
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrasound-driven fabrication of hybrid magnetic tryptophan nanoparticles†
Anshul
Baral
 a,
Haiyan
Zhu
a,
Bradford A.
Moffat
b,
Santanu
Chattopadhyay
c,
Francesca
Cavalieri
*de and
Muthupandian
Ashokkumar
*a
a,
Haiyan
Zhu
a,
Bradford A.
Moffat
b,
Santanu
Chattopadhyay
c,
Francesca
Cavalieri
*de and
Muthupandian
Ashokkumar
*a
aSchool of Chemistry, The University of Melbourne, Parkville, VIC 3010, Australia. E-mail: masho@unimelb.edu.au
bThe Melbourne Brain Centre Imaging Unit, Department of Radiology, The University of Melbourne, Parkville, Victoria 3010, Australia
cRubber Technology Centre, Indian Institute of Technology, Kharagpur 721302, India
dSchool of Science, RMIT University, Melbourne, VIC 3000, Australia
eDepartment of Chemical Sciences and Technologies, University of Rome “Tor Vergata”, 00133, Rome, Italy
First published on 2nd September 2023
Abstract
A facile sonochemical method to fabricate magnetic tryptophan-iron oxide hybrid nanoparticles was established in this study. First, superparamagnetic iron oxide nanoparticles (SPIONs) of an average size of 5 nm were synthesised using hydrothermal processes, followed by surface modification using tryptophan to improve their colloidal stability. Then, the surface-modified iron oxide nanoparticles were subjected to 490 kHz ultrasound treatment in tryptophan solution to fabricate magnetic tryptophan nanoparticles (MTNs) with an average size of 167 ± 29 nm. A comprehensive analysis of the synthesised nanocomposite was performed to describe the possible mechanism of MTNs formation. We show that high-frequency ultrasound transforms tryptophan into hydroxylated, oligomeric, and hybrid SPIONs-Trp products, forming magnetic tryptophan hybrid nanoparticles. These hybrid MTNs showed fluorescence properties that could be utilised to probe the dissolution of MTNs inside the cells. In addition, MTNs also exhibited sensitivity in magnetic resonance imaging (MRI) analysis which could be further exploited for diagnostic applications.
Introduction
Magnetic hybrid nanostructures have been recently applied in many biomedical applications like targeted drug delivery,1–5 magnetic resonance imaging (MRI)6–9 and hyperthermia therapy.10–13 In particular, superparamagnetic iron-oxide nanoparticles (SPIONs) exhibit magnetic properties only under an external magnetic field without retaining any residual magnetism after removing the magnetic field,14 making them an appropriate fit for these biomedical applications. Furthermore, their magnetic properties can be remotely guided to a specific target inside the body, making them an excellent tool for targeted drug delivery applications. However, the successful use of SPIONs in biomedical applications is significantly determined by their structural characteristics, exceptionally uniform shape, nanometric size and narrow size distribution, high magnetisation values and precise surface chemistry.14 In addition, pristine SPIONs generally lose their intrinsic superparamagnetic properties because of their susceptibility to form aggregates in solution. Therefore, most of these applications require surface modification of SPIONs, which involves two steps: surface coating of SPIONs and functionalisation with specific biomolecules for targeting specific cells or tissues.15,16 Furthermore, these hybrid nanostructures should be non-toxic and biocompatible while retaining their superparamagnetic nature and targeting capability.14 The general strategy for the bio-functionalisation of SPIONs is to attach layers of specific biomolecules on the surface of pristine SPIONs, which should hinder their tendency to aggregate and promote the bio-conjugation of active biomolecules onto these layers.In particular, natural amino acids represent a versatile set of biomolecules to stabilise MNPs. Nosrati et al.17 reported the stabilisation of magnetic nanoparticles by simple amino acids, including L-tyrosine, L-phenylalanine, L-arginine, glycine and L-lysine. To stabilise the SPIONs, the electrostatic interactions between the negatively charged carboxyl group of amino acids and the positively charged surface of nanoparticles at pH 6 are exploited.17,18 Ebrahiminezhad et al.19 reported a simple two-step method for preparing fluorescent iron oxide nanoparticles using amino acids as linkers and spacers with potential bioimaging applications. Galhoum et al.20 demonstrated the formation of chitosan-magnetite nanocomposite functionalised with amino acids, viz., alanine and serine. Mathur et al.21 reported a tryptophan-conjugated magnetic nanoparticle targeting tumours using overexpressed LAT1 transporters and showed promising results for imaging applications. In addition, Tokmedash et al.22 synthesised a targeted drug delivery system for treating breast cancer cells by tagging tryptophan to MNPs using 3-aminopropyl triethoxysilane as the crosslinker. In general, the preparation of all these hybrid magnetic nanoparticles typically requires multiple reaction and purification steps and the use of toxic coupling agents, which could compromise the biocompatibility of the final products.
Recently ultrasound technology has been identified as the green technology to efficiently generate tunable multifunctional nanoparticles from biomolecules.23–26 Most importantly, the ultrasound technique does not typically require any external toxic reagent or solvent, and can induce crosslinking of biomolecules in water, one pot and at ambient conditions. Of note, we have reported that the ultrasonic synthesis of nanoparticles is a versatile method that enables the control of nanoparticles size by regulating the acoustic power. A reduction in nanoparticle size is typically observed at higher power levels.24 The reason is that by applying higher acoustic power the number of transient and active bubbles increases. More transient energy input due to the collapse of acoustic bubbles would increase the rate of nucleation. A higher nucleation rate would lead to a reduction in the size of nanoparticles as the rate of nucleation and radius are inversely related to each other.27 For instance, Cavalieri et al.28 introduced the mechanism of ultrasound-induced oxidative coupling and self-assembly of tyrosine derivative molecules to form well-defined nanoparticles. Bhangu et al.23,24 further exploited the mechanism for sono-responsive aromatic amino acids like phenylalanine and tryptophan to manufacture tunable multifunctional nanoparticles. Furthermore, Baral et al.26 showed that ultrasound could also influence the self-assembling of oligopeptides to fabricate well-defined biofunctional nanoparticles. In this study, we sought to apply the developed methodology to manufacture multifunctional hybrid magnetic nanoparticles. Herein, we demonstrated the formation of hybrid magnetic tryptophan nanoparticles using high-frequency ultrasound of 490 kHz. The selection of tryptophan is influenced by the fact that they are sono-responsive and possess excellent inherent fluorescence properties which could be utilised in bioimaging applications.23 Firstly, SPIONs were synthesised using a hydrothermal technique and surface-modified using tryptophan. Then, a tryptophan solution was sonicated in the presence of surface-modified SPIONs, forming well-defined magnetic tryptophan nanoparticles (MTNs). After a comprehensive analysis of MTNs, a possible mechanism describing their formation pathway has been proposed. The mechanism involves the transformation of tryptophan into hydroxylated, oligomeric, and SPIONs -Trp products, which self-assembled to form magnetic tryptophan hybrid nanoparticles. MTNs were readily internalized by breast cancer cells and only marginal cytotoxicity was observed. The optical properties of MTNs can be used to track the dissolution of MTNs inside cells. In addition, we show that MTNs retained their superparamagnetic properties and can be exploited as MRI contrast agents owing to their uniform morphology, and colloidal stability.
