DOI:
10.1039/D2MA01066F
(Paper)
Mater. Adv., 2023,
4, 1354-1362
Effective regulation on catalytic performance of nickel–iron–vanadium layered double hydroxide for urea oxidation via sulfur incorporation†
Received
1st December 2022
, Accepted 25th January 2023
First published on 14th February 2023
Abstract
The effective regulation of catalytic active sites and reaction kinetics has been the key to promoting an efficient urea oxidation reaction (UOR). Herein, well-defined nickel–iron–vanadium layered double hydroxide nanosheets modified by sulfur incorporation (S-NiFeV LDH) on a nickel foam substrate are synthesized by a facile two-step hydrothermal method. Benefiting from the improved intrinsic activity and electrical conductivity derived from sulfur doping, and the large specific surface area of nanosheet architectures, the as-prepared S-NiFeV LDH catalyst shows a superior electrocatalytic performance with a low potential of 1.38 V at the current density of 100 mA cm−2 and the Tafel slope of 30.1 mV dec−1 in 1.0 M KOH and 0.33 M urea electrolyte. In addition, it displays robust stability while operating sustainably for 25 h at 50 mA cm−2 without any distinct activity attenuation. The results of density functional theory (DFT) calculations further indicate that the introduction of sulfur is more conducive to the adsorption of urea molecules on the catalyst surface, and the optimized Gibbs free energy of CO(NH2)2* decomposition and desorption of CO* and NH* in the S-NiFeV LDH catalyst facilitate accelerating the reaction kinetics of the UOR. Accordingly, this work provides a potential strategy for developing highly-efficient electrocatalysts for the UOR.
1. Introduction
With the rapid development of advanced new energy technology, the electrocatalytic urea oxidation reaction (UOR) has emerged as a competitive alternative for several crucial energy-related technologies, involving urea-assisted water splitting, urea-assisted rechargeable Zn–air batteries, and direct urea fuel cells (DUFCs), by virtue of the low thermodynamic potential (0.37 V), abundance and ready availability of the raw materials in wastewater and as by-products of industrial activities.1–5 Unfortunately, the UOR process remains drastically limited by constitutionally slow kinetics stemming from the complex six-electron reaction path (CO(NH2)2 + 6OH− → N2 + 5H2O + CO2+ 6e−) in an alkaline medium and diversified intermediate adsorbed species.6,7 Therefore, it is vital to develop efficient electrocatalysts to drive the UOR process. Currently, noble metal-based oxide catalysts (especially RuO2 and IrO2) are widely employed for the UOR process due to their superior intrinsic performance.8–10 Nevertheless, their practical application in the UOR is seriously restricted by the low global reserves and exorbitant prices of noble metals. Consequently, extensive effort has been devoted to searching for efficient replaceable noble metal oxide electrocatalysts intended to promote the complete oxidation of urea.11,12
As is known to all, it is well demonstrated that NiFe-based catalysts, relating to the corresponding oxides, (oxy)hydroxides, sulfides, selenides, phosphides, and assorted hybrids,13–19 have been explored as excellent substituted materials by virtue of their impressive catalytic performance toward the UOR in recent years. In particular, Ni–Fe layered double hydroxide (LDH) has received more research interest in the UOR due to its unique lamellar structure, tunable intercalation spacing and metal composition, and abundant active sites, in addition to the indispensable synergistic effect of Ni and Fe.20–22 Nevertheless, the obstructions of poor conductivity, limited specific surface area, and low electron transfer rate in the NiFe LDH for the UOR process require further research. A potential improvement strategy is to introduce a third metal into the nanoarchitectures to construct ternary LDH electrocatalysts, such as Cr, Co, Mo, Mn, and V.23–27 These can effectively modulate the d-band density of states (DOS) of intrinsic atoms in favor of intermediates binding, thereby accelerating the sluggish UOR kinetics. For instance, Wang and co-workers reported the Mo and V engineering of a hierarchical ternary NiMoV LDH nanosheet array on NF for an efficient UOR.25 The prepared NiMoV LDH/NF only required a low potential of 1.40 V to deliver 100 mA cm−2, benefiting from the modulated electronic structure of Ni active sites and optimizing the adsorption energy of urea molecules by introducing Mo and V. Moreover, Fan et al. presented Ce modified NiFe LDH nanosheets for urea oxidation.28 The obtained ternary Ce-NiFe LDH catalyst exhibited a superior electrocatalytic performance with a potential of 1.49 V at 10 mA cm−2, which was attributed to the contribution from the regulating lattice and the local electron configuration and the improved electron transfer rate of the ternary LDH. Particularly, it is worth noting that the element vanadium has been explored as a pivotal aspect for improving the UOR performance due to a wide range of valence states, strong electronic interactions, and synergistic effects with other metals in LDH laminates.29–33 Besides, the nickel-based LDH incorporating high-valence vanadium would well stabilize the surface oxygen species and maintain the surface configuration to produce robust stability.31,33 Consequently, there is potential to explore the properties of V-introduced Ni-based LDH catalysts for application in the UOR.
In addition to this, nonmetallic heteroatom doping strategies (such as N, P, and S) have also been evidenced as a promising way for further optimizing the adsorption ability of intermediate species via regulating the electronic structure of metal active sites.34–37 Particularly, it is well known that the NiFe LDH contains a large amount of oxygen, while sulfur (as one of the anion dopants) originates from the same group and possesses similar ionic radii, easing the doping process and anion regulation.38 More importantly, the introduction of sulfide into the NiFe LDH would regulate the covalent and ionic properties of the interaction between the cation and anion, and further modify the overall electronic structure of the active site due to the polarization of the anion.39 A parallel example was provided from Zhang and co-workers, who reported the synthesis of sulfur-doped NiCo carbonate hydroxide (SS-NiCo and S-NiCo) for an efficient UOR process.40 The incorporation of sulfur could modulate the electronic structure of NiCo(OH)2CO3, and the sulfate groups yielding on the catalyst surface could effectively accelerate the interface dynamics of the charge transfer and reaction, which could synergistically promote the intrinsically catalytic performance of the SS-NiCo and S-NiCo. Although heterogeneous atom doping strategies offer potential for improving the performance of the catalysts toward the UOR, the advances in sulfur modulating ternary LDH electrocatalysts for urea oxidation have not yet been specifically studied.
