
Vanillin-derived α,ω-diene monomer for thermosets preparation via thiol–ene click polymerization†
Zijun
Gao
a,
Yang
You
a,
Qin
Chen
*a,
Michael
North
 b and
Haibo
Xie
*a
b and
Haibo
Xie
*a
aDepartment of Polymeric Materials & Engineering, College of Materials & Metallurgy, Guizhou University, Huaxi District, Guiyang 550025, P.R. China. E-mail: hbxie@gzu.edu.cn; qchen6@gzu.edu.cn
bGreen Chemistry Centre of Excellence, Department of Chemistry, University of York, York, YO10 5DD, UK
First published on 24th October 2022
Abstract
It is of great significance for sustainable development to produce degradable, thiol–ene networks obtained from bio-based monomers using environment-friendly processes. Hence, we have prepared tunable and degradable thiol–ene crosslinked networks from renewable vanillin and multifunctional thiols. Vanillin was converted into an α,ω-diene monomer (1a) with degradable ester group via Williamson and Tishchenko reactions. UV-thermally induced thiol–ene click polymerizations between 1a and thiols with various functionalities were performed to prepare thiol–ene networks. 1,2-Ethanedithiol and 2,4,6,8-tetramethyl-2,4,6,8-tetravinylcyclotetrasiloxane were also incorporated into the networks to further regulate their properties. The curing behaviours were studied by FT-IR and DSC to determine the optimal curing conditions. The thermomechanical, thermal and mechanical characteristics of the networks were investigated by DMA, DSC, TGA, and tensile strength tests to understand the influence of different crosslinkers on the material's properties. By varying the amount of co-monomer, the networks exhibited glass transition temperature, tensile strength, and elongations at break in the ranges of 14.5–43.3 °C, 2.6–24.8 MPa, and 29.1–510.2%, respectively. All the thiol–ene networks had good transparency with transmittance between 85 and 90% at 600 nm. Moreover, the networks could be degraded in mild alkaline aqueous solution owing to their inherent ester groups.
Introduction
Polymers play very important roles in our daily lives and are industrially produced on a very large scale. Based on the changes they undergo on heating, polymers can be classified as thermoplastics or thermosets. Thermoplastics tend to be linear polymers whilst thermosets are permanently cross-linked by covalent bonds, which gives them higher shape stability, better solvent resistance, and enhanced thermal stability.1,2 As a result, they find diverse applications as coatings, and electronics and aerospace parts in the form of epoxy resins, vulcanized rubber, and phenolic resin.3–5 However, the permanent 3D-network structure also prevents thermosets from decomposing by melting or by processing in solution,6 resulting in waste of resources and environmental pollution.7,8 Commercial thermosets are currently prepared from non-renewable petroleum-based resources. Furthermore, modelling of most thermosetting materials requires complex curing processes.9 Therefore, replacing petroleum-based feedstocks with renewable resources to prepare degradable thermosets using mild and efficient process is one of the most promising strategies to overcome the above-mentioned problems.10,11Lignin is, by far, the most abundant natural poly-aromatic chemical, with an annual output of about 300 billion tons.12 Various aromatic platform chemicals can be obtained by depolymerization of lignin, such as vanillin, vanillic acid, guaiacol and eugenol.12 Due to their unique aromatic structure, polymers based on lignin depolymerization products usually have good heat resistance and high Young's modulus. Vanillin is the most available aromatic compound currently produced from lignin at an industrial scale.13 The presence of reactive aldehyde and phenolic groups makes vanillin a highly versatile feedstock. For example, the aldehyde groups can react with hydroxyl groups to form acetals and produce Schiff bases with amines; whilst the phenolic hydroxyl groups can react with halogen-containing organics to prepare compounds with a variety of functional groups.14–16 Thus, vanillin has the potential to be a key renewable aromatic building block in biobased polymers. In the past few years, vanillin and its derivatives have been used in the synthesis of new bio-based polymers including epoxy resins,15,17 polycarbonates18 and polyesters.19
