
Sequential chemo–biocatalytic synthesis of aroma compounds†
Romina D.
Ceccoli
 a,
Dario A.
Bianchi
*b and
Daniela V.
Rial
*a
a,
Dario A.
Bianchi
*b and
Daniela V.
Rial
*a
aÁrea Biología Molecular, Departamento de Ciencias Biológicas, Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario (UNR), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Suipacha 531, S2002LRK Rosario, Argentina. E-mail: rial@inv.rosario-conicet.gov.ar; drial@fbioyf.unr.edu.ar
bInstituto de Química Rosario (IQUIR, CONICET-UNR), Departamento de Química Orgánica, Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario (UNR), Suipacha 531, S2002LRK Rosario, Argentina. E-mail: bianchi@iquir-conicet.gov.ar; dbianchi@fbioyf.unr.edu.ar
First published on 8th December 2022
Abstract
A large number of multienzymatic reactions allow access to chemical compounds with variable structures for diverse applications. One of the most attractive targets is the production of small bioactive compounds, such as aroma and flavour compounds. 2-phenylethyl acetate (2-PEA) and 2-phenylethanol (2-PE) are two rose-like aroma compounds massively used by the industry as fragrance ingredients in cosmetic and non-cosmetic products, as well as flavours and aromas in food and drink preparations. Although there have been several approaches to produce them biotechnologically, the vast majority of these compounds is obtained by chemical synthesis. In this work, we propose a new eco-friendly route towards 2-PEA and eventually 2-PE from simple starting materials. The route involves a solvent-free aldol condensation reaction, followed by a biocatalytic cascade, which involves a Baeyer–Villiger monooxygenase and other redox biocatalysts.
Introduction
The combination of different enzymatic activities for synthetic purposes is an actively growing field, in which a large number of cascades or multienzymatic reactions allow access to chemical compounds with variable structures for diverse applications. Within this topic, one of the most attractive targets is the production of small bioactive compounds, such as aroma and flavour compounds.2-Phenylethyl acetate (2-PEA, also known as phenethyl acetate) is an ester with rose scent and honey notes1 used yearly in hundreds of tons all over the world by different industrial sectors.2,3 It is a component of several fragrances in cosmetic products such as shampoos and soaps, and in non-cosmetic products such as detergents and cleaners.2 This ester is also used in food and drinks as an aroma and flavour ingredient4 and it is considered generally recognized as safe (GRAS).2,5 The vast majority of 2-PEA is produced chemically by esterification of 2-phenylethanol (2-PE, also known as phenethyl alcohol) with acetic acid or by transesterification of 2-PE with acetate esters.4,5 2-PE has a rose-like odour and is considered as a GRAS flavouring compound.5–7 It is very commonly used as an ingredient in the cosmetic, pharmaceutical and food industries, with a world production of approximately 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons per year. Besides, 2-PE exhibits antimicrobial and antifungal activities.5,6 Most of the 2-PE is synthesised by different chemical procedures, including the Friedel–Crafts reaction of ethylene oxide with benzene in the presence of aluminium chloride and the hydrogenation of styrene oxide, and as a by-product of the production of propylene oxide.5,8 These procedures have some drawbacks since they involve chemical catalysts and strong temperature and/or pressure conditions and the grade of purity of 2-PE and 2-PEA can be affected by the presence of undesirable by-products derived from synthetic routes.
000 tons per year. Besides, 2-PE exhibits antimicrobial and antifungal activities.5,6 Most of the 2-PE is synthesised by different chemical procedures, including the Friedel–Crafts reaction of ethylene oxide with benzene in the presence of aluminium chloride and the hydrogenation of styrene oxide, and as a by-product of the production of propylene oxide.5,8 These procedures have some drawbacks since they involve chemical catalysts and strong temperature and/or pressure conditions and the grade of purity of 2-PE and 2-PEA can be affected by the presence of undesirable by-products derived from synthetic routes.
Another source of these aroma compounds is nature. 2-PEA is present in several plants, being particularly abundant in evergreen trees (Cinnamomum species) and cloves.2 It is a major aromatic volatile ester emitted from rose flowers.4 2-PE is present in the essential oils of some flowers and plants, such as hyacinths, jasmine, narcissi and lilies. It can also be found in a variety of food products that involve a fermentation process, including wine, cheese and beer.8 However, the amount of 2-PE and 2-PEA obtained via extraction from plants together with the increased consumers’ preference for natural products and the limitations imposed to the use of chemically synthesised flavours have stimulated the development of eco-friendly alternatives towards 2-PE and 2-PEA. These methods include the biotechnological production of 2-PE and 2-PEA based on yeast metabolism and the enzymatic production of 2-PEA based on lipase activity.4,5
2-PE can be produced by microbial metabolism, especially by some yeasts, but in concentrations insufficient to satisfy the demand. However, the addition of L-phenylalanine (L-phe) to the growth medium can stimulate the production of 2-PE via the Ehrlich pathway.5,6,8 In addition, 2-PE can also be obtained by de novo synthesis through the metabolic shikimate pathway from simple sugars to phenylpyruvate, which then enters the Ehrlich pathway.5,6,8 There is a third pathway, the phenylethylamine pathway, mainly used by plants.5
Kim et al. designed a biotechnological approach that involved genetically modified Saccharomyces cerevisiae and a two-phase fermentation method to obtain 2-PE. With this strategy, they improved the yield of 2-PE about nine-fold with respect to wild-type yeast.9 The production of 2-PE and 2-PEA by several non-Saccharomyces strains was also reported.10–16 In addition, different bacterial and yeast strains have been engineered to improve this metabolism in order to favour 2-PE and 2-PEA formation.5,6,17–20 The metabolic formation of 2-PEA from 2-PE is a transesterification reaction mediated by an alcohol acyl transferase.5 In a different approach, 2-PEA was detected after 72 h of biotransformation with the fungus Beauveria bassiana, but only in very low levels.21 In another report, the formation of 2-PEA was investigated by evaluating the enzymatic transesterification of vinyl acetate with 2-PE catalysed by an immobilized lipase from Candida antarctica.4 The limitations of the microbial production of 2-PE are mostly related to the high cost of using L-phe as the starting material, protein expression drawbacks, cellular toxicity and the scarce amount of achievable products.17,22
In a previous work of our group, we explored the substrate profile of a type I Baeyer–Villiger monooxygenase from Leptospira biflexa (LbBVMO) expressed in Escherichia coli and unveiled its ability to produce 2-PEA in vivo by oxygenation of benzylacetone (IUPAC: 4-phenylbutan-2-one).23 Type I BVMOs are FAD-dependent flavoenzymes that catalyse the Baeyer–Villiger oxidation of linear ketones to esters or cyclic ketones to lactones, among other substrates, in a NADPH-dependent manner.24,25 Therefore, LbBVMO appeared to be an obvious alternative for the synthesis of 2-PEA.
Coupling two or more consecutive reactions, in a one-pot or tandem sequence, has become a method of choice to achieve the (chemo)enzymatic synthesis of both simple and value-added chemical compounds, as it has been extensively reviewed in the last few years.26–30 Cascade reactions offer substantial advantages over sequential single-step synthesis since there is no need to purify reaction intermediates, saving time and resources, and the production of by-products and waste can be significantly reduced. In addition, in vivo biocatalytic cascade reactions take advantage of cellular metabolism to recycle enzyme cofactors. In 2019, Vorster et al. reported a biocatalytic route using cinnamaldehyde as a starting material to obtain 2-PE after hydrolysis of a formate ester intermediate.31
In this work, we propose a new eco-friendly route towards 2-PEA and eventually 2-PE from simple starting materials. The route involves a solvent-free aldol condensation reaction followed by a biocatalytic cascade. For the biocatalytic steps, we coupled the reaction catalysed by the LbBVMO with other redox biocatalysts in an artificial cascade.