Based on the previously mentioned considerations, in this work, we report the preparation of advanced sulfur doped ternary NiFeV LDH nanosheets (S-NiFeV LDH) on nickel foam (NF) for efficient UORs based on a facile two-step hydrothermal process. Impressively, the obtained self-supported S-NiFeV LDH electrode exhibits an exceptional catalytic performance for the UOR with a potential as low as 1.38 V to produce 100 mA cm−2 current density in 1.0 M KOH and 0.33 M urea electrolyte. Furthermore, the incorporation of sulfur increases the electrical conductivity of the S-NiFeV LDH, which is reflected by the relatively low charge transfer resistance value in comparison to NiFeV undoped with sulfur. In addition, no noticeable performance attenuation is detected during a 25 h chronopotentiometry (CP) stability test at a current density of 50 mA cm−2. The improved catalytic performance could be attributed to the favorable adsorption for CO(NH2)2*, optimized Gibbs free energy for CO* and NH* desorption and increased electrical conductivity of the S-NiFeV LDH, rendering the process a possible competitive alternative for energy-related technologies.
2. Experimental
2.1 Chemicals and materials
Nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O, AR), iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O, AR), vanadium(III) chloride (VCl3, AR), urea (CO(NH2)2, AR), sodium sulfide (Na2S, AR), hydrochloric acid (HCl) and potassium hydroxide (KOH, AR) and nickel foam (NF) were purchased from Shanghai Aladdin and Kunshan Guangjiayuan Co, Ltd. All chemicals and materials were used without any further purification.
2.2 Preparation of S-NiFeV/NiFeV/NiFe LDH on NF
Prior to synthesizing the targeted catalyst, the NF (2.5 × 3 cm−2) was initially pretreated with 1 M HCl in an ultrasonic water bath for 15 min to remove the surface oxide layer and was then rinsed with absolute ethanol and ultrapure water for 5 min each. For a typical fabrication of the NiFeV LDH: 0.70 g Ni(NO3)2·6H2O, 0.16 g Fe(NO3)3·9H2O, 0.06 g VCl3 and 0.3 g urea were dissolved in 35 mL of water in turn under intense ultrasound for 20 min. Subsequently, the acquired clear solution coupled with the pretreated NF was transferred into a 50 mL Teflon-lined stainless steel autoclave, which was sealed and maintained at 120 °C for 12 h in an oven until the end of the reaction and then cooled naturally. The obtained NiFeV LDHs were then subjected to washing with ultrapure water several times, and dried in an oven at 60 °C for 12 h. To prepare the S-NiFeV LDH: 0.114 g of Na2S was dissolved in 35 mL of ultrapure water with intense ultrasound for 15 min. The homogeneous solution coupled with the NiFeV LDH-coated NF was then transferred into a 50 mL Teflon-lined stainless steel autoclave, which was sealed and maintained at 90 °C for 8 h in the oven. After cooling to room temperature, the obtained S-NiFeV LDH was washed with ultrapure water and ethanol several times then dried at 60 °C for 8 h. In comparison, the NiFe LDH was synthesized using the same procedure as for the NiFeV LDH, except without the addition of vanadium sources in the hydrothermal process. The mass loadings of the S-NiFeV LDH, NiFeV LDH and NiFe LDH were determined to be 1.34, 1.30, and 1.03 mg cm−2, respectively.
2.3 Physical characterization
The morphology and structural features of the as-obtained samples were investigated using scanning electron microscopy (SEM, JSM-7800F), transmission electron microscopy and high-resolution transmission electron microscopy images (TEM and HRTEM, JEOL-2100F, 200 kV) coupled with high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDX). In addition, X-ray diffraction (XRD, CuKα, λ = 1.54 Å) patterns were obtained to investigate the crystal information of the as-obtained catalysts at 10° min−1 under 40 kV. X-Ray photoelectron spectroscopy (XPS, Thermo Fisher Nexsa) was employed to analyze the surface elemental states. Inductively coupled plasma–mass spectroscopy (ICP–MS) was used to determine the precise contents of Ni, Fe, V and S.
2.4 Electrochemical measurements
The electrochemical measurements of the as-prepared catalysts were obtained using an electrochemical workstation (CHI660E, Shanghai Chenhua) in a three-electrode system at room temperature. Catalysts directly grown on NF were utilized as the working electrode, while the Ag/AgCl (saturated KCl) and Pt mesh served as the reference and counter electrodes, respectively. All the potentials in this work were calibrated by the reversible hydrogen electrode (RHE) as follows: ERHE = E + EAg/AgCl + 0.059pH. Before the performance evaluation, cyclic voltammetry (CV) was conducted in 1 M KOH and 0.33 M urea solution for 50 cycles with a scan rate of 50 mV s−1 to achieve a stable state. The linear sweep voltammetry (LSV) curves were recorded at a scan rate of 5 mV s−1 in 1.0 M KOH with and without the 0.33 M urea solution. In particular, the results of the LSV curves were exhibited with iR-corrected values unless otherwise stated. Electrochemical impedance spectroscopy (EIS) was performed in the frequency range from 10−1 Hz to 105 Hz. The electrochemical surface area (ECSA) was detected from the electrochemical double layer capacitance (Cdl) values using CV measurements with various scan rates (20, 40, 60, 80, 100 mV s−1) in the non-faradaic region. By plotting the current density at 0.923 V vs. RHE against the scan rate, the obtained Cdl values were further applied to represent the ECSAs as follows: ESCA = Cdl/Cs (Cs = 0.04 mF cm−2). The stability performance of the catalyst was investigated by chronopotentiometry (CP) curves recorded at 50 mA cm−2 in 1.0 M KOH and 0.33 M urea electrolyte for 25 h. Similarly, a commercial RuO2 catalyst supported on NF and bare NF served as a comparison in all tests.
2.5 Computational details
The results of the density functional theory (DFT) calculations were obtained using the Vienna ab initio simulation package (VASP) with the projector-augmented wave (PAW) method.41,42 Moreover, the exchange and correlation terms were described by generalized gradient approximations (GGA) and the Perdew–Burke–Ernzerhof (PBE) functional.8 The DFT+U method (Hubbard-U correction) with Ueff (Coulomb U – exchange J) values of 3.8, 4.3, and 3.4 for Ni, Fe, and V was adopted to describe the S-NiFeV LDH system.29 The cut-off energy was set as 500 eV in the plane-wave expansion and a vacuum thickness of 15 Å was added in the z-direction of the surface. The geometric optimizations were fully relaxed unless the forces and energy converged to −0.02 eV Å−1 and 10−5 eV, respectively. The Gibbs free energy (ΔG) of each reaction step was calculated according to the following expression: ΔG = ΔE + ΔEZPE − TΔS.43
The urea oxidation reaction (UOR) in the alkaline medium would undergo the following reaction:31
| CO(NH2)2* + 2OH− → CO* + 2NH* + 2H2O(l) + 2e− |
| CO* + 2OH− → CO2(g) + H2O(l) + 2e− |
| 2NH* + 2OH−→ N2(g) + 2H2O(l)+ 2e− |
where Δ
E represents the adsorption energy, and Δ
EZPE and
TΔ
S (298.15 K) represent the corrections of the zero-point energy and entropy contributions in each reaction step, respectively.
3. Results and discussion
3.1 Structural characterizations
The commercial NF with an abundant 3D skeleton and superior electrical conductivity is selected as the substrate for synthesizing the self-standing S-NiFeV LDH nanosheets, as schematically illustrated in Fig. 1. The S-NiFeV LDH is grown on the NF using a facile two-step hydrothermal method. Firstly, the NiFeV LDH was synthesized with Ni(NO3)2, Fe(NO3)3, VCl3, and urea as the reactants, and then an anion exchange process occurred in the sulfurization process to obtain the S-NiFeV LDH through partially replacing oxygen with sulfur from Na2S. The crystal structures and the composition of the as-prepared LDH catalysts are initially verified by the XRD patterns, as displayed in Fig. 2. For the NiFe LDH and NiFeV LDH, the distinct diffraction peaks of (006), (012), (015), (110) and (013) are detected following hydrothermal preparation on the NF substrate, which can be well indexed into the standard crystal card (JCPDS no. 40-0215),44,45 demonstrating that the introduction of vanadium would not cause any variations in the original layered structure.29 After sulfurization, in addition to a series of characteristic peaks from the NF substrate, the diffraction peaks of the S-NiFeV LDH are in good agreement with the NiFeV LDH and no other reflections are observed, suggesting the incorporation of sulfur into the crystal and that no phase change is induced by sulfur doping.
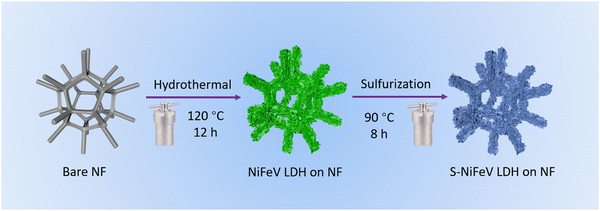 |
| | Fig. 1 Schematic illustration of the preparation process for the S-NiFeV LDH sample. | |
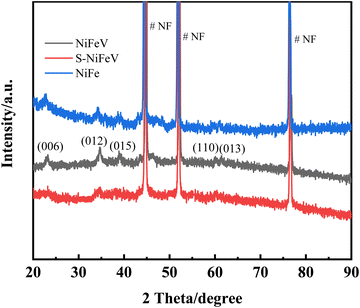 |
| | Fig. 2 XRD patterns of the as-prepared NiFe LDH, NiFeV LDH and S-NiFeV LDH catalysts. | |
The morphology information of the obtained LDH catalysts is revealed by SEM and TEM analysis. As displayed in Fig. 3(a and b) and Fig. S2 (ESI†), both the S-NiFeV LDH and NiFeV LDH show obvious interconnected nanosheet structures growing vertically on the Ni foam substrate, which is associated with the typical morphology of LDH.46 It is worth noting that the S-NiFeV LDH exhibits a rougher surface after hydrothermal sulfurization in comparison to the pure NiFeV LDH, indicating that more active sites can be exposed and a larger specific surface area can be generated, which is conducive to accelerating the kinetic process of the UOR. In addition, the EDS mapping results in Fig. S1 and S2 (ESI†) verify that the corresponding elements (Ni, Fe, V, O, and S) are homogeneously distributed on the as-grown LDH catalysts. The concrete morphological features are further demonstrated by TEM as shown in Fig. 3(c). The typical interconnected nanosheet architectures can be observed and the average thickness of the S-NiFeV LDH nanosheet is determined as approximately 20 nm. The inset of Fig. 3(c) displays the SAED patterns of the S-NiFeV nanosheets with the clear diffraction rings, representing a polycrystalline structure. Furthermore, as observed in the HRTEM image in Fig. 3(d), the S-NiFeV nanosheets show a distinct lattice fringe with an interplanar spacing of 0.268 nm, which is consistent with the interplanar distance of (101) in the NiFe LDH.47 In addition, the STEM image and the corresponding EDX mappings in Fig. 3(e and f) confirm the presence of uniformly distributed Ni, Fe, V and S elements in the S-NiFeV LDH nanosheet sample, suggesting that sulfur and vanadium are successfully incorporated into the NiFe LDH crystal structures. The quantitative atomic ratio of Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe:V:S in the S-NiFeV LDH sample is determined to be 8.93:1.43:1:2.69 using ICP–MS.
:Fe:V:S in the S-NiFeV LDH sample is determined to be 8.93:1.43:1:2.69 using ICP–MS.
 |
| | Fig. 3 (a and b) SEM images of the S-NiFeV LDH catalysts. (c and d) TEM and HRTEM images of the S-NiFeV LDH catalysts; the inset illustrates the SAED pattern. (e and f) HAADF-STEM image and the corresponding EDX mappings of Ni, Fe, S, and V elements in the S-NiFeV LDH. | |
To investigate the surface element valence states and their composition in the as-synthesized S-NiFeV LDH, full-scan and high resolution XPS spectra are performed. As displayed in Fig. 4(a), it confirms the existence of all Ni, Fe, V, S, and O elements in the S-NiFeV LDH, which is well consistent with the EDS mapping results. In the XPS of Ni 2p in Fig. 4(b), the Ni 2p spectrum can be deconvoluted into four clearly characteristic peaks. The binding energies at 856.5 and 874.2 eV are assigned to Ni 2p3/2 and Ni 2p1/2 and two satellite peaks are observed at 862.2 and 880.1 eV, suggesting the presence of Ni2+ in the S-NiFeV LDH.48 For the Fe 2p XPS spectra in Fig. 4(c), the characteristic peaks located at 715.1 and 727.5 eV are attributed to Fe 2p3/2 and Fe 2p1/2, respectively, indicating the existence of Fe3+.29 In the XPS spectra of V 2p (Fig. 4(d)), three different characteristic peaks can be deconvoluted into V 2p3/2 (518.1 eV) and V 2p1/2 (524.6 eV), which are assigned to the V5+ state, while the V 2p3/2 with the binding energy of 516.6 eV is attributed to the V4+ state.49Fig. 4(e) demonstrates the presence of S in the as-synthesized S-NiFeV LDH. The curve-fitting high-resolution S 2p spectra with the binding energy of 163.1 eV is identified as metal–S bonding, indicating that some of the OH− ions are substituted by S2− ions.50 The other peaks with the binding energy of 168.8 eV can be attributed to the oxidized sulphates species.50,51 As for the XPS spectra of O 1s in Fig. 4(f), they can be fitted into two different components. The characteristic peaks located at 531.5 and 533.6 eV are ascribed to the O–H bond and surface-adsorbed oxygen in the S-NiFeV LDH, respectively.38 It is worth noting that since the electronegativity of sulfur is lower than that of oxygen, the substitution of hydroxyl anions by sulfur anions would increase the electron donor ability of the surrounding anionic environment, which means that the introduction of sulfur substances can adjust the electronic structure and further boost the intrinsic activity.38,52,53
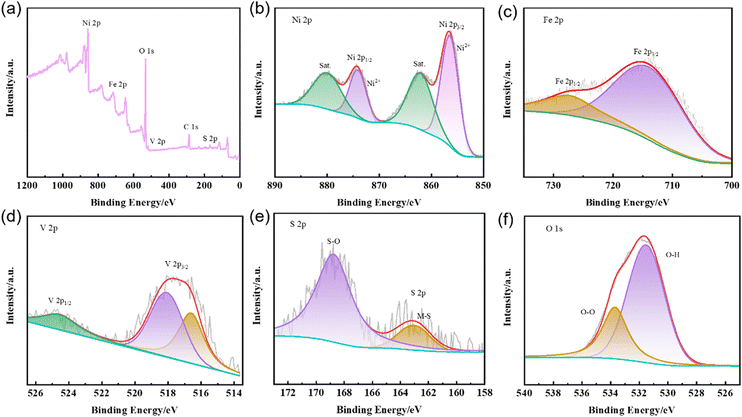 |
| | Fig. 4 (a) Full-scan XPS spectra of the S-NiFeV LDH. High resolution XPS spectra of (b) Ni 2p, (c) Fe 2p, (d) V 2p, (e) S 2p and (f) O 1s in the S-NiFeV LDH. | |
3.2 Electrochemical performance
The electrochemical performance of the as-prepared S-NiFeV LDH catalyst toward the UOR is initially investigated in 1 M KOH and 0.33 M urea electrolyte with a three-electrode system at room temperature as displayed in Fig. 5(a). For comparison purposes, the LSV curve recorded in the absence of urea is also provided. It can be clearly observed that there is an obvious response to the oxygen evolution reaction process for the S-NiFeV LDH electrode in 1.0 M KOH solution without urea, which originates from the oxidation peak of the Ni species (Ni2+/Ni3+).54–57 Depicted in Fig. 5(b) are the LSV curves of the S-NiFeV LDH, NiFeV LDH, and NiFe LDH for the UOR, along with those of the commercial RuO2 and bare NF as a contrast. It is evident that the elaborate S-NiFeV LDH demonstrates a significantly improved UOR performance with a low onset potential of 1.34 V (vs. RHE), beyond which a dramatic increase in current density is determined. The S-NiFeV LDH only requires a potential of 1.38 V to deliver the current density of 100 mA cm−2, which is lower than those of the NiFeV LDH (1.42 V), and NiFe LDH (1.46 V), commercial RuO2 (1.62 V), and some previously reported heterogeneous atom doping systems, as summarized in Table S1 (ESI†), indicating the notably accelerated kinetic process for the UOR. Moreover, the S-NiFeV catalyst growing vertically on the NF substrate reveals an enhanced UOR current density 1.4–2.5-fold higher than those of the NiFeV LDH, and NiFe LDH, RuO2 and bare NF at an applied potential of 1.38 V, which confirms the incorporation of sulfur atoms into the LDH induces a positive catalytic impact on the UOR process. In addition, the improved UOR kinetics can be supported by the determined values of the Tafel slopes, which are derived from the corresponding UOR polarization curves. As revealed in Fig. 5(c), the S-NiFeV LDH catalyst shows the smallest Tafel slope value of 30.1 mV dec−1, against the NiFeV LDH (40.2 mV dec−1), NiFe LDH (66.5 mV dec−1), RuO2 (69.8 mV dec−1), and bare NF (77.1 mV dec−1), suggesting its superior reaction kinetics toward the UOR. In addition, the EIS spectra for various catalysts are also recorded to gain more insight into the charge transfer dynamics during the UOR process. The Nyquist plots and charge transfer resistances (Rct) of the five different specimens are presented in Fig. 5(d), revealing that Rct increases in the order of S-NiFeV LDH (0.54 Ω) < NiFeV LDH (0.68 Ω) < NiFe LDH (0.85 Ω) < RuO2 (1.79 Ω) < bare NF (3.96 Ω). The lowest Rct value of the S-NiFeV LDH is in accordance with its smallest Tafel slope and facilitates boosting the efficiency of electron transfer at the electrode–electrolyte interface. Furthermore, the nanosheet arrays with large surface areas can create more active sites, which also facilitates the diffusion of adsorbed intermediates and gaseous products, thus suppressing the accumulation of evolutionary bubbles during the UOR.58–61
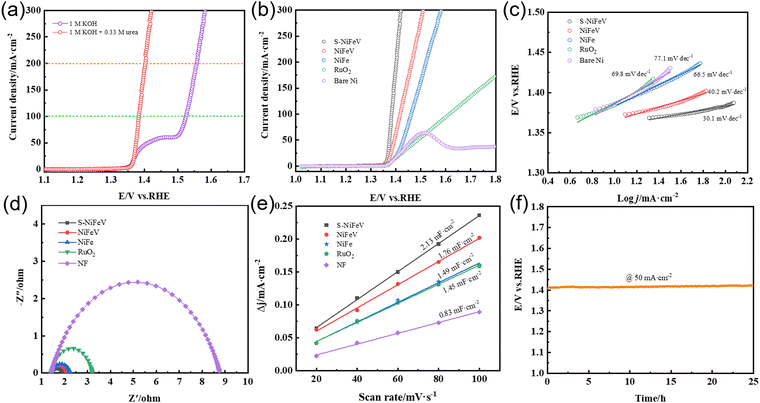 |
| | Fig. 5 (a) Polarization curves for the S-NiFeV LDH measured in 1.0 M KOH with and without 0.33 M urea at a scan rate of 5 mV s−1. (b) LSV curves of various catalysts for the UOR recorded in 1.0 M KOH with 0.33 M urea at a scan rate of 5 mV s−1. (c) Tafel slopes. (d) EIS spectra. (e) Cdl values. (f) Stability measurement. | |
The electrochemical Cdl, which is a crucial response to the real electrochemically active surface area (ECSA) of the prepared catalysts, is acquired by CV curves at scan rates from 20 to 100 mV s−1 as shown in Fig. S3 (ESI†). As demonstrated in Fig. 5(e), the obtained S-NiFeV LDH electrode exhibits the highest Cdl value of 2.13 mF cm−2, with the NiFeV LDH (1.76 mF cm−2) coming next, followed by the NiFe LDH (1.49 mF cm−2) and RuO2 (1.45 mF cm−2), and bare NF (0.83 mF cm−2) is the last. This indicates that the S-NiFeV LDH possesses more active sites than the sulfur-undoped samples along with RuO2 and bare NF. This could be attributed to the formation of a hierarchical nanosheet array morphology with a rougher surface and synergistic effect induced by the introduction of sulfur atoms. To further evaluate the intrinsic activities of the catalysts, the LSV curves of the UOR normalized by the ECSAs are presented in Fig. S4 (ESI†). It is evident from the figure that the current density of the S-NiFeV LDH is still higher in comparison to the NiFeV LDH, NiFe LDH, RuO2, and bare NF. This result further proves the superiority of the as-constructed S-NiFeV LDH nanoarrays in ameliorating the electrochemical performance of the UOR. Long-term stability was another pivotal indicator for evaluating the catalytic performance of the as-synthesized catalysts toward the UOR. Fig. 5(f) presents the chronoamperometry (CP) curve of the S-NiFeV LDH in an alkaline electrolyte at 50 mA cm−2 for 25 h. Here, it is clear that no obvious degradation in potential can be observed for the S-NiFeV LDH after continuously operating for 25 h, suggesting the superior stability toward the UOR. The LSV curve of the S-NiFeV LDH is conducted in fresh electrolyte after the 25 h stability measurement. As displayed in Fig. S5 (ESI†), only a slight difference is evident in the LSV curves. More importantly, the well-preserved nanosheet architectures can be clearly observed from the SEM image in Fig. S6(d) (ESI†), further certifying its excellent mechanical stability. In addition, another XPS analysis is performed to study the changes in surface active sites after the stability test. Fig. S6(a) (ESI†) displays the XPS spectra of Ni 2p in the S-NiFeV LDH. By comparing it with the original NiFeV LDH catalyst, the binding energy of Ni 2p exhibits a small positive shift, and two new characteristic peaks at 856.8 eV and 974.9 eV appear as a result of Ni3+, which is assigned to the formation of NiOOH.62 In addition, the near disappearance of the M–S bonds in S 2p (Fig. S6(b), ESI†) and the sharp increase of the M–O bonds in O 1s (Fig. S6(c), ESI†) imply that the in situ formation of Ni–OOH on the catalyst surface is the real active site for the UOR process.17,57,63
3.3 Theoretical analysis
To gain further insights into the influence of S2− on the S-NiFeV LDH during the UOR electrocatalytic process, Hubbard-U corrected density functional theory (DFT+U) calculations have been performed. As illustrated in Fig. 6(a), the mechanism of action for the S-NiFeV LDH toward the UOR is proposed based on previously reported literature,64,65 and mainly involves the adsorption and activation of urea molecules, the breaking of C–N bonds and the generation of CO2 and N2 molecules. Compared to the original NiFeV LDH surface, a smaller adsorption energy of urea molecules on the S-NiFeV LDH surface is acquired, as shown in Fig. 6(b). This reveals that the surface of the S-NiFeV catalyst adsorbs urea molecules more easily and is conducive to the UOR process. The important role of sulfur in the S-NiFeV LDH is further demonstrated by calculating the Gibbs free energy (ΔG) of each step in the UOR process. As displayed in Fig. 6(c), the decomposition of CO(NH2)2* into CO* and NH2* with the largest ΔG change corresponds to the rate determining step (RDS). In comparison to the NiFeV LDH (5.72 eV), the S-NiFeV LDH shows a relatively low ΔG value of 4.40 eV, indicating that the introduction of sulfur can dramatically promote the desorption of CO* and NH* intermediates and can accelerate the slow kinetics of the UOR process.31 Moreover, the densities of states (DOS) of both the pristine NiFeV LDH and the sulfur-doped NiFeV LDH are calculated to investigate the effect of sulfur on the electronic structure in Fig. 6(d) and Fig. S7 (ESI†). The Fermi surface of the NiFeV LDH with sulfur modification obviously shifts to the conduction band edge relative to the pristine NiFeV LDH, suggesting a higher conductivity for inducing a faster electron transfer and a better electrochemical performance toward the UOR.8,66 All above results indicate that the introduction of sulfur can regulate the adsorption of urea molecules and the desorption ability of the CO* and NH* intermediates, and improve the electrical conductivity, further enhancing the retarded kinetics and catalytic properties of the UOR.