Conventional polymerization technologies may lead to limitations in terms of slow production, labour intensiveness and high cost of the process.20 In recent years, thiol–ene click polymerization has been widely utilized to prepare bio-based thiol–ene networks. Compared with traditional polymerization, thiol–ene polymerization has advantages including high reaction rates, simple procedures and formation of uniformly cross-linked structures21 which has attracted great attention in the fields of coatings,22,23 optics,24 electronic equipment,25 3D printing,26 and biomedical implants.27 In addition, the properties of thiol–ene networks are tunable by selection of appropriate thiols. Recently, many biomass resources have been used to prepare thiol–ene cross-linked materials, including soybean oil,28 isosorbide,29 eugenol,30,31 resveratrol32 and ferulic acid.33 Generally, the thermal and mechanical properties of those bio-based thiol–ene networks are lower than bio-based epoxy resins due to the presence of flexible thioether bonds. It is reasonable to speculate that introducing vanillin into thiol–ene networks is beneficial to improve their thermal and mechanical properties owing to the rigid benzene structure of vanillin. However, there are few reports on vanillin-based thiol–ene polymer networks.34,35
As with all crosslinked polymers, thiol–ene networks have drawbacks of being difficult to recycle, so they are mostly landfilled or incinerated,36 resulting in waste of resources and environmental pollution. An effective way to solve this problem is to develop new recyclable thiol–ene networks by introducing degradable groups into the cross-linked networks.35 The introduction of dynamic covalent bonds can also facilitate the degradation of polymers, but the existence of dynamic covalent bonds will lead to the materials with poor dimensional stability.37–39 Ester groups have attracted extensive attention from the point of view of degradability as they can be easily cleaved under acidic or alkaline conditions.40,41 For example, aliphatic polyesters, such as polylactic acid, can be depolymerized to small molecules and this has been widely used in medical and packaging materials.42–44 Thermosetting polyesters are commonly prepared through polycondensation of diols and diacids and then cured by a crosslinker.45–47 However, the synthesis of these polyesters needs complex procedures in a special device, which leads to disadvantages of slow polymerization rates and high energy consumption.48,49 Therefore, to meet the demand for sustainable development, it is of great importance to develop a new strategy to introduce ester groups into polymerizable monomers and construct cross-linked polymer networks by more efficient and green polymerization methods.
In our previous work, we have demonstrated that lignin-based aromatics, such as vanillin and syringaldehyde, could be converted into α,ω-diene functionalized carboxylic ester monomers with high yields via a 100% atom economic Tishchenko reaction.50 A series of linear polyesters with high molecular weights and excellent thermal properties were obtained through thiol–ene click and acyclic diene metathesis polymerizations. The existence of α,ω-diene group makes such monomers suitable for polymerization, and the inherent ester bond provide degradable group in the final polymer. Therefore, those novel monomers have great potential to prepare degradable crosslinked polymers. Herein, a vanillin-based α,ω-diene monomer containing ester group (1a) was synthesized according to our previous work. Monomer 1a was then cured with multi-functional thiols through UV-initiated thiol–ene click polymerizations to form thiol–ene networks. In addition, commercial 1,2-ethanedithiol and tetravinyl siloxane monomers were employed as co-monomers to systematically explore the relationship between structure and properties. The degradation behaviour of these thiol–ene networks was investigated under mild alkaline conditions.
Materials and experimental
Materials
Vanillin, allyl bromide, 2,4,6,8-tetravinyl-2,4,6,8-tetramethylcyclotetrasiloxane (1b), 1,2-ethanedithiol (2a), trimethylolpropane tris(3-mercaptopropionate) (2b), pentaerythritol tetrakis(3-mercaptopropionate) (2c), pentaerythritol tetrakis(mercaptoacetate) (2d), 1-dodecanethiol (1-DT) and 2,2-dimethoxy-2-phenylacetophenone (DMPA) were purchased from Aladdin (Shanghai, China). Other chemicals were supplied by local chemical companies and used as received.Methods
1H NMR and 13C NMR spectra were recorded on JNM-ECZ-400S/L1 NMR spectrometer (JEOL, Japan) using CDCl3 with TMS as internal standard. The conversion of the allyl double bond in 1a was calculated as follows: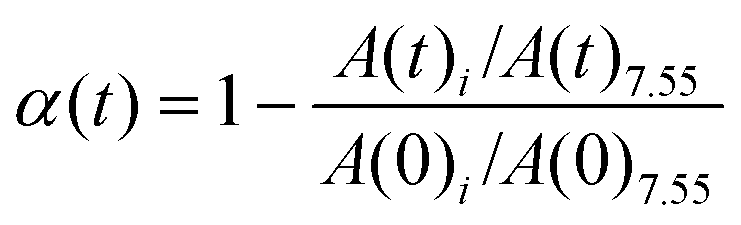 | (1) |
Fourier transform infrared (FT-IR) spectra were obtained using a Nicolet iS50 FT-IR (Thermo Nicolet Corporation, America) instrument under Attenuated Total Reflectance (ATR) mode, with 32 scans over a wavenumber range of 550 to 4000 cm−1. The glass transition temperature (Tg) of polymers was measured by differential scanning calorimetry (Q2000, TA Instruments, USA) under a nitrogen atmosphere with a heating rate of 10 °C min−1 from −20 °C to 120 °C. The value of Tg was obtained from the second heating cycle. Dynamic mechanical properties of the bio-based polymers were determined by a dynamic mechanical analyser (DMA Q800, TA Instruments, USA). All samples were prepared as a rectangle of 30 × 6 mm and tested using the film tension mode with a heating rate of 3 °C min−1 from −50 to 100 °C under the frequency of 1 Hz. Thermal stability was measured using a thermogravimetric analyser (209F1 TG, Netzsch, Germany), from room temperature to 800 °C with a heating rate of 10 °C min−1 under a nitrogen atmosphere. Tensile tests were conducted on an electronic universal testing machine (TSE104B, Shenzhen, China) at an elongation rate of 10 mm min−1 at room temperature. All tensile data were obtained on the average of five effective measurements. The transparency properties of cross-linked networks were acquired using a UV-visible spectrophotometer (UV-2700, Shimadzu, Japan). The gel content of the cured films was measured by methanol extraction. Samples after extraction with methanol for 48 h were allowed to dry at 60 °C under vacuum for 24 h. The gel content (GC) was calculated as follow:
 | (2) |
Synthesis of 4-(allyloxy)-3-methoxybenzaldehyde (AM)
Vanillin (100 mmol, 1.0 equiv.), K2CO3 (110 mmol, 1.1 equiv.) and anhydrous ethanol (100 mL) were added to a 250 mL two-necked round bottom flask equipped with magnetic stirrer and heated at 40 °C. Then, allyl bromide (110 mmol, 1.1 equiv.) was added dropwise, and the mixture was refluxed at 80 °C for 48 hours. After cooling to room temperature, the precipitate was removed by filtration and the filtrate was concentrated in vacuum. Ethyl acetate (200 mL) was added, then the solution was washed with deionized water and the organic layer was dried with anhydrous Na2SO4. The crude product was purified by silica gel column chromatography (ethyl acetate/petroleum ether = 1/3) to afford AM in 96% yield. 1H NMR (400 MHz, CDCl3) δ: 9.81 (s, 1H), 7.41 (d, J = 8.0 Hz, 1H), 7.38 (s, 1H), 6.94 (d, J = 8.0 Hz, 1H), 6.10–6.01 (m, 1H), 5.41 (ddd, J = 13.9 Hz, 11.7 Hz, 1.3 Hz, 2H), 4.67 (dt, J = 5.2, 1.2 Hz, 2H), 3.90 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 191.01, 153.51, 149.89, 132.26, 130.20, 126.72, 118.90, 111.91, 109.23, 76.87, 69.84, 56.07.Synthesis of 4-(allyloxy)-3-methoxybenzyl 4-(allyloxy)-3-methoxybenzoate (1a)
Sodium hydride (60% dispersion in mineral oil, 0.12 g, 3 mmol, 10 mol%) was added to AM (30 mmol) in a Schlenk-flask at 110 °C. The mixture was stirred under vacuum for 4 h, then charged with nitrogen and further stirred for 24 h at 110 °C. After the reaction, the mixture was diluted with ethyl acetate (30 mL), the aqueous phase was extracted with ethyl acetate. The combined organic layers were dried over anhydrous Na2SO4 and concentrated. The crude product was purified by silica gel column chromatograph (ethyl acetate /petroleum ether = 1/10) to afford 1a in 94% yield. 1H NMR (400 MHz, CDCl3) δ: 7.67 (dd, J = 8.4 Hz, 2.0 Hz, 1H), 7.57 (d, J = 2.0 Hz, 1H), 7.00–6.96 (m, 2H), 6.88 (d, J = 1.2 Hz, 1H), 6.86 (d, J = 1.2 Hz, 1H), 6.13–6.02 (m, 2H), 5.43 (dd, J = 3.8 Hz, 1.6 Hz, 1H), 5.39 (dd, J = 3.8 Hz, 1.6 Hz, 1H), 5.31 (d, J = 10.4, 2H), 5.27 (s, 2H), 4.67 (d, J = 5.6 Hz, 2H), 4.62 (d, J = 5.6 Hz, 2H), 3.91 (s, 3H), 3.89 (s, 3H). 13C NMR (100 MHz, DMSO) δ: 166.33, 152.05, 149.41, 148.93, 148.03, 133.23, 132.54, 129.11, 123.54, 122.86, 121.09, 118.63, 118.15, 113.12, 112.39, 112.11, 111.88, 69.87, 66.68, 56.00.Thiol–ene reaction of 1a and 1-DT
1a and 1-DT were uniformly mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of allyl double bonds and thiol groups, then photoinitiator (DMPA) was added into reaction tube, and samples were removed every 2 min.
:1 ratio of allyl double bonds and thiol groups, then photoinitiator (DMPA) was added into reaction tube, and samples were removed every 2 min.
Preparation of the thiol–ene polymer network
As an example, the preparation of P1a2b system is described here. 1a was melted at 100 °C, 2b (1 equiv. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/–SH) and DMPA (2 wt% of the total weight of 1a and 2b) were added and fully mixed at 100 °C to obtain a clear liquid. The liquid was then transferred to a culture plate and cured under UV irradiation at 365 nm for 10 min and then heated at 120 °C for 30 min. Other thiol–ene networks with different formulations were prepared according to Table 1. Since monomer 1b is not miscible with thiols, ethyl acetate (3 mL) was used to dissolve all components in the preparation of P2d1a/1b.
C/–SH) and DMPA (2 wt% of the total weight of 1a and 2b) were added and fully mixed at 100 °C to obtain a clear liquid. The liquid was then transferred to a culture plate and cured under UV irradiation at 365 nm for 10 min and then heated at 120 °C for 30 min. Other thiol–ene networks with different formulations were prepared according to Table 1. Since monomer 1b is not miscible with thiols, ethyl acetate (3 mL) was used to dissolve all components in the preparation of P2d1a/1b.
| Sample | CC |
SH | Gel content |
|---|---|---|---|
|
a Molar ratio of S–H from 2a and 2b–d.
b Molar ratio of CC from 1a and 1b.
|
|||
| P1a2b | 1a | 2b | 99% |
| P1a2c | 1a | 2c | 99% |
| P1a2d | 1a | 2d | 98% |
| P1a2a/2b r1 | 1a |
2a/2b = 2:8a |
98% |
| P1a2a/2c r1 | 1a |
2a/2c = 2:8a |
99% |
| P1a2a/2d r1 | 1a |
2a/2d = 2:8a |
99% |
| P1a2a/2d r2 | 1a |
2a/2d = 4:6a |
96% |
| P1a2a/2d r3 | 1a |
2a/2d = 6:4a |
97% |
| P1a2a/2d r4 | 1a |
2a/2d = 8:2a |
95% |
| P2d1a/1b r1 |
1a/1b = 2:8b |
2d | 97% |
| P2d1a/1b r2 |
1a/1b = 4:6b |
2d | 97% |
| P2d1a/1b r3 |
1a/1b = 6:4b |
2d | 95% |
| P2d1a/1b r4 |
1a/1b = 8:2b |
2d | 95% |
Degradation behaviour of thermoset films
Thermoset films of P1a2n and P1a2a/2n were cut into small square and immersed in NaOH solution (1 mol L−1) at 60 °C for degradation. After complete dissolution, the pH of the formed homogeneous solution was adjusted to 6 with acetic acid and extracted with ethyl acetate. The aqueous phase was freeze-dried to obtain DP-H2O and the organic phase was dried at 60 °C under vacuum to obtain DP-EA.Results and discussion
Synthesis and characterization of 1a
AM and 1a were prepared as shown in Scheme 1 and characterized by NMR techniques. As shown in the 1H and 13C NMR spectra (Fig. S1 and S2†), the structure of 1a was confirmed with the appearance of new signals at 6.13–6.02 ppm, 5.43–5.31 ppm, and 5.27 ppm. | ||
| Scheme 1 Synthesis of 1a. | ||
Reaction efficiency of 1a and 1-DT
To verify the efficiency of the thiol–ene UV photopolymerization, 1a and a mono-thiol monomer (1-DT) were used under click-reaction conditions and the progress of the reaction was monitored by 1H NMR spectroscopy. As shown in Fig. S3a,† on increasing reaction time between 1a and 1-DT, the resonances at 6.12–6.01 ppm and 5.45–5.28 ppm corresponding to the allyl groups in 1a gradually decrease in intensity. The reaction is almost 100% complete after 10 minutes, calculated according to eqn (1).UV-curing behaviour and structure of network
FT-IR spectroscopy was used to monitor the thiol–ene “click” polymerizations (Scheme 2) by observing the variation of the CC and S–H absorption peaks. 1a, 2b and DMPA were uniformly blended and cast onto a KBr crystal plate for FT-IR analysis. As shown in Fig. 1, the absorption peaks at 2570 and 1638 cm−1 are ascribed to S–H and CC bonds, respectively.51 After 10 minutes of irradiation, the intensity of these peaks decreased sharply, indicating the success of thiol–ene polymerization. Further increase of the irradiation time hardly changed the intensity of these peaks. It has been reported that during UV curing, the conversion of S–H groups could only reach 80–90% with various diene monomers.31,52,53 This phenomenon may be caused by the decreased mobility of molecules at high conversion which inhibits access to reactive groups. Therefore, isothermal post-curing was commonly applied to achieve quantitative conversion.54,55 In this work, the appropriate isothermal post-curing for thiol–ene network was determined by DSC after UV irradiation. The thiol–ene network derived from 1a and 2b was first irradiated under UV for 10 min, and then encapsulated into an aluminium pan for DSC testing. The DSC testing was performed through three cycles of heating and cooling scans at a heating rate of 10 °C min−1. It is apparent that the Tg of the UV-cured sample was increased from 13.5 °C (first heating scan) to 17.9 °C (second heating scan) after heating to 120 °C. The Tg remained unchanged after the third heating scan. From Fig. 1, the characteristic FT-IR peaks of S–H and CC completely disappeared after 10 minutes of UV irradiation followed by isothermal curing at 120 °C for 30 minutes. This is due to heating promoting the movement of chain segments, allowing the remaining alkene and thiol groups to come into close contact and thus contributing to the improvement of the polymeric network. Based on these results, UV irradiation for 10 min followed by isothermal treatment at 120 °C for 30 min was chosen for the preparation of all thiol–ene networks in this study (Table 1).