Experimental
General
Ketones, esters and alcohols used in this study were either from commercial sources or synthesised in our lab. Enzymes and reagents for molecular biology as well as other chemicals were purchased from commercial sources (Promega Corp., Madison, WI, USA; Invitrogen Corp., Carlsbad, CA, USA; Sigma-Aldrich Corp., St Louis, MO, USA; Merck KGaA, Darmstadt, Germany; Genbiotech S.R.L., CABA, Argentina; BD (Becton, Dickinson and Company), Franklin Lakes, NJ, USA; Cicarelli Laboratorios, San Lorenzo, Argentina; Bio Basic Inc., Markham, ON, Canada; Thermo Fisher Scientific, MP Biomedicals, Santa Ana, CA, USA; Alfa Aesar, Tewksbury, MA, USA).Synthesis of benzalacetone
Benzalacetone was synthesised by a solvent-free Claisen–Schmidt condensation reaction. In procedure A, benzaldehyde (1.5706 g, 14.80 mmol, 1 equiv.) and acetone (7.91 g, 136.191 mmol, 9.2 equiv.) were mixed at room temperature and powdered sodium hydroxide (0.2027 g, 5.1 mmol) was added. Sodium hydroxide beads were ground with a mortar and pestle just before addition to the reaction mixture. The reaction was allowed to proceed for 5 min with gentle agitation and then 0.5 mL of 3 M hydrochloric acid solution was added. After 5 min, excess acetone was removed using a rotavapor, resulting in an oily product. Subsequently and without organic solvent extraction, the crude reactant was adsorbed on silica gel and purified by standard flash chromatography using hexane/ethyl acetate and identified by NMR. Benzalacetone, 1.1 g, 51% yield, a yellow oil which solidified upon standing overnight in a refrigerator. 1H NMR (300 MHz, CDCl3): δ 7.56–7.53 (m, 2H), 7.51 (d, J = 16.3 Hz, 1H), 7.42–7.35 (m, 3H), 6.72 (d, J = 16.3 Hz, 1H), 2.38 (d, 3H). 13C NMR (75 MHz, CDCl3): δ 198.40, 143.44, 134.44, 130.53, 128.98 (2C), 128.26 (2C), 127.17, 27.52. Yellow solid mp: 39–41 °C. 1H NMR and 13C NMR were recorded using a Bruker Avance II 300 spectrometer with CDCl3 as the solvent. Tetramethylsilane was used as the internal standard. Chemical shifts (δ) are reported in ppm and the coupling constants are in Hz.Procedure B is based on procedure A, with some modifications. Benzaldehyde (0.4976 g, 4.689 mmol, 1 equiv.) and acetone (4.07 g, 70.076 mmol, 14.95 equiv.) were mixed at room temperature and powdered sodium hydroxide (0.043 g, 1.1 mmol) was added. Sodium hydroxide beads were ground with a mortar and pestle just before addition to the reaction mixture. The reaction was allowed to proceed for 5 min with gentle agitation and then 0.5 mL of 3 M hydrochloric acid solution was added. After 5 min, excess acetone was removed using a rotavapor. Subsequently, organic solvent extraction was carried out with a mixture of hexane/ethyl acetate (80:20), and the solvent was removed using a rotavapor. The crude reactant was adsorbed on silica gel and purified by flash chromatography using hexane/ethyl acetate, and the product was identified by NMR. For the purification, the amounts of silica gel and the solvents used were ca. 3.4 and 2.6 times lower, respectively, compared to those in procedure A. Benzalacetone, 0.350 g, 51% yield, a yellow oil which solidified upon standing overnight in a refrigerator.
In procedure C, benzaldehyde (0.567 g, 5.343 mmol, 1 equiv.) and acetone (1.12 g, 19.284 mmol, 3.61 equiv.) were mixed at room temperature and 10% (w/v) sodium hydroxide solution (0.55 g) was added dropwise. The reaction was allowed to proceed for 140 min with gentle agitation and then 1 mL of 3 M hydrochloric acid solution was added. After 5 min, the crude reactant was extracted with a mixture of hexane/ethyl acetate (80:20) and dried with Na2SO4, and the solvent was removed using a rotavapor. The crude reactant was adsorbed on silica gel and purified by flash chromatography using hexane/ethyl acetate, and the product was identified by NMR. For the purification, the amounts of silica gel and the solvents used were ca. 5 and 5.4 times lower, respectively, compared to those in procedure A. Benzalacetone, 0.436 g, 56% yield, a yellow oil which solidified upon standing overnight in a refrigerator.
Enzyme sequences
The sequence of the putative alcohol dehydrogenase was selected by NCBI tblastn analysis against the Bradyrhizobium diazoefficiens USDA 110 genome (https://www.ncbi.nlm.nih.gov), and we named it BdADH2. The gene coding for this putative alcohol dehydrogenase is identified as blr4378 in the genome of B. diazoefficiens USDA 110. The gene coding for LbBVMO was selected and cloned by our group previously.23Bacterial strains and culture conditions
B. diazoefficiens USDA 110 was grown at 30 °C in liquid yeast extract-mannitol broth (YEM, 0.5 g L−1 K2HPO4, 0.2 g L−1 MgSO4, 0.1 g L−1 NaCl, 10 g L−1 mannitol and 0.4 g L−1 yeast extract, pH 6.8) at 150 rpm for 5–7 days. E. coli strains were cultured at 37 °C in lysogeny broth (LB, 5 g L−1 yeast extract, 10 g L−1 peptone, and 10 g L−1 NaCl) or LB-agar medium (20 g L−1 agar). When necessary, the media were supplemented with 50 μg mL−1 kanamycin (LB–kanamycin).Genomic DNA extraction, cloning and gene expression
Total genomic DNA was isolated as described previously.32 The blr4378 gene from B. diazoefficiens USDA 110 was amplified using primers 5′-catgcatatgaacatggcagggcaggt-3′ and 5′-aatactcgagcgaggagtgaaggctgt-3′ containing the restriction recognition sites for NdeI and XhoI, respectively. PCR amplification of genomic DNA was performed using the Phusion High-Fidelity DNA polymerase (New England Biolabs, MA, USA), according to the manufacturer's protocol and it was supplemented with 5% (v/v) DMSO. After digestion with the corresponding restriction enzymes, the DNA fragment was ligated into the compatible sites of the pET-TEV vector to produce the plasmid pHBd12, which codes for the BdADH2. The pHLb01 plasmid containing the LbBVMO was previously constructed by our group.23 The blr4378 gene was digested from the pHBd12 vector and subcloned into the second multiple cloning site of pRSFDuet-1 (Novagen, USA) using NdeI and XhoI to obtain pDBd12. Subsequently, the BVMO gene from L. biflexa was amplified using primers 5′-gactgaattcgatgacaacatcaggttttag-3′ and 5′-actgaagcttttattgggtggtgagac-3′ containing the restriction recognition sites for EcoRI and HindIII, respectively, and the Phusion High-Fidelity DNA polymerase (New England Biolabs, MA, USA) according to the manufacturer's protocol. After digestion with the corresponding restriction enzymes, the DNA fragment was cloned into the compatible sites of the first multiple cloning site of pDBd12 to produce the pDLbBd112 vector. Therefore, the plasmid pDLbBd112 codes for both LbBVMO and BdADH2.All DNA fragments were purified using the Wizard® SV Gel and PCR Clean-Up System (Promega Corp., Madison, WI, USA). Recombinant plasmids were isolated using the Wizard Plus SV Miniprep DNA Purification System (Promega Corp., Madison, WI, USA) and their sequences were confirmed by DNA sequencing. E. coli strains were chemically transformed with the plasmids by standard procedures.33
Protein expression and purification
The pre-cultures of E. coli BL21(DE3) cells harbouring recombinant plasmids were grown overnight in an LB–kanamycin medium. Then, a fresh LB–kanamycin medium was inoculated with the overnight pre-culture (2% (v/v)) and incubated at 37 °C. When the bacterial density reached an OD600 = 0.4–0.6, IPTG was added at a final concentration of 0.3 mM and the culture was incubated at 24 °C for 16 h at 200 rpm. Then, the E. coli cells were harvested by centrifugation and resuspended in buffer A (50 mM Tris-HCl, pH 8.0 and 150 mM NaCl) containing 0.05 mg mL−1 lysozyme, 0.1 mM benzamidine and 0.5% (v/v) Triton X-100. The presence of each recombinant protein in the soluble and insoluble fractions was analysed by SDS-PAGE on 12% polyacrylamide gels, followed by coomassie brilliant blue staining.To purify BdADH2 or LbBVMO, resuspended cells expressing the His-tagged proteins were lysed by sonication and, subsequently, subjected to centrifugation at 12000g and 4 °C for 45 min. Ni-NTA agarose (Qiagen) resin was equilibrated with buffer A. For LbBVMO purification, buffer A also contained 10% (v/v) glycerol. The cleared cell extract was incubated with the equilibrated resin for 1 h at 4 °C. Then, this material was packed in an empty column and washed with fifteen column volumes of buffer A supplemented with 10 mM or 20 mM imidazole to remove nonspecifically bound proteins. The ADH or BVMO protein was eluted with two column volumes of buffer A supplemented with 200 mM imidazole. Eluted fractions with purified ADH were pooled and desalted by dialysis against buffer A. Eluted fractions of the BVMO purification that contained FAD-bound proteins were pooled, precipitated with (NH4)2SO4 and desalted using a Sephadex G-25 column. The purified proteins were stored at 4 or −80 °C.
Spectrophotometric analysis and enzyme activity
Alcohol dehydrogenase activity was measured spectrophotometrically by monitoring the absorbance at 340 nm (NADH, ε340 = 6220 M−1 cm−1) after enzyme addition at 24 °C. The reaction mixtures (0.5 mL) typically contained 50 mM Tris-HCl (pH 9.0) or 100 mM sodium phosphate (pH 8.0) buffer, 100 μM NADH or 1 mM NAD+, 6 mM benzylacetone or 6 mM 4-phenyl-2-butanol and 0.45 μM enzyme. The concentration of the purified ADH was determined by the Sedmak and Grossberg assay.34The concentration of the purified BVMO was estimated based on the FAD content. The molar absorption coefficient of BVMO-bound FAD was determined by the comparison of the UV-vis spectrum of the native protein with the spectrum of free FAD (ε450 = 11300 M−1 cm−1) obtained after the addition of 0.2% (w/v) sodium dodecyl sulphate to the sample.35In vitro BVMO activity was determined spectrophotometrically at 340 nm (NADPH, ε340 = 6220 M−1 cm−1). The reaction mixtures (0.5 mL) typically contained 50 mM Tris-HCl (pH 9.0) or 100 mM sodium phosphate (pH 8.0) buffer, 100 μM NADPH, 6 mM benzylacetone and 0.45 μM enzyme.
The UV-visible absorption spectra and all measurements were performed using an Agilent Cary-60 UV-vis spectrophotometer (Agilent, Santa Clara, CA, USA) and quartz cuvettes of 1 cm path length. One unit of BVMO is defined as the amount of protein that oxidises 1 μmol NADPH per minute and one unit of ADH is defined as the amount of protein that oxidises or reduces 1 μmol NADH or NAD+ per minute.
Whole-cell biotransformations
Biotransformations with yeasts contained 0.1 g wet weight of commercially available Baker's yeast (Calsa®, Argentina), 50 mM Tris-HCl (pH 9.0) or 100 mM potassium phosphate (pH 8.0), 200 mM glucose and 100 mM glycerol in a total volume of 1 mL. Benzalacetone (IUPAC: 4-phenylbut-3-en-2-one) was added as the substrate at a final concentration of 1 mg mL−1. The reactions were stopped after 0 h, 4 h, 8 h, 16 h and 24 h of incubation at 24 °C.To perform biotransformations with resting E. coli cells harbouring recombinant plasmids, each culture was prepared inoculating a fresh LB–kanamycin medium with 2% (v/v) of an overnight grown pre-culture of E. coli BL21(DE3) recombinant cells. The culture was incubated at 37 °C and 200 rpm until the bacterial density reached OD600 = 0.4–0.6. Then, IPTG was added at a final concentration of 0.3 mM and the culture was incubated at 24 °C and 200 rpm for 16 h. The cells were collected by centrifugation. Biotransformation assays were performed using 0.1 g wet weight of E. coli BL21(DE3) cells expressing either BdADH2 or LbBVMO, or co-expressing both enzymes resuspended in 50 mM Tris-HCl (pH 9.0) or 100 mM potassium phosphate (pH 8.0), 200 mM glucose and 100 mM glycerol in a total volume of 1 mL. The corresponding substrate (benzylacetone or 4-phenyl-2-butanol) was added at a final concentration of 1 mg mL−1. Alternatively, the supernatant of the Baker's yeast biotransformation supplemented with glucose was used to resuspend the E. coli cells. This supernatant contained a mixture of benzylacetone and 4-phenyl-2-butanol. The reactions were stopped after 0 h, 0.25 h, 0.5 h, 1 h, 2 h, 5 h, 8 h or 24 h of incubation at 24 °C, according to each assay. Control experiments were carried out with non-recombinant E. coli cells.
Analytical samples were extracted with two volumes of ethyl acetate supplemented with 0.1 mg mL−1 1,3,5-trimethylbenzene as the internal standard. The organic phase was removed, dried with anhydrous Na2SO4, and analysed by gas chromatography-mass spectrometry (GC-MS; Shimadzu GCMS-QP2010 Plus from Shimadzu Corporation, Kyoto, Japan) using an achiral (Zebron ZB-1 GC Column from Phenomenex, Torrance, CA, USA and SPB-1 Capillary GC Column from Supelco, Bellefonte, PA, USA, 30 m × 0.25 mm ID, 0.25 μm film) or a chiral capillary column (β-DEX 325 column from Supelco, Bellefonte, PA, USA, 30 m × 0.25 mm ID, 0.25 μm film). The reaction products were predicted by GC-MS or by comparison with pure standards. All compounds were quantified using GC standard curves and, the relative concentration and selectivity, when applicable, were reported. In the graphs, error bars indicate the standard deviation of the mean of replicates.
Biocatalytic cascade on a preparative scale
Biotransformations were scaled up to 75 mL in flasks. They were carried out according to the procedures established for small-scale experiments, with minor modifications.In a typical experiment designed to evaluate the whole biocatalytic sequence, yeasts (7.5 g) were suspended in 75 mL of 50 mM Tris-HCl (pH 9.0), 200 mM glucose and 100 mM glycerol, and 75 mg of benzalacetone was added as the substrate. After 16 h of biotransformation, the cells were separated by centrifugation and, the supernatant was supplemented with glucose and used to resuspend 7.5 g of E. coli cells co-expressing BdADH2 and LbBVMO. After 8 h of biotransformation, the cells were separated by centrifugation, and a minor aliquot of the supernatant was extracted for GC-MS analysis, as described above. Additionally, the remaining amount of the supernatant was extracted with ethyl acetate, purified by flash chromatography and analysed by NMR.
To evaluate the yeast bioreduction step, yeasts (7.5 g) were suspended in 75 mL of 50 mM Tris-HCl (pH 9.0), 200 mM glucose and 100 mM glycerol, and 75 mg of benzalacetone was added as the substrate. After 16 h of biotransformation, the cells were separated by centrifugation and a minor aliquot of the supernatant was extracted for GC-MS analysis, as described above. Additionally, the remaining amount of the supernatant was extracted with ethyl acetate, purified by flash chromatography and analysed by NMR.
Results and discussion
Proposed strategy towards 2-PEA and 2-PE
We designed our strategy so as to meet the following conditions: (i) starting materials should be cheap and easily accessible and/or available from natural and renewable resources; (ii) the route should contain biocatalytic steps combined in an in vivo cascade reaction; and (iii) the operational time should be convenient and the product yield should meet the expectations.Our proposed route towards 2-PEA and 2-PE is depicted in Scheme 1. The first reaction is an aldol condensation reaction between benzaldehyde (1) and acetone (2) to obtain benzalacetone (3) (Scheme 1, reaction a). The biocatalytic cascade includes the reduction of α,β-unsaturated ketone 3 by Baker's yeast to benzylacetone (4) (Scheme 1, reaction b). This biocatalytic reaction could proceed further to 4-phenyl-2-butanol (IUPAC: 4-phenylbutan-2-ol) (5), consuming ketone 4 to produce the secondary alcohol 5 as a side product. To counteract this conversion, we included an alcohol dehydrogenase-mediated step in order to oxidize 4-phenyl-2-butanol (5) back to benzylacetone (4) (Scheme 1, reaction c). In this way, we aimed at recovering the saturated ketone 4 as much as possible to make it available for the subsequent reaction. Next, benzylacetone (4) was oxidised by a BVMO-mediated reaction to 2-PEA (6) (Scheme 1, reaction d). Eventually, 2-PEA (6) could hydrolyse spontaneously in aqueous solution to 2-PE (7) (Scheme 1, reaction e).
 | ||
| Scheme 1 The proposed route for the synthesis of 2-phenylethyl acetate (2-PEA) and 2-phenylethanol (2-PE). Aldol condensation of acetone (1) and benzaldehyde (2) provided benzalacetone (3) (reaction a). Multiple biocatalysts were coordinated: Baker's yeast (reaction b, bioreduction of 3 towards benzylacetone (4); 4-phenyl-2-butanol (5) was detected as the side product); E. coli expressing LbBVMO and BdADH2 (reaction c: biooxidation of 5 towards 4 and reaction d: biooxidation of 4 to 2-PEA (6)). Final deacetylation of 2-PEA (6) to 2-PE (7) (reaction e). | ||
With this strategy, we aimed at adding value to simple carbonyl compounds that can be generated by an appropriate treatment of biomass. Benzaldehyde can be obtained after some simple chemical modifications of xylose, a sugar derived from the acid treatment of hemicellulosic biomass.36–38 Acetone is a product of fermentation of some bacterial species grown in glucose, which can be derived from biomass treatments.39 In the proposed biocatalytic route towards 2-PEA, 2-PE could be formed spontaneously, thus giving access to these two compounds widely used as aroma compounds.2,7 Currently, the focus and main interest of our work is on 2-PEA.
Synthesis of benzalacetone (3) (Scheme 1, reaction a)
In order to contribute to environmentally benign procedures, we focused on solvent-free reactions. The synthesis of benzalacetone was carried out by the Claisen–Schmidt condensation reaction of benzaldehyde and acetone using NaOH as an inorganic base catalyst. In a previous report, benzalacetone was obtained by a solvent-free Claisen–Schmidt condensation reaction.40 We designed the synthetic procedure in a different way and, three classical solvent-free reactions were carried out as described in the Experimental section. Variations in the reaction, work-up and purification steps were analysed in order to make a comprehensive comparison of the environmental impact of our experimental set-ups. Benzalacetone was obtained as a yellow solid. The NMR spectral data are provided in the Experimental section and are consistent with the information from the literature.40 Procedure A utilised freshly ground NaOH as the catalyst and skipped the organic solvent extraction step after the reaction work-up. In fact, after the evaporation of the excess acetone, the crude reactant was directly adsorbed on silica gel. Procedure B was similar to procedure A, but a higher concentration of acetone was included and lower amounts of silica and solvents were used for purification. In procedure C, a 3.61:1 molar ratio of acetone:benzaldehyde was used for condensation, the catalyst was added as a solution and the purification was carried out using the least possible amounts of silica and solvents.
In order to assess the environmental impact of our procedures, we calculated the green metrics parameters: atom economy and E-factor. Based on the reaction equation, the condensation of benzaldehyde and acetone shows an atom economy of 89%. The E-factor was determined as the mass of waste produced per mass of the desired product.41 We decided to evaluate the reaction, work-up and purification separately so as to show the individual contribution of each step to the environmental footprint. Our calculations were based on a previously published template.42 The results are shown in Table S1.† Remarkably, the three procedures described in this work showed very low E-factor values for the reaction (7.8 procedure A, 12.2 procedure B, 4.1 procedure C), excluding the work-up and purification steps. Such small E-factors indicate a very low environmental impact. Even taking into account the reaction work-up, the E-factors remained low. The E-factor increased when the contribution of the flash chromatography step was considered in the equation. Therefore, to reduce the environmental impact of the purification step, we significantly reduced the amounts of silica and the solvents used for flash chromatography (as described in procedures B and C), which produced a meaningful decrease in the E-factor (Table S1†).
We compared our procedures with several reports that included the same catalyst we used (Table S1†). In general, detailed data of work-up and product isolation conditions are not available in the original publications. In consequence, some assumptions43 were required to be made for a rough comparison of these procedures. We considered two reports that included a purification step by flash chromatography.44,45 Although the E-factor values of these reactions were similar to the ones calculated for our procedures, the authors applied extensive work-up, which increased the E-factor values. We estimated the flash chromatography step to be similar to the one applied in our standard procedure A. Then, the total E-factor values calculated were similar to that in our procedure A. We also considered two reports where purification of the product was carried out by (re)crystallisation.46,47 Once again, since the information about the procedures is limited, we based our conclusions on assumptions and approximations.43 These procedures avoid the standard flash chromatography step, but they used solvents in the reaction, in the work-up and for recrystallisation. A rough estimation would allow us to speculate that these procedures would have environmental impacts comparable to our procedures B and C.
In conclusion, since our procedures B and C provided lower E-factor values than the standard procedure A, we suggest procedure C as a greener alternative for this reaction. It is worth mentioning that, in our hands, the grade of purity of benzalacetone achieved before purification allowed us to consider eliminating the purification step in the future.
In vivo biocatalytic approach towards 2-PEA and 2-PE
Baker's yeast (S. cerevisiae) has long been used as a catalyst for the reduction of a great variety of ketones, giving access to valuable building blocks for academic and commercial applications.48–50 Baker's yeast can catalyse the stereoselective reduction of carbonyl groups to produce alcohols, and therefore, is considered as a particularly key biocatalyst for the production of chiral alcohols starting from prochiral ketones and other dicarbonyl compounds. Additional relevant substrates for Baker's yeast are α,β-unsaturated ketones, such as α-haloenones, which allow access to chiral haloketones and halohydrins as building blocks for chemical synthesis,51 and (4R)-carvone, which yields dihydrocarvone and carveol,52 and a variety of other compounds.53 Poor chemoselectivity has been reported for the whole-cell bioreduction of enones and enals due to the competing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO reduction catalysed by native enone reductases and dehydrogenases.48 However, the conjugated CC and the CO bonds of α,β-unsaturated ketones can be selectively reduced, successfully producing saturated ketones based on careful reaction control to avoid over-reduction to sec-alcohols.48,49 In fact, Baker's yeast can catalyse the reduction of oxoisophorone to (R)-levodione in kg scale, a valuable intermediate in the industrial synthesis of 3-hydroxycarotenoids.48,49
C and CO reduction catalysed by native enone reductases and dehydrogenases.48 However, the conjugated CC and the CO bonds of α,β-unsaturated ketones can be selectively reduced, successfully producing saturated ketones based on careful reaction control to avoid over-reduction to sec-alcohols.48,49 In fact, Baker's yeast can catalyse the reduction of oxoisophorone to (R)-levodione in kg scale, a valuable intermediate in the industrial synthesis of 3-hydroxycarotenoids.48,49
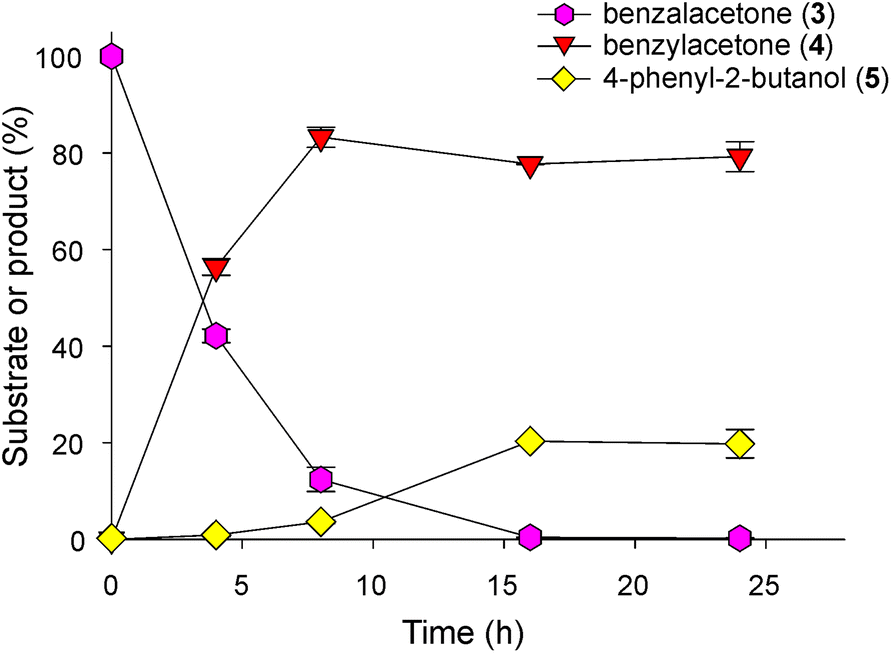 | ||
| Fig. 1 Baker's yeast biotransformation of benzalacetone. Time-course analysis of the biotransformation at pH 8.0. The reactions were stopped at different times (0 h, 4 h, 8 h, 16 h and 24 h). 3, benzalacetone; 4, benzylacetone; 5, 4-phenyl-2-butanol. | ||
After 24 h, benzylacetone and 4-phenyl-2-butanol were the major and minor products, respectively, while benzalacetone was detected only as traces. Based on the previous reports,6,8,55 we explored the production of metabolism-derived 2-PE in yeast. Under our experimental conditions, only traces of this primary alcohol were detected both in the presence and in the absence of benzalacetone, thus being negligible for the quantification of benzalacetone bioreduction (not shown).
By using bioinformatic tools, we selected the sequence of the BdADH2 for cloning purposes. The sequence was amplified from genomic DNA, and the product was digested with restriction enzymes and cloned into the pET-TEV vector to yield the plasmid pHBd12, as described in the Experimental section. Recombinant gene expression was induced with IPTG to produce N-terminally His-tagged fusion proteins. Bacterial cultures were collected and lysed, and the soluble and insoluble protein extracts were analysed by SDS-PAGE and coomassie brilliant blue staining (Fig. 2). The recombinant BdADH2 (∼30.5 kDa) was detected in a soluble form and migrated as a single band with an estimated molecular mass that matched with its calculated mass.
 | ||
| Fig. 2 Recombinant expressions of individual BdADH2 and LbBVMO in E. coli cells. Soluble (S) and insoluble (I) fractions of the protein extracts of E. coli BL21(DE3) transformed with the pHBd12 or pHLb01 plasmid, and expressing the recombinant BdADH2 (yellow asterisk) or LbBVMO (red asterisk), respectively, which were subjected to 12% SDS-PAGE, followed by coomassie brilliant blue staining. M, molecular mass marker. | ||
In order to gain insight into the redox reaction of BdADH2 on benzylacetone and 4-phenyl-2-butanol, we tested these pure compounds as substrates in parallel biotransformation assays using resting cells. Both compounds were readily accepted by the biocatalyst. Indeed, the reversible nature of this reaction was evident since almost similar percentages of benzylacetone and 4-phenyl-2-butanol were found when the starting material was benzylacetone, 4-phenyl-2-butanol or a mixture of benzylacetone and 4-phenyl-2-butanol obtained after 16 h of Baker's yeast biotransformation on benzalacetone (Fig. 3). The same experiments were carried out at pH 8.0 and 9.0. When we increased the pH, we observed higher levels of benzylacetone and lower levels of the sec-alcohol compared to the values obtained at pH 8.0, independent of the starting material (benzylacetone, 4-phenyl-2-butanol or a mixture of benzylacetone and 4-phenyl-2-butanol obtained after 16 h of Baker's yeast biotransformation on benzalacetone). Since we implemented our cascade in recombinant whole cells, we challenged non-recombinant E. coli cells with benzylacetone or 4-phenyl-2-butanol at pH 8.0 or 9.0 as controls in order to evaluate possible background reactions (Fig. S3†). When benzylacetone was used as the substrate, about 11% of 4-phenyl-2-butanol was detected at 8 h of biotransformation. In parallel, no reaction was observed on 4-phenyl-2-butanol. Therefore, we confirmed that the oxidation of the sec-alcohol is catalysed by the recombinant BdADH2. We suspected that the higher levels of benzylacetone detected at pH 9.0 could be due to different BdADH2-oxidation and reduction rates. To corroborate this hypothesis, we purified the BdADH2 to homogeneity (Fig. S4†) and measured its activity in vitro as described in the Experimental section. We determined the specific activities for both the oxidation of 4-phenyl-2-butanol and the reduction of benzylacetone at pH 8.0 or pH 9.0 by BdADH2. We obtained the values 3.149 ± 0.245 U mg−1 at pH 8.0 and 3.279 ± 0.232 U mg−1 at pH 9.0 for the oxidation of the sec-alcohol, and 2.728 ± 0.068 U mg−1 at pH 8.0 and 1.932 ± 0.123 U mg−1 at pH 9.0 for the reduction of benzylacetone. Therefore, the enzyme activity for the reduction reaction decreases significantly at a high pH, thus favouring the oxidation reaction. This result is in agreement with the outcomes of the biotransformations shown in Fig. 3. A similar behaviour regarding the influence of the pH on ADH activity has been reported previously for other ADHs.61–63
 | ||
| Fig. 3 Biotransformation of benzylacetone and 4-phenyl-2-butanol by E. coli cells expressing BdADH2. The starting material was benzylacetone (4), 4-phenyl-2-butanol (5) or the supernatant of Baker's yeast biotransformation containing a mixture of 4 and 5. The effect of two different pH conditions (8.0 and 9.0) was evaluated. | ||
To carry out the next step and couple it to the ADH reaction, we designed two strategies. In the first approach, a culture of E. coli BL21(DE3) cells expressing the BdADH2 and a culture of E. coli BL21(DE3) cells expressing the LbBVMO were grown and subjected to centrifugation, and the cells were resuspended and mixed in a buffer solution, as described in the Experimental section. The biotransformations were carried out in resting cells. The starting material for these assays was the product of a 16 h-biotransformation of benzalacetone catalysed by Baker's yeast, composed mainly of benzylacetone and 4-phenyl-2-butanol. Time-course analyses of the successive reactions are shown in Fig. 4. Since several enzymatic activities had to take place in a one-pot format, we analysed the influence of pH on the outcome of the successive reactions. Under the conditions tested, a sharp decay in the amount of benzylacetone occurred, and at 30 min of reaction, ca. 27% of benzylacetone remained in the mixture at pH 8.0 (Fig. 4A). This decrease in benzylacetone correlated with the detection of 2-PEA and 4-phenyl-2-butanol. Thus, benzylacetone was readily oxidised to 2-PEA by LbBVMO and the product was detected at 30 min of reaction, already. Besides, we proposed that the increase in the amount of 4-phenyl-2-butanol observed at the beginning of the biotransformation was due to the reduction of benzylacetone by BdADH2. After 8 h of biotransformation, the amount of 2-PEA increased up to ca. 39% and the sec-alcohol decreased to ca. 25%, 2-PE accounted for ca. 12% of the total product and the amount of benzylacetone remained almost stable at ca. 24%. In all samples, only traces of benzalacetone could be detected and this trend remained constant throughout the experiments. Representative chromatograms are shown in Fig. S5.† We observed a similar global behaviour at pH 9.0, but with higher amounts of 2-PEA and 2-PE found at the end of the assay (Fig. 4B). In other words, after 8 h of biotransformation at pH 9.0, we obtained ca. 47% of 2-PEA and 12% of 2-PE, whereas only 18.7% of 4-phenyl-2-butanol and 17.4% of benzylacetone were detected. Representative chromatograms are shown in Fig. S5.† In parallel, we purified the LbBVMO to homogeneity (Fig. S6†) and evaluated its activity in vitro at pH 8.0 and 9.0 with benzylacetone as the substrate, as described in the Experimental section. At pH 8.0, the specific activity calculated for LbBVMO was 0.616 ± 0.160 U mg−1, while a significantly higher value was obtained at pH 9.0 (1.402 ± 0.187 U mg−1). This result together with the outcome of the evaluation of the ADH activity at both pH values (Fig. 3) justify the higher levels of 2-PEA detected at pH 9.0.
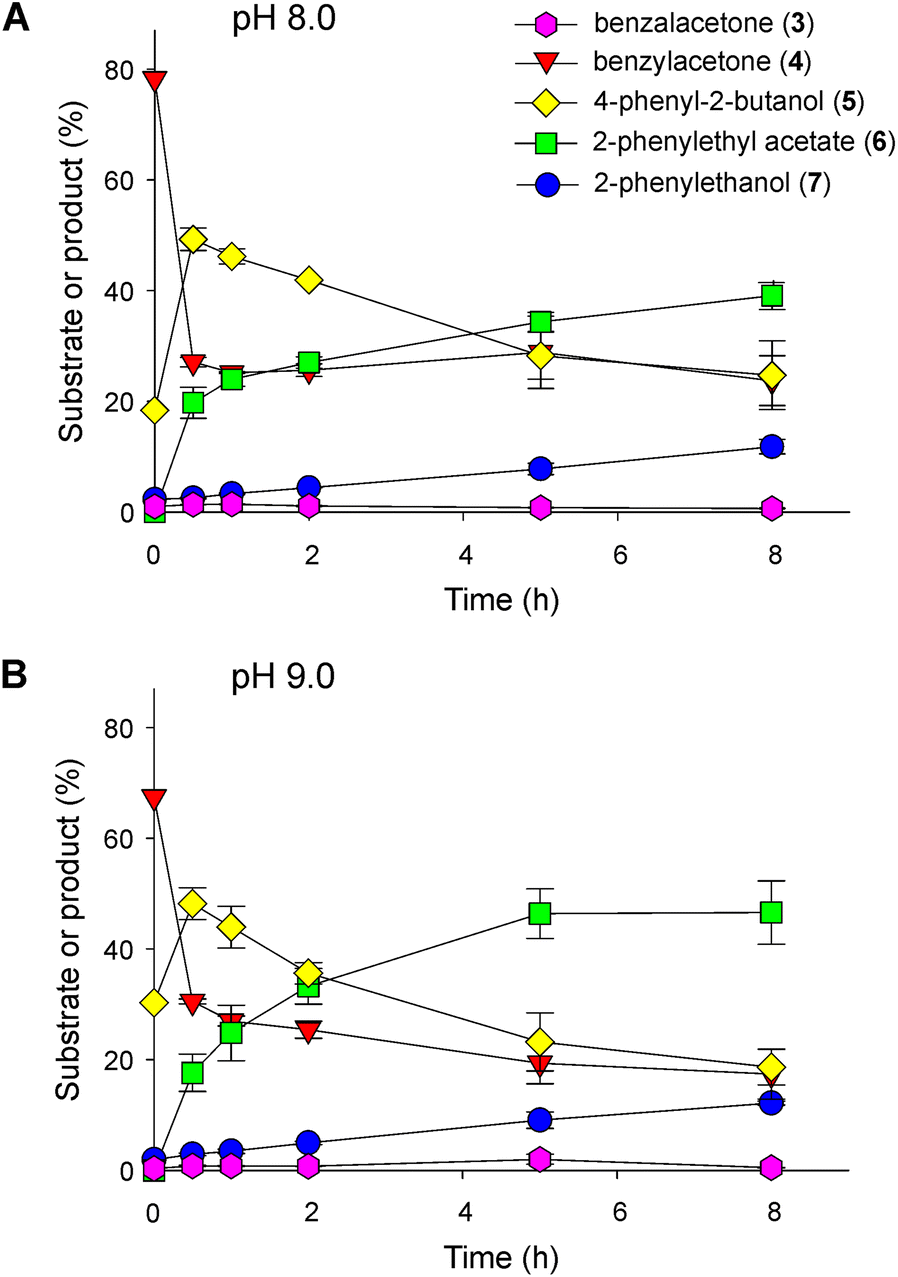 | ||
| Fig. 4 Biotransformation catalysed by E. coli cells expressing LbBVMO and BdADH2 individually. Reactions were incubated for different time periods (0 h, 0.5 h, 1 h, 2 h, 5 h and 8 h) at either pH 8.0 (A) or 9.0 (B). The starting material was the supernatant obtained after 16 h of Baker's yeast biotransformation of benzalacetone at the corresponding pH condition. 3, benzalacetone; 4, benzylacetone; 5, 4-phenyl-2-butanol; 6, 2-phenylethyl acetate; 7, 2-phenylethanol. | ||
At this point, we have proved the concept and feasibility of this biocatalytic sequential reaction. Nevertheless, we decided to carry out further attempts in order to achieve higher amounts of products.
Biocatalytic route using yeasts and E. coli cells co-expressing LbBVMO and BdADH2
We designed a second strategy in order to force the biocatalytic cascade to completion and improve the performance of the cascade. We cloned the coding sequence of the LbBVMO and the coding sequence of the BdADH2 into the first and the second multiple cloning sites of the plasmid pRSFDuet-1, respectively, obtaining the pDLbBd112 vector. After the transformation into E. coli BL21(DE3) cells and upon IPTG induction, LbBVMO and BdADH2 were expressed from independent promoters. Protein extracts were analysed by SDS-PAGE and coomassie brilliant blue staining and both recombinant proteins were detected in the soluble fraction (Fig. 5A, red and yellow asterisks). Hence, this biocatalyst was able to co-express the LbBVMO and the BdADH2. Thus, we included it in the cascade in order to replace the use of LbBVMO and BdADH2 as separate biocatalysts. Once again, we assayed the cascade as a function of time at pH 9.0. We observed an almost full conversion of benzylacetone and 4-phenyl-2-butanol after 8 h of biotransformation with a concomitant production of ∼79% of 2-PEA and ∼19.5% of 2-PE (Fig. 5B and Fig. S7†). | ||
| Fig. 5 Biooxidation reactions catalysed by E. coli cells co-expressing BdADH2 and LbBVMO. (A) Soluble (S) and insoluble (I) fractions of the protein extracts of E. coli BL21(DE3) transformed with pDLbBd112 and co-expressing the recombinant BdADH2 (yellow asterisk) and LbBVMO (red asterisk), which were subjected to 12% SDS-PAGE, followed by coomassie brilliant blue staining. M, molecular mass marker. (B) Biotransformation of a mixture of benzylacetone and 4-phenyl-2-butanol. Reactions were incubated for different time periods (0 h, 0.5 h, 1 h, 2 h, 5 h and 8 h). The starting material was the supernatant obtained after 16 h of Baker's yeast biotransformation of benzalacetone at pH 9.0. 3, benzalacetone; 4, benzylacetone; 5, 4-phenyl-2-butanol; 6, 2-phenylethyl acetate; 7, 2-phenylethanol. | ||
We also investigated the up-scaling of the cascade in an attempt to assess it further. We performed the bioreduction step by suspending 7.5 g of yeasts in 75 mL of 50 mM Tris-HCl (pH 9.0), 200 mM glucose and 100 mM glycerol, and 75 mg of benzalacetone was added as the substrate. After 16 h of biotransformation, the cells were separated by centrifugation, and the supernatant was used to resuspend bacterial cells co-expressing LbBVMO and BdADH2. After 8 h of bioconversion, the cells were removed by centrifugation and the supernatant was subjected to extraction with an organic solvent and GC-MS analysis. Representative GC chromatograms are shown in Fig. S8.† Upon product purification by flash chromatography and NMR analysis, we achieved 2-PEA and 2-PE in 43.8–44.6% and 10.8–15.5% yields, respectively. In this way, we were able to obtain ∼36.9–37.6 mg of 2-PEA and ∼8.3–9.7 mg of 2-PE from 75 mg of benzalacetone as the starting material. Additionally, we scaled up the yeast bioreduction step in parallel. After 16 h of biotransformation on benzalacetone, the cells were removed by centrifugation and the products were extracted and analysed by GC as well as isolated and analysed by NMR. After flash chromatography purification, we obtained 40 mg of benzylacetone and 12.8 mg of 4-phenyl-2-butanol, in 52.6% and 16.6% yields, respectively. By comparing the yield of the complete sequence (yeast bioreduction followed by biooxidation) to the yield of the yeast bioreduction step, we determined that the use of native yeasts limited the yield of the whole sequence.
Native microorganisms have been used in the production of volatile compounds for many years. As summarised in Table S2,† the yeast Hanseniaspora guilliermondii has been utilised for the synthesis of 2-PEA from 2-PE,10 and different strains of Kluyveromyces marxianus have been exploited for the production of 2-PEA and 2-PE from L-phe.11,12,14,64 The yeast Candida glabrata was able to produce 2-PEA using a medium supplemented with 7 g L−1L-phe or tequila vinasse as the substrate.13 Solid-state fermentation has also been applied to the production of 2-PEA and 2-PE using native K. marxianus on a substrate containing sugarcane bagasse – an agro-industrial waste – supplemented with L-phe (Table S2†).15,16
The biosynthesis of 2-PE from L-phe has been intensively studied and improved by using recombinant yeast or E. coli strains.9,22,65,66 However, the synthesis of 2-PEA by recombinant cells has been addressed more recently. Rodriguez et al. reported the use of the branched-chain keto acid-based alcohol pathway combined with alcohol acyltransferase I from S. cerevisiae (ATF1) in an engineered E. coli strain deficient of the major fermentation pathways. In this route, individually fed phenylpyruvate was decarboxylated to the corresponding aldehyde in a reaction catalysed by KivD from Lactococcus lactis and subsequently reduced to 2-PE by an endogenous aldehyde reductase, and 2-PE was esterified by ATF1 using acetyl-CoA, obtaining about 500 and 300 mg L−1 2-PEA and 2-PE, respectively.67 In the same work, Rodriguez et al. obtained about 300 mg L−1 2-PEA from 2-PE in recombinant E. coli cells expressing chloramphenicol acetyltransferase that functioned as acetyl-CoA transferase67 (Table 1).
| Biocatalyst | Substrate | Products | Ref. | |
|---|---|---|---|---|
| 2-PEA (mg L−1) | 2-PE (mg L−1) | |||
| L-phe, L-phenylalanine; n.a., not applicable. | ||||
| Engineered E. coli: | ||||
| (i) JCL260 strain, kivD, ATF1 | (i) Phenylpyruvate | (i) ∼500 | (i) ∼300 | 67 |
| (ii) AL704 strain, cat | (ii) 2-PE | (ii) ∼300 | (ii) n.a. | 67 |
| Engineered E. coli: kdc, yjgB, aro8, atf1 | L-phe | 268 | 277 | 68 |
| Engineered E. coli: | ||||
| (i) aroGfbr, pheAfbr, kdc, yjgB | Glucose | (i) 188 | (i) 578 | 17 |
| (ii) aroGfbr, pheAfbr, kdc, yjgB, aro8 | (ii) 246 | (ii) 1016 | 17 | |
| (iii) aroGfbr, pheAfbr, kdc, yjgB, aro8, atf1 | (iii) 687 | (iii) 46 | 17 | |
| Baker's yeast + engineered E. coli: LbBVMO, BdADH2 | Benzalacetone | 501 | 129 | This study |
A mixture of 2-PEA (268 mg L−1) and 2-PE (277 mg L−1) was produced from L-phe in E. coli cells expressing a 2-keto acid decarboxylase, an aldehyde reductase, and ATF1 from S. cerevisiae68 (Table 1). In 2018, Guo et al. reported a fermentative route to 2-PE in recombinant E. coli cells directly from glucose. They obtained 188 mg L−1 2-PEA and 578 mg L−1 2-PE in a four-enzyme biosynthetic pathway or 246 mg L−1 2-PEA and 1016 mg L−1 2-PE when they included an aminotransferase in a five-enzyme pathway17 (Table 1). Since 2-PEA can be obtained by esterification of 2-PE, the authors also added ATF1 to their route in an attempt to improve the production of 2-PEA, thus achieving 687 mg L−1 2-PEA and 46 mg L−1 2-PE in a six-enzyme pathway17 (Table 1). The concentration of 2-PEA obtained in our work is in the same range as that reported previously (Table 1); however, our route is completely different.
To evaluate the environmental impact of the biocatalytic steps of our route, we also quantified the E-factor, a relevant parameter when considering future bioprocess optimization. We based our approximations on non-volatile input materials43 and used the template published by Andraos and Hent.42 The high volume of water usually involved in bioprocesses contributes strongly to the E-factor. Therefore, the exclusion of water from E-factor calculation could enable a meaningful comparison between different biotechnological approaches.69 It is worth noting that all our calculations were focused on our target product 2-PEA. Initially, we considered the biomass as dry weight and considered only the biosynthetic reaction; thus, the waste generated during the upstream and downstream processing was excluded. Under these conditions, our biocatalytic route from benzalacetone to 2-PEA showed an estimated E-factor of 186.9. However, when we took into account the solid waste generated for biomass production, this value increased to 632.4. Additionally, these E-factor values were ∼13 and ∼43-fold higher, respectively, when water was considered and increased even further after purification. Consequently, there is a clear need for extra efforts to minimise waste (including water) in future bioprocess developments. In terms of eco-friendliness, the biocatalytic strategy reported here is easily achievable from benzalacetone using readily available yeasts and recombinant E. coli cells co-expressing two enzymes only.
Conclusions
In this work, we presented an environmentally friendly strategy for the synthesis of two extensively used aroma compounds, 2-PEA and 2-PE. The experimental approach towards 2-PEA included a solvent-free aldol condensation reaction, followed by a biocatalytic cascade, combining a reduction step catalysed by Baker's yeast, followed by a Baeyer–Villiger biooxidation reaction mediated by the LbBVMO and an alcohol oxidation reaction catalysed by the BdADH2. A final spontaneous deacetylation step could give access to 2-PE. We proposed the biocatalytic synthesis of 2-PEA from an α,β-unsaturated ketone that can be produced from simple starting materials. Our aim was to select starting materials that could be obtained from transformations of renewable resources and the whole strategy was designed to add value to biomass-derived products. The Claisen–Schmidt condensation reaction towards benzalacetone showed a very good yield and the biocatalytic cascade allowed the production of 2-PEA at a concentration similar to the results reported for former 2-PEA bioproduction strategies, in which 2-PE was an intermediate. However, our route is completely different and original. In our work, the formation of 2-PEA is independent of the previous synthesis of 2-PE, since 2-PEA is formed by the oxidation of benzylacetone.We evaluated the environmental footprint of our route based on E-factor values. We presented three procedures for the synthesis of benzalacetone by aldol condensation of benzaldehyde and acetone using NaOH as the catalyst and evaluated the contribution of the reaction, the work-up and the purification to the total E-factor. In all three methods, the reaction and work-up steps showed very low E-factors, indicative of small environmental impact. Although the flash chromatography purification increased this value markedly, by using the least possible amount of silica gel and solvent, the environmental impact can be significantly reduced. A satisfactory E-factor was also found for the biocatalytic steps towards 2-PEA when we considered the reaction only (E-factor: 186.9). In our sequential design, we detected the yeast-catalysed reaction as the yield-limiting step. Therefore, we are conducting further investigations in order to relieve this bottleneck and additionally diminish the environmental impact. To the best of our knowledge, this work is the first approach that describes such a synergistic combination between green chemistry and biotechnology for the synthesis of 2-PEA.
Author contributions
Romina D. Ceccoli: methodology, validation, formal analysis, investigation, resources, writing – original draft, writing – review and editing, and visualization. Dario A. Bianchi: conceptualization, methodology, investigation, resources, writing – review and editing, and funding acquisition. Daniela V. Rial: conceptualization, methodology, resources, writing – original draft, writing – review and editing, visualization, project administration, funding acquisition, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Agencia Nacional de Promoción de la Investigación, el Desarrollo Tecnológico y la Innovación (Agencia I+D+i; grant number: PICT 2018-04264), Argentina, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET; grant numbers: PIP 11220150100934CO and PIP 11220200103142CO), Argentina, and Universidad Nacional de Rosario (UNR; grant numbers: 80020180300138UR and 80020190300235UR), Argentina, for financial support. We thank Prof. Dr Anibal Lodeiro from Instituto de Bioquímica y Biología Molecular (IBBM), Departamento de Ciencias Biológicas, Facultad de Ciencias Exactas, Universidad Nacional de La Plata, Centro Científico Tecnológico CONICET La Plata, La Plata, Argentina for kindly providing the B. diazoefficiens USDA 110 strain. We would also like to thank Guillermo Marcuzzi (CCT-Rosario CONICET) for his assistance with the GC measurements. DVR, DAB and RDC are staff members of CONICET, Argentina. DVR and DAB are Assistant Professors and RDC is a Teaching Assistant at Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Rosario, Argentina.References
- H. Surburg and J. Panten, Common fragrance and flavor materials: preparation, properties and uses, John Wiley & Sons, 2016 Search PubMed.
- D. McGinty, D. Vitale, C. S. Letizia and A. M. Api, Food Chem. Toxicol., 2012, 50, S491–S497 CrossRef CAS PubMed.
- D. Belsito, D. Bickers, M. Bruze, P. Calow, M. L. Dagli, A. D. Fryer, H. Greim, Y. Miyachi, J. H. Saurat and I. G. Sipes, Food Chem. Toxicol., 2012, 50, S269–S313 CrossRef CAS PubMed.
- C. H. Kuo, S. H. Chiang, H. Y. Ju, Y. M. Chen, M. Y. Liao, Y. C. Liu and C. J. Shieh, J. Sci. Food Agric., 2012, 92, 2141–2147 CrossRef CAS PubMed.
- O. Martínez-Ávila, A. Sánchez, X. Font and R. Barrena, Appl. Microbiol. Biotechnol., 2018, 102, 9991–10004 CrossRef PubMed.
- S. Mitri, M. Koubaa, R. G. Maroun, T. Rossignol, J. M. Nicaud and N. Louka, Foods, 2022, 11, 109 CrossRef CAS PubMed.
- J. Scognamiglio, L. Jones, C. S. Letizia and A. M. Api, Food Chem. Toxicol., 2012, 50, S224–S239 CrossRef CAS PubMed.
- M. Etschmann, W. Bluemke, D. Sell and J. Schrader, Appl. Microbiol. Biotechnol., 2002, 59, 1–8 CrossRef CAS PubMed.
- B. Kim, B. R. Cho and J. S. Hahn, Biotechnol. Bioeng., 2014, 111, 115–124 CrossRef CAS PubMed.
- V. Rojas, J. V. Gil, F. Piñaga and P. Manzanares, Int. J. Food Microbiol., 2001, 70, 283–289 CrossRef CAS PubMed.
- M. M. W. Etschmann and J. Schrader, Appl. Microbiol. Biotechnol., 2006, 71, 440–443 CrossRef CAS PubMed.
- M. M. W. Etschmann, D. Sell and J. Schrader, Biotechnol. Bioeng., 2005, 92, 624–634 CrossRef CAS PubMed.
- J. D. J. Rodríguez-Romero, C. A. Aceves-Lara, C. F. Silva, A. Gschaedler, L. Amaya-Delgado and J. Arrizon, Biotechnol. Rep., 2020, 25, e00420 CrossRef PubMed.
- P. J. Adame-Soto, E. T. Aréchiga-Carvajal, M. G. López, S. M. González-Herrera, M. R. Moreno-Jiménez, N. Urtiz-Estrada and O. M. Rutiaga-Quiñones, Ann. Microbiol., 2019, 69, 989–1000 CrossRef CAS.
- O. Martínez, A. Sánchez, X. Font and R. Barrena, Appl. Microbiol. Biotechnol., 2018, 102, 4703–4716 CrossRef PubMed.
- O. Martínez-Ávila, A. Sánchez, X. Font and R. Barrena, J. Agric. Food Chem., 2019, 67, 3389–3399 CrossRef PubMed.
- D. Guo, L. Zhang, S. Kong, Z. Liu, X. Li and H. Pan, J. Agric. Food Chem., 2018, 66, 5886–5891 CrossRef CAS PubMed.
- S. Kong, H. Pan, X. Liu, X. Li and D. Guo, Enzyme Microb. Technol., 2020, 133, 109459 CrossRef CAS PubMed.
- H. Zhang, M. Cao, X. Jiang, H. Zou, C. Wang, X. Xu and M. Xian, BMC Biotechnol., 2014, 14, 1–7 CrossRef PubMed.