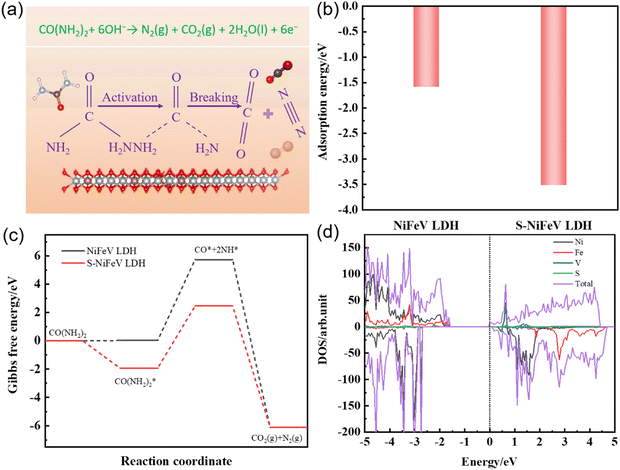 |
| | Fig. 6 (a) Proposed mechanism of action for the as-prepared catalysts toward the UOR. (b) Adsorption energy of urea molecules on the NiFeV LDH and the S-NiFeV LDH catalysts. (c) Reaction free energy of the UOR on the NiFeV LDH and the S-NiFeV LDH surface. (d) Density of states (DOS) of the S-NiFeV LDH sample. | |
4. Conclusions
In conclusion, the self-standing S-NiFeV LDH nanosheet electrocatalyst on an NF substrate is prepared by a facile two-step hydrothermal method. Such an electrode displays an excellent electrocatalytic performance with a very low potential of 1.38 V at 100 mA cm−2 and a Tafel slope of 30.1 mV dec−1, outperforming numerous previously reported UOR electrocatalysts in 1.0 M KOH and 0.33 M urea electrolyte. This superior property can be attributed to the robust nanosheet array architecture, abundant active sites, rapid mass transport, modified electronic structures and improved electrical conductivity, which is achieved by incorporating sulfur. The DFT calculation results also reveal that the presence of sulfur reduces the adsorption energy of the urea molecules on the catalyst surface, promotes the desorption of the CO* and NH* intermediates, and further accelerates the slow kinetics of the UOR process. Therefore, this work provides new insights for rationally designing highly effective non-noble metal catalysts for the UOR through a nonmetallic heteroatom doping strategy.
Author contributions
Kai Peng: original draft, formal analysis, methodology, software. Liyan Liu: editing, formal analysis. Narayanamoorthy Bhuvanendran: reviewing and editing, data curation. Fen Qiao: reviewing and editing, software. Guangping Lei: reviewing and editing, software. Sae Youn Lee: reviewing and editing. Qian Xu: formal analysis, reviewing. Huaneng Su: reviewing, supervision, funding acquisition.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2018YFE0121200), the National Natural Science Foundation of China (No. 21676126), and the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions.
References
- S. Ni, H. Qu, Z. Xu, X. Zhu, H. Xing, L. Wang, J. Yu, H. Liu, C. Chen and L. Yang, Appl. Catal., B, 2021, 299, 120638 CrossRef CAS.
- H. Xu, K. Ye, K. Zhu, Y. Gao, J. Yin, J. Yan, G. Wang and D. Cao, Inorg. Chem. Front., 2021, 8, 2788–2797 RSC.
- Q. Li, N. Li, J. An and H. Pang, Inorg. Chem. Front., 2020, 7, 2089–2096 RSC.
- B. Huang, Z. Sun and G. Sun, eScience, 2022, 2, 243–277 CrossRef.
- Y. Yang, D. Wu, R. Li, P. Rao, J. Li, P. Deng, J. Luo, W. Huang, Q. Chen, Z. Kang, Y. Shen and X. Tian, Appl. Catal., B, 2022, 317, 121796 CrossRef CAS.
- G. Ma, Q. Xue, J. Zhu, X. Zhang, X. Wang, H. Yao, G. Zhou and Y. Chen, Appl. Catal., B, 2020, 265, 118567 CrossRef CAS.
- L. Zhang, L. Wang, H. Lin, Y. Liu, J. Ye, Y. Wen, A. Chen, L. Wang, F. Ni, Z. Zhou, S. Sun, Y. Li, B. Zhang and H. Peng, Angew. Chem., Int. Ed., 2019, 58, 16820–16825 CrossRef CAS PubMed.
- Y. Tong, P. Chen, M. Zhang, T. Zhou, L. Zhang, W. Chu, C. Wu and Y. Xie, ACS Catal., 2018, 8, 1–7 CrossRef.
- S.-K. Geng, Y. Zheng, S.-Q. Li, H. Su, X. Zhao, J. Hu, H.-B. Shu, M. Jaroniec, P. Chen, Q.-H. Liu and S.-Z. Qiao, Nat. Energy, 2021, 6, 904–912 CrossRef CAS.
- B. Zhu, Z. Liang and R. Zou, Small, 2020, 16, 1906133 CrossRef CAS PubMed.
- Q. Zhang, F. M. D. Kazim, S. Ma, K. Qu, M. Li, Y. Wang, H. Hu, W. Cai and Z. Yang, Appl. Catal., B, 2021, 280, 119436 CrossRef CAS.
- M. P. Browne, Z. Sofer and M. Pumera, Energy Environ. Sci., 2019, 12, 41–58 RSC.
- H. Sun, W. Zhang, J.-G. Li, Z. Li, X. Ao, K.-H. Xue, K. K. Ostrikov, J. Tang and C. Wang, Appl. Catal., B, 2021, 284, 119740 CrossRef CAS.
- P. Babar, A. Lokhande, V. Karade, I. J. Lee, D. Lee, S. Pawar and J. H. Kim, J. Colloid Interface Sci., 2019, 557, 10–17 CrossRef CAS PubMed.
- L. Chen, H. Wang, L. Tan, D. Qiao, X. Liu, Y. Wen, W. Hou and T. Zhan, J. Colloid Interface Sci., 2022, 618, 141–148 CrossRef CAS PubMed.
- W. Zhu, Z. Yue, W. Zhang, N. Hu, Z. Luo, M. Ren, Z. Xu, Z. Wei, Y. Suo and J. Wang, J. Mater. Chem. A, 2018, 6, 4346–4353 RSC.
- K. Wang, M. Hou, W. Huang, Q. Cao, Y. Zhao, X. Sun, R. Ding, W. Lin, E. Liu and P. Gao, J. Colloid Interface Sci., 2022, 615, 309–317 CrossRef CAS PubMed.
- M. Sun, C. Yuan, R.-T. Gao, R. Zhang, X. Liu, T. Nakajima, X. Zhang, Y. Su and L. Wang, Chem. Eng. J., 2021, 426, 131062 CrossRef CAS.
- S. Feng, J. Luo, J. Li, Y. Yu, Z. Kang, W. Huang, Q. Chen, P. Deng, Y. Shen and X. Tian, Mater. Today Phys., 2022, 23, 100646 CrossRef CAS.
- S. A. Lee, J. W. Yang, T. H. Lee, I. J. Park, C. Kim, S. H. Hong, H. Lee, S. Choi, J. Moon, S. Y. Kim, J. Y. Kim and H. W. Jang, Appl. Catal., B, 2022, 317, 121765 CrossRef CAS.
- Y. Wang, Y. Liu, M. Zhang, B. Liu, Z. Zhao and K. Yan, Sci. China Mater., 2022, 65, 1805–1813 CrossRef CAS.
- X. Zhao, Y. Wang, Y. Zhang, S. Luo, H. Zhang and D. Y. C. Leung, ChemSusChem, 2022, 15, e202102614 CAS.
- Z. Ji, Y. Song, S. Zhao, Y. Li, J. Liu and W. Hu, ACS Catal., 2022, 12, 569–579 CrossRef CAS.
- Y. Zheng, K. Sun, J. Pang, J. Hou, G. Wang, W. Guo, L. Wang, X. Guo and L. Chen, J. Alloys Compd., 2022, 925, 166754 CrossRef CAS.
- Z. Wang, W. Liu, J. Bao, Y. Song, X. She, Y. Hua, G. Lv, J. Yuan, H. Li and H. Xu, Chem. Eng. J., 2022, 430, 133100 CrossRef CAS.
- Z. Wang, W. Liu, Y. Hu, M. Guan, L. Xu, H. Li, J. Bao and H. Li, Appl. Catal., B, 2020, 272, 118959 CrossRef CAS.
- H. Sun, L. Chen, Y. Lian, W. Yang, L. Lin, Y. Chen, J. Xu, D. Wang, X. Yang, M. H. Rümmerli, J. Guo, J. Zhong, Z. Deng, Y. Jiao, Y. Peng and S. Qiao, Adv. Mater., 2020, 32, 2006784 CrossRef PubMed.
- J. Fan and X. Du, Dalton Trans., 2022, 51, 8240–8248 RSC.
- P. Li, X. Duan, Y. Kuang, Y. Li, G. Zhang, W. Liu and X. Sun, Adv. Energy Mater., 2018, 8, 1703341 CrossRef.
- K. Fan, H. Chen, Y. Ji, H. Huang, P. M. Claesson, Q. Daniel, B. Philippe, H. Rensmo, F. Li, Y. Luo and L. Sun, Nat. Commun., 2016, 7, 11981 CrossRef CAS PubMed.
- H. Sun, L. Li, H.-C. Chen, D. Duan, M. Humayun, Y. Qiu, X. Zhang, X. Ao, Y. Wu, Y. Pang, K. Huo, C. Wang and Y. Xiong, Sci. Bull., 2022, 67, 1763–1775 CrossRef CAS PubMed.
- Q. Cao, Y. Yuan, K. Wang, W. Huang, Y. Zhao, X. Sun, R. Ding, W. Lin, E. Liu and P. Gao, J. Colloid Interface Sci., 2022, 618, 411–418 CrossRef CAS PubMed.
- R.-Q. Li, Q. Liu, Y. Zhou, M. Lu, J. Hou, K. Qu, Y. Zhu and O. Fontaine, J. Mater. Chem. A, 2021, 9, 4159–4166 RSC.
- D. Zhu, H. Zhang, J. Miao, F. Hu, L. Wang, Y. Tang, M. Qiao and C. Guo, J. Mater. Chem. A, 2022, 10, 3296–3313 RSC.
- J. Zhang, S. Huang, P. Ning, P. Xin, Z. Chen, Q. Wang, K. Uvdal and Z. Hu, Nano Res., 2022, 15, 1916–1925 CrossRef CAS.
- J. Kang, F. Yang, C. Sheng, H. Xu, J. Wang, Y. Qing, Y. Wu and X. Lu, Small, 2022, 18, 2200950 CrossRef CAS PubMed.
- Y. Yu, Q. Chen, J. Li, P. Rao, R. Li, Y. Du, C. Jia, W. Huang, J. Luo, P. Deng, Y. Shen and X. Tian, J. Colloid Interface Sci., 2022, 607, 1091–1102 CrossRef CAS PubMed.