Experimental
Materials
Iron(III) acetylacetonate (C15H21FeO6, 97%), L-Tryptophan (C11H12N2O2, reagent grade, ≥98%), 1-diphenyl-2-picrylhydrazyl (DPPH), Dulbecco's phosphate-buffered saline (D-PBS), bovine serum albumin (BSA), hydrogen peroxide (30%), and EIPA (ethyl isopropyl amiloride) were procured from Sigma Aldrich (St. Louis, MO, USA). Methanol (99.9%) and HPLC grade formic acid were bought from Fisher Chemicals (Hampton, NH, USA). Phalloidin Alexa 480, rabbit anti-EEA-1 monoclonal antibody and rabbit anti-Rab7 monoclonal antibodies were purchased from Cell Signalling Technology (Beverly, MA, USA). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) reagent, Rabbit LAMP-1 and goat anti-rabbit IgG secondary antibody Alexa Fluor 647 were provided by Invitrogen (Carlsbad, CA, USA). Fetal bovine serum (FBS) was procured from Bovogen (Victoria, Australia). Dulbecco's Modified Eagle's medium (DMEM) was obtained from Lonza (Basel, Switzerland). All solutions were prepared in Milli-Q water with a resistivity of 18.2 MΩ cm unless otherwise stated.Preparation and surface modification of SPIONs
SPIONs were synthesised using the hydrothermal process. Iron(III) acetylacetonate (1.4 g, 4 mmol) was dispersed in 20 mL of N-methyl pyrrolidone. The mixture was purged with N2 gas for 30 min to remove the dissolved O2 inside the solution and heated under constant vigorous stirring until it reached the refluxing temperature of 220 °C and maintained under the same conditions for 1 h. Afterwards, 20 mL of methanol was added to the reaction mixture as soon as the temperature cooled down to room temperature. Then the reaction mixture was centrifuged (3000 rpm for 5 min) to collect the precipitated particles. The precipitated particles were washed four times with acetone and dried in a vacuum oven at 40 °C. The as-prepared particles were analysed using Fourier-transform infrared spectroscopy (FTIR), high-resolution transmission electron microscopy (HRTEM), and vibrating sample magnetometer (VSM), then referred to as the superparamagnetic iron oxide nanoparticles (SPIONs). SPIONs were dispersed in Milli-Q water at a concentration of 0.5 mg mL−1 with constant ultrasonication in an ultrasound bath for 1 h and subsequently evaluated for their colloidal stability using a dynamic light scattering (DLS) analyser.The as-prepared SPIONs were surface-modified using tryptophan to improve their colloidal stability. To achieve the surface modification, 5 mL of tryptophan solution (1 mg mL−1) was mixed with 5 mL of pristine SPIONs solution (0.5 mg mL−1). Afterwards, the reaction mixture was ultrasonicated for 1 h in the ultrasonic bath and kept overnight on an orbital shaker. Next, the reaction mixture was centrifuged (3000 rpm, 5 min) to remove the unreacted tryptophan, and the surface modified-SPIONs were separated from the pristine SPIONs in the mixture solution using a strong magnet and further centrifugation (500 rpm, 1 min) for 2–3 times. Finally, the surface modified-SPIONs were freeze-dried and resuspended in Milli-Q water at a concentration of 0.5 mg mL−1. The colloidal stability of the surface modified-SPIONs was measured using the DLS analyser. Furthermore, the morphology of surface modified-SPIONs was analysed using HRTEM and scanning electron microscopy (SEM).
Preparation of magnetic tryptophan nanoparticles (MTNs)
Magnetically functionalised tryptophan nanoparticles were obtained using a high-frequency ultrasound setup consisting of a double-walled glass cell connected to a water bath mounted on an ELAC Nautik USW 51-052 transducer of diameter 5.4 cm powered by T&C Power Conversion, Inc. (Rochester, NY, USA). Initially, 10 mg of tryptophan was dissolved in the 10 mL of surface modified-SPIONs solution (0.5 mg mL−1) in a 15 mL glass vial and then subjected to a high-frequency ultrasound of 490 kHz and 60 W acoustic power for 5 h at a temperature of 37 ± 2 °C. The sonicated sample was centrifuged (7000 rpm, 5 min) to separate the synthesised nanoparticles and washed several times before characterisation. Then, a comprehensive characterisation of the synthesised nanoparticles was performed using FTIR, DLS, fluorescence spectroscopy, VSM, TEM, mass spectroscopy (MS), high-performance liquid chromatography (HPLC), and SEM coupled with Energy Dispersive Spectroscopy (EDS), to establish the possible mechanism driving the formation of MTNs.Characterisation techniques
Fourier-transform infrared spectroscopy (FTIR) spectra were acquired on SPIONs and MTNs samples from 4000 to 400 cm−1 with 128 scans and 4 cm−1 resolution using PerkinElmer Spectrum two FTIR spectroscopy in the attenuated total reflection (ATR) mode using a diamond crystal (Medtech, Waltham, MA, USA). ZEN0040, Malvern Instruments (Malvern, UK) was used to evaluate the zeta potential and hydrodynamic diameter of the SPIONs and MTNs with a measurement angle of 173°. The hydrodynamic diameter of particles was estimated using the cumulant fit method. DLS measurements were performed using a volume of 80 μL suspensions of 0.5 mg mL−1 concentration in Milli-Q at pH 6. Magnetic properties of SPIONs and MTNs powder samples were measured using a superconducting quantum interference device (SQUID) (MPMS; Quantum Design) under a magnetic field of 20 kOe (2 Tesla) at 298 K. Samples were weighed between 10 to 15 mg, wrapped with Teflon tape, and placed in a spherically designed sample holder. The M–H loop measurement was initiated from 0 T and evaluated the magnetisation up to +2 T. Then, the magnetic field was decreased from +2 T to −2 T to determine the hysteresis loop. A PerkinElmer AxION® 2 ToF quadrupole mass spectrometer (PerkinElmer, Waltham, MA, USA) was utilised in positive ion mode to determine and compare the molecular weight fragments of the sonicated solutions and compared with unsonicated tryptophan. Gradient elution was carried out with solvent A (0.1% formic acid) and solvent B (acetonitrile with 0.1% formic acid) at 30 °C. The optical properties of MTNs are investigated using a Shimadzu RF-5301PC fluorescence spectrophotometer (Shimadzu, Kyoto, Japan) equipped with a xenon lamp and a 1.0 cm quartz cell. Fluorescence emission spectra were recorded at different wavelengths. Shimadzu SCL-10AVP HPLC (Agilent Technologies, Santa Clara, CA, USA) equipped with the Phenomenex “Jupiter 5u C18 300A” column (Phenomenex, Torrance, CA, USA) and a UV detector set at 205 nm was employed to carry out the high-performance liquid chromatography (HPLC) analysis of tryptophan and sonicated MTNs. The injection volume was 20 μL, and the flow rate was 1 mL min−1 with methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water:formic acid (70:30:0.5) used as an eluent. MTNs morphologies were analysed using an FEI Teneo VolumeScope (Thermo Fisher Scientific, Waltham, MA, USA) scanning electron microscope (SEM) at an accelerating voltage of 10 kV and current of 50 pA coupled with an Oxford energy dispersive X-ray spectroscopy. Transmission electron microscopy (TEM) was also employed to investigate the morphology MTNs using an FEI Tecnai F20 TEM instrument (Thermo Fisher Scientific, Waltham, MA, USA). The stability of MTNs was investigated in Milli-Q, PBS and 100% FBS solutions for 24 h.
:water:formic acid (70:30:0.5) used as an eluent. MTNs morphologies were analysed using an FEI Teneo VolumeScope (Thermo Fisher Scientific, Waltham, MA, USA) scanning electron microscope (SEM) at an accelerating voltage of 10 kV and current of 50 pA coupled with an Oxford energy dispersive X-ray spectroscopy. Transmission electron microscopy (TEM) was also employed to investigate the morphology MTNs using an FEI Tecnai F20 TEM instrument (Thermo Fisher Scientific, Waltham, MA, USA). The stability of MTNs was investigated in Milli-Q, PBS and 100% FBS solutions for 24 h.
The cell studies of MTNs were investigated using MDA-MB-231 cells, which were seeded at a density of 40000 cells per well in NuncTM Labtek 8-well chamber cover glass slides (ThermoFisher Scientific, Scoresby, Australia) and incubated overnight at 37 °C with 5% CO2. First, the medium was replaced, and the diluted MTNs were added to cells at a final concentration of 0.075 mg mL−1. Next, the MTNs were incubated for 6 h, the medium was replaced, and cells were washed three times with PBS-BSA (1% BSA in PBS) to remove unbound particles and further incubated for 12 h and 24 h. Next, the cells were fixed with 4% paraformaldehyde for 15 min at room temperature, washed, and permeabilised with 0.1% TritonX-100 solution in PBS for 5 min and washed three times with PBS. Finally, the samples were blocked with 2.5% PBS-BSA for 1 h. Cells were then imaged using Nikon A1R confocal microscope with a 60 × 1.4NA oil immersion objective. A similar study was performed without washing out the MTNs and analysed under confocal microscopy. In addition, a dissolution study was conducted using 0.075 mg mL−1 MTNs in PBS solution (pH 7.4) at 37 °C. The particles were centrifuged out, and the fluorescent peaks at 450 nm were measured for the supernatant, followed by mixing the centrifuged MTNs into the supernatant solution. The procedure was repeated at different intervals of time till 24 h.
For MRI relaxivity measurements, 500 μL of synthesised SPIONs and MTNs were placed in a 48-well plate at 100 μg mL−1 concentration. The 48 well-plate was imaged at room temperature (approximately 21 °C) in a Siemens (Germany) 7T Magnetom MRI scanner using an eight-channel transmit and 32-channel radio-frequency receiver coil array (Nova Medical, NY). For quantifying T2 relaxivity, a Carr-Purcell Meiboom and Gill multi-echo spin echo sequence,29 was used to acquire 12 images at echo times (TE) ranging from 7 to 84 ms with a repetition time (TR) of 5 s. All images were acquired with a 2 mm slice thickness, 128 mm FOV, 128 × 128 matrix size. Zero filling was used to reconstruct all images to a matrix size of 256 × 256. The mean signal from regions of interest (25 voxels) centred within each well was averaged and plotted as a function of TE to evaluate T2. The T2 value was then estimated (as the decay constants) by numerically fitting (using a nonlinear least-squares algorithm (Matlab, MA)) the data to a monoexponential equation as follows:
| S = S0exp(−TE/T2) |
Results and discussions
Pristine SPIONs and surface modification
Superparamagnetic iron oxide nanoparticles (SPIONs) were prepared and characterised by FTIR, VSM and HRTEM techniques. The FTIR spectra (Fig. S1, ESI†) showed the characteristic peak for iron oxide nanoparticles at 590 cm−1 that corresponds to the Fe–O stretching vibration and the wide band around 3430 cm−1 corresponding to the surface –OH stretching frequency of the pristine SPIONs.30,31 The average diameter of SPIONs was 5 ± 2 nm, as shown in Fig. 1a, and the superparamagnetic nature of the particles was confirmed by the VSM analysis, as exhibited in Fig. 1b. The particles’ saturation magnetisation (Ms) and retentivity (Mr) were 31.25 emu g−1 and 0.354 emu g−1, respectively. The nanoparticles did not exhibit hysteresis loop and coercivity, due to the superparamagnetic nature of the synthesised magnetic nanoparticles.32 However, the pristine SPIONs (0.5 mg mL−1) in MilliQ solution at pH 7.4 tend to form aggregates of around 500 nm, as depicted in Fig. S2 (ESI†), and therefore, surface modification of pristine SPIONs was performed using tryptophan to minimise the aggregation. After surface modification, the average particle size of surface-modified SPIONs was reduced to 18 ± 4 nm (Fig. 1c and 1d), suggesting that tryptophan acts as a stabilizer agent.33 The size of surface-modified SPIONs was much larger when compared to the pristine SPIONs, which could be due to the aggregation of 2–3 pristine SPIONs in proximity by tryptophan during surface modification. The ζ-potential was measured to be −29 ± 5 mV, possibly due to carboxyl groups of tryptophan exposed on the surface of SPIONs. In addition, a significant improvement in the colloidal stability of tryptophan-modified SPIONs suspension in MilliQ was observed with colloids of sizes around 70 nm (Fig. S3, ESI†).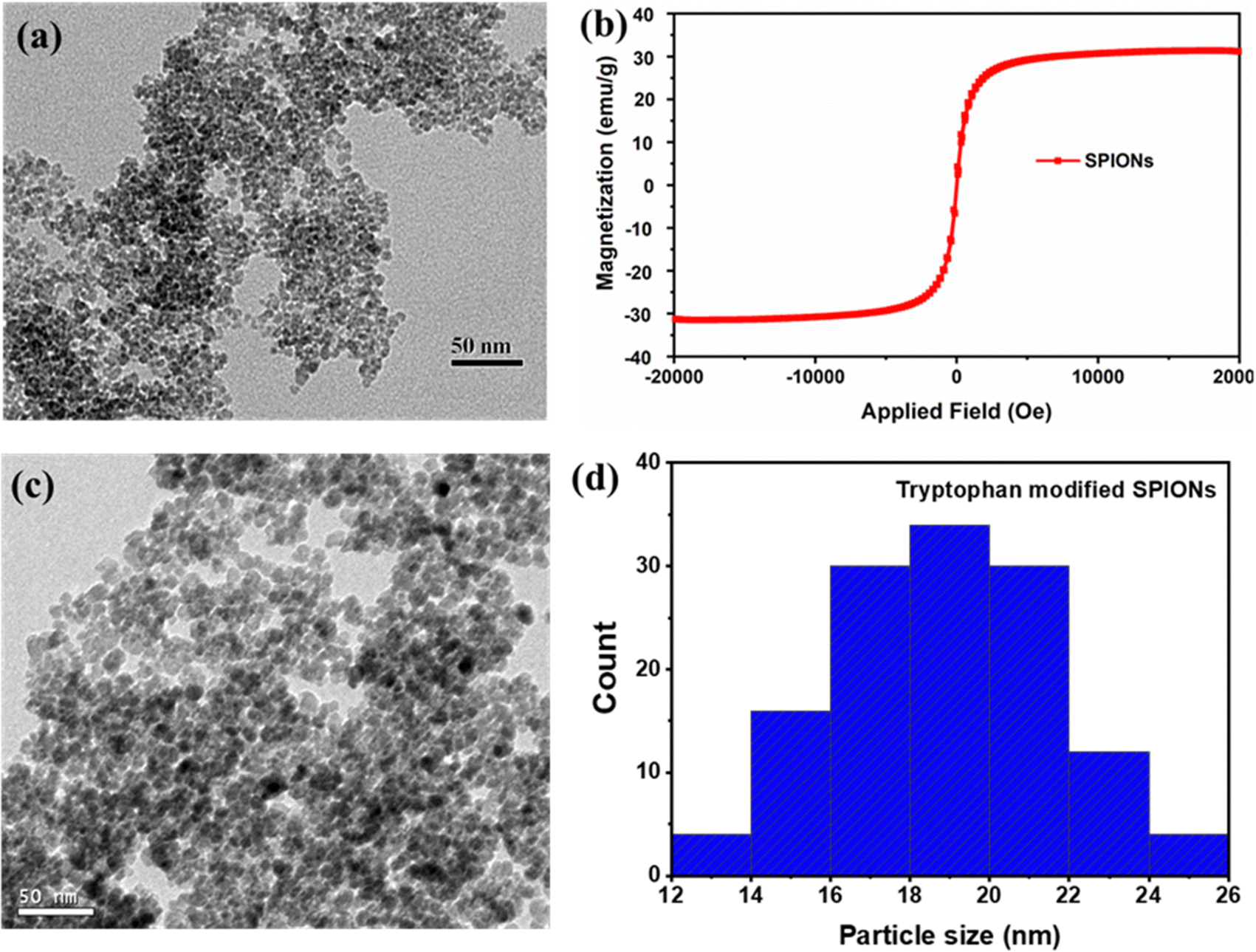 | ||
| Fig. 1 (a) TEM image of pristine SPIONs, (b) magnetisation curve of pristine SPIONs studied at room temperature using VSM, (c) TEM image and (d) histogram of the particle size distribution of surface modified SPIONs using tryptophan. | ||
Magnetic tryptophan nanoparticles (MTNs)