- Y. Zhan, M. Zhou, H. Wang, L. Chen, Z. Li, D. Cai, Z. Wen, X. Ma and S. Chen, Appl. Microbiol. Biotechnol., 2020, 104, 7507–7520 CrossRef CAS PubMed.
- C. Fuganti, J. Minut, G. P. Fantoni and S. Servi, J. Mol. Catal. B: Enzym., 1998, 4, 47–52 CrossRef CAS.
- Y. Wang, H. Zhang, X. Lu, H. Zong and B. Zhuge, Biotechnol. Adv., 2019, 37, 403–409 CrossRef CAS PubMed.
- R. D. Ceccoli, D. A. Bianchi, M. J. Fink, M. D. Mihovilovic and D. V. Rial, AMB Express, 2017, 7, 87 CrossRef PubMed.
- R. D. Ceccoli, D. A. Bianchi and D. V. Rial, Front. Microbiol., 2014, 5, 25 Search PubMed.
- H. Leisch, K. Morley and P. C. Lau, Chem. Rev., 2011, 111, 4165–4222 CrossRef CAS PubMed.
- J. Muschiol, C. Peters, N. Oberleitner, M. D. Mihovilovic, U. T. Bornscheuer and F. Rudroff, Chem. Commun., 2015, 51, 5798–5811 RSC.
- G. de Gonzalo and C. E. Paul, Curr. Opin. Green Sustainable Chem., 2021, 32, 100548 CrossRef CAS.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed.
- Z. Wang, B. S. Sekar and Z. Li, Bioresour. Technol., 2021, 323, 124551 CrossRef CAS PubMed.
- E. Ricca, B. Brucher and J. H. Schrittwieser, Adv. Synth. Catal., 2011, 353, 2239–2262 CrossRef CAS.
- A. Vorster, M. S. Smit and D. J. Opperman, Org. Lett., 2019, 21, 7024–7027 CrossRef CAS PubMed.
- W. P. Chen and T. T. Kuo, Nucleic Acids Res., 1993, 21, 2260–2260 CrossRef CAS PubMed.
- J. Sambrook, T. Maniatis and E. F. Fritsch, Molecular Cloning. A Laboratory Manual, Cold Spring Harbor Laboratory Press, New York, 2nd edn, 1989 Search PubMed.
- J. J. Sedmak and S. E. Grossberg, Anal. Biochem., 1977, 79, 544–552 CrossRef CAS PubMed.
- R. D. Ceccoli, D. A. Bianchi, M. A. Carabajal and D. V. Rial, Mol. Catal., 2020, 486, 110875 CrossRef CAS.
- S. K. Green, R. E. Patet, N. Nikbin, C. L. Williams, C. C. Chang, J. Yu, R. J. Gorte, S. Caratzoulas, W. Fan, D. G. Vlachos and P. J. Dauenhauer, Appl. Catal., B, 2016, 180, 487–496 CrossRef CAS.
- F. Wang, J. Xu, X. Li, J. Gao, L. Zhou and R. Ohnishi, Adv. Synth. Catal., 2005, 347, 1987–1992 CrossRef CAS.
- E. R. Sacia, M. Balakrishnan, M. H. Deaner, K. A. Goulas, F. D. Toste and A. T. Bell, ChemSusChem, 2015, 8, 1726–1736 CrossRef CAS PubMed.
- P. Anbarasan, Z. C. Baer, S. Sreekumar, E. Gross, J. B. Binder, H. W. Blanch, D. S. Clark and F. D. Toste, Nature, 2012, 491, 235 CrossRef CAS PubMed.
- A. F. M. M. Rahman, R. Ali, Y. Jahng and A. A. Kadi, Molecules, 2012, 17, 571–583 CrossRef CAS PubMed.
- R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC.
- J. Andraos and A. Hent, J. Chem. Educ., 2015, 92, 1820–1830 CrossRef CAS.
- J. H. Schrittwieser, F. Coccia, S. Kara, B. Grischek, W. Kroutil, N. d'Alessandro and F. Hollmann, Green Chem., 2013, 15, 3318–3331 RSC.
- S. J. Nam, S.-Y. Ham, H. Kwon, H.-S. Kim, S. Moon, J.-H. Lee, T. Lim, S.-H. Son, H.-D. Park and Y. Byun, J. Med. Chem., 2020, 63, 8388–8407 CrossRef CAS PubMed.
- Z.-F. Xiao, T.-H. Ding, S.-W. Mao, Z. Shah, X.-S. Ning and Y.-B. Kang, Org. Lett., 2016, 18, 5672–5675 CrossRef CAS PubMed.
- G. L. Kad, K. P. Kaur, V. Singh and J. Singh, Synth. Commun., 1999, 29, 2583–2586 CrossRef CAS.
- J. Tatsuzaki, K. F. Bastow, K. Nakagawa-Goto, S. Nakamura, H. Itokawa and K.-H. Lee, J. Nat. Prod., 2006, 69, 1445–1449 CrossRef CAS PubMed.
- R. Stuermer, B. Hauer, M. Hall and K. Faber, Curr. Opin. Chem. Biol., 2007, 11, 203–213 CrossRef CAS PubMed.
- B. Pscheidt and A. Glieder, Microb. Cell Fact., 2008, 7, 25 CrossRef PubMed.
- F. Hollmann, I. W. C. E. Arends and D. Holtmann, Green Chem., 2011, 13, 2285–2314 RSC.
- D. S. Zampieri, L. A. Zampieri, J. A. R. Rodrigues, B. R. S. de Paula and P. J. S. Moran, J. Mol. Catal. B: Enzym., 2011, 72, 289–293 CrossRef CAS.
- V. D. Silva, B. U. Stambuk and M. da Graça Nascimento, J. Mol. Catal. B: Enzym., 2012, 77, 98–104 CrossRef CAS.
- W. G. Birolli, I. M. Ferreira, N. Alvarenga, D. D. A. Santos, I. L. de Matos, J. V. Comasseto and A. L. M. Porto, Biotechnol. Adv., 2015, 33, 481–510 CrossRef CAS PubMed.
- C. Cheng and Y.-C. Nian, J. Chromatogr. A, 2015, 1380, 1–10 CrossRef CAS PubMed.
- N. Eshkol, M. Sendovski, M. Bahalul, T. Katz-Ezov, Y. Kashi and A. Fishman, J. Appl. Microbiol., 2009, 106, 534–542 CrossRef CAS PubMed.
- J. Liu, S. Wu and Z. Li, Curr. Opin. Chem. Biol., 2018, 43, 77–86 CrossRef CAS PubMed.
- T. Matsuda, R. Yamanaka and K. Nakamura, Tetrahedron: Asymmetry, 2009, 20, 513–557 CrossRef CAS.
- W. Kroutil, H. Mang, K. Edegger and K. Faber, Curr. Opin. Chem. Biol., 2004, 8, 120–126 CrossRef CAS PubMed.
- H. Puetz, E. Puchľová, K. Vranková and F. Hollmann, Catalysts, 2020, 10, 952 CrossRef CAS.
- A. A. Koesoema, D. M. Standley, T. Senda and T. Matsuda, Appl. Microbiol. Biotechnol., 2020, 104, 2897–2909 CrossRef CAS PubMed.
- V. Höllrigl, F. Hollmann, A. C. Kleeb, K. Buehler and A. Schmid, Appl. Microbiol. Biotechnol., 2008, 81, 263–273 CrossRef PubMed.
- C. Tse, N. E. Ibrahim and K. Ma, J. Appl. Microbiol. Biochem., 2017, 2, 2 Search PubMed.
- K. Napora-Wijata, G. A. Strohmeier, M. N. Sonavane, M. Avi, K. Robins and M. Winkler, Biomolecules, 2013, 3, 449–460 CrossRef PubMed.
- J.-J. Chang, C.-Y. Ho, C.-C. Huang, M.-C. Shih and W.-H. Li, US Pat., 8703474 B2, 2014 Search PubMed.
- J.-Y. Hwang, J. Park, J.-H. Seo, M. Cha, B.-K. Cho, J. Kim and B.-G. Kim, Biotechnol. Bioeng., 2009, 102, 1323–1329 CrossRef CAS PubMed.
- D. Koma, H. Yamanaka, K. Moriyoshi, T. Ohmoto and K. Sakai, Appl. Environ. Microbiol., 2012, 78, 6203–6216 CrossRef CAS PubMed.
- G. M. Rodriguez, Y. Tashiro and S. Atsumi, Nat. Chem. Biol., 2014, 10, 259–265 CrossRef CAS PubMed.
- D. Guo, L. Zhang, H. Pan and X. Li, MicrobiologyOpen, 2017, 6, e00486 CrossRef PubMed.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc02866b |
| This journal is © The Royal Society of Chemistry 2023 |