
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanochemical synthesis of Li-rich (Li2Fe)SO cathode for Li-ion batteries†
M. A. A.
Mohamed
 *ab,
H. A. A.
Saadallah
ab,
I. G.
Gonzalez-Martinez
a,
M.
Hantusch
a,
M.
Valldor
c,
B.
Büchner
a,
S.
Hampel
a and
N.
Gräßler
*a
*ab,
H. A. A.
Saadallah
ab,
I. G.
Gonzalez-Martinez
a,
M.
Hantusch
a,
M.
Valldor
c,
B.
Büchner
a,
S.
Hampel
a and
N.
Gräßler
*a
aLeibniz Institute for Solid State and Materials Research (IFW) Dresden e.V., Helmholtzstraße 20, D-01069 Dresden, Germany. E-mail: n.graessler@ifw-dresden.de; m.a.a.mohamed@ifw-dresden.de
bDepartment of Physics, Faculty of Science, Sohag University, 82524 Sohag, Egypt
cDepartment of Chemistry, University of Oslo, Postbox 1033, Blindern, N - 0315 Oslo, Norway
First published on 31st March 2023
Abstract
Li-rich antiperovskite (Li2Fe)SO with its high specific capacity is an attractive cathode material for Li-ion battery applications. While many battery materials depend on hazardous substances and their production is also rarely sustainable, we present an environmentally friendly and sustainable approach for the synthesis of Li-rich (Li2Fe)SO using mechanochemistry based on ball milling. This one step process enables preparing a large quantity of phase-pure (Li2Fe)SO using low-cost and non-toxic precursors, making it a viable alternative to current solid state synthetic method in terms of simplicity, laboratory safety and scalability. The obtained micro-sized particles are nearly spherical and have a small size distribution. To control the crystallinity and reduce the intrinsic defects of the ball-milled (Li2Fe)SO material, a post-heat treatment procedure was tested. Thermodynamic measurements confirmed the high thermal stability of the ball-milled (Li2Fe)SO material. Increasing the ball to powder weight ratio was found to be an effective strategy to decrease the milling time required for the synthesis, thus promoting energy saving. Overall, this work provides a practical guide for the green and scalable production of (Li2Fe)SO cathode material, as well as a method for particle modification for improved electrochemical properties.
1. Introduction
The demand for Li-ion batteries (LIBs) in the market of portable electronics and electric vehicles is steadily growing due to their high energy density.1–3 For the further development of LIBs, research is being conducted on cathode materials that combine low cost, sustainability and high performance.1,3 Despite their great commercial success, current cathode materials such as LiCoO2 (LCO), LiNi1/3Mn1/3Co1/3O2, (NMC 111), LiNi0.8Co0.15Al0.05O2 (NCA), LiMn2O4 (LMO) and LiFePO4 (LFP) have specific limitations.1,3 The recently reported (Li2Fe)SO cathode, which belongs to the Li-rich antiperovskites with the general formula (Li2TM)ChO (TM = Fe, Mn and Ch = S, Se), is a promising alternative with superior theoretical and practical specific capacities due to its multi-electron storage through cationic and anionic redox processes.4–7 In addition, the antiperovskite (Li2Fe)SO has low cost and toxicity compared to Co-containing cathode materials, which can reduce the cost and environmental footprint of Li-ion batteries.8–11 Numerous studies show approaches for recycling of Li-ion batteries including sulfur-containing materials as well.12–14 The preparation of Li-rich (Li2Fe)SO using non-toxic and low-cost raw materials such as lithium oxide, sulfur, and iron in a one-step solid-state reaction (SSR) has been reported.4,5 This process is characterized by its simplicity and the absence of hazardous chemicals or solvents.15 However, the solid-state method has two major disadvantages. The high temperature required for the reaction results in high energy consumption, and it is difficult to control the morphology and size distribution of the particles, which affect the electrochemical properties. Herein, we propose a new approach to prepare Li-rich (Li2Fe)SO material using mechanochemistry (MC) by ball-milling (BM) to overcome the existing limitations in the SSR method.16–18 In general, MC synthesis is recognized for its sustainability that can satisfy several points of the twelve principles of Green Chemistry.19 In particular, the high laboratory safety, low production cost, high reproducibility, easily accessible and scalable equipment make it attractive for both laboratory and industrial applications.19 We have monitored the mechanochemical synthesis of (Li2Fe)SO by ex situ XRD measurements. It was found that several grams of phase-pure (Li2Fe)SO, consisting of particles with primary micrometric size, can be obtained directly by ball milling. In contrast to the SSR method, the reaction is performed at room temperature, which is beneficial for energy saving and safety aspects. Ex situ XRD measurements showed also low crystallinity of the ball-milled (Li2Fe)SO sample, which comes from residual defects and amorphization caused by mechanical energy input.15 To tune the structural and morphological properties of (Li2Fe)SO cathode material, we investigated the effect of post-heat treatment as a guideline for improved electrochemical properties.2. Results and discussion
2.1. One-step mechanochemical synthesis
Previously, the preparation of (Li2Fe)SO was carried out via SSR method, requiring several steps as seen Fig. 1a.5 In SSR method, the raw materials (Li2O, Fe, and S) are mixed in an Ar-filled glovebox using an agate mortar and filled in a corundum crucible. The crucible is placed inside a silica tube which is temporarily closed by a rubber stopper. Afterwards, the silica tube is transferred outside the glovebox, evacuated, refilled with Ar gas to adjust the internal pressure to 200 mbar, and finally melt-sealed by a gas burner. The sealed ampoule is slowly heated up in a furnace (heating rate: 50 °C h−1) to 750 °C and held at this temperature for 2–10 h. After the reaction, the ampoule is quenched in water which is necessary to prevent the formation of impurities. Finally, the silica ampule is inserted again in the glovebox and broken to extract the final product. | ||
| Fig. 1 Schematic diagram of the synthesis procedures of (Li2Fe)SO by the reported solid state reaction (a) and the novel mechanochemical (b) methods. | ||
In contrast, MC synthesis of (Li2Fe)SO involves the steps shown in Fig. 1b. The raw powders (Li2O, Fe, and S) together with the stainless steels balls are loaded to the jar (milling reactor) inside the glovebox. Then, the closed jar is transferred to the milling machine and the reaction is performed. Eventually, the jar is opened inside the glovebox and the product is extracted. In MC synthesis, the mechanical energy due to collision events between balls and powder precursors results in continuous deformation, flattening, fracture and welding of the raw materials.20,21 As a result, the nano-scaled composite material, formed from the selected constituents, contains defects, amorphous and/or metastable phases.20 In the nano-scaled composite of powders, chemical reactions are induced across the grain boundaries of adjacent reactants due to local and temporary instants of mechanical friction.20 These circumstances enhance the reaction kinetics by (i) reduction of the atoms diffusion length through formation of nanoscale grain size (ii) decreasing the reaction threshold energy induced by high residual energy in the form of structural defects (iii) continuous generation of fresh reactive surfaces by repeated fraction and welding of the raw materials.20,21 As a result, MC approach can permit chemical reactions that normally happens at elevated temperature to be induced near-room temperature. There are a lot of variables that can influence the milling process such as the kind of the mill, the characteristics of the milling media (size, mass, mechanical strength and composition), milling media to sample ratio, filling degree of the milling chamber, milling atmosphere, milling speed, and milling time.15 These parameters can change the nature of collision such as the stress frequency, stress energy and the quantity of trapped powder between the colliding particles, which eventually determine the amount of energy transfer.15 The total amount of transferred energy (Et) during milling process can be given as Et = SN ×  , where SN is the number of stress events and
, where SN is the number of stress events and 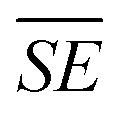 is the average stress energy.15 The average stress energy
is the average stress energy.15 The average stress energy  represents the maximum transferred energy to the reactants during a single collision and can be calculated as
represents the maximum transferred energy to the reactants during a single collision and can be calculated as  , where VR and (m1 and m2) are the relative velocity and the masses of the colliding bodies, respectively.15 The MC synthesis of (Li2Fe)SO offers significant advantages in various aspects compared to SSR as follows:
, where VR and (m1 and m2) are the relative velocity and the masses of the colliding bodies, respectively.15 The MC synthesis of (Li2Fe)SO offers significant advantages in various aspects compared to SSR as follows:
(i) Safety and environmental impact: MC reaction is performed in a tight stainless-steel chamber without usage of any external heating, which guarantees safe handling together with lower risk of explosion or gas release during the reaction compared to SSR method. Possible explosion of the silica tube used in SSR before completion of the reaction may be accompanied by release of volatile S gas (and possibly toxic H2S because of a reaction with moist air at high reaction temperature) which is a risk on both the human health and environment. Besides, additional thermal quenching in water at high temperature (750 °C) is required to reduce the formed impurities during synthesis by SSR.5 This sudden thermal perturbation exaggerates the risk of explosion.
(ii) Energy consumption: In this work, (Li2Fe)SO is synthesised directly by MC without the need for any external thermal energy. This advantageous and not common feature allows controlling and reducing the consumed electrical energy by controlling the milling conditions such as the BPR or rotation velocity.22 In contrast, SSR includes heating the sample to high temperature (750 °C) for long time with difficulty to optimize or reduce the consumed electrical energy.5 Moreover, additional amount of energy is required in SSR to adjust the internal pressure by rotary pump.5 It is worth mentioning that quantitative comparison of the consumed energy between MC (new approach) and reported SSR (highly optimized) approaches would not be fair due to (i) the different degree of optimizations, (ii) the different mass production (∼1 g in SSR and ∼24 g in MC), and (iii) the overestimation of the milling time by cooling of the reaction and relaxation of active amorphous phases during the here adopted step-by-step XRD protocol.
(iii) Monitoring of the reaction: The possibility of in situ and ex situ monitoring of the chemical reaction in MC method by several techniques gives opportunity to understand the intermediate chemical steps and mechanism.19 These techniques can provide valuable information about potential over-pressure inside the milling container and the volatile gases, which can be helpful in increasing the safety issue.19 Although, ex situ technique is only applied here, our findings open the door for in situ investigations such as synchrotron X-ray diffraction, Raman spectroscopy, and real-time temperature sensing for more understanding of the reaction mechanism and better safety. In contrast, it is difficult to monitor the reaction in SSR method due to the used high temperature.
2.2. Monitoring of the mechanochemical reaction by ex situ XRD studies
The progress of the mechanochemical synthesis was monitored through periodic interruption of the grinding process, followed by extracting a small amount from the sample in the glovebox for XRD analysis, as shown in Fig. 2. The initial ball-to-powder weight ratio (BPR) was set at 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. After 8 hours of milling, XRD analysis revealed the presence of the diffraction peaks of (Li2Fe)SO and some other secondary phases. These secondary phases were labelled from p1 to p9, although some peaks remained unidentified due to the possibility of multiple phases existence at the same Bragg position (see Fig. S1†). With increasing the milling time, a progressive conversion to (Li2Fe)SO phase takes place as indicated by the increase in the relative intensity of the (Li2Fe)SO diffraction peaks. The improvement in the phase purity of (Li2Fe)SO over time can be attributed to the increase in the energy transfer to the reactants.15 Moving from 85 h to 110 h milling time, no noticeable change of the XRD pattern can be observed, which suggests a completion of the reaction. Nevertheless, a small peak (label p4) with a small shoulder is still visible. The minor secondary phase (p4) is determined as metallic iron (Fe) (1.2 vol%) by the Rietveld analyses (see Fig. S2†) which confirms the successful synthesis of almost pure (Li2Fe)SO (space group: Pm
:1. After 8 hours of milling, XRD analysis revealed the presence of the diffraction peaks of (Li2Fe)SO and some other secondary phases. These secondary phases were labelled from p1 to p9, although some peaks remained unidentified due to the possibility of multiple phases existence at the same Bragg position (see Fig. S1†). With increasing the milling time, a progressive conversion to (Li2Fe)SO phase takes place as indicated by the increase in the relative intensity of the (Li2Fe)SO diffraction peaks. The improvement in the phase purity of (Li2Fe)SO over time can be attributed to the increase in the energy transfer to the reactants.15 Moving from 85 h to 110 h milling time, no noticeable change of the XRD pattern can be observed, which suggests a completion of the reaction. Nevertheless, a small peak (label p4) with a small shoulder is still visible. The minor secondary phase (p4) is determined as metallic iron (Fe) (1.2 vol%) by the Rietveld analyses (see Fig. S2†) which confirms the successful synthesis of almost pure (Li2Fe)SO (space group: Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). The origin of this metallic Fe may come from either the unreacted Fe from the raw powders, a slight contamination from the stainless-steel vial and/or balls, or a simultaneous contribution of both mentioned sources. Metallic Fe is electrochemically inactive and no significant effect of such a negligible amount on the electrochemical performance is expected. Despite applying such long milling time, (Li2Fe)SO phase is still apparent, indicating its high mechanical stability. In the following discussions, it is referred to the sample obtained after 110 h as ball-milled (Li2Fe)SO.
m). The origin of this metallic Fe may come from either the unreacted Fe from the raw powders, a slight contamination from the stainless-steel vial and/or balls, or a simultaneous contribution of both mentioned sources. Metallic Fe is electrochemically inactive and no significant effect of such a negligible amount on the electrochemical performance is expected. Despite applying such long milling time, (Li2Fe)SO phase is still apparent, indicating its high mechanical stability. In the following discussions, it is referred to the sample obtained after 110 h as ball-milled (Li2Fe)SO.
 | ||
| Fig. 2 XRD patterns of the milled powder as a function of the milling time using 3:1 as BPR. The labels (p1–p9) refer to the secondary phases, while the red marks (as reference) refer to the ideal Bragg positions of (Li2Fe)SO. | ||
From the diffraction data a cubic lattice parameter for ball-milled (Li2Fe)SO a = 3.935(5) Å is obtained, which is the comparable with the previously reported value (3.9139 Å).5 By considering the peak broadening for the most intense diffraction peaks for the ball-milled (Li2Fe)SO, the average crystallite size (DScherrer) was calculated from Debye Scherrer equation to be 9.98(8) nm.23 In order to explore the effect of lattice strain contribution to the observed peak broadening, the average crystallite size (DW–H) was recalculated again from Williamson–Hall (W–H) model (Fig. S3†).24 W–H model yields DW–H of 12(1) nm comparable with DScherrer, which suggests minor contribution of the lattice strain to the peak broadening.
Interestingly, around 24 g is produced at the end of the milling, confirming the effectiveness of MC approach as a scalable technique. However, as expected by using dry milling, a minor amount of the powder (∼3%) is stuck on the balls and the wall of the milling container which hinders complete removal of the powder.25 Nevertheless, some studies confirm that the sticky powder can beneficially act as a protective layer to prevent the contamination from the milling medium and container.26
2.3. Effect of post-heat treatment
27 and FeS,28 their observed amounts in the heat-treated (at 500 °C) ball-milled (Li2Fe)SO (0.15 vol%, and 0.2 vol%, respectively) suggest negligible contribution to the delivered capacity and the overall electrochemical performance. The decrease of XRD intensity of peak p3 at 500 °C suggests unstable thermal behaviour for its corresponding phases (FeS and LiFeO2 based on Rietveld analysis).
 | ||
| Fig. 3 Influence of the heat treatment temperature on the XRD of ball-milled (Li2Fe)SO (BM) (a) and on the XRD intensity of (111) and (200) peaks (b). The red marks in (a) refer to the ideal Bragg positions of (Li2Fe)SO as a reference. | ||
The average crystallite size (DScherrer) and dislocation density (δ) of the heat-treated (Li2Fe)SO samples were calculated and the results are shown in Fig. S5a.†29 The DScherre increases from 11.1(1) nm at 100 °C to reach its maximum value of 0.11(01) μm at 500 °C which agrees with expectation on thermal grain-size growth. In addition, the lattice parameter (Fig. S5b†) slightly decreases from 3.921(3) Å at 100 °C to reach 3.915 (3) Å at 500 °C which is very close to the ideal reported value (3.9139 Å).5 This trend of the lattice parameter is attributed to strain relaxation and reduction of the internal structural defects such as lattice dislocations (as seen in Fig. S5a†), stacking faults, vacancies, and grain boundaries. Noteworthy, optimizing the internal lattice strain, which is feasible for (Li2Fe)SO by post-heat treatment strategy, was recently reported to have strong influence on the electrochemical performance for other cathode materials.30 This observation also confirms the possibility of using post-milling heat treatment to control the internal structure and crystallinity of (Li2Fe)SO. Changing the material's crystallinity was reported to have a strong influence on the transport properties of Li+ inside the material, with no general rule determining which has the higher ionic conductivity.15
 | ||
| Fig. 4 SEM images at low and high magnifications for ball-milled (Li2Fe)SO (a and d) and ball-milled (Li2Fe)SO followed by post-heat treating at 300 °C (b and e) and 500 °C (c and f). | ||
TEM was used to further explore the effect of heat treatment on the internal structure and crystallinity of the ball-milled (Li2Fe)SO. Fig. 5a–c reveal the HRTEM images at different magnifications of ball-milled (Li2Fe)SO. Crystalline domains are clearly visible in Fig. 5b and c, highlighting the crystalline nature of the synthesized ball-milled (Li2Fe)SO sample which is also supported by appearance of dotted rings (evidence of polycrystalline domains) in the selected area electron diffraction (SEAD) image (inset of Fig. 5a). Inverse Fourier transformation (FT) analysis and its corresponding plot profile (see Fig. S10a†) yield an average interplanar distance of ∼0.2741 nm which in good agreement with d spacing for (110) plane of (Li2Fe)SO. The appearance of flake-like nanostructures in Fig. 5 and 6 agree well with the SEM observation. The gradual change in color from black to gray, in Fig. 6b, indicates different thickness of the adjacent grains/flakes within the same particle due to cold welding effect.
 | ||
| Fig. 5 TEM images for ball-milled (Li2Fe)SO (a–c) and ball-milled (Li2Fe)SO followed by post-heat treatment at 500 °C (d–f). The insets in Fig. 5a and d present the SEAD images. | ||
 | ||
| Fig. 6 TEM image without (a) and with (b) marking the grain boundaries (yellow dashed lines) between the agglomerated grains/flakes of ball-milled (Li2Fe)SO. | ||
Heat treatment of the ball-milled (Li2Fe)SO at 500° C increases the crystallinity, as seen in Fig. 5e and f at different magnifications. This is confirmed by appearance of aligned dots (evidence of single crystalline domain) in the SEAD data (inset of Fig. 5d) and further supported by increasing the crystalline domain size (Fig. 5e). Inverse FT and its corresponding plot profile (Fig. S10b†) produces an average interplanar distance of 0.2735 nm consistent with the (110) lattice plane of (Li2Fe)SO. Interestingly, this value of lattice spacing is slightly lower than the value obtained for ball-milled (Li2Fe)SO which is in good agreement with the XRD data (Fig. S5b†). A few crystalline domains with relatively high inter-planar spacing (∼0.47 nm) are visible in the TEM image for the heat-treated (at 500 °C) ball-milled (Li2Fe)SO (Fig. S11†), which coincides with the (101) lattice plane of hexagonal FeS.34 This agrees well with Rietveld analysis (Fig. S4†) of existence of minor FeS impurity phase (0.15 vol%).
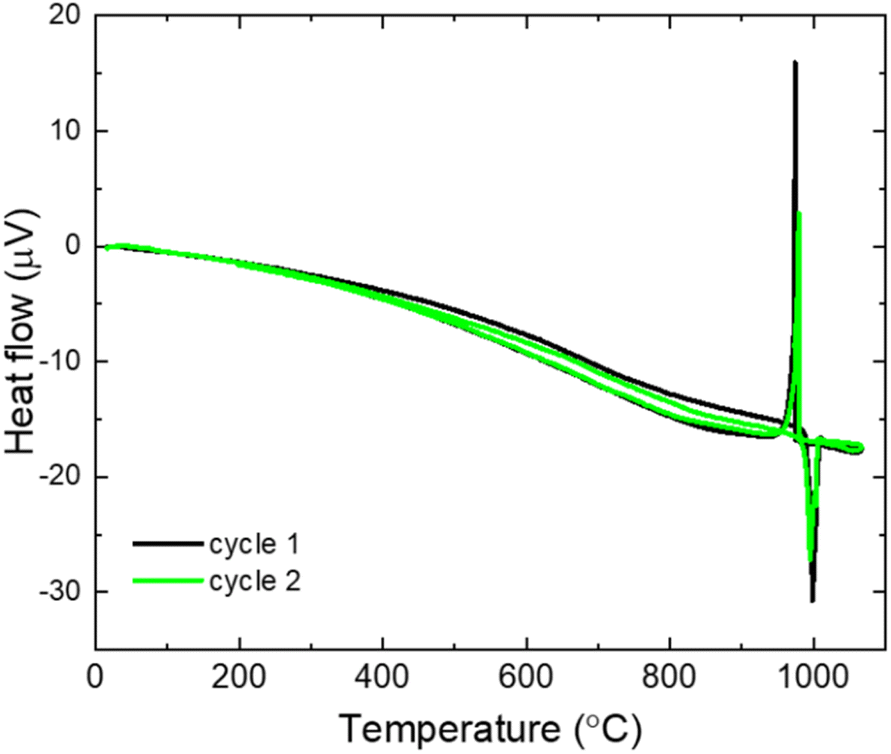 | ||
| Fig. 7 DTA for ball-milled (Li2Fe)SO. | ||
 | ||
| Fig. 8 XPS high-resolution spectrum of (a) Li 1s/Fe 3p, (b) Fe 2p, (c) O 1s, and (d) S 2p for (Li2Fe)SO prepared by direct ball-milling (BM) and ball-milling followed by post-heat treatment at 500 °C (BM + HT). | ||
 | ||
| Fig. 9 XRD patterns of the milled powder as a function of the milling time using 7:1 as BPR (a) and comparison between different BPRs (3:1 and 7:1) at two milling times (b and c). The labels (p1–p9) refer to the secondary phases, while the red short lines in (a) represent the ideal Bragg positions for the (Li2Fe)SO phase. | ||
On the other hand, other authors ascribed this signal to a contamination by chemisorbed oxygen species.37Fig. 8d presents S 2p peak for ball-milled (Li2Fe)SO in the range 158.5–163.5 eV which consists of two overlapped parts (2p1/2 and 2p3/2) due to spin–orbit coupling. Overall, no significant change in the binding energies of (Li 1s/Fe 3p, Fe 2p, O 1s and S 2p peaks was observed upon subsequent heat treatment at 500 °C of the ball-milled (Li2Fe)SO. However, an increase of the OI (lattice oxygen) contribution at the expense of OII is remarked upon heat treatment. This may be attributed to either a reduction of defects and enhancement of the lattice oxygen upon heat treatment or, most probably, decreasing of the chemisorbed oxygen species. Overall, the XPS studies indicate no change in the oxidation states of the constituent elements upon post-heat treatment after milling. This means that the crystallinity and morphology of ball-milled (Li2Fe)SO can be easily controlled without affecting the electronic structure of the involved elements.
2.4. Optimization of the ball milling conditions
In section 2.1, we mentioned that different parameters affect the MC process such as a higher BPR. To verify this, we used a higher BPR (7:1 instead of 3:1) to increase the energy transfer and thus reduce the milling time, which further decreases the energy consumption. The ex situ XRD patterns at different milling times using 7:1 BPR are shown in Fig. 9a. Note, BPR was controlled in this study by changing the number of balls while maintaining the same ball size. For comparison, the XRD of two selected milling times (35 h and 55 h) for the two samples milled with different BPR (3:1 and 7:1) are displayed in Fig. 9b and c, respectively. It is evident that at a given milling time, the increased BPR speeds up the conversion of the milled reactants into (Li2Fe)SO phase. This comes from the improvement in the reaction kinetics by increasing the energy transfer.22 Clearly, a similar XRD pattern to the ball-milled (Li2Fe)SO (3:1 BPR and 110 h) was obtained after only 65 h milling time using 7:1 as BPR. This result confirms that the milling time used in producing (Li2Fe)SO can be significantly reduced by increasing the BPR, which is a beneficial for energy saving. This also opens the way to further reduce the consumed energy by optimizing the milling parameters. The sustainable synthesis presented here, combined with the promising reported EC properties for antiperovskite (Li2Fe)SO compared to other commercial cathodes (see Table S3†), pouches its progress as a potential cathode candidate.4 Studying the effect of the synthesis conditions on the electrochemical performance of ball-milled (Li2Fe)SO is planned for the future work.
3. Conclusions
We propose a facile, green, and scalable mechanochemical synthesis to prepare (Li2Fe)SO cathode material. This novel approach can overcome the current obstacles of solid-state reaction method due to its simplicity, environmentally benign, and scalability. The ex situ XRD data confirms the successful preparation of (Li2Fe)SO by direct mechanochemistry. The obtained nearly spherical particles are in the micrometer range with a small size distribution. Subsequent post-heat treatment strategy after ball-milling was found to be an effective strategy to control the internal structure, crystallinity, and morphology of the ball-milled (Li2Fe)SO. Thermodynamic investigations confirmed the congruent melting with high thermal stability for the prepared samples. A strategy to significantly reduce the mechanochemical reaction time by increasing the ball-to-powder ratio was also introduced, which is beneficial for energy saving. Our findings highlight the advantages of mechanochemical synthesis in scaling up and controlling the structure and morphology of (Li2Fe)SO cathode for tunable electrochemical properties.4. Experimental details
4.1. Materials synthesis
Stoichiometric amounts of Li2O, Fe and S powders (as bought from Alfa Aesar) were loaded into a stainless-steel jar in an argon-filled glovebox with controlled atmosphere (O2 and H2O < 1 ppm) from MBraun. The mixture was ball-milled using high energy SPEX SamplePrep 8000D with a rotation speed of 875 rpm at room temperature and using hardened stainless-steel balls with a size of 10 mm. Similar material (stainless steel) for grinding medium and milling container was chosen to prevent the contamination that might come from the abrasion and damage of the milling vial.26 At the end of each milling process, the jar was opened inside the glovebox and the powder was extracted for further characterization. For the heat treatment of the samples after ball-milling, a certain amount of the sample was filled in a corundum crucible (Aliaxis, Frialit-Degussite, AL23) in glovebox. The crucible was placed into a silica tube (QSILAG; Quarzschmelze, Ilmenau) and temporarily closed with a rubber stopper. Outside the glovebox, the tube was evacuated to a pressure of less than 10−3 mbar, refilled with Ar to adjust the internal pressure to 0.2 bar, and sealed with a gas burner. The closed ampoule was placed into a furnace and heated at various temperatures (heating rate: 50 °C h−1) for 3 h.4.2. Characterization
cosθ, where K is the shape factor (approximated to be 1), λ is the wavelength of the XRD source, β is the full width at half maximum obtained from Voigt fitting of the XRD peaks and subtracting the instrumental broadening, and θ is the Bragg angle.23 The dislocation density (δ), which indicates the number of defects in the sample, was calculated using the formula δ = 1/(DScherrer).29
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Regional Development Fund through Sächsische Aufbaubank and project LUKSIAK (project number 100350438). M. Valldor thanks DFG (German Science Foundation) for the financial support via project number 388667006. M. A. A. Mohamed thanks the IFW excellence program for the financial support. N. Gräßler thanks DFG (German Science Foundation) for the financial support (project number: 516651026). The authors thank A. Voss and A. Voidel (IFW Dresden) for performing ICP-OES measurements. The authors thank R. Klingeler and L. Singer (Kirchhoff Institute for Physics, Heidelberg University) for helpful discussion.References
- A. Manthiram, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed.
- D. Deng, Energy Sci. Eng., 2015, 3, 385–418 CrossRef.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- D. Mikhailova, L. Giebeler, S. Maletti, S. Oswald, A. Sarapulova, S. Indris, Z. Hu, J. Bednarcik and M. Valldor, ACS Appl. Energy Mater., 2018, 1, 6593–6599 CrossRef CAS.
- K. T. Lai, I. Antonyshyn, Y. Prots and M. Valldor, J. Am. Chem. Soc., 2017, 139, 9645–9649 CrossRef CAS PubMed.
- M. A. A. Mohamed, M. V. Gorbunov, M. Valldor, S. Hampel, N. Gräßler and D. Mikhailova, J. Mater. Chem. A, 2021, 9, 23095–23105 RSC.
- M. A. A. Mohamed, L. Singer, H. Hahn, D. Djendjur, A. Özkara and E. Thauer, J. Power Sources, 2023, 558, 232547 CrossRef CAS.
- B. E. Murdock, K. E. Toghill and N. Tapia-Ruiz, Adv. Energy Mater., 2021, 11, 2102028 CrossRef CAS.
- A. Butt, G. Ali, K. Tul Kubra, R. Sharif, A. Salman, M. Bashir and S. Jamil, Energy Technol., 2022, 10, 2100775 CrossRef CAS.
- H. Munir, R. R. Srivastava, H. Kim, S. Ilyas, M. K. Khosa and B. Yameen, J. Chem. Technol. Biotechnol., 2020, 95, 2286–2294 CrossRef CAS.
- S. Ilyas, R. Ranjan Srivastava, V. K. Singh, R. Chi and H. Kim, Waste Manage., 2022, 154, 175–186 CrossRef CAS PubMed.
- L. Schwich and B. Friedrich, Metals, 2022, 12, 1108 CrossRef CAS.
- L. Schwich, P. Sabarny and B. Friedrich, Metals, 2020, 10, 1513 CrossRef CAS.
- Y. Li, W. Lv, H. Huang, W. Yan, X. Li, P. Ning, H. Cao and Z. Sun, Green Chem., 2021, 23, 6139–6171 RSC.
- A. Banik, T. Famprikis, M. Ghidiu, S. Ohno, M. A. Kraft and W. G. Zeier, Chem. Sci., 2021, 12, 6238–6263 RSC.
- A. Perona, P. Hoyos, Á. Farrán and M. J. Hernáiz, Green Chem., 2020, 22, 5559–5583 RSC.
- J. Andersen and J. Mack, Green Chem., 2018, 20, 1435–1443 RSC.
- J. L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 1–19 CrossRef PubMed.
- T. Tsuzuki, Commun. Chem., 2021, 4, 143 CrossRef CAS PubMed.
- P. F. M. De Oliveira, R. M. Torresi, F. Emmerling and P. H. C. Camargo, J. Mater. Chem. A, 2020, 8, 16114–16141 RSC.
- J. Jepsen, G. Capurso, J. Puszkiel, N. Busch, T. Werner, C. Milanese, A. Girella, J. B. von Colbe, M. Dornheim and T. Klassen, Metals, 2019, 9, 349 CrossRef CAS.
- Y. Y. Kim, A. S. Schenk, J. Ihli, A. N. Kulak, N. B. J. Hetherington, C. C. Tang, W. W. Schmahl, E. Griesshaber, G. Hyett and F. C. Meldrum, Nat. Commun., 2014, 5, 4341 CrossRef CAS PubMed.
- P. Muhammed Shafi and A. Chandra Bose, AIP Adv., 2015, 5, 057137 CrossRef.
- C. Liu, M. Wu, Y. Liu, Z. Lu, Y. Yang, S. Shi and G. Yang, Mater. Res. Bull., 2018, 99, 436–443 CrossRef CAS.
- I. Lahiri and K. Balasubramanian, Bull. Mater. Sci., 2007, 30, 157–161 CrossRef CAS.
- Y. Hu, X. Liu and N. Tapia-Ruiz, J. Mater. Sci., 2020, 55, 8651–8664 CrossRef CAS.
- C. Li, A. Sarapulova, K. Pfeifer and S. Dsoke, ChemSusChem, 2020, 13, 986–995 CrossRef CAS PubMed.
- M. Dongol, A. El-Denglawey, M. S. Abd El Sadek and I. S. Yahia, Optik, 2015, 126, 1352–1357 CrossRef CAS.
- S. S. Kim, D. N. Agyeman-Budu, J. J. Zak, A. Dawson, Q. Yan, M. Cában-Acevedo, K. M. Wiaderek, A. A. Yakovenko, Y. Yao, A. Irshad, S. R. Narayan, J. Luo, J. Nelson Weker, S. H. Tolbert and K. A. See, Chem. Mater., 2022, 34, 3236–3245 CrossRef CAS.
- W. Xie, E. Polikarpov, J. Choi, M. E. Bowden, K. Sun and J. Cui, J. Alloys Compd., 2016, 680, 1–5 CrossRef CAS.
- J. Zhang, J. Qiao, K. Sun and Z. Wang, Particuology, 2022, 61, 18–29 CrossRef CAS.
- M. Ullah, M. E. Ali and S. B. A. Hamid, Rev. Adv. Mater. Sci., 2014, 37, 1–14 CAS.
- X. Wang, Q. Xiang, B. Liu, L. Wang, T. Luo, D. Chen and G. Shen, Sci. Rep., 2013, 3, 1–8 Search PubMed.
- R. Dedryvère, M. Maccario, L. Croguennec, F. Le Cras, C. Delmas and D. Gonbeau, Chem. Mater., 2008, 20, 7164–7170 CrossRef.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- J. C. Dupin, D. Gonbeau, P. Vinatier and A. Levasseur, Phys. Chem. Chem. Phys., 2000, 2, 1319–1324 RSC.
- H. Idriss, Surf. Sci., 2021, 712, 2–7 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00861d |
| This journal is © The Royal Society of Chemistry 2023 |