Next, the tryptophan-modified SPIONs were used to obtain the hybrid MTNs. MTNs were formed by subjecting the tryptophan-modified SPIONs to 490 kHz ultrasound treatment in a tryptophan solution (0.5 mg of tryptophan-modified SPIONs/mg of tryptophan per mL of MilliQ) for 5 h. MTNs showed an average particle size of 167 ± 29 nm, as depicted in the SEM image of dried samples (Fig. 2a). In addition, the hydrodynamic diameter and ζ-potential of the hydrated hybrid MTNs were 230 ± 32 nm and −27 ± 6 mV, respectively. The particle showed excellent colloidal stability, with only a tiny trace of particle settling down when kept undisturbed for 24 h in Milli-Q. The negative surface charge of the MTN is due to the deprotonation of the aromatic hydroxyl groups in water at pH 7. Therefore, the electrostatic repulsion imparts colloidal stability to the nanoparticles. The hydrodynamic diameter of MTNs in PBS and 100% FBS were found to be 235 ± 29 nm and 254 ± 45 nm, respectively. There was a negligible change in the hydrodynamic diameter of MTNs in Milli-Q and PBS solutions after 24 h of incubation. However, a slight increase in MTNs size was observed from 254 ± 45 nm to 287 ± 56 nm, which could be induced by the adsorption of protein corona on the surface of nanoparticles over time, whereas no aggregation or disassembly of MTNs were observed.23 The particles were stored for over 6 months and retained their colloidal stability with a hydrodynamic diameter of 242 ± 28 nm after dispersing in MilliQ water at a concentration of 0.5 mg mL−1. The morphological structure remains unaltered, as shown in SEM images (Fig. S4, ESI†). Notably, the hybrid MTNs maintained their superparamagnetic nature with a slight reduction in the magnetisation value compared to the pristine SPIONs, as shown in Fig. 2b.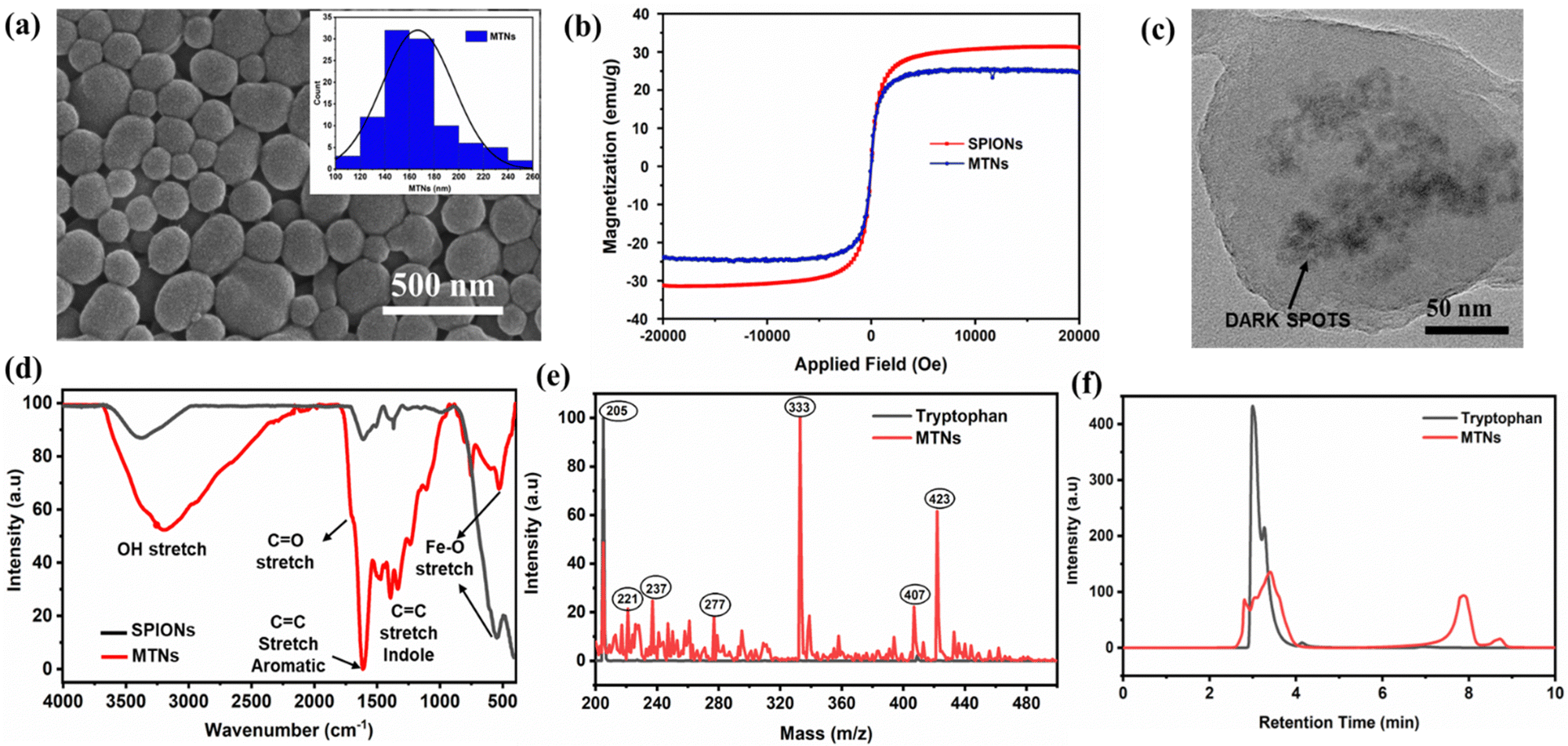 | ||
| Fig. 2 (a) SEM image of the MTNs with particle size distribution inset, (b) comparison of the magnetisation curve of pristine SPIONs and MTNs studied at room temperature using VSM, (c) TEM image of a single MTN where dark spots indicate the presence of iron-oxide nanoparticles inside tryptophan matrix, and (d) FTIR spectra of SPIONs and MTNs. (e) Mass spectroscopic and (f) HPLC analysis of the MTNs after dissolving in MilliQ water at pH 2.5, depicting the formation of different species during the sonication process. | ||
The successful incorporation of iron-oxide nanoparticles within MTNs was confirmed using TEM, SEM-EDS and FTIR analysis. The appearance of dark spots in the TEM image (Fig. 2c) could be attributed to the presence of heavy metallic iron-oxide nanoparticles within tryptophan nanoassemblies, which was further confirmed by the common Fe–O stretch (560 cm−1 wavenumber) present in the FTIR spectra of both SPIONs and MTNs as depicted in Fig. 2d. In addition, the peaks at 1700 cm−1 could be due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch, 1601 cm−1 resembles the CC aromatic stretch, 1342 cm−1 may be attributed to the CC indole stretch, and the broad peak around 3250 cm−1 corresponds to the O–H stretching.23 The incorporation of SPIONs within the matrix of tryptophan assembly was further validated by the iron (Fe) signal peak in SEM-EDS spectra, as shown in Fig. S5 (ESI†). Subsequently, the atomic and weight ratio of Carbon to Iron (C:Fe) content in MTNs was measured using SEM-EDS from different portions of the sample. The atomic and weight ratio was consistent all over the sample and was found to be 6.9:1 (C:Fe) and 1.5:1 (C:Fe), respectively. The weight percentage of the Fe content was also measured in the sample and found to be 33.2%, which could provide the approximate content of SPIONs in MTNs. In addition, the fluorescence properties of MTNs were studied to gain further insight into the multi-functionalities of MTNs. Fig. S6 (ESI†) describes the fluorescence emission spectra of the sonicated solution containing MTNs and other soluble products compared to untreated tryptophan. A new emission peak was observed at 450 nm (Fig. S6a, ESI†), which could be ascribed to the formation of tryptophan dimers, as previously reported.23,24 The phenomenon involves the sono-induced oxidative coupling of tryptophan molecules to form hydroxylated monomers dimers. In addition, a shift in emission peaks (Fig. S6b, ESI†) was detected when excited between 400 to 460 nm indicating the presence of various species formed upon sonication. In contrast, the MTNs suspension obtained after centrifugation and resuspension exhibited a weak signal at 450 nm compared to the dissolved MTNs, as shown in Fig. S7a (ESI†). The weaker signal could be likely due to the quenching of signals by magnetic nanoparticles.34–36 SPIONs were added to sonicated tryptophan solution to confirm the quenching of the fluorescence signal, and we observed a significant decrease of dimer signal at 450 nm, as shown in Fig. S7b (ESI†).
