Gabidullin, A. Hu, F. Zhang and M. Murugesu, 2,3,5,6-Tetra(1H-tetrazol-5-yl)pyrazine: A Thermally Stable Nitrogen-Rich Energetic Material, ACS Appl. Energy Mater., 2018, 1, 589–593 CrossRef CAS.
- R. E. Del Sesto, A. M. Arif, J. J. Novoa, I. Anusiewicz, P. Skurski, J. Simons, B. C. Dunn, E. M. Eyring and J. S. Miller, Chemical Reduction of 2,4,6-Tricyano-1,3,5-triazine and 1,3,5-Tricyanobenzene. Formation of Novel 4,4′,6,6′-Tetracyano-2,2‘-bitriazine and Its Radical Anion, J. Org. Chem., 2003, 68, 3367–3379 CrossRef CAS PubMed.
- Bruker, APEX2, SAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2009 Search PubMed.
- G. M. Sheldrick, SHELXT – Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- C. Steinbeck, S. Krause and S. Kuhn, NMRShiftDB Constructing a Free Chemical Information System with Open-Source Components, J. Chem. Inf. Comput. Sci., 2003, 43, 1733–1739 CrossRef CAS PubMed.
- A. M. Castillo, L. Patiny and J. Wist, Fast and accurate algorithm for the simulation of NMR spectra of large spin systems, J. Magn. Reson., 2011, 209, 123–130 CrossRef CAS PubMed.
- D. McAteer and J. Akhavan, Nitrogen-14 NMR Spectroscopic Detection of Explosophores in Solution, Propellants, Explos., Pyrotech., 2016, 41, 367–370 CrossRef CAS.
- H.-L. Deng, X.-S. Luo, Z. Lin, J. Niu and M.-H. Huang, 14N NMR as a General Tool to Characterize the Nitrogen-Containing Species and Monitor the Nitration Process, J. Org. Chem., 2021, 86, 16699–16706 CrossRef CAS PubMed.
- Y. Tang, H. Yang, X. Ju, H. Huang, C. Lu and G. Cheng, A novel N–N bond cleavage in 1,5-diaminotetrazole: synthesis and characterization of 5-picrylamino-1,2,3,4-tetrazole (PAT), J. Mater. Chem. A, 2014, 2, 4127–4131 RSC.
- Y. Guo, G.-H. Tao, Z. Zeng, H. Gao, D. A. Parrish and J. M. Shreeve, Energetic Salts Based on Monoanions of N,N-Bis(1H-tetrazol-5-yl)amine and 5,5′-Bis(tetrazole), Chem. – Eur. J., 2010, 16, 3753–3762 CrossRef CAS PubMed.
- Q. Zhang, J. Zhang, D. A. Parrish and J. M. Shreeve, Energetic N-Trinitroethyl-Substituted Mono-, Di-, and Triaminotetrazoles, Chem. – Eur. J., 2013, 19, 11000–11006 CrossRef CAS PubMed.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- A. Hammerl, M. A. Hiskey, G. Holl, T. M. Klapötke, K. Polborn, J. Stierstorfer and J. J. Weigand, Azidoformamidinium and Guanidinium 5,5′-Azotetrazolate Salts, Chem. Mater., 2005, 17, 3784–3793 CrossRef CAS.
- M. L. Gettings, M. T. Thoenen, E. F. C. Byrd, J. J. Sabatini, M. Zeller and D. G. Piercey, Tetrazole Azasydnone (C2N7O2H) And Its Salts: High-Performing Zwitterionic Energetic Materials Containing A Unique Explosophore, Chem. – Eur. J., 2020, 26, 14530–14535 CrossRef CAS PubMed.
- J. N. Johnson, C. L. Mader and S. Goldstein, Performance Properties of Commercial Explosives, Propellants, Explos., Pyrotech., 1983, 8, 8–18 CrossRef CAS.
- C. He, Y. Tang, L. A. Mitchell, D. A. Parrish and J. M. Shreeve, N -Oxides light up energetic performances: synthesis and characterization of dinitraminobisfuroxans and their salts, J. Mater. Chem. A, 2016, 4, 8969–8973 RSC.
- T. G. Witkowski, P. Richardson, B. Gabidullin, A. Hu and M. Murugesu, Synthesis and Investigation of 2,3,5,6-Tetra-(1H-tetrazol-5-yl)pyrazine Based Energetic Materials, ChemPlusChem, 2018, 83, 984–990 CrossRef CAS PubMed.
- J. S. Murray, P. Lane and P. Politzer, Relationships between impact sensitivities and molecular surface electrostatic potentials of nitroaromatic and nitroheterocyclic molecules, Mol. Phys., 1995, 85, 1–8 CrossRef CAS.
- B. M. Rice and J. J. Hare, A Quantum Mechanical Investigation of the Relation between Impact Sensitivity and the Charge Distribution in Energetic Molecules, J. Phys. Chem. A, 2002, 106, 1770–1783 CrossRef CAS.
- D. S. Kretić, J. I. Radovanović and D. Ž. Veljković, Can the sensitivity of energetic materials be tuned by using hydrogen bonds? Another look at the role of hydrogen bonding in the design of high energetic compounds, Phys. Chem. Chem. Phys., 2021, 23, 7472–7479 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- L. A. Curtiss, K. Raghavachari, P. C. Redfern and J. A. Pople, Assessment of Gaussian-2 and density functional theories for the computation of enthalpies of formation, J. Chem. Phys., 1997, 106, 1063–1079 CrossRef CAS.
- B. M. Rice, S. V. Pai and J. Hare, Predicting heats of formation of energetic materials using quantum mechanical calculations, Combust. Flame, 1999, 118, 445–458 CrossRef CAS.
- E. F. C. Byrd and B. M. Rice, Improved Prediction of Heats of Formation of Energetic Materials Using Quantum Mechanical Calculations, J. Phys. Chem. A, 2006, 110, 1005–1013 CrossRef CAS PubMed.
- F. Trouton, IV. On molecular latent heat, London, Edinburgh Dublin Philos. Mag. J. Sci., 1884, 18, 54–57 CrossRef.
- H. D. B. Jenkins, D. Tudela and L. Glasser, Lattice Potential Energy Estimation for Complex Ionic Salts from Density Measurements, Inorg. Chem., 2002, 41, 2364–2367 CrossRef CAS PubMed.
- L. Fried and P. Souers, CHEETAH: A next generation thermochemical code, Lawrence Livermore National Lab, Livermore CA, 1994 Search PubMed.
- M. Hara and W. Trzciński, Experimental and Theoretical Investigation of the Heat of Combustion of RDX-based Propellants, Cent. Eur. J. Energ. Mater., 2019, 16, 399–411 CrossRef CAS.
- I. A. Johnston, Understanding and Predicting Gun Barrel Erosion, Weapons Systems Division – Defence Science and Technology Organisation (DSTO), Edinburgh, Australia, 2005 Search PubMed.
- X. Li, Y. Zang, L. Mu, Y. Lian and Q. Qin, Erosion analysis of machine gun barrel and lifespan prediction under typical shooting conditions, Wear, 2020, 444–445, 203177 CrossRef CAS.
- J. Lavoie, C.-F. Petre, S. Durand and C. Dubois, Stability and performance of gun propellants incorporating 3,6-dihydrazino-s-tetrazine and 5-aminotetrazolium nitrate, J. Hazard. Mater., 2019, 363, 457–463 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic details, IR and NMR spectra, TGA and DSC data. CCDC number 2271491. For crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ma00410d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |