
Heteropolyacid catalyzed O-alkylation of oximes with alcohols via a carbocation in dimethyl carbonate and mechanism insight†
Hongfeng
Zhuang
,
Qin
Hou
 ,
Feng
Han
*,
Haotian
Lv
and
Chengxia
Miao
*
,
Feng
Han
*,
Haotian
Lv
and
Chengxia
Miao
*
Key Laboratory of Agricultural Film Application of Ministry of Agriculture and Rural Affairs, College of Chemistry and Material Science, Shandong Agricultural University, Tai'an 271018, Shandong, China. E-mail: fenghan@sdau.edu.cn; chxmiao@sdau.edu.cn
First published on 5th December 2022
Abstract
The C–O bond as a crucial structural skeleton exhibits high bioactivities in organic chemistry. There are few reports about the direct construction of C–O bonds by O-alkylation of oximes with alcohols, particularly the development of catalytic systems. Herein, a simple catalytic system only including H3PW12O40·xH2O was established for the O-alkylation of oximes with alcohols in dimethyl carbonate as the green solvent, which simplified the reaction systems, realized catalytic transformation, and improved the yields and selectivities. The developed system has broad substrate scope and good functional group tolerance. It is suitable for the O-alkylation of aromatic or aliphatic ketoximes and aldoximes, and oximes bearing heteroatoms with benzyl alcohols, aliphatic alcohols, allylic alcohols, and so on, affording up to 98% yield. And the gram-scale reaction and further transformations of oxime ethers have also been carried out well. Triphenylmethyl tetrafluoroborate instead of triphenylmethanol still generated the desired product, which indicated that the Ph3C+ would be captured by the oxime and the reaction went through a carbocation process. The appearance and weakness of a new band at 404 and 433 nm in UV-Vis spectroscopy further proved a carbocation formation during the reaction. The changes of the absorption peak in FT-IR spectra and control experiments of different acids demonstrated the hydrogen bonding effect between the catalyst and the hydroxyl of triphenylmethanol.
Introduction
The construction of the C–O bond is an essential field in organic chemistry because of its high bioactivities and wide existence in many pharmaceuticals, agricultural chemicals and advanced materials.1 Currently, C–O bond construction via C–H activation is widely concerned with the high atom economy.2 Due to the low reactivity of the C–H bond, expensive transition metal catalysts combined with ligands, strong oxidants, strong bases, or other conditions are generally required to achieve the acyloxylation,3 alkoxylation,4 hydroxylation5etc.6 to build the C–O bond with the formation of esters, ethers, and hydroxy compounds. In addition, cross-coupling reactions, substitutions and additions are also the classical methods for the construction of C–O bonds catalyzed by Cu, Pd, Ni, Fe, etc. or in the presence of metal-free catalysts.7–16 In this way, significant progress has been made in the construction of C–O bonds, but harsh conditions such as a stoichiometric amount of base, high reaction temperature, or the production of lots of wastes still exist in the developed systems. Therefore, it is vital to put forward an environmentally friendly, efficient, and high atom-economy catalytic system to construct the C–O bond.Additionally, O-alkylation of oximes with alcohols is another good strategy for the formation of the C–O bond. Since oximes and oxime ethers as vital and versatile structural moieties not only widely exist in pharmaceuticals including cymoxanil, fenpyroximate and so on (Scheme 1),17 but also could be converted into isoxazoles, amines, nitrile lactams and other heterocyclic compounds containing nitrogen atoms in organic synthesis.18 And the direct substitution of alcohols was recognized as one of the 10 Key Green Chemistry Research Areas.19 Nevertheless, there are only a few reports for the O-alkylation reaction from alcohols (Scheme 2, previous systems). Initially, an O-alkylated oxime from a primary alcohol and benzophenone oxime was afforded under the effect of stoichiometric triphenylphosphine-diethyl azodiformate.20 And the reactions of alcohols with an oxime catalyzed by tetrabutylammonium iodide (TBAI) in the presence of triphenylphosphine, CCl4, and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) furnished the desired product in moderate yields.21 Besides, treatment with excess BF3·(C2H5)2O is another way to construct oxime-substituted derivatives from the reactions of an oxime and tertiary alcohols in CH2Cl2 containing molecular sieves.22 Furthermore, in the presence of stoichiometric Cs2CO3 and Sc(OTf)3, the intramolecular O-allylation of oximes was achieved by the formation of the C–O bond with an Ir complex as the catalyst.23 Obviously, the problems of previously reported systems were complicated components, lots of by-products, low yields and low selectivity due to the formation of an amide by Beckman rearrangement. Meanwhile, previous research mainly focused on constructing C–O bonds, and there are few investigations on the reaction mechanism. Therefore, it is necessary to develop a simple, green, and efficient catalytic system with broad substrate scope for the C–O bond construction from various oximes and all kinds of alcohols, and investigate the corresponding reaction mechanism in detail.
 | ||
| Scheme 1 Several examples of oxime ethers with pharmacological activities. | ||
 | ||
| Scheme 2 Different systems for the formation of oxime ethers from alcohols and oximes. | ||
A heteropolyacid such as phosphotungstic acid (H3PW12O40·xH2O, HPWA), phosphomolybdic acid (H3PMo12O40·xH2O) or silicotungstic acid (H4SiW12O40·xH2O) as a kind of green solid acid is stable, non-toxic, efficient and clean in homogeneous or heterogeneous catalysis.24 And dimethyl carbonate (DMC), as a green and versatile solvent, has drawn more and more attention owing to its low toxicity, low viscosity, and high biodegradability.25 Continuing our research on the transformation of alcohols catalyzed by Brønsted acids,26 we propose an environmentally friendly and efficient strategy only employing a heteropolyacid as the catalyst for the O-alkylation of oximes with alcohols (Scheme 2, this work). Catalyzed by H3PW12O40·xH2O, tertiary aromatic and aliphatic alcohols, and secondary aromatic alcohols were suitable for the O-alkylation of aromatic or aliphatic ketoximes and aldoximes in DMC as the green solvent, giving 10–98% yields. In the systems of the C–O bond formation, it was the first time that the Ph3C+ derived from triphenylmethyl tetrafluoroborate was captured by the oxime, which indirectly proved the carbocation process of the developed system. Besides, the carbocation intermediate was further illustrated by UV-Vis. And the stretching vibration change in FT-IR spectra and control experiments also demonstrated the hydrogen bonding effect between the catalyst and the hydroxyl of alcohol.
Results and discussion
Initially, we started our investigation of the O-alkylation reaction to construct a new C–O bond using benzophenone oxime (1a) and triphenylmethanol (2a) as the substrates (see Table 1 and Tables S1–S4†). The reaction catalyzed by HPWA using DMC as a green solvent without any additives afforded oxime ether in 84% yield (Table S1, entry 19†). No improvement in yield was obtained after testing the effect of reaction time, temperature, solvent, and the ratio of the substrates (Table S1†). The addition of 4A molecular sieves, DCC (N,N’-dicyclohexylcarbodiimide), or MgSO4 had an apparent influence on the reaction (Table S2†). To our delight, adding 1.2 eq. of MgSO4 gave the best yield (up to 96%) (Table S2, entry 18†). And the result should be ascribed to water absorption of MgSO4 to move the reaction to the right. The yield was raised from 76% to 96% by increasing the ratio of 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a from 1:1 to 1:3, and there was only a trace increase in yield upon further increasing the ratio to 1:4 or 1:5 (Table S3, entries 1–7†). And increasing the amount of 1a to 2 or 3 eq. of 2a led to 93% yield (Table S3, entries 8 and 9†). Next, the influence of reaction time was examined with a 1:3 ratio of 1a:2a (Table S2, entries 3 and 10–14†). And 2 h was the ideal time because a longer time led to the formation of benzophenone as a by-product (Table S3, entry 3†). Exploring the amount of catalyst (Table S3, entries 15–21†), the desired product could be obtained in 98% yield when using 1 mol% HPWA as the catalyst (Table S3, entry 18†), while the reaction was inert without a catalyst (Table S3, entry 21†). Surprisingly, the yield would decrease, when the catalyst amount was increased from 2 to 10 mol%. The phenomenon may be attributed to the fact that phosphotungstic acid containing too much crystal water would inhibit the reaction (Table S3, entry 15 vs. 16†). Other heteropolyacids such as H3PMo12O40·xH2O and H4SiW12O40·xH2O also showed excellent activities, providing oxime ether in 95% and 94% yields, respectively (Table 1, entries 2 and 3). Additionally, some simple organic Brønsted acids, such as dodecylbenzenesulfonic acid, p-TsOH, and so on, were used to catalyze the reaction and they exhibited inert activities (Table 1, entries 4–8). Finally, the influence of solvents on the reaction is depicted in Table S4.† Toxic polar aprotic solvents such as ethyl acetate, dichloromethane, acetonitrile, and acetone were advantageous to the reaction (Table S4, entries 2–5†). In addition, the reaction could also perform well when using diethyl ether or toluene as the solvent (Table S4, entries 6 and 7†). Unfortunately, 1a was nearly recovered in other solvents such as hexane, tetrahydrofuran, ethanol, and so on (Table S4, entries 8–13†).
a
:2a from 1:1 to 1:3, and there was only a trace increase in yield upon further increasing the ratio to 1:4 or 1:5 (Table S3, entries 1–7†). And increasing the amount of 1a to 2 or 3 eq. of 2a led to 93% yield (Table S3, entries 8 and 9†). Next, the influence of reaction time was examined with a 1:3 ratio of 1a:2a (Table S2, entries 3 and 10–14†). And 2 h was the ideal time because a longer time led to the formation of benzophenone as a by-product (Table S3, entry 3†). Exploring the amount of catalyst (Table S3, entries 15–21†), the desired product could be obtained in 98% yield when using 1 mol% HPWA as the catalyst (Table S3, entry 18†), while the reaction was inert without a catalyst (Table S3, entry 21†). Surprisingly, the yield would decrease, when the catalyst amount was increased from 2 to 10 mol%. The phenomenon may be attributed to the fact that phosphotungstic acid containing too much crystal water would inhibit the reaction (Table S3, entry 15 vs. 16†). Other heteropolyacids such as H3PMo12O40·xH2O and H4SiW12O40·xH2O also showed excellent activities, providing oxime ether in 95% and 94% yields, respectively (Table 1, entries 2 and 3). Additionally, some simple organic Brønsted acids, such as dodecylbenzenesulfonic acid, p-TsOH, and so on, were used to catalyze the reaction and they exhibited inert activities (Table 1, entries 4–8). Finally, the influence of solvents on the reaction is depicted in Table S4.† Toxic polar aprotic solvents such as ethyl acetate, dichloromethane, acetonitrile, and acetone were advantageous to the reaction (Table S4, entries 2–5†). In addition, the reaction could also perform well when using diethyl ether or toluene as the solvent (Table S4, entries 6 and 7†). Unfortunately, 1a was nearly recovered in other solvents such as hexane, tetrahydrofuran, ethanol, and so on (Table S4, entries 8–13†).
a
|
|
||
|---|---|---|
| Entry | Cat. (mol%) | Yieldb (%) |
| a Reaction conditions: benzophenone oxime 1a (0.3 mmol), triphenylmethanol 2a (0.9 mmol), catalyst (α mol% amount refers to 1a), MgSO4 (1.2 eq., 0.36 mmol), dimethyl carbonate (DMC) (2 mL), at room temperature (RT) for 2 h. b Isolated yield. c No reaction (NR). | ||
| 1 | H3PW12O40·xH2O (1) | 98 |
| 2 | H3PMo12O40·xH2O (1) | 95 |
| 3 | H4SiW12O40·xH2O (1) | 94 |
| 4 | Dodecylbenzenesulfonic acid (1) | NRc |
| 5 | p-TsOH (1) | NRc |
| 6 | (±)-10-Camphorsulfonic acid (10) | NRc |
| 7 | Bu4NHSO4 (10) | NRc |
| 8 | N-Dodecylphosphonic acid (10) | NRc |

With the optimized reaction conditions established, a series of oximes were employed as substrates to achieve the O-alkylation of oximes with triphenylmethanol (Table 2). Catalyzed by 1 mol% H3PW12O40·xH2O at room temperature, benzophenone oximes (1a–1p) containing electron-donating or -withdrawing groups were all suitable for the formation of new C–O bonds in excellent yields. And the results indicated that the yields have nearly no correlation with the number, electronic properties, and positions of substituted groups. The oximes (1m–1p) with methyl or Cl at the 2- or 3-position of the phenyl ring were tolerated well and the yields of the desired products were above 95%. The crystal structure of compound 3g showed that the absolute configuration was the higher stability of the Z-isomer (Fig. 1), which may be attributed to the low energy of calculated formation enthalpy.21 In addition, acetophenone oximes were beneficial for the construction of new C–O bonds by O-alkylation. The substrates 1q–1z could react with 2a to provide the corresponding oxime ethers 3q–3z in up to 98% yield. And the electronic properties and the steric hindrance also slightly affected this transformation. Furthermore, 3,4-dihydronaphthalen-1(2H)-one oxime (1aa) and 9H-fluoren-9-one oxime (1ab) were compatible for the O-alkylation. Surprisingly, the reaction could proceed smoothly with 1ac as the substrate, affording the product in 87% yield. Moreover, the oximes bearing heteroatoms such as N and O could generate the corresponding products, although the yield was only 24% with (E)-1-(pyridin-3-yl)ethan-1-one oxime as the substrate, and 3ae was obtained in 80% yield. Aliphatic oximes were also tested for the preparation of oxime ethers, and target products such as 3af, 3ag, 3ah, 3ai, and 3aj were generated in moderate to excellent yields. To our delight, various aldoximes were also suitable substrates for the developed system. The target product 3ak was obtained in 86% yield, while 1al and 1am containing heteroatoms were smoothly converted into the desired products 3al and 3am in 24% and 76% yields, respectively. However, 3ad and 3al containing nitrogen atoms gave poorer results. There may be two reasons for the activities of the two substrates. First, due to the existence of the pyridine ring, 1ad and 1al showing alkalinity may react with the acidic sites of heteropolyacids, reducing the catalytic activity. On the other hand, pyridine is less aromatic than benzene and the oxime is not easy to form a nitrone isomer to generate the desired product.27 In addition, the aliphatic aldoximes were compatible for O-alkylation with triphenylmethanol, furnishing the corresponding products in moderate to good yields (3an, 3ao and 3ap). Besides, the allylic aldoxime (1aq) was suitable for constructing the C–O bond, releasing 3aq in 65% yield.


Next, the scope of alcohols was investigated with 1a, 1q or 1ak as the oximes (Table 3). In the case of the reactions between triphenylmethanols and 1a, the activities of 2a–2l as the substrates have nearly no relationship with the electronic properties and the steric hindrance for the preparation of oxime ethers to construct new C–O bonds, and excellent yields were afforded. 4j and 4k containing two electron-donating groups and electron-withdrawing groups were obtained in 93% and 95% yields, respectively. And the other tertiary benzyl alcohols, such as 1,1-diphenylethan-1-ol and 2-phenylpropan-2-ol, were also able to achieve the O-alkylation reaction with 1a to generate the corresponding products 4l and 4m accompanied by small amounts of olefins as the by-products derived from the dehydration of the corresponding alcohols. Meanwhile, the reaction of 1a with 1,1-diphenylethylene was inert, which demonstrated that the reaction would not go through the olefin intermediate. Unfortunately, 4n could not be acquired by column chromatography, while 4o was obtained in 17% yield by using 2-methyl-4-phenylbutan-2-ol and 1a as the substrates. It is worth noting that the aliphatic tertiary alcohols as less active substrates, including tert-butanol, tert-amyl alcohol and 1-methylcyclohexan-1-ol, could provide the desired products 4p–4r in low to moderate yields. Interestingly, 1-adamantanol and 1a as the substrates could produce the corresponding oxime ether 4s in 92% yield. Moreover, the activities of a series of secondary alcohols with 1a were tested. Diphenylmethanol and 1-phenylethan-1-ol were applied to the system with good to excellent yields (4t, 97% yield; 4u, 78% yield). Allylic alcohol such as (E)-1,3-diphenylprop-2-en-1-ol as the material was effective to provide 4v in 94% yield. However, 4w could not be obtained under the optimized reaction conditions. And the desired products could not be obtained using primary alcohols (such as benzyl alcohol, 4-methoxybenzyl alcohol, phenylethanol, trans-2-hexenol, cis-2-hexenol and isobutanol) and 1a as the substrates. Besides, 33% yield of 4x was obtained from the reaction between tert-butanol and 1q. To our delighted, the reactions between aldoxime 1ak and aliphatic tertiary alcohols (tert-butanol and 1-adamantanol) could also provide 4y and 4z in 35% or 48% yields, respectively.

Furthermore, the gram-scale reaction for the synthesis of oxime ether by the construction of a new C–O bond proceeded well (Scheme 3). When 3 mmol of benzophenone oxime was used as the substrate at room temperature, the yield reached 94% after 2 h. And still 82% yield of 3a was obtained with 12 mmol of 1a as the substrate under the same conditions. Additionally, the reaction could successfully provide 4.8737 g (92%) of 3a in the presence of 1.2 eq. MgSO4 as the additive and 80 mL DMC as the solvent by prolonging the reaction time to 24 h.
 | ||
| Scheme 3 The gram-scale reaction for the synthesis of oxime ether. | ||
In order to further demonstrate the transformation of the oxime ether as the substrate, aldol condensation between 4x and benzaldehyde was carried out smoothly in the presence of n-BuLi, giving 35% yield of T1 (Scheme 4, (1)).28 Additionally, T2 as a secondary amine could be provided through the reaction between 4y and allylmagnesium bromide in 96% yield (Scheme 4, (2)).29
 | ||
| Scheme 4 The transformation of 4x and 4y. | ||
As an attempt to get more insights into the reaction mechanism, some control experiments and in situ detection were conducted. According to the reported studies,30 the triphenylmethyl carbocation (Ph3C+) in solution showed a yellow colour, which was consistent with our experimental phenomenon (Fig. S1†). To verify the process of O-alkylation to construct new C–O bonds through a carbocation intermediate, triphenylmethyl tetrafluoroborate 5 instead of 2a was reacted with 1a under the optimal reaction conditions (Scheme 5, (1)), which provided only 64% yield of the product 3a. The low yield was probably due to the influence of the produced HBF4 that increased the acidity of the system. Then, we investigated the effects of NaBF4 and HBF4 (Scheme 5, (2)–(6)). Obviously, the yield would decrease when increasing the amount of HBF4 (Scheme 5, (2)–(4)). Excellent yield was provided in the absence of HBF4 (Scheme 5, (5)). And the presence of NaBF4 had no significant effect on the reaction (Scheme 5, (6)). The results proved that the adverse effects of hydrogen ions in HBF4 led to low yield and the O-alkylation went through a carbocation process rather than other path to form a C–O bond. In addition, the outcomes were further supported by UV-Vis spectroscopy (Fig. 2). The absorption of the substrates, catalyst, or product, respectively, was observed before 300 nm (Fig. S2†). When the catalyst was added to triphenylmethanol, new bands appeared at 404 and 433 nm, which belonged to the triphenylmethyl carbocation.31 Moreover, 1a was added multiple times to the mixture of triphenylmethanol and catalyst, showing absorption at 404 and 433 nm. As the usage of 1a increased, the concentration of the carbocation decreased, which well exhibited that the reaction really went through the process of Ph3C+.
 | ||
| Scheme 5 Control experiments for the influence of HBF4. | ||
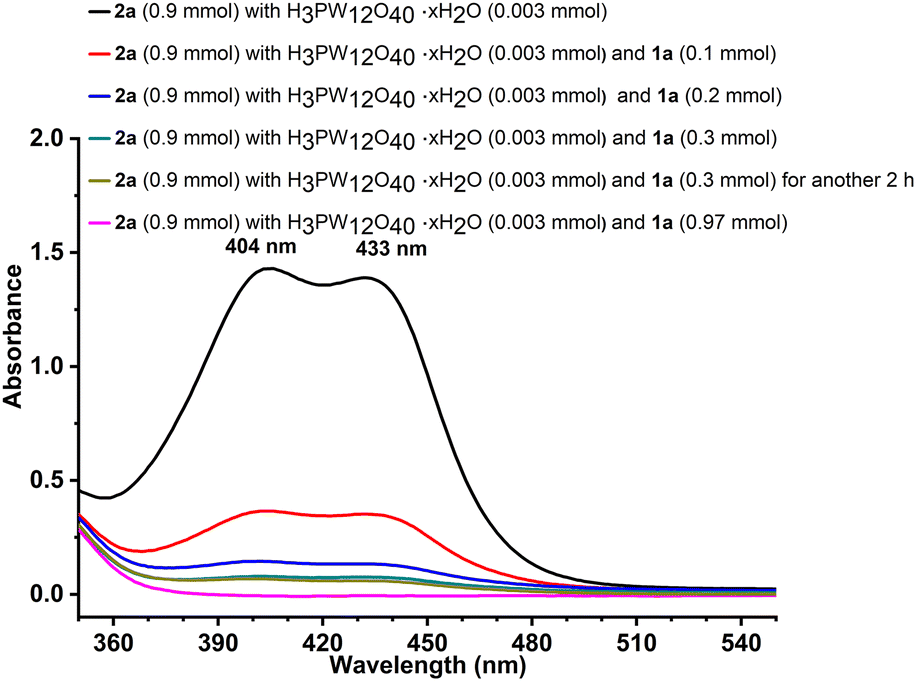 | ||
| Fig. 2 Reaction phenomena detected by UV-Vis spectroscopy. | ||
Besides, to prove the interaction of the catalyst and the substrates, some other experiments were carried out. H3PO4 instead of H3PW12O40·xH2O as the catalyst showed poor activity for the O-alkylation, suggesting that the anion, particularly W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of H3PW12O40·xH2O played an indispensable role (Scheme 6, (1) and (2)). The results indicated that there might be interactions such as the hydrogen bonding effect between triphenylmethanol and H3PW12O40·xH2O. Then, the FT-IR spectra of triphenylmethanol, H3PW12O40·xH2O, or the mixture of 2a and HPWA further confirmed the interaction between the substrate and catalyst (Fig. 3).32 The band at 1158 cm−1 assigned to the C–O bond of triphenylmethanol weakened when H3PW12O40·xH2O was added to the solution of triphenylmethanol. Besides, the stretching vibration of W–Oc–W at 796 cm−1 belonging to H3PW12O40·xH2O also shifted to the left to 808 cm−1 accompanied by negligible changes of P–Oa at 1076 cm−1, WOt at 975 cm−1, and W–Ob–W at 894 cm−1 when H3PW12O40·xH2O was mixed with triphenylmethanol. The stretching vibration change of FT-IR spectra further suggested the effect of hydrogen bonding between WO and O–H of triphenylmethanol.
O of H3PW12O40·xH2O played an indispensable role (Scheme 6, (1) and (2)). The results indicated that there might be interactions such as the hydrogen bonding effect between triphenylmethanol and H3PW12O40·xH2O. Then, the FT-IR spectra of triphenylmethanol, H3PW12O40·xH2O, or the mixture of 2a and HPWA further confirmed the interaction between the substrate and catalyst (Fig. 3).32 The band at 1158 cm−1 assigned to the C–O bond of triphenylmethanol weakened when H3PW12O40·xH2O was added to the solution of triphenylmethanol. Besides, the stretching vibration of W–Oc–W at 796 cm−1 belonging to H3PW12O40·xH2O also shifted to the left to 808 cm−1 accompanied by negligible changes of P–Oa at 1076 cm−1, WOt at 975 cm−1, and W–Ob–W at 894 cm−1 when H3PW12O40·xH2O was mixed with triphenylmethanol. The stretching vibration change of FT-IR spectra further suggested the effect of hydrogen bonding between WO and O–H of triphenylmethanol.
 | ||
| Scheme 6 Control experiments using H3PO4 instead of H3PW12O40·xH2O. | ||
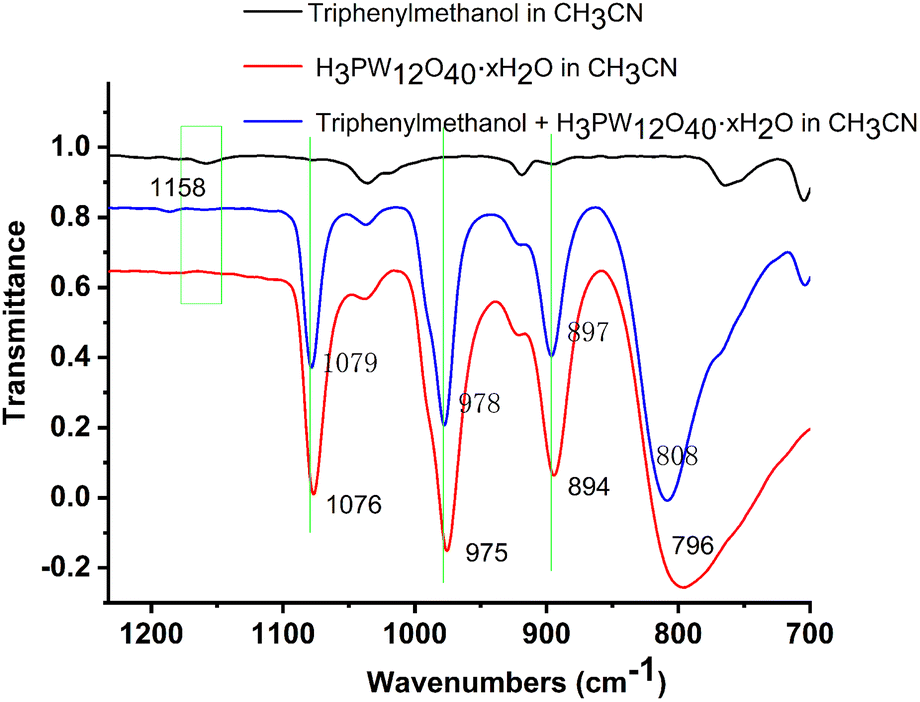 | ||
| Fig. 3 The interaction between the catalyst and triphenylmethanol monitored by FT-IR. | ||
Based on the above evidence and previous reports,33 a possible mechanism involving the carbocation and hydrogen bonding effect has been proposed (Scheme 7). HPWA as the catalyst plays double roles, acting as an acid to provide the H+ and forming hydrogen bonds with the substrate. At first, the triphenylmethyl carbocation is released by the dehydration of triphenylmethanol, which can be activated by hydrogen bonding with HPWA (intermediate I). Then the nitrone tautomer 1a′,34 which is in equilibrium with benzophenone oxime, can react with II to provide intermediate III through the nucleophilic attack. Finally, the desired product 3a is afforded by the release of H, and HPWA as the catalyst is regenerated simultaneously.
 | ||
| Scheme 7 Proposed mechanism for the C–O bond construction catalyzed by HPWA. | ||
Conclusions
In conclusion, we have developed an environmentally friendly, mild, and efficient strategy to construct C–O bonds via O-alkylation of oximes with alcohols using H3PW12O40·xH2O as the catalyst and DMC as the solvent. In the O-alkylation, not only tertiary or secondary benzyl alcohols and aliphatic alcohols could react well with oximes, but also aromatic or aliphatic ketoximes and aldoximes with triphenylmethanol were compatible for constructing oxime ethers, generating up to 98% yield of the target products. Moreover, the O-alkylation reaction could occur with oximes containing a heteroatom. In addition, the gram-scale reaction could be conducted smoothly. A secondary amine and β-oxime ether alcohol were successfully generated by further transformation of the oxime ether. The use of triphenylmethyl tetrafluoroborate instead of triphenylmethanol as the substrate generated the desired product indicating that the triphenylmethyl carbocation was the possible intermediate. The peak of the triphenylmethyl carbocation detected by UV-Vis would disappear upon adding an oxime, which further proved the carbocation process. Moreover, the stretching vibration change in FT-IR spectra revealed the effect of hydrogen bonding between the anion of the heteropolyacid and the hydroxyl of alcohol. And a mechanism involving the triphenylmethyl carbocation derived from the action of the H+ and the hydrogen bonding effect between H3PW12O40·xH2O and triphenylmethanol was proposed. Further studies on activating the alcohols to construct other new C–X bonds are ongoing in our lab.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Fluorosilicone Materials Joint Funds of Shandong Provincial Natural Science Foundation (ZR2019LFG009), National Natural Science Foundation of China (21972079 and 22272094), Natural Science Foundation of Shandong Province (ZR2019MB002), and Incubation Program of Youth Innovation in Shandong Province.References
- (a) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318–5365 CrossRef CAS; (b) S. Enthaler and A. Company, Chem. Soc. Rev., 2011, 40, 4912–4924 RSC; (c) G. Evano, J. Wang and A. Nitelet, Org. Chem. Front., 2017, 4, 2480–2499 RSC.
- (a) Q. Zheng, J. Chen and G.-W. Rao, Russ. J. Org. Chem., 2019, 55, 569–586 CrossRef CAS; (b) K. Peng and Z. B. Dong, Adv. Synth. Catal., 2021, 363, 1185–1201 CrossRef CAS.
- J. Wu, K. L. M. Hoang, M. L. Leow and X.-W. Liu, Org. Chem. Front., 2015, 2, 502–505 RSC.
- (a) B. D. Barve, Y. C. Wu, M. El-Shazly, M. Korinek, Y. B. Cheng, J. J. Wang and F. R. Chang, Org. Lett., 2014, 16, 1912–1915 CrossRef CAS; (b) H. Wang, K. Liang, W. Xiong, S. Samanta, W. Li and A. Lei, Sci. Adv., 2020, 6, eaaz0590 CrossRef CAS PubMed.
- (a) R. C. Betori, C. M. May and K. A. Scheidt, Angew. Chem., Int. Ed., 2019, 58, 16490–16494 CrossRef CAS PubMed; (b) D. Saha, P. Das, P. Biswas and J. Guin, Chem. – Asian J., 2019, 14, 4534–4548 CrossRef CAS.
- (a) Q. Wang, H. Zheng, W. Chai, D. Chen, X. Zeng, R. Fu and R. Yuan, Org. Biomol. Chem., 2014, 12, 6549–6553 RSC; (b) J. Borgel, L. Tanwar, F. Berger and T. Ritter, J. Am. Chem. Soc., 2018, 140, 16026–16031 CrossRef.
- (a) F. Ullmann and P. Sponagel, Ber. Dtsch. Chem. Ges., 1905, 38, 2211–2212 CrossRef CAS; (b) S. Bhunia, G. G. Pawar, S. V. Kumar, Y. Jiang and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 16136–16179 CrossRef CAS.
- (a) N. A. Butt and W. Zhang, Chem. Soc. Rev., 2015, 44, 7929–7967 RSC; (b) J. Moran, M. Dryzhakov and E. Richmond, Synthesis, 2016, 935–959 CrossRef.
- (a) G. Fang and X. Bi, Chem. Soc. Rev., 2015, 44, 8124–8173 RSC; (b) R. Blieck, M. Taillefer and F. Monnier, Chem. Rev., 2020, 120, 13545–13598 CrossRef CAS PubMed.
- (a) A. J. Chinn, B. Kim, Y. Kwon and S. J. Miller, J. Am. Chem. Soc., 2017, 139, 18107–18114 CrossRef CAS; (b) R. Mao, J. Balon and X. Hu, Angew. Chem., Int. Ed., 2018, 57, 13624–13628 CrossRef CAS PubMed.
- (a) S. Gowrisankar, A. G. Sergeev, P. Anbarasan, A. Spannenberg, H. Neumann and M. Beller, J. Am. Chem. Soc., 2010, 132, 11592–11598 CrossRef CAS; (b) K. M. Morrison, R. T. McGuire, M. J. Ferguson and M. Stradiotto, ACS Catal., 2021, 11, 10878–10884 CrossRef CAS; (c) Q. K. Kang, Y. Lin, Y. Li, L. Xu, K. Li and H. Shi, Angew. Chem., Int. Ed., 2021, 60, 20391–20399 CrossRef CAS.
- (a) J. M. Lee, E. J. Park, S. H. Cho and S. Chang, J. Am. Chem. Soc., 2008, 130, 7824–7825 CrossRef CAS; (b) B. Tan, N. Toda and C. F. Barbas III, Angew. Chem., Int. Ed., 2012, 51, 12538–12541 CrossRef CAS PubMed; (c) A. O. Terenťev, I. B. Krylov, V. P. Timofeev, Z. A. Starikova, V. M. Merkulova, A. I. Ilovaisky and G. I. Nikishin, Adv. Synth. Catal., 2013, 355, 2375–2390 CrossRef.
- (a) Y. Sun, A. Abdukader, H. Zhang, W. Yang and C. Liu, RSC Adv., 2017, 7, 55786–55789 RSC; (b) Z. Y. Chen, H. J. Liang, R. X. Chen, L. Chen, X. Z. Tang, M. Yan and X. J. Zhang, Adv. Synth. Catal., 2019, 361, 3324–3330 CrossRef CAS; (c) Z. Mirjafary, M. Abdoli, H. Saeidian, A. Kakanejadifard and S. M. F. Farnia, RSC Adv., 2016, 6, 17740–17758 RSC.
- (a) L. Zhang, G. Zhang, M. Zhang and J. Cheng, J. Org. Chem., 2010, 75, 7472–7474 CrossRef CAS; (b) M. K. Arachchi and H. M. Nguyen, Adv. Synth. Catal., 2021, 363, 4239–4246 CrossRef CAS.
- (a) J. Hu, M. Zhu, Y. Xu and J. Yan, Synthesis, 2012, 1226–1232 CAS; (b) G. S. Kumar, C. U. Maheswari, R. A. Kumar, M. L. Kantam and K. R. Reddy, Angew. Chem., Int. Ed., 2011, 50, 11748–11751 CrossRef CAS.
- (a) Q. Xu, H. Xie, P. Chen, L. Yu, J. Chen and X. Hu, Green Chem., 2015, 17, 2774–2779 RSC; (b) J. Madasu, S. Shinde, R. Das, S. Patel and A. Shard, Org. Biomol. Chem., 2020, 18, 8346–8365 RSC; (c) S. Estopina-Duran and J. E. Taylor, Chemistry, 2021, 27, 106–120 CrossRef CAS; (d) A. Jalil, Y. O. Yang, Z. Chen, R. Jia, T. Bi, X. Li and Y. Du, Mini-Rev. Org. Chem., 2021, 18, 540–605 CrossRef CAS.
- (a) F. Tellier, R. Fritz, L. Kerhoas, P.-H. Ducrot, A. Carlin-Sinclair, J. Einhorn and P. Leroux, Pest Manage. Sci., 2009, 65, 129–136 CrossRef CAS; (b) I. Ayed-Boussema, H. Hamdi, H. Chaabani, A. M′nassri, M. Mokni and S. Abid, Neurotoxicology, 2022, 91, 177–187 CrossRef CAS; (c) J. Zhu, J. Mo, H. Lin, Y. Chen and H. Sun, Bioorg. Med. Chem., 2018, 26, 3065–3075 CrossRef CAS.
- (a) A. Y. Sukhorukov and S. L. Ioffe, Chem. Rev., 2011, 111, 5004–5041 CrossRef CAS; (b) Z. Mirjafary, M. Abdoli, H. Saeidian, S. Boroon and A. Kakanejadifard, RSC Adv., 2015, 5, 79361–79384 RSC; (c) C. Chen, J. Zhao, X. Shi, L. Liu, Y.-P. Zhu, W. Sun and B. Zhu, Org. Chem. Front., 2020, 7, 1948–1969 RSC.
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082–5103 RSC.
- S. Bittner and S. Grinberg, J. Chem. Soc., Perkin Trans. 1, 1976, 1708–1711 RSC.
- M. N. S. Rad, A. Khalafi-Nezhad, F. Karimitabar and S. Behrouz, Synthesis, 2010, 1724–1730 CAS.
- P. Brown, S. H. Calvert, P. C. A. Chapman, S. C. Cosham, A. J. Eglington, R. L. Elliot, M. A. Harris, J. D. Hinks, J. Lowther, D. J. Merrikin, M. J. Pearson, R. J. Ponsford and J. V. Syms, J. Chem. Soc., Perkin Trans. 1, 1991, 881–891 RSC.
- T. Sandmeier and E. M. Carreira, Org. Lett., 2021, 23, 2643–2647 CrossRef PubMed.
- (a) M. Egi, T. Kawai, M. Umemura and S. Akai, J. Org. Chem., 2012, 77, 7092–7097 CrossRef CAS PubMed; (b) N. Narkhede, S. Singh and A. Patel, Green Chem., 2015, 17, 89–107 RSC; (c) L. Lian, H. Zhang, S. An, W. Chen and Y.-F. Song, Sci. China: Chem., 2021, 64, 1117–1130 CrossRef CAS; (d) G. P. Yang, X. Wu, B. Yu and C. W. Hu, ACS Sustainable Chem. Eng., 2019, 7, 3727–2732 CrossRef CAS.
- (a) P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS; (b) P. Tundo, M. Musolino and F. Aricò, Green Chem., 2018, 20, 28–85 RSC.
- (a) F. Han, L. Yang, Z. Li and C. Xia, Adv. Synth. Catal., 2012, 354, 1052–1060 CrossRef CAS; (b) F. Han, L. Yang, Z. Li, Y. Zhao and C. Xia, Adv. Synth. Catal., 2014, 356, 2506–2516 CrossRef CAS; (c) C. X. Miao, X.-X. Li, Y.-M. Lee, C. Xia, Y. Wang, W. Nam and W. Sun, Chem. Sci., 2017, 8, 7476–7482 RSC; (d) C. X. Miao, H. Zhuang, Y. Wen, F. Han, Q.-F. Yang, L. Yang, Z. Li and C. Xia, Eur. J. Org. Chem., 2019, 3012–3021 CrossRef CAS; (e) C. Miao, Q. Hou, Y. Wen, F. Han, Z. Li, L. Yang and C.-G. Xia, ACS Sustainable Chem. Eng., 2019, 7, 12008–12013 CAS; (f) Z. Li, Y. Wen, N. Wang, F. Han, Y. Li, H. Zhuang and C. Miao, Eur. J. Org. Chem., 2021, 3819–3826 CrossRef CAS.
- (a) I. K. Song, M. S. Kaba and M. A. Barteau, J. Phys. Chem., 1996, 100, 17528–17534 CrossRef CAS; (b) A. Vimont, A. Travert, C. Binet, C. Pichon, P. Mialane, F. Sécheresse and J.-C. Lavalley, J. Catal., 2006, 241, 221–224 CrossRef CAS.
- Y. Jiang, W. C. Chan and C.-M. Park, J. Am. Chem. Soc., 2012, 134, 4104–4107 CrossRef CAS.
- (a) P. P. Mukhopadhyay, O. Miyata and T. Naito, Synlett, 2007, 1403–1406 CAS; (b) Y. Nishida, M. Ueda, M. Hayashi, N. Takeda and O. Miyata, Eur. J. Org. Chem., 2016, 22–25 CrossRef CAS.
- N. Bartalucci, F. Marchetti, S. Zacchini and G. Pampaloni, Dalton Trans., 2019, 48, 5725–5734 RSC.
- (a) H. J. Shine, D. H. Bae, A. K. M. M. Hoque, A. Kajstura, W. K. Lee, R. W. Shaw, M. Soroka, P. S. Engel and D. E. Keys, Phosphorus, Sulfur Silicon Relat. Elem., 1985, 23, 111–141 CrossRef CAS; (b) Y. Sadaoka, M. Matsuguchi and Y. Sakai, J. Electrochem. Soc., 1991, 138, 614–615 CrossRef CAS.
- (a) M. Misono, Catal. Rev. Sci. Eng., 1987, 29, 269–321 CrossRef CAS; (b) P. Zhao, Y. Zhang, Y. Wang, H. Cui, F. Song, X. Sun and L. Zhang, Green Chem., 2018, 20, 1551–1559 RSC; (c) F. C. Oliveira, A. M. L. Denadai, F. H. Fulgêncio, W. F. Magalhães, A. F. C. Alcântara, D. Windmöller and J. C. Machado, J. Mol. Struct., 2012, 1017, 84–89 CrossRef CAS.
- (a) G.-W. Wang, Y.-B. Shen and X.-L. Wu, Eur. J. Org. Chem., 2008, 4999–5004 CrossRef CAS; (b) R. Grigg, Chem. Soc. Rev., 1987, 16, 89–121 RSC; (c) F. Wu, Y. Zhao, Y. Wang, Y. Xu, M. Tang, Z. Wang, B. Han and Z. Liu, Green Chem., 2022, 24, 3137–3142 RSC.
- D. Roca-López, A. Darù, T. Tejero and P. Merino, RSC Adv., 2016, 6, 22161–22173 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, optimization of reaction conditions, the experiments for mechanism investigation, characterization data, the NMR spectra of the products and X-ray crystallographic data (CIF). CCDC 2193227. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2gc03214g |
| This journal is © The Royal Society of Chemistry 2023 |