O stretch, 1601 cm−1 resembles the CC aromatic stretch, 1342 cm−1 may be attributed to the CC indole stretch, and the broad peak around 3250 cm−1 corresponds to the O–H stretching.23 The incorporation of SPIONs within the matrix of tryptophan assembly was further validated by the iron (Fe) signal peak in SEM-EDS spectra, as shown in Fig. S5 (ESI†). Subsequently, the atomic and weight ratio of Carbon to Iron (C:Fe) content in MTNs was measured using SEM-EDS from different portions of the sample. The atomic and weight ratio was consistent all over the sample and was found to be 6.9:1 (C:Fe) and 1.5:1 (C:Fe), respectively. The weight percentage of the Fe content was also measured in the sample and found to be 33.2%, which could provide the approximate content of SPIONs in MTNs. In addition, the fluorescence properties of MTNs were studied to gain further insight into the multi-functionalities of MTNs. Fig. S6 (ESI†) describes the fluorescence emission spectra of the sonicated solution containing MTNs and other soluble products compared to untreated tryptophan. A new emission peak was observed at 450 nm (Fig. S6a, ESI†), which could be ascribed to the formation of tryptophan dimers, as previously reported.23,24 The phenomenon involves the sono-induced oxidative coupling of tryptophan molecules to form hydroxylated monomers dimers. In addition, a shift in emission peaks (Fig. S6b, ESI†) was detected when excited between 400 to 460 nm indicating the presence of various species formed upon sonication. In contrast, the MTNs suspension obtained after centrifugation and resuspension exhibited a weak signal at 450 nm compared to the dissolved MTNs, as shown in Fig. S7a (ESI†). The weaker signal could be likely due to the quenching of signals by magnetic nanoparticles.34–36 SPIONs were added to sonicated tryptophan solution to confirm the quenching of the fluorescence signal, and we observed a significant decrease of dimer signal at 450 nm, as shown in Fig. S7b (ESI†).
Mass spectroscopy and HPLC characterisation were performed to establish the possible mechanism involved in forming MTNs. The mass spectroscopic analysis confirmed the transformation of tryptophan in the presence of SPIONs into different products upon sonication, as shown in Fig. 2e, with some significant peaks ascribed to the possible sonicated product formed depicted in Table 1. Furthermore, it was evident that tryptophan (m/z = 205) transformed into some hydroxylated species ascribed from the peaks at 221 and 237. In addition, a strong signal was observed at an m/z value of 333, unlike the mass spectroscopic result of sonicated tryptophan in the absence of SPIONs,23,24 which could correspond to the fragment of Fe–O attached to tryptophan (SPIONs-Trp) during the high-frequency sonication. However, peaks at m/z values of 407 and 423 were assigned to tryptophan dimers and hydroxylated tryptophan dimers obtained during the process, in agreement with previous studies.
| m/z | Possible molecular weight fragments of sonicated products |
|---|---|
| 205 | Tryptophan (Trp) |
| 221 & 237 | Trp + OH & Trp + 2OH |
| 277 & 333 | Trp + Fe–O & Trp + 2Fe–O |
| 407 | 2Trp |
| 423 | 2Trp + OH |
However, it was observed that a higher proportion of dimerised tryptophan was produced when it was sonicated in the presence of SPIONs compared to the sonication process carried out without SPIONs. The HPLC result also exhibited the transformation of tryptophan (retention time = 3.2 min) into different products, as depicted by two peaks, as shown in Fig. 2f. The first peak at the retention time of 3.5 min could be attributed to the parent tryptophan and other hydroxylated tryptophans. In contrast, the other peak at the retention time of ∼8 min corresponds to the higher molecular weight species formed, including the different dimerised products of tryptophan and SPIONs -Trp products.
After the comprehensive analysis of MTNs, a mechanism for forming MTNs was hypothesised, as shown in Fig. 3. Initially, SPIONs were surface-modified with tryptophan (Step-1) for better colloidal stability, followed by sonication in tryptophan solution (Step-2). During sonication, the surface-modified SPIONs and tryptophan encounter at the interface of the cavitating bubbles (Step-3), triggering the transformation of tryptophan into various products, i.e. dimers and hydroxylated species, as discussed earlier. Some of these sonicated products remain attached to the SPIONs surface (Step-4), forming intermediate hybrid SPIONs, and the remaining SPIONs remain suspended in the reaction mixture. During continuous sonication, the intermediate hybrid SPIONs and the dimers further interact at the bubble interface (Step-5) to form hybrid MTNs by co-assembly upon bubble collapse. Although nanobubbles are less reactive than microbubbles upon sonication,37 in principle, both micro and nano-bubbles can grow under acoustic cavitation and collapse to contribute to nanoparticles formation.