- Z. Wan, Z. Ma, H. Yuan, K. Liu and X. Wang, ACS Appl. Energy Mater., 2022, 5, 4603–4612 CrossRef CAS.
- B.-Q. Li, S.-Y. Zhang, C. Tang, X. Cui and Q. Zhang, Small, 2017, 13, 1700610 CrossRef PubMed.
- Z. Zhang, J. Yang, J. Liu, Z.-G. Gu and X. Yan, Electrochim. Acta, 2022, 426, 140792 CrossRef CAS.
- X. Jia, H. Kang, X. Yang, Y. Li, K. Cui, X. Wu, W. Qin and G. Wu, Appl. Catal., B, 2022, 312, 121389 CrossRef CAS.
- H. Jiang, M. Sun, S. Wu, B. Huang, C.-S. Lee and W. Zhang, Adv. Funct. Mater., 2021, 31, 2104951 CrossRef CAS.
- G. Wang, J. Chen, Y. Li, J. Jia, P. Cai and Z. Wen, Chem. Commun., 2018, 54, 2603–2606 RSC.
- H. Zhang, X. Li, A. Hähnel, V. Naumann, C. Lin, S. Azimi, S. L. Schweizer, A. W. Maijenburg and R. B. Wehrspohn, Adv. Funct. Mater., 2018, 28, 1706847 CrossRef.
- Y. Liang, J. Wang, D. Liu, L. Wu, T. Li, S. Yan, Q. Fan, K. Zhu and Z. Zou, J. Mater. Chem. A, 2021, 9, 21785–21791 RSC.
- D. P. Sahoo, K. K. Das, S. Mansingh, S. Sultana and K. Parida, Coord. Chem. Rev., 2022, 469, 214666 CrossRef CAS.
- S. Y. Jung, S. Kang, K. M. Kim, S. Mhin, J. C. Kim, S. J. Kim, E. Enkhtuvshin, S. Choi and H. Han, Appl. Surf. Sci., 2021, 568, 150965 CrossRef CAS.
- Y. Li and C. Zhao, ACS Catal., 2017, 7, 2535–2541 CrossRef CAS.
- M. Arif, G. Yasin, L. Luo, W. Ye, M. A. Mushtaq, X. Fang, X. Xiang, S. Ji and D. Yan, Appl. Catal., B, 2020, 265, 118559 CrossRef CAS.
- L.-M. Cao, J.-W. Wang, D.-C. Zhong and T.-B. Lu, J. Mater. Chem. A, 2018, 6, 3224–3230 RSC.
- W. Xu, J. Chen, M. Yu, Y. Zeng, Y. Long, X. Lu and Y. Tong, J. Mater. Chem. A, 2016, 4, 10779–10785 RSC.
- X. Zhu, X. Dou, J. Dai, X. An, Y. Guo, L. Zhang, S. Tao, J. Zhao, W. Chu, X. C. Zeng, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2016, 55, 12465–12469 CrossRef CAS PubMed.
- L. Yang, H. Qin, Z. Dong, T. Wang, G. Wang and L. Jiao, Small, 2021, 17, 2102027 CrossRef CAS PubMed.
- C. Chen, L. Jin, L. Hu, T. Zhang, J. He, P. Gu, Q. Xu and J. Lu, J. Colloid Interface Sci., 2022, 628, 1008–1018 CrossRef CAS PubMed.
- Y. Ma, D. Leng, X. Zhang, J. Fu, C. Pi, Y. Zheng, B. Gao, X. Li, N. Li, P. K. Chu, Y. Luo and K. Huo, Small, 2022, 2203173 CrossRef CAS PubMed.
- Y. Gan, Z. Li, Y. Ye, X. Dai, F. Nie, X. Yin, Z. Ren, B. Wu, Y. Cao, R. Cai, X. Zhang and W. Song, ChemSusChem, 2022, 15, e202201205 CrossRef CAS PubMed.
- N. Chen, Y.-X. Du, G. Zhang, W.-T. Lu and F.-F. Cao, Nano Energy, 2021, 81, 105605 CrossRef CAS.
- X. Xu, J. Li, C. Zhang, S. Zhang, G. Su, Z. Shi, H. Wang and M. Huang, Appl. Catal., B, 2022, 319, 121949 CrossRef CAS.
- X. Xu, X. Hou, P. Du, C. Zhang, S. Zhang, H. Wang, A. Toghan and M. Huang, Nano Res., 2022, 15, 7124–7133 CrossRef CAS.
- C. Liang, P. Zou, A. Nairan, Y. Zhang, J. Liu, K. Liu, S. Hu, F. Kang, H. J. Fan and C. Yang, Energy Environ. Sci., 2020, 13, 86–95 RSC.
- L. Jiang, N. Yang, C. Yang, X. Zhu, Y. Jiang, X. Shen, C. Li and Q. Sun, Appl. Catal., B, 2020, 269, 118780 CrossRef CAS.
- X. Gao, X. Liu, W. Zang, H. Dong, Y. Pang, Z. Kou, P. Wang, Z. Pan, S. Wei, S. Mu and J. Wang, Nano Energy, 2020, 78, 105355 CrossRef CAS.
- T. Yu, Q. Xu, J. Chen, G. Qian, X. Zhuo, H. Yang and S. Yin, Chem. Eng. J., 2022, 449, 137791 CrossRef CAS.
- L. Lv, Z. Li, H. Wan and C. Wang, J. Colloid Interface Sci., 2021, 592, 13–21 CrossRef CAS PubMed.
- C. Wang, H. Lu, Z. Mao, C. Yan, G. Shen and X. Wang, Adv. Funct. Mater., 2020, 30, 2000556 CrossRef CAS.
- R. Tian, S. Zhao, J. Li, Z. Chen, W. Peng, Y. He, L. Zhang, S. Yan, L. Wu, R. Ahuja and H. Gou, J. Mater. Chem. A, 2021, 9, 6469–6475 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Fen
Qiao
b,
Fen
Qiao





