
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpeeding up sustainable solution-phase peptide synthesis using T3P® as a green coupling reagent: methods and challenges†
Alexia
Mattellone
 ,
Dario
Corbisiero
,
Lucia
Ferrazzano
,
Paolo
Cantelmi
,
Giulia
Martelli
,
Chiara
Palladino
,
Alessandra
Tolomelli
* and
Walter
Cabri
*
,
Dario
Corbisiero
,
Lucia
Ferrazzano
,
Paolo
Cantelmi
,
Giulia
Martelli
,
Chiara
Palladino
,
Alessandra
Tolomelli
* and
Walter
Cabri
*
P4i Lab, Department of Chemistry “Giacomo Ciamician”, Alma Mater Studiorum University of Bologna, Via Selmi 2, 40126, Bologna, Italy. E-mail: alessandra.tolomelli@unibo.it; walter.cabri@unibo.it
First published on 2nd March 2023
Abstract
In peptide synthesis, the issues related to poor sustainability, long reaction times and high process mass intensity (PMI) are necessary to promote actions aimed at redefining procedural aspects projected towards more sustainable synthetic processes. Herein, we report a fast, widely applicable and green solution-phase peptide synthesis (GSolPPS) via a continuous protocol using propylphosphonic anhydride T3P® as the coupling reagent and N-benzyloxycarbonyl-protecting group (Z), which is easily removed by hydrogenation. Because N,N-dimethylformamide (DMF) replacement was a priority, the iterative process was performed in EtOAc, pushing further on overall sustainability. The efficiency of the synthetic protocol in terms of conversion, racemization and reaction times allowed extending the scope of the work to the synthesis of the standard peptide Leu-enkephalin as a proof of concept. Among the various explored procedures, the one-pot protocol (Acont plus), avoiding work-ups, intermediate purification and any dispersion effect, allowed the achievement of PMI = 30 for each deprotection/coupling sequence necessary to introduce a single amino acid in the iterative process, without considering the possibility of solvent and base recovery. This value is the lowest reported for an oligopeptide synthesis protocol to date.
Peptide modality represents one of the emerging areas in drug discovery, as witnessed by the increasing number of peptide therapeutics approved by regulatory agencies1 and entering different stages of clinical trial routes owing to their proven efficacy in interfering with upregulated or unwanted biological processes.2
The amide bond plays a key role in medicinal chemistry; therefore, various methods for its formation have been developed.3 Moving to peptide chemistry, the methodologies have to be compatible with iterative synthesis, minimizing work-up and side reactions.
Therefore, the identification of a simple and sustainable iterative methodology that can induce the complete formation of the peptide bond in a short time at room temperature using easily removable N-protecting groups, affording oligopeptides with pharmaceutical grade purity, represents an important challenge for the sustainable production of this emerging class of therapeutics.
Short peptides (3–8 amino acids) used as cosmeceuticals owing to the scientific data, unlike cosmetics, support their role as active principles.4
After the identification and introduction of hormone peptides in the market in the early twentieth century, limited attention has been paid to the investigation of novel peptide drugs because of their difficult isolation from natural sources and limited bioavailability. Moreover, the short half-life due to enzyme degradation susceptibility strongly reduced the efficacy of peptides as therapeutics, requiring their administration via the intravenous route. The discovery of solid-phase synthetic technologies in the sixties5 and their optimization in the last six decades allowed us to overcome the previous bias and open access to the preparation of peptides on a large scale and to the introduction of non-coded residues, which can increase resistance to hydrolysis.6
Critical issues in peptide synthesis are poor greenness scores, such as the process mass intensity (PMI), and the common use of solvents and reagents, which are major concerns in large excess.
Dimethylformamide (DMF) is the best solvent for peptide synthesis, both in the solid- and solution-phase, because of its great performance in terms of the solubilization of reagents and by-products. It is reprotoxic and labelled as a hazardous solvent; accordingly, its replacement has been considered “advisable or requested” by the ACS Green Chemistry Institute® Pharmaceutical Round Table (GCIPR), which defined this goal as one of the hot topics in the list of the 12 green chemistry key research areas (KRAs).7 In addition, strong restrictions on its use in the industry will be applied from December 2023.8
Many efforts have been devoted in the last few years to greening solid-phase peptide synthesis (SPPS)9 by searching for alternatives to DMF.10
Moreover, piperidine,11 the base of choice in Fmoc-protection removal in SPPS, is a substance under strict regulation used for the production of illegal narcotics,12 and the need for a large excess of reagents and the amount of solvents required in washing steps to remove the coupling reagent's by-products represent weaknesses of this methodology.
With the introduction of SPPS, the traditional solution-phase approach was dismissed because a more complex impurity profile was generally obtained from the same synthetic sequence, making the isolation of intermediates by chromatography a key step to reaching acceptable purity for biomedical applications. Recently, liquid phase peptide synthesis (LPPS) has been reconsidered owing to the introduction of innovative technologies, which allow the precipitation of growing peptides or isolating them via membrane technology.13 Therefore, because of the great interest in reaching sustainable and efficient processes, LPPS recently received new attention in the synthesis of bioactive molecules, such as peptides14 and oligonucleotides.15 In particular, extensive improvements could arise from continuous approaches because few examples have been reported to date.9,16 An important requirement that would make solution-phase peptide synthesis (SolPPS) a much more “solid” technique to rely on, by combining efficiency and greenness score, would be the identification of greener effective coupling reagents,17 which can induce the complete formation of the peptide bond in a short time at room temperature.
Propylphosphonic anhydride (T3P®), first reported by Wissmann & Kleiner in 1980,18 has been documented in the literature to be a suitable reagent in the formation of peptide bonds in solution19 and several other transformations.20
This reagent has never been used for iterative continuous processes in solution because of its high sensitivity to water. All the methodologies reported to date perform only a single coupling step at a time and cannot be applied to a continuous deprotection-coupling iterative process.21 Recently, Albericio et al.22 reported the use of T3P® in SPPS by screening various solvents and temperatures to confirm the versatility of this coupling system. Under these conditions, where the use of water is not needed, this coupling reagent was insufficient to have the complete formation of the peptide bond and had to be combined with Oxyma Pure to afford excellent coupling reagent couples, taking the role usually played by carbodiimides.
T3P® has several practical advantages over other coupling reagents. It is not inflammable, not toxic (LD50 in rats >2000 mg kg−1)23 and the side product propylphosphonic acid can be easily removed by water washings. Moreover, this reagent is actually commercialized in solution in several solvents, such as ethyl acetate and alkyl carbonates, with a stability of 2 years.24
In addition to the above reported characteristics of T3P®, its effect on preventing racemization in peptide bond formation has been particularly addressed. All the reported applications displayed some drawbacks caused by long reaction times, low yields, and the formation of by-products. More recently, Dunetz et al.25 confirmed the prevention of racemization in T3P®-mediated amide formation, selecting pyridine as the most suitable base.
We explored, herein, the possibility of applying this reagent to continuous solution-phase synthesis, eliminating the need for other coupling reagents and carbodiimides, which display several disadvantages in terms of toxicity and safety,26 to develop a fast and green protocol for oligopeptide synthesis.
Consequently, we needed to avoid intermediate isolation, in particular with water involving work-ups, both in the coupling and protecting group removal steps. We identified the N-benzyloxycarbonyl (Z) group as the most suitable protection for the possibility of simple removal by hydrogenation without generating by-products that could affect the following steps. Recently, Lipshutz et al.27 reported a single Z-deprotection/coupling 2-steps continuous reaction performed in water using oligopeptide fragments, adding a surfactant to create an efficient nano-micellar environment. From the above cited methodologies, any attempt to extend this procedure to a deprotection/coupling iterative process did not succeed.
First, we optimized the general conditions for solution-phase peptide synthesis using a standard reaction of the coupling between (L)-phenylglycine and (L)-prolinamide in DMF because this peptide bond is considered difficult and prone to racemization28 (Table 1).
|
|
||||
|---|---|---|---|---|
| Entrya | T3P® (equiv.) | Base | Conversionc (%) | |
| (Equiv.) | Greenness scoreb | |||
| a Reactions were performed by dissolving Z-Phg in DMF (0.1 M conc) under nitrogen atmosphere and adding reagents to the following order: Pro-NH2, DIPEA and finally T3P®. b According to the GSK base selection guide, see ref. 29. c Conversion was determined by LC-MS (see ESI for details†). d Reaction was performed at 0 °C. e Pre-activation of the acid was performed by adding T3P® and DIPEA before Pro-NH2. The presence of Z-(D)-Phg-Pro-NH2 was detected in a 5% amount. | ||||
| 1 | 1 | DIPEA (1) | 6.5 | 81 |
| 2 | 1 | DIPEA (2) | 6.5 | 88 |
| 3 | 1.5 | DIPEA (3) | 6.5 | >99 |
| 4d | 1.5 | DIPEA (3) | 6.5 | 93 |
| 5e | 1.5 | DIPEA (3) | 6.5 | 80 |
| 6 | 1.5 | 2,6-Lutidine (3) | 8.3 | 56 |
| 7 | 1.5 | Pyridine (3) | 7.5 | 42 |
| 8 | 1.5 | TEA (3) | 6.9 | 94 |
| 9 | 1.5 | NMM (3) | 6.9 | 90 |
| 10 | 1.5 | t BuNH2 (3) | 6.5 | 93 |
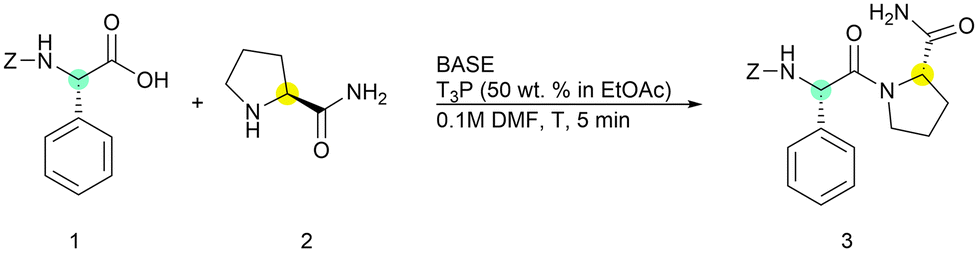
The coupling reaction was performed under nitrogen atmosphere to reduce the presence of air moisture, which could eventually degrade T3P®. The selected results are reported in Table 1.
The attention was initially focused on the T3P®/base ratio and on the order of the addition of reagents to the vessel. To our surprise, the coupling reaction turned out to be very fast and afforded excellent conversions after only five minutes, supporting the idea of exploiting this protocol for iterative solution-phase peptide synthesis.
Moreover, when incomplete conversion was observed, little increase in dipeptide 1 formation occurred even after longer reaction times, suggesting that the acid activation by T3P® is almost instantaneous as well as the nucleophilic attack of the free amino ester.
Aiming at setting up the greenest possible protocol, we started adding only one equivalent for each reagent (entry 1), choosing DIPEA as the base. However, incomplete formation of the dipeptide was observed. With the only exception of entry 5, the base and T3P® were sequentially added to the mixture of the two amino acids. An increase in the conversion was observed by adding two equivalents of the base to neutralize both triphosphate salt functions (entries 2 and 3). The best results were obtained using a 1.5/3 ratio of the two reagents by achieving complete conversion in 5 minutes at room temperature (entries 3). The presence of 0.5% and 0.4% of Z-(D)-Phg-Pro-NH2 was observed when the reaction was performed at rt and 0 °C, respectively (entries 3 and 4, see ESI for details†). Finally, when the preactivation of the acid was performed by adding T3P® and DIPEA before Pro-NH2, only 80% conversion could be obtained, probably for the lability of the activated mixed anhydride (entry 5). Under these conditions, a 5% amount of racemized product was also detected (see ESI for details†). Having established optimized conditions, we screened a small library of bases and selected among those reported to be compatible with T3P® and those having a good greenness score according to the GSK selection guide. Although 2,6-lutidine and pyridine did not afford satisfactory results (entries 6 and 7) when the reaction was performed with triethylamine (TEA), N-methylmorpholine (NMM) or tBuNH2, peptide 3 was isolated with better yields (entries 8–10) even if slightly lower compared to DIPEA. This was confirmed to be the best base for obtaining a trade-off between greenness and peptide formation.
Concerning the solvent of choice, we then focused on replacing DMF with greener solvents29 under the above identified optimized conditions. The selected results are reported in Table 2.
| Entrya | Solvent | Coupling reagents | Temperature | 3 (%) | (D)/(L + D) (%) |
|---|---|---|---|---|---|
| a The reactions were performed under the conditions used in entry 3, Table 1, and conversion evaluated after 5 minutes. b n.d. = not detected. c Conversion was 90–93% after 1 h. | |||||
| 1 | NBP | T3P® | r.t. | 56 | 0.6 |
| 2 | NOP | T3P® | r.t. | 56 | n.d.b |
| 3 | DMC | T3P® | r.t. | 93 | 0.2 |
| 4 | GVL | T3P® | r.t. | 89 | 0.7 |
| 5 | ACN | T3P® | r.t. | 96 | 0.2 |
| 6 | THF | T3P® | r.t. | 93 | 0.1 |
| 7 | 2-MeTHF | T3P® | r.t. | 80 | 0.3 |
| 8 | DCM | T3P® | r.t. | 98 | 0.2 |
| 9 | iPrOAc | T3P® | r.t. | 69 | 1.1 |
| 10 | EtOAc | T3P® | r.t. | 94 | 0.5 |
| 11 | EtOAc | T3P® | 0 °C to r.t. | 96 | 0.3 |
| 12 | DMF | T3P® | r.t. | >99 | 0.5 |
| 13 | DMF | T3P® | 0 °C to r.t. | 93 | 0.4 |
| 14 | DMF | Oxyma pure/DIC | r.t. | 73c | 1.0 |
| 15 | DMF | Oxyma pure/DIC | 0 °C to r.t. | 70c | 1.0 |
| 16 | EtOAc | Oxyma pure/DIC | r.t. | 96 | 1.0 |
| 17 | EtOAc | Oxyma pure/DIC | 0 °C to r.t. | 86 | 0.9 |
The screening confirmed the compatibility of T3P® with almost all the tested solvents even if the reactions that were performed in N-alkyl-pyrrolidones, such as N-octylpyrrolidone (NOP) and N-butylpyrrolidone (NBP), afforded limited conversions (entries 1 and 2) because of more difficult dissolution of activated mixed anhydride in low polar lipophilic solvents. Moreover, for all the solvents, the degree of racemization was very low if compared, for instance, with the commonly used coupling system Oxyma Pure/DIC (entries 10 and 11 vs. entries 16 and 17 or entries 12 and 13 vs. entries 14 and 15).
Among the solvents with the best green score, EtOAc, THF and ACN exhibited the highest conversions. However, EtOAc has a lower affinity for water. This aspect needs to be particularly underlined considering the sensitivity to water displayed by T3P®. The reaction in EtOAc was also performed on a gram scale, affording excellent results (see ESI†).
EtOAc was selected as the solvent of choice to proceed with the investigation because, in addition to its technical characteristics, it is readily available at a competitive price with respect to DMF.
The exceptional speed of the T3P® that promoted the coupling reaction prompted us to apply the optimized protocol to the difficult reactions of the sterically hindered Aib amino acid (Table 3).
| Entry | H2N-AA-OPg | Z-AA-OH | T3P®/DIPEA (equiv.) | C (%) |
|---|---|---|---|---|
| a Conversion calculated by HPLC. b By adding H2N-L-OtBu after 5 minutes, the conversion of the remaining activated Z-Aib afforded 92% of Z-Aib-L OtBu dipeptide was detected. | ||||
| 1 | H2N-Pro-NH2 | Z-Aib | 2/4 | 92 |
| 2 | H2N-Phe-OtBu | Z-Aib | 2/4 | 96 |
| 3 | H2N-FL-OtBu | Z-Aib | 2.5/5 | 98 |
| 4 | H2N-Aib-OMe | Z-Phe | 2/4 | 78 |
| 5 | H2N-Aib-OMe | Z-Aib | 2/4 | 4 |
| 6 | H2N-Aib-OMe (+H2N-L-OtBu) | Z-Aib | 2/4 | 4 (92)b |
Thus, Z-Aib was activated under previously optimized conditions and added to prolinamide (entry 1) or phenylalanine ester (entry 2).
In both cases, the coupling gave satisfactory results using a slight excess of the coupling mixture (2/4 equivalents), affording the dipeptides in 92–96% conversion.
These results are of remarkable interest considering that in SPPS, the introduction of the Aib amino acid generally requires a double coupling step.30
An even better conversion was obtained in the reaction of Z-Aib with the dipeptide H-Phe-Leu-OtBu 5 (98%, entry 3). On the contrary, when Aib was used as the nucleophilic amino acid and it reacted with activated Z-Phe, lower conversion was observed (78%, entry 4), suggesting that the hindrance of the triphosphate chain does not allow the approach of the bulky Aib nucleophile.
The coupling system turned out to be completely ineffective in the Aib–Aib dipeptide formation because only 4% conversion was obtained (entry 5).
However, when H2N-Leu-OtBu was added to the reaction mixture after 5 minutes, a mixture of 4% of Aib–Aib and 96% of Leu-Aib was obtained, showing that the low reactivity was not caused by difficult Aib activation but by only the clash between the hindering moieties when Aib is also the nucleophile (entry 6).
With the target of performing the deprotection/coupling iterative process, the Z-removal step was explored via hydrogenation in EtOAc as well (Table 4). To this purpose, we set up two different protocols, (i) performing the reaction in a vessel using Pd/C10% catalyst powder at atmospheric pressure (method A) and (ii) with the H-Cube® flow system,31 that exploit a reusable cartridge containing the Pd/C10% heterogeneous catalyst (method B). Method B has the advantage of in situ generating endogenous on-demand hydrogen from water, further increasing the greenness score of the synthetic process. The optimal conditions were identified for both systems, as reported in Table 3, by performing Z-group removal from dipeptide Z-F-L-OtBu 4, which is the initial fragment of the pentapeptide Leu-enkephalin (H-YGGFL-OH), commonly used to test the efficiency of a new protocol for peptide synthesis.
|
|
||||
|---|---|---|---|---|
| Entry | Contaminant (equiv.) | Method | Time (min) | 5 (%) |
| a Reaction performed with Pd/C10% (10% w/w) not previously dried under vacuum. b Injection of 4 mL of 0.1 M sample. The H-Cube® setup generated 6 mL of death volume solvent and a complete recovery of 5 was obtained by collecting the solution after 18 minutes. | ||||
| 1 | — | Aa | 300 | >99 |
| 2 | — | A | 180 | >99 |
| 3 | DIPEA (0.1 eq.) | A | 60 | >99 |
| 4 | DIPEA (3 eq.) | A | 30 | >99 |
| 5 | T3P®![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1,5 eq.) :H2O (1,5 eq.) |
A | 240 | >99 |
| 6 | DIPEA:T3P® (1.5/3) |
A | 120 | >99 |
| 7 | DIPEA:HCl (3 eq.) |
A | 240 | >99 |
| 8 | — | Bb | 18 | >99 |
| 9 | DIPEA (0.1 eq.) | B | 16 | >99 |
| 10 | DIPEA (1 eq.) | B | 18 | >99 |
| 11 | DIPEA:T3P® (1.5/3) |
B | 18 | >99 |
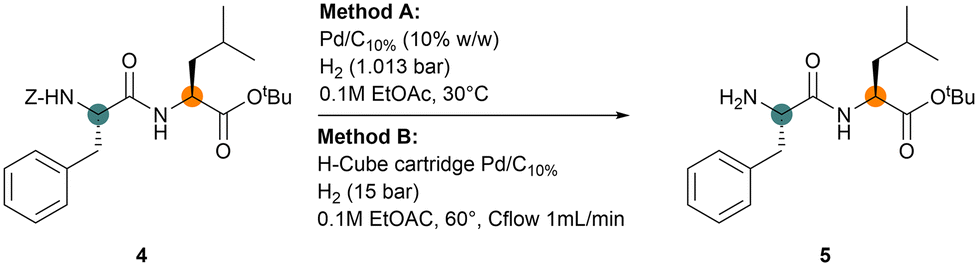
To mimic continuous processes, we removed the amine protective group in the presence of possible contaminants derived from a previous coupling step through their direct addition.
The mild conditions applied in method A and the use of Pd/C10% catalyst, without any pre-treatment, allowed us to obtain the complete conversion of 4 into 5 in five hours (entry 1). Using a dehydrated catalyst, the reaction time decreased to 3 hours (entry 2). The presence of DIPEA in large excess afforded a much more reactive system (entry 4) but even the addition of 0.1 equivalent of base allowed to completely remove the Z protecting group in one hour (entry 3). On the contrary, when the triphosphate salt derived from T3P® treatment with water was added to the mixture, simulating the reacted coupling reagent, the transformation was strongly decreased (entry 5).
By adding both DIPEA and T3P®, in the same amount and ratio used in the optimized coupling step, the reaction slightly accelerated compared with the one performed on pure 4 (entry 6 vs. entry 2). The reaction was also tested using the hydrochloride salt of DIPEA to verify the effect of the co-products derived from a coupling step involving the use of amino ester hydrochloride. In this case, a decrease in reaction speed was observed, and complete conversion was achieved in four hours. The free tertiary amine could speed up the reaction by favoring the displacement of the final product from the catalyst surface.32 To perform hydrogenation in the H-Cube® flow system (method B), we had to apply stronger conditions to increase speed and decrease time. The model reaction was performed on 4 mL of 0.1 M solution of 4 in EtOAc. The content of the Pd/C10% catalyst charged in the cartridge was about twice the amount used in the reaction in the batch. However, the flow system allows for a high catalyst/substrate local concentration in the hydrogenation cell (roughly 2/1 ratio) and transforms high quantities of flowing substrate, thereby decreasing the impact of the catalyst. Excellent and rapid conversion was achieved under H2 pressure (15–20 bar) at 60 °C with a flow of 1 mL min−1, collecting compound 5 in 18 minutes (entry 8), but completeness was achieved after the addition of DIPEA to the flowing solution (entries 9 and 10). The effect of additives was similar to the results observed before (entry 11).
Based on the previous information, we decided to perform the solution-phase continuous synthesis (GSolPPS) of Leu-Enkephalin by testing both hydrogenation methods in the removal of the Z-protecting group (Fig. 1 and Table 5).
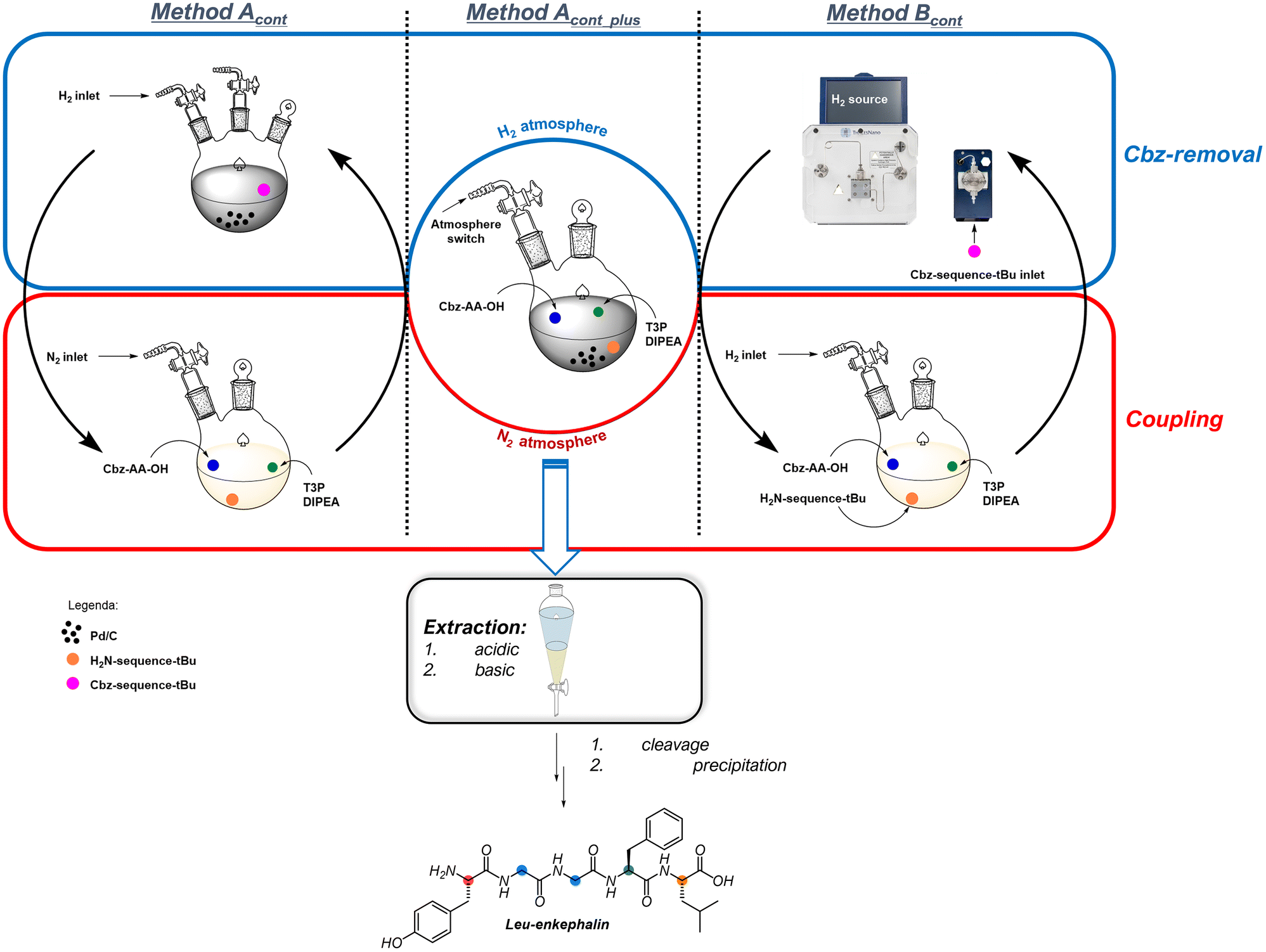 | ||
| Fig. 1 Method Acont, Bcont and Acont_plus general procedures. | ||
| Entry | Method | Boc-Leu-Enk-OtBua (purity %) | Yieldb (%) | Leu-Enkc (purity %) | PMI | Recoveryd (yield %) | PMIr |
|---|---|---|---|---|---|---|---|
| a Calculated by HPLC (area % uncorrected with RRF). b Yield related only to Boc-Leu-Enk-OtBu in the crude after washings. c Calculated by HPLC (area % uncorrected with RRF). d EtOAC and DIPEA were recovered by distillation of the organic phases after acid/basic water washings. | |||||||
| 1 | Acont | 92 | 76 | 91 | 299 | EtOAc (90) | 180 |
| DIPEA (80) | |||||||
| 2 | Bcont | 86 | 81 | 85 | 552 | EtOAc (90) | 200 |
| DIPEA (80) | |||||||
| 3 | Acont_plus | 86 | 78 | 86 | 197 | EtOAc (90) | 129 |
| DIPEA (80) | |||||||
We first matched the coupling steps with DIPEA and T3P® with the “in flow” deprotection through the H-Cube® system (method Bcont). We performed the iterative process without isolating the intermediate peptides but checked conversion and crude composition by HPLC analysis for each step. The first coupling step to produce 4 achieved >99% conversion in 5 minutes, and the crude solution was directly introduced into the hydrogenating flow system.
As reported above, treatment of 4 under these conditions easily afforded 5 with complete conversion.
Owing to the dispersion effect related to HCube® instrument setup, the volume of the collected solution was quadrupled compared to the starting solution.
Recently, practical issues regarding product diffusion in bolus injection mode for this system have been investigated, and stacked injections have been proposed as an alternative, but still dilution of the final product was observed.33 Several factors can affect the diffusion such as the reactor geometry, the affinity of the reactant and product for the catalyst, the particle size of the catalyst and the ability of the system to reach a steady state. However, oligopeptides have multiple binding sites for the Pd catalyst, and the design of the iterative process does not allow them to reach a steady state, thus limiting diffusion.
Therefore, before performing the following coupling step, EtOAc was distilled under an inert atmosphere to achieve a 0.1 M concentration, which is suitable for a further coupling step. Moreover, the recovery of EtOAc after each deprotection step reduced the impact of solvent volume on the PMI of the process.
In the following iterative coupling/deprotection steps, to ensure complete conversions and solvent dryness, a slightly higher excess of coupling reagents (2 equiv. of T3P® and 4 equiv. of DIPEA) was used. The Z-protection removal steps were always complete under the optimized conditions, but the dilution of the deprotected peptide always occurred, requiring partial concentration of the solution under vacuum before residue addition. At the end of this continuous synthesis, Boc-Leu-Enk-OtBu was isolated in 81% yield with 86% purity. In the last step, Boc-Tyr(OtBu)-OH was used to obtain the contemporary cleavage of all the protecting groups in a single reaction. For the entire reaction sequence, the solution remained clear and the DIPEA and T3P® deriving salts did not interfere with the multistep process completion or give backpressure failures related to increased viscosity of the solvent flow entering the H-Cube® catalyst cartridge.
To contain the PMI, we explored the deprotection/coupling iterative process in batches. We performed the solution-phase synthesis of the peptide by alternating the coupling reaction under nitrogen with the hydrogenation performed in a round bottom flask using an internal filtering septum that offers the possibility to remove Pd/C10% under nitrogen atmosphere, thus avoiding air moisture contamination (method Acont). Monitoring by HPLC each coupling and deprotection step revealed that under these conditions, each hydrogenation was completed in 1 hour as the couplings were performed in 5 minutes. The Pd catalyst was filtered and dried under a vacuum to be recycled for further use. The minimal amount of EtOAc added to wash the catalyst was removed under nitrogen flow to return to the standard volume. As for method Bcont, the last step was carried out using Boc-Tyr(OtBu)-OH, giving Boc-Leu-Enk-OtBu in 76% isolated yield with 92% purity. The synthesis of Boc-Leu-Enk-OtBu was also achieved with a [2 + 2 + 1] sequence using method Acont and by coupling the dipeptides Z-Gly–Gly and H-Phe-Leu-OtBu before adding the last Boc-Tyr-OH residue. The satisfactory results obtained (98% conversion and 93% purity, see ESI for details†) confirmed that T3P®is an excellent reagent for coupling oligomers obtained by parallel synthesis, which is a common approach applied in medicinal chemistry for difficult sequence production.
At the end of the synthetic sequence, Pd/C10% catalyst was filtered and recovered. By submitting the organic phase containing the peptide to acid washing (HCl 0.1 M), all the unreacted amines were removed, and the unreacted acids were eliminated by basic washing (NaHCO3 0.05 M). DIPEA was then recovered by turning the pH of the acid water phase to basic and extracting it with EtOAc. The yield of the protected peptide was calculated after distillation and the recovery of the organic phase under vacuum.
A comparison of the two methodologies did not display great advantages of one over the other because the shorter reaction time in the deprotection for method Bcont was compensated for by the need for a larger amount of solvent and related distillation.
The data obtained from both methods represented the starting point for the development of a really green and sustainable solution-phase peptide synthesis (GSolPPS).
To decrease the amount of solvent and simplify the operations, allowing the decrease in investment for industrial scale application, we used a one-pot procedure.
Because the side products and unreacted reagents/catalysts of the two steps were proven not to interfere with the desired reactivity, we performed the synthesis of the pentapeptide using Method Acont without any intermediate work-up, just adding the reagent sequentially in the proper order (method Acont_plus).
During the multistep synthesis, no addition of solvent was required, and no addition of the Pd/C10% catalyst was performed. This is because the activity remained unaffected in every hydrogenation step, and the atmosphere was simply switched from hydrogen to nitrogen and vice versa.
Therefore, after the first peptide coupling with amino acids, DIPEA and T3P® (1:1:4:1.5 ratio) in EtOAc (0.1 M) for 5 minutes, Pd/C10% (10% w/w) with respect to the dipeptide, corresponding to 4.4 mol%, was directly added into the flask, and the atmosphere was switched from nitrogen to hydrogen.
When the deprotection step was complete after 1 hour, hydrogen was removed, and the new Z-amino acid (1 equiv.) was followed by DIPEA (4 equiv.) and T3P® (2 equiv.) were added and stirred for 5 minutes. This iterative sequence of additions was repeated until the pentapeptide was finally synthesized.
A 0.1 equiv. amount of DIPEA was added before each hydrogenation to reproduce the optimal conditions (see Table 3 entry 3). The recovery of Pd and base was performed as reported above for Method Acont. From this continuous approach, peptide Boc-Leu-Enk-OtBu was obtained in 86% purity and 78% yield (Table 5 entry 3).
This last protocol could be very useful for the synthesis of short peptides. For example, those commonly used in the cosmetic industry.34 Thus, the synthesis of Pentapeptide-18 (Leuphasyl), composed of the sequence H-tyr-Ala-Gly-Phe-Leu-OH (H-YAGFL-OH), was performed following method Acont. This peptide is an active peptide ingredient present in anti-aging formulations owing to its effectiveness in wrinkles reduction, which decreases the ACh secretion in the synaptic cleft similar to enkephalins. Under the one-pot synthetic conditions, H-YAGFL-OH was obtained at 89% purity. In the field of the green synthesis of oligopeptides, several strategies have been applied to improve the environmental footprint of these molecules.
To further confirm the sustainability of the synthetic process, we decided to calculate the process mass intensity and the recovery of the reaction solvent and base (Table 5, PMI and PMIr).
As reported above, EtOAc was recovered by simple distillation under vacuum, while DIPEA was recovered after extraction from the basified water phase and distillation. The reported values include the deprotection step using typical TFA/TIS/water cocktail cleavage and the precipitation of the free peptide with diisopropyl ether (DIPE). Detailed calculations are reported in the ESI.† The downstream process for the isolation of pure peptides to achieve pharmaceutical-grade purity is not reported herein. However, the described coupling conditions were able to minimize the formation of diastereoisomers, which are the most critical impurities to be removed in the downstream process.35
Among the three methodologies, the one-pot method Acont_plus is certainly the greenest because a lower PMI was obtained (197), and owing to the recovery of solvent and base, the value of PMIr was further decreased to 129 (Table 5, entry 3). For methods Acont and Bcont, the PMI values were higher (entries 1 and 2) but still very convenient, as the PMIr values were <200.
To correctly compare these methodologies with SPPS and LPPS,10a,36,37 we calculated the average PMI for the introduction of a single amino acid in the iterative process (Table 6), considering only solvents, reagents and water, being all the contribution of other factors (resin, tag and cleavage cocktail) depending on the length of the synthesized peptide. Among the LPPS technologies, Ajiphase® was selected, being the most efficient, even though it uses toxic solvents (if chloroform and DMF) and potentially explosive coupling reagents, such as HOBT.9a
| Entry | Method | PMI/aa |
|---|---|---|
| a Data calculated from the average PMI/aa in the synthesis of Octreotide (see ref. 10a). b Data calculated from the synthesis of Bivaluridin with LPPS using Ajiphase® technology (see ref. 36). c Data calculated from the synthesis of 65–74ACP under microwave-assisted SPPS (see ref. 37). | ||
| 1 | Acont | 46 |
| 2 | Bcont | 89 |
| 3 | Acont plus | 30 |
| 4 | GSPPSa | 86 |
| 5 | LPPSb | 200 |
| 6 | MW-SPPSc | 180 |
The one-pot method Acont plus (entry 3) was confirmed to be more sustainable (PMI/aa = 30). The GSPPS (entry 4), the LPPS, performed with Ajiphase® tag technology (entry 5), and the MW-assisted SPPS (entry 6) have higher PMI/aa values of 86, 200 and 180, respectively.
On these bases, to the best of our knowledge, the one-pot iterative deprotection/coupling method Acont plus represents the first example of a totally green solution-phase approach competitive to SPPS.
Conclusions
Greening peptide synthesis represents an important research goal owing to the growing number of peptide drugs on the market. The reported processes were designed using solvent, reagents and reaction conditions with low environmental (PMI and ecotoxicity), health (human toxicity) and safety impact, requiring low energy usage. To complete the puzzle of sustainable reagents in this field, we explored the reactivity of T3P® as a coupling reagent in EtOAc solution, optimizing three methodologies that allowed the synthesis of peptides with a very good purity grade in a remarkably short time. Moreover, owing to the presence of a limited number of impurities, an easy downstream process can be foreseen. The use of the Z-protecting group was guaranteed to maintain dryness during all the steps, affording conditions suitable for using T3P® as a coupling reagent in iterative processes. Any other deprotection methodology using bases or acids, which require water washing, is not compatible with this green reagent.The main characteristics of the described process are simplicity, short reaction time, and the use of green reagents and solvents at room temperature. Solvent and base can be easily recovered without great energy usage. Finally, the comparison of the PMI values for a single amino acid introduced with other iterative methodologies confirms the remarkable advancement in terms of sustainability for one-pot oligopeptide synthesis.
Author contributions
Alexia Mattellone and Dario Corbisiero equally contributed to the execution and analysis of the experiments. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We kindly acknowledge Alma Mater Studiorum University of Bologna for financial support. Fondazione del Monte is gratefully acknowledged for funding the project prot. N°702bis/2019. Curia Global, Inc. is gratefully acknowledged for the gift of T3P® samples in different solvents. We also thank Faggi Enrico SpA for the precious technical support in Pd/C10% recovery and analysis, as well as for financial support in the acquisition of H-Cube® instrument. The Italian Ministry of University and Research (MIUR) is kindly acknowledged for the PhD fellowship to Chiara Palladino, sponsored with PON program FSE REACT-EU – A.A. 2021/2022 fundings. We thank Dr Christian Hornung (CSIRO Manufacturing) for useful discussions on flow hydrogenation reactors.References
- D. Al Shaer, O. Al Musaimi, F. Albericio and B. de la Torre, Pharmaceutical, 2022, 15, 222 CAS.
- W. Cabri, P. Cantelmi, D. Corbisiero, T. Fantoni, L. Ferrazzano, G. Martelli, A. Mattellone and A. Tolomelli, Front. Mol. Biosci., 2021, 8, 697586 CrossRef CAS PubMed.
- (a) S. Kumari, A. V. Carmona, A. K. Tiwari and P. C. Trippier, J. Med. Chem., 2020, 63, 12290 CrossRef CAS PubMed; (b) S. M. Akondi, P. Gangireddy, T. C. Pickel and L. S. Liebeskind, Org. Lett., 2018, 20, 538 CrossRef CAS PubMed; (c) L. S. Liebeskind, P. Gangireddy and M. G. Lindale, J. Am. Chem. Soc., 2016, 138, 6715 CrossRef CAS PubMed.
- F. Errante, P. Ledwon, R. Latajka, P. Rovero and A. M. Papini, Front. Chem., 2020, 8, 572923 CrossRef CAS PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149 CrossRef CAS.
- A. Abdildinova, M. J. Kurth and Y.-D. Gong, Asian J. Org. Chem., 2021, 10, 2300 CrossRef CAS.
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082 RSC.
- Commission Regulation (EU) 2021/2030 of 19 November 2021; https://eur-lex.Europa.eu/eli/reg/2021/2030/oj.
- (a) L. Ferrazzano, M. Catani, A. Cavazzini, G. Martelli, D. Corbisiero, P. Cantelmi, T. Fantoni, A. Mattellone, C. De Luca, S. Felletti, W. Cabri and A. Tolomelli, Green Chem., 2022, 24, 975 RSC; (b) J. Pawlas and J. H. Rasmussen, Green Chem., 2019, 21, 5990 RSC.
- (a) G. Martelli, P. Cantelmi, A. Tolomelli, D. Corbisiero, A. Mattellone, A. Ricci, T. Fantoni, W. Cabri, F. Vacondio, F. Ferlenghi, M. Mor and L. Ferrazzano, Green Chem., 2021, 23, 4095 RSC; (b) V. Martin, S. Jadhav, P. H. G. Egelund, R. Liffert, H. J. Castro, T. Krüger, K. F. Haselmann, S. Thordal Le Quement, F. Albericio, F. Dettner, C. Lechner, R. Schönleber and D. S. Pedersen, Green Chem., 2021, 23, 3295 RSC; (c) A. Kumar, Y. E. Jad, A. El-Faham, B. G. de la Torre and F. Albericio, Tetrahedron Lett., 2017, 58, 2986 CrossRef CAS; A. Kumar, A. Sharma, B. G. de la Torre and F. Albericio, Green Chem. Lett. Rev., 2021, 14, 545 Search PubMed; (d) A. Kumar, M. Alhassan, J. Lopez, F. Albericio and B. G. de la Torre, ChemSusChem, 2020, 13, 5288 CrossRef CAS PubMed; (e) L. Ferrazzano, D. Corbisiero, G. Martelli, A. Tolomelli, A. Viola, A. Ricci and W. Cabri, ACS Sustainable Chem. Eng., 2019, 7, 12867 CrossRef CAS.
- (a) W. Cabri, P. Cantelmi, D. Corbisiero, T. Fantoni, L. Ferrazzano, M. Macis, G. Martelli, A. Mattellone, C. Palladino, A. Ricci and A. Tolomelli, Green Chem., 2021, 23, 8096 RSC; (b) F. Dettner, P. H. G. Egelund, S. Jadhav, H. Johansson Castro, C. Lechner, S. T. Le Quement, V. Martin, D. S. Pedersen, F. Richner and R. Schoenleber, ACS Sustainable Chem. Eng., 2021, 9, 14202 CrossRef; (c) B. Biondi, W. Cabri, F. Formaggio, I. Guryanov, A. Orlandin, A. Ricci and A. Viola, Org. Process Res. Dev., 2020, 24, 274 CrossRef.
- Drug of Abuse, A DEA resource Guide, 2020 Ed.
- (a) D. Takahashi, T. Inomata and T. Fukui, Angew. Chem., 2017, 129, 7911 CrossRef; (b) Y. Okada, R. Takasawa, D. Kubo, N. Iwanaga, S. Fujita, K. Suzuki, H. Suzuki, H. Kamiya and K. Chiba, Org. Process Res. Dev., 2019, 23, 2576 CrossRef CAS; (c) J. Yeo, L. Peeva, S. Chung, P. Gaffney, D. Kim, C. Luciani, S. Tsukanov, K. Seibert, M. Kopach, F. Albericio and A. Livingston, Angew. Chem., Int. Ed., 2021, 60, 7786 CrossRef CAS PubMed.
- A. Sharma, A. Kumar, B. G. de la Torre and F. Albericio, Chem. Rev., 2022, 122, 13516 CrossRef CAS PubMed.
- L. Ferrazzano, D. Corbisiero, A. Tolomelli and W. Cabri, Green Chem., 2023, 25, 1217 RSC.
- (a) A. I. Alfano, M. Brindisi and H. Lange, ChemSusChem, 2022, 15, e202102708 CrossRef CAS PubMed; (b) F. T. Bakelaar, I. F. Eggen, A. Petersen and P. B. W. Ten Kortenaar, Org. Process Res. Dev., 2005, 9, 98 CrossRef; (c) F. Albericio, P. Cherkupally, B. G. de la Torre, T. Govender, H. G. Kruger and S. Ramesh, Sci. Synth., 2017, 357 Search PubMed.
- F. Albericio and A. El-Faham, Org. Process Res. Dev., 2018, 22, 760 CrossRef CAS.
- H. Wissmann and H. J. Kleiner, Angew. Chem., Int. Ed. Engl., 1980, 19, 133 CrossRef.
- (a) R. Escher and P. Bünning, Angew. Chem., Int. Ed. Engl., 1986, 25, 277 CrossRef; (b) J. Klose, M. Bienert, C. Mollenkopf, D. Wehle, C.-W. Zhang, L. A. Carpino and P. Henklein, Chem. Commun., 1999, 1847 RSC.
- B. T. M. Vishwanatha, N. R. Panguluri and V. V. Sureshbabu, Synthesis, 2013, 1569 Search PubMed.
- J. K. Augustine, A. Bombrun and S. Venkatachaliah, Tetrahedron Lett., 2011, 52, 6814 CrossRef CAS.
- O. Al Musaimi, R. Wisdom, P. Talbiersky, B. G. De La Torre and F. Albericio, ChemistrySelect, 2021, 6, 2649 CrossRef CAS.
- See ECHA web site for T3P®: https://echa.Europa.eu/substance-information/-/substanceinfo/100.102.078 CrossRef; A. A. Waghmare, R. M. Hindupur and H. N. Pati, Rev. J. Chem., 2014, 4, 53 CrossRef.
- A. A. Waghmare, R. M. Hindupur and H. N. Pati, Rev. J. Chem., 2014, 4, 53 CrossRef.
- J. R. Dunetz, Y. Xiang, A. Baldwin and J. Ringling, Org. Lett., 2011, 13, 5048 CrossRef CAS PubMed.
- S. R. Manne, B. G. de la Torre, A. El-Faham and F. Albericio, Synthesis, 2020, 3189 CAS; ECHA web site for DIC: https://echa.europa.eu/substance-information/-/substanceinfo/100.010.677.
- M. Cortes-Clerget, S. E. Spink, G. P. Gallagher, L. Chaisemartin, E. Filaire, J.-Y. Berthon and B. H. Lipshutz, Green Chem., 2019, 21, 2610 RSC.
- M. A. Elsawy, C. Hewage and B. Walker, J. Pept. Sci., 2012, 18, 302 CrossRef CAS PubMed.
- Green Solvent guides: (a) A. Amelio, G. Genduso, S. Vreysen, P. Luis and B. Van der Bruggen, Green Chem., 2014, 16, 3045 RSC; (b) D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546 RSC; (c) A. Benazzouz, L. Moity, C. Pierlot, M. Sergent, V. Molinier and J. M. Aubry, Ind. Eng. Chem. Res.,, 2013, 52, 16585 CrossRef CAS; (d) D. Prat, O. Pardigon, H. W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517 CrossRef CAS; (e) L. Bergkamp and N. Herbatschek, RECIEL, 2014, 23, 221–245 CrossRef; (f) F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef; (g) D. S. MacMillan, J. Murray, H. F. Sneddon, C. Jamieson and A. J. B. Watson, Green Chem., 2013, 15, 596 RSC.
- (a) Y. E. Jad, G. A. Acosta, S. N. Khattab, B. G. de la Torre, T. Govender, H. G. Kruger, A. El-Faham and F. Albericio, Org. Biomol. Chem., 2015, 13, 2393 RSC; (b) V. Martin, P. H. G. Egelund, H. Johansson, S. Thordal Le Quement, F. Wojcik and D. S. Pedersen, RSC Adv., 2020, 10, 42457 RSC.
- https://thalesnano.com/products-and-services/h-cube-mini-plus/ .
- J. Muzart, J. Mol. Catal. A: Chem., 2009, 308, 15 CrossRef CAS.
- J. W. Eschelbach, D. Wernick, M. C. Bryan and E. M. Doherty, Aust. J. Chem., 2013, 66, 165 CrossRef CAS.
- F. Errante, P. Ledwon, R. Lataika, P. Rovero and A. M. Papini, Front. Chem., 2020, 8, 57923 Search PubMed.
- C. De Luca, G. Lievore, D. Bozza, A. Buratti, A. Cavazzini, A. Ricci, M. Macis, W. Cabri, S. Felletti and M. Catani, Molecules, 2021, 26, 4688 CrossRef CAS PubMed.
- D. Takahashi, T. Inomata and T. Fukui, Angew. Chem., 2017, 129, 7911 CrossRef.
- J. M. Collins, K. A. Porter, S. K. Singh and G. S. Vanier, Org. Lett., 2014, 16, 940 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00431g |
| This journal is © The Royal Society of Chemistry 2023 |