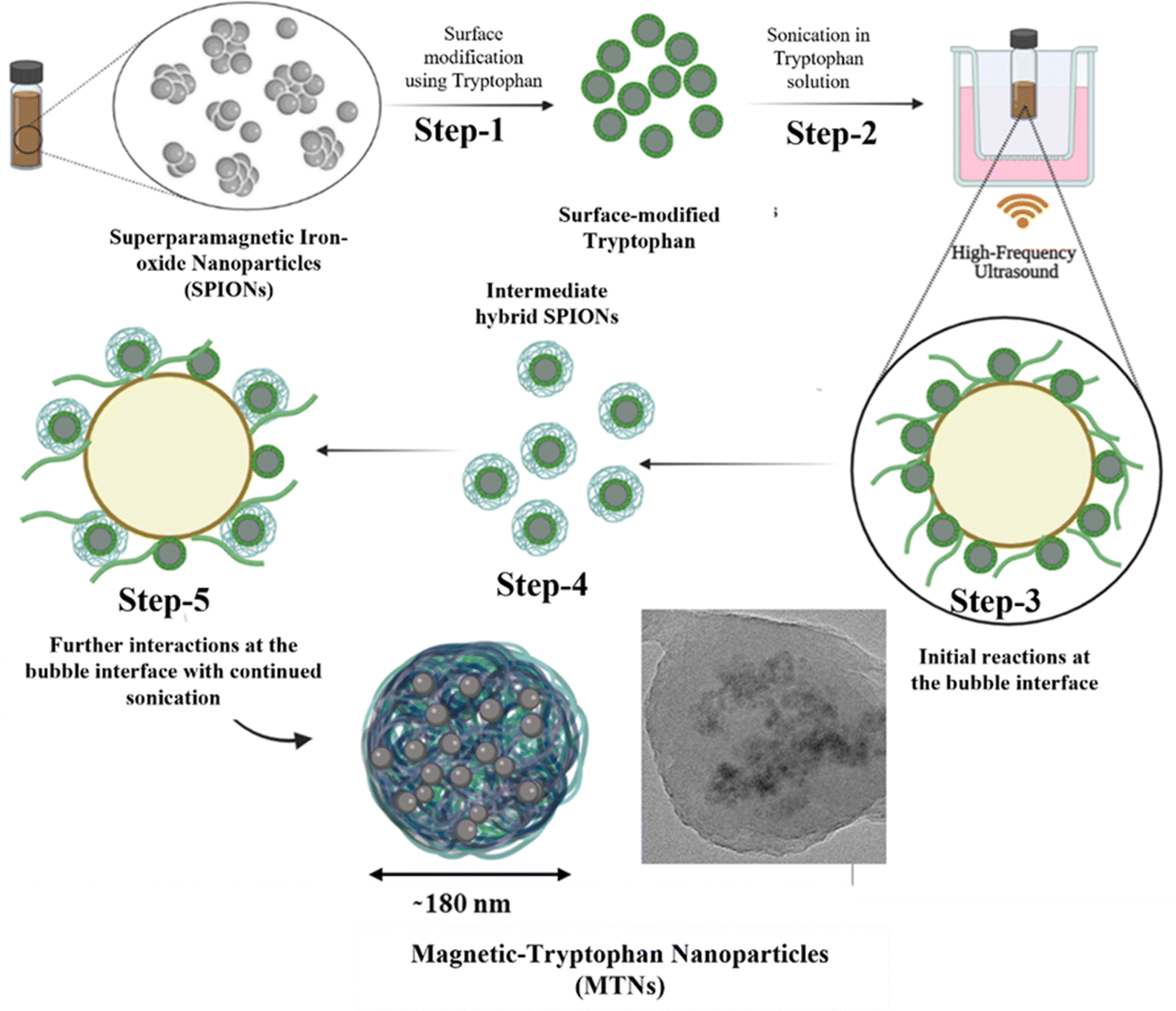 | ||
| Fig. 3 Schematic representing the possible mechanism of formation of magnetic tryptophan nanoparticles starting from surface modification of SPIONs. | ||
Intracellular behaviour of MTNs
Estimating the safety and non-toxicity of all nanoparticle-based drug systems is critical for biomedical applications. Therefore, cell viability was initially analysed using the MTT assay with an MDA-MB-231 cell line after 24 h incubation at different concentrations. MTNs showed minimal cytotoxicity up to 100 μg mL−1 with cell viability of 90%, as shown in Fig. S8a (ESI†). Next, MTNs suspension was analysed under laser confocal microscopy to study the fluorescent signals emitted by MTNs and compared with the tryptophan nanoparticles (TNs), as shown in Fig. S8b and c (ESI†). Upon irradiation, the MTNs showed a weak fluorescence signal (Fig. S8b-I, ESI†) compared to the TNs (Fig. S8c-I, ESI†), which confirmed the quenching effects induced by iron-oxide nanoparticles discussed earlier.33–35To give a deeper insight into the intracellular behavior of MTNs, MDA-MB-231 cells were incubated with MTNs, for 6 h. Then, the non-internalised particles were washed out, the culture medium was replaced, and the incubation continued for 12 h and 24 h. The cells were fixed and analysed under laser confocal microscopy after 6 h, 12 h and 24 h treatment (Fig. S8d–f-II, ESI†). Initially, limited fluorescence emissions were observed from MTNs at an incubation time of 6 h (Fig. S8d, ESI†). However, the intensity of fluorescent emissions gradually increased with incubation time, as shown in Fig. 4a and Fig. S8e and f-II (ESI†). As the intact MTN appeared not fluorescent (Fig. S8b-II, ESI†), the increase in intracellular fluorescence signal may be ascribed to the progressive dissolution of MTNs during intracellular trafficking. To confirm this, a dissolution study was carried out in a test tube by dissolving the MTNs at pH 7.4 in PBS solution at a concentration of 75 μg mL−1. It was found (Fig. 4b) that MTNs emitted fluorescent signals during their dissolution. A progressive increase in fluorescence emission over time was observed, suggesting that the static quenching of tryptophan fluorescence could be attributed to the intermolecular forces of attraction between SPIONs and tryptophan nanoassemblies.38
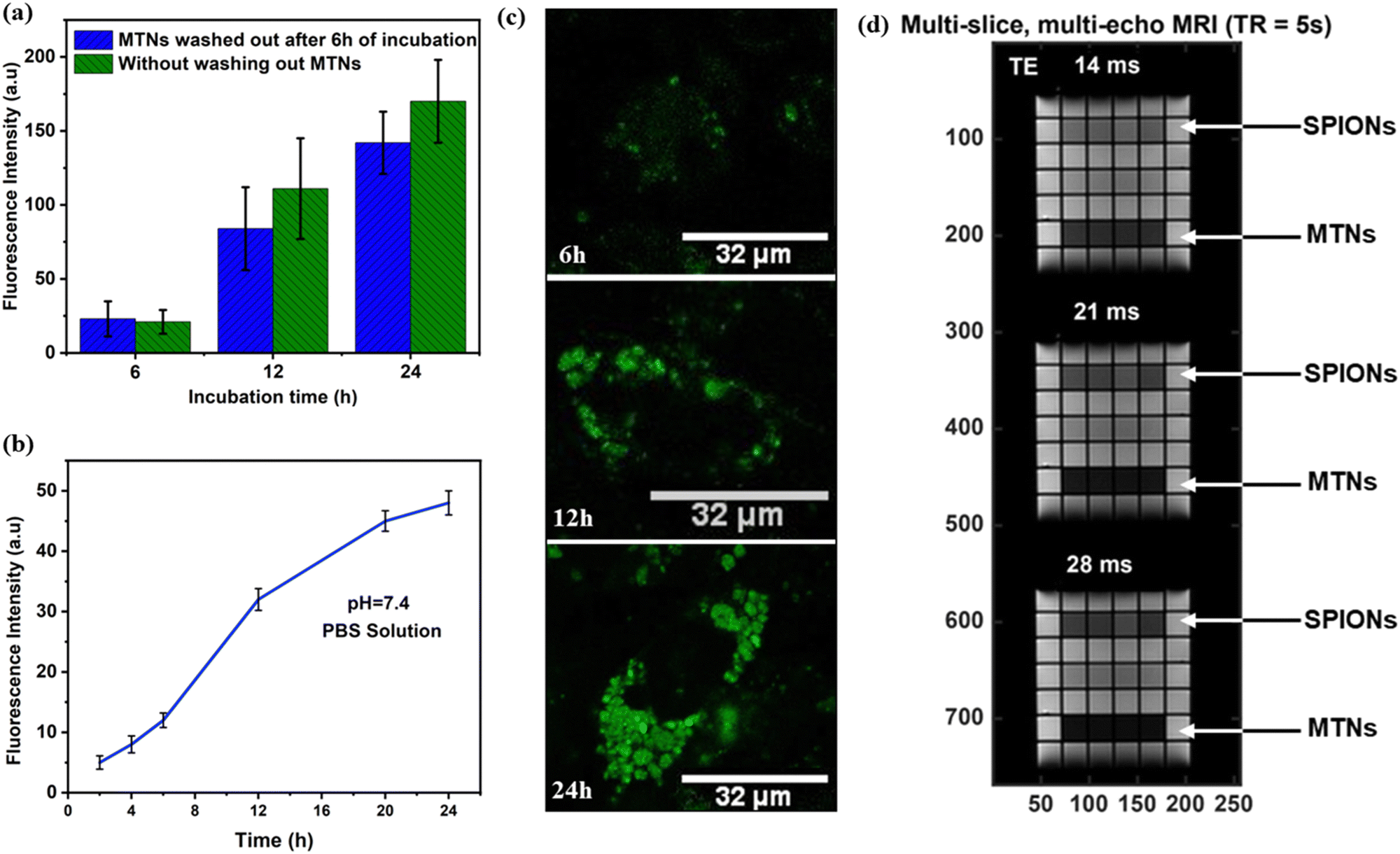 | ||
| Fig. 4 (a) Fluorescence intensity measured from confocal images for MTNs at different incubation times with MDA-MB-231 cell line using two protocols as shown in the graph, and (b) dissolution kinetics of MTNs at 37 °C at pH 7.4 as a function of time suggests that the fluorescent emission increases with the dissolution of MTNs. Confocal microscopy images (Green Channel) of MTNs incubated with MDA-MB-231 cell line (without washing out unbounded MTNs) was acquired at (c) 6 h, (d) 12 h, and (e) 24 h of incubation time. (d) A multi-slice multi-echo (MSME) sequence MRI image with TE from 14 ms to 28 ms with a repetition time (TR) of 5 s, exhibiting the darker images created by MTNs compared to the SPIONs at the same concentration. | ||
In addition, MDA-MB-231 cells were incubated with MTNs continuously without washing out the MTNs from the cell medium and analysed after 6 h, 12 h and 24 h incubation. Similar results were obtained, and an increased intracellular fluorescence intensity with incubation time was observed (Fig. 4c). However, the intensities measured were higher at 12 h and 24 h compared to the experiment set (Fig. 4a), where MTNs were washed out from the cell culture. It could be attributed to more MTNs taken up by cells during incubation and the increasing fluorescence emission during dissolution. Owing to these peculiar fluorescence properties, MTNs could be utilised to monitor the dissolution of MTNs within cells and subsequently could help probe the sustained release of drugs encapsulated inside the MTNs.
MRI analysis of MTNs
SPIONs-based contrast agents are widely used for T2-weighted MRI in medical diagnosis,39–41 where T2 is referred to as the transverse relaxation of water in tissues caused by the loss of phase coherence or order among the protons in the transverse plane. Therefore, due to their superparamagnetic nature, MTNs aqueous suspension at 100 μg mL−1 was used to investigate transverse relaxation (T2) and examine their suitability in MRI applications. MTNs conferred a shorter T2 relaxation of water (12 ms) compared to SPIONs (26 ms) at the same concentration, as shown in Fig. S9 (ESI†). Accordingly, the transverse relaxivity (r2) of MTNs was found to be 83.3 s−1. A multi-slice multi-echo (MSME) sequence with TE from 14 ms to 28 ms is also demonstrated in Fig. 4d, exhibiting the darker images created by MTNs compared to the SPIONs at the same concentration. The shortening of proton T2 for MTNs generated by entrapping SPIONs with tryptophan nanoassemblies could be attributed to several physical phenomena. Firstly, the MTNs presumably tumble in solution slower than monodisperse SPIONs. Therefore, rotational correlation effects could reduce the relaxation time when SPIONs are dispersed in the tryptophan nanoassemblies.42 Secondly, the observed trend may be partially due to the aggregation and ordering of the SPIONs inside the tryptophan matrix of the MTNs.43–45 The encapsulation of SPIONs within the tryptophan matrix considerably lowers the diffusion coefficient of water near the particles. Thus, the matrix allows water protons to interact longer with the strong magnetic field at the particle's surface than the dispersed pristine SPIONs.43–45 However, the mechanism of the interaction is complicated and has not been fully elucidated, but Monte Carlo simulations have demonstrated that the proximity of SPIONs within a cluster, their shape, size and polydispersity can influence the magnetisation processes.46–48 Another possible reason for the shortening of T2 in MTNs could be the inhomogeneous distribution of SPIONs within the tryptophan matrix and variations in the cluster sizes and shapes, as shown in Fig. S10 (ESI†). It would cause magnetisation differences in discrete regions in the MTNs, which may lead to local field gradients that quicken the loss of phase coherence of the spins contributing to the MRI signal producing a decrement in the T2 value.42,49,50These results suggest that hybrid MTNs could be used as a nanocarrier of therapeutic agents due to their non-toxicity, stability, nanometric size and superparamagnetic nature. In addition, the fluorescent properties exhibited by MTNs could be utilised to monitor their dissolution and, thereby, the sustained release of encapsulated drug molecules. MRI analysis demonstrated the suitability of MTNs as T2-based MRI contrast agents for better T2-weighed magnetic resonance images.
Conclusions
We have demonstrated the formation of multifunctional hybrid magnetic tryptophan nanoparticles (MTNs) using high-frequency ultrasound. The mechanism of MTNs formation was elucidated. MTNs showed promising optical and biological properties as potential nanocarriers for hydrophobic and hydrophilic drugs that could be further embedded into the MTNs or readily adsorbed on the surface of the particles. In addition, MTNs exhibited increased image contrast in T2-weighed MRI images, which could be further explored and fine-tuned for advanced MRI and theragnostic applications. Furthermore, MTNs could be investigated for suitability in hyperthermia and targeted drug delivery applications due to their excellent properties and superparamagnetic nature.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Anshul Baral acknowledges the University of Melbourne for offering the Melbourne Research Scholarship (MRS). Francesca Cavalieri acknowledges the RMIT Vice-Chancellor Senior Research Fellowship. The authors also acknowledge the facilities (7T MRI scanner) and scientific and technical assistance of the National Imaging Facility, a National Collaborative Research Infrastructure Strategy (NCRIS) capability, at the Melbourne Brain Centre Imaging Unit and Bio21 Ian Holmes Imaging Centre at The University of Melbourne.References
- S. Rudge, C. Peterson, C. Vessely, J. Koda, S. Stevens and L. Catterall, J. Controlled Release, 2001, 74, 335–340 CrossRef CAS PubMed.
- T. K. Jain, M. A. Morales, S. K. Sahoo, D. L. Leslie-Pelecky and V. Labhasetwar, Mol. Pharmaceutics, 2005, 2, 194–205 CrossRef CAS PubMed.
- C. Alexiou, R. Jurgons, R. J. Schmid, C. Bergemann, J. Henke, W. Erhard, E. Huenges and F. Parak, J. Drug Targeting, 2003, 11, 139–149 CrossRef CAS PubMed.
- W. H. De Jong and P. J. Borm, Int. J. Nanomed., 2008, 3, 133 CrossRef CAS PubMed.
- L. Zhao, H. Yang, T. Amano, H. Qin, L. Zheng, A. Takahashi, S. Zhao, I. Tooyama, T. Murakami and N. Komatsu, J. Mater. Chem. B, 2016, 4, 7741–7748 RSC.
- Y. M. Huh, E. S. Lee, J. H. Lee, Y. W. Jun, P. H. Kim, C. O. Yun, J. H. Kim, J. S. Suh and J. Cheon, Adv. Mater., 2007, 19, 3109–3112 CrossRef CAS.
- Y.-W. Jun, Y.-M. Huh, J.-s Choi, J.-H. Lee, H.-T. Song, S. Kim, S. Yoon, K.-S. Kim, J.-S. Shin and J.-S. Suh, J. Am. Chem. Soc., 2005, 127, 5732–5733 CrossRef CAS PubMed.
- A. Avasthi, C. Caro, E. Pozo-Torres, M. P. Leal and M. L. García-Martín, Surface-modified Nanobiomaterials for Electrochemical and Biomedicine Applications, 2020, pp. 49–91 Search PubMed.
- Y. Lin, S. Wang, Y. Zhang, J. Gao, L. Hong, X. Wang, W. Wu and X. Jiang, J. Mater. Chem. B, 2015, 3, 5702–5710 RSC.
- D. Shigeoka, H. Katayanagi, Y. Moro, S. Kimura, T. Mashino and Y. Ichiyanagi, 2010.
- L. Stanciu, Y.-H. Won, M. Ganesana and S. Andreescu, Sensors, 2009, 9, 2976–2999 CrossRef CAS PubMed.
- J.-K. Yang, J.-H. Yu, J. Kim and Y.-H. Choa, Mater. Sci. Eng., A, 2007, 449, 477–479 CrossRef.
- Y. Xiao and J. Du, J. Mater. Chem. B, 2020, 8, 354–367 RSC.
- M. M. Lin, D. K. Kim, A. J. El Haj and J. Dobson, IEEE Trans. NanoBiosci., 2008, 7, 298–305 Search PubMed.
- C. C. Berry, S. Wells, S. Charles, G. Aitchison and A. S. Curtis, Biomaterials, 2004, 25, 5405–5413 CrossRef CAS PubMed.
- F. Sonvico, S. Mornet, S. Vasseur, C. Dubernet, D. Jaillard, J. Degrouard, J. Hoebeke, E. Duguet, P. Colombo and P. Couvreur, Bioconjugate Chem., 2005, 16, 1181–1188 CrossRef CAS PubMed.
- H. Nosrati, M. Salehiabar, E. Attari, S. Davaran, H. Danafar and H. K. Manjili, Appl. Organomet. Chem., 2018, 32, e4069 CrossRef.
- S. P. Schwaminger, P. F. García, G. K. Merck, F. A. Bodensteiner, S. Heissler, S. Günther and S. Berensmeier, J. Phys. Chem. C, 2015, 119, 23032–23041 CrossRef CAS.
- A. Ebrahiminezhad, Y. Ghasemi, S. Rasoul-Amini, J. Barar and S. Davaran, Colloids Surf., B, 2013, 102, 534–539 CrossRef CAS PubMed.
- A. A. Galhoum, M. G. Mahfouz, A. A. Atia, S. T. Abdel-Rehem, N. A. Gomaa, T. Vincent and E. Guibal, Ind. Eng. Chem. Res., 2015, 54, 12374–12385 CrossRef CAS.
- R. Mathur, R. P. Chauhan, G. Singh, S. Singh, R. Varshney, A. Kaul, S. Jain and A. K. Mishra, J. Mater. Sci.: Mater. Med., 2020, 31, 87 CrossRef CAS PubMed.
- M. A. Tokmedash, E. Seyyedi Zadeh, E. N. Balouchi, Z. Salehi and M. S. Ardestani, Biomed. Mater., 2022, 17, 045026 CrossRef PubMed.
- S. K. Bhangu, G. Bocchinfuso, M. Ashokkumar and F. Cavalieri, Nanoscale Horiz., 2020, 5, 553–563 RSC.
- S. K. Bhangu, M. Ashokkumar and F. Cavalieri, Ultrason. Sonochem., 2020, 63, 104967 CrossRef CAS PubMed.
- S. K. Bhangu, A. Baral, H. Zhu, M. Ashokkumar and F. Cavalieri, Nanoscale Adv., 2021, 3, 4907–4917 RSC.
- A. Baral, S. K. Bhangu, R. Cimino, J. N. B. D. Pelin, W. A. Alves, S. Chattopadhyay, M. Ashokkumar and F. Cavalieri, Nanomaterials, 2020, 10, 1772 CrossRef CAS PubMed.
- M. Ashokkumar, Ultrasonic synthesis of functional materials, Springer, 2016, pp. 17–40 Search PubMed.
- F. Cavalieri, E. Colombo, E. Nicolai, N. Rosato and M. Ashokkumar, Mater. Horiz., 2016, 3, 563–567 RSC.
- S. Meiboom and D. Gill, Rev. Sci. Instrum., 1958, 29, 688–691 CrossRef CAS.
- S. Panja, S. Maji, T. K. Maiti and S. Chattopadhyay, ACS Appl. Mater. Interfaces, 2015, 7, 24229–24241 CrossRef CAS PubMed.
- E. D. Smolensky, H.-Y. E. Park, Y. Zhou, G. A. Rolla, M. Marjańska, M. Botta and V. C. Pierre, J. Mater. Chem. B, 2013, 1, 2818–2828 RSC.
- P. Dong, T. Zhang, H. Xiang, X. Xu, Y. Lv, Y. Wang and C. Lu, J. Mater. Chem. B, 2021, 9, 958–968 RSC.
- T. Yildirim, M. Pervez, B. Li and R. K. O’Reilly, J. Mater. Chem. B, 2020, 8, 5330–5335 RSC.
- P. P. Macaroff, A. R. Simioni, Z. G. M. Lacava, E. C. D. Lima, P. C. Morais and A. C. Tedesco, J. Appl. Phys., 2006, 99, 08S102 CrossRef.
- B.-F. Pan, F. Gao and L.-M. Ao, J. Magn. Magn. Mater., 2005, 293, 252–258 CrossRef CAS.
- J. Li, Y. Hu, J. Yang, W. Sun, H. Cai, P. Wei, Y. Sun, G. Zhang, X. Shi and M. Shen, J. Mater. Chem. B, 2015, 3, 5720–5730 RSC.
- B. Helfield, Y. Zou and N. Matsuura, Front. Phys., 2021, 9, 654374 CrossRef.
- X. Zhuang, T. Ha, H. D. Kim, T. Centner, S. Labeit and S. Chu, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 14241–14244 CrossRef CAS PubMed.
- H. Yang, H. Wang, C. Wen, S. Bai, P. Wei, B. Xu, Y. Xu, C. Liang, Y. Zhang, G. Zhang, H. Wen and L. Zhang, J. Nanobiotechnol., 2022, 20, 98 CrossRef CAS PubMed.
- L. Feng, D. Yang, F. He, S. Gai, C. Li, Y. Dai and P. Yang, Adv. Healthcare Mater., 2017, 6, 1700502 CrossRef PubMed.
- S. Dong, Y. Dong, T. Jia, F. Zhang, Z. Wang, L. Feng, Q. Sun, S. Gai and P. Yang, Chem. Mater., 2020, 32, 9868–9881 CrossRef CAS.
- D. P. Acharya, B. A. Moffat, A. Polyzos, L. Waddington, G. Coia, D. K. Wright, H. X. Wang, G. F. Egan, B. W. Muir and P. G. Hartley, RSC Adv., 2012, 2, 6655–6662 RSC.
- H. Ai, C. Flask, B. Weinberg, X. T. Shuai, M. D. Pagel, D. Farrell, J. Duerk and J. Gao, Adv. Mater., 2005, 17, 1949–1952 CrossRef CAS.
- Y. Zhang, J. Cheng and W. Liu, Sensors, 2019, 19, 3396 CrossRef CAS PubMed.
- C. Paquet, H. W. de Haan, D. M. Leek, H.-Y. Lin, B. Xiang, G. Tian, A. Kell and B. Simard, ACS Nano, 2011, 5, 3104–3112 CrossRef CAS PubMed.
- V. Schaller, G. Wahnström, A. Sanz-Velasco, P. Enoksson and C. Johansson, 2010.
- N. Usov and O. Serebryakova, AIP Adv., 2016, 6, 075315 CrossRef.
- S.-M. Lai, J.-K. Hsiao, H.-P. Yu, C.-W. Lu, C.-C. Huang, M.-J. Shieh and P.-S. Lai, J. Mater. Chem., 2012, 22, 15160–15167 RSC.
- K. Wasser, A. Schnitzer, J. Brade and S. Schoenberg, Der Radiologe, 2010, 50, 1022–1029 CrossRef CAS PubMed.
- C. Yang, G. Lin, C. Zhu, X. Pang, Y. Zhang, X. Wang, X. Li, B. Wang, H. Xia and G. Liu, J. Mater. Chem. B, 2018, 6, 2528–2535 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00137g |
| This journal is © The Royal Society of Chemistry 2023 |