 | ||
| Scheme 2 Formation and tunable structures of the networks. | ||
 | ||
| Fig. 1 FT-IR spectra of P1a2b before and after curing (a), and the DSC curves of P1a2b in the first, second, and third heating process (b). | ||
1,2-Ethanedithiol and 1b were employed as co-monomers to prepare mixed thiol–ene networks (Tables 2 and 3). The purpose of the 1,2-ethanedithiol and 1b used in the system is to create thiol–ene network with different crosslinking densities. Therefore, the ratio of 1,2-ethanedithiol and 1b loading was systematically studied to illuminate the effect of this variable on the thermal and mechanical properties. Scheme 2 shows the thiol–ene reaction process and the cross-linked structure of polymer networks. With the increase of thiol functionality or added tetravinyl siloxane 1b, the distance between cross-linking points becomes shorter, so the cross-linking density should increase. In contrast, introduction of ethanedithiol, with fewer thiol functionalities, the distance between cross-linking points becomes longer, so the cross-linking density decreases. These conjectures have been confirmed by the thermal calculations.
| Sample | T gDSC (°C) | T gDMA (°C) | E′ (25 °C) (MPa) | T d5% (°C) | T d10% (°C) | T max (°C) | Char yield (%) | E′ at TgDMA + 40 °C (MPa) | υ e (mol m−3) |
|---|---|---|---|---|---|---|---|---|---|
| 1a | — | — | — | 248.8 | 256.7 | 272.7 | 12.8 | — | — |
| P1a2b | 17.9 | 23.3 | 20.1 | 279.6 | 316.7 | 382.3 | 15.6 | 2.1 | 284.0 |
| P1a2c | 25.3 | 32.9 | 609.4 | 274.9 | 322.6 | 393.9 | 19.0 | 6.7 | 877.7 |
| P1a2d | 32.7 | 37.1 | 1340.6 | 269.1 | 302.9 | 395.9 | 21.0 | 6.0 | 775.4 |
| P1a2a/2b r1 | 16.3 | 27.5 | 105.4 | 322.3 | 336.2 | 352.7 | 11.3 | 2.1 | 247.2 |
| P1a2a/2c r1 | 17.6 | 30.3 | 237.9 | 286.2 | 322.9 | 353.2 | 16.2 | 2.3 | 268.5 |
| P1a2a/2d r1 | 22.9 | 32.6 | 1183.1 | 289.8 | 323.4 | 392.9 | 20.4 | 1.5 | 173.9 |
| P1a2a/2d r2 | 20.1 | 30.9 | 1124.7 | 279.2 | 320.7 | 352.1 | 20.1 | — | — |
| P1a2a/2d r3 | 19.5 | 34.8 | 669.5 | 293.4 | 321.9 | 346.1 | 18.5 | — | — |
| P1a2a/2d r4 | 14.5 | — | — | 275.7 | 303.9 | 351.3 | 15.5 | — | — |
| P2d1a/1b r1 | 43.3 | 48.3 | 1449.7 | 346.7 | 363.6 | 369.4 | 37.2 | 21.4 | 2373.7 |
| P2d1a/1b r2 | 41.2 | 51.9 | 2172.2 | 347.6 | 361.1 | 374.1 | 38.7 | 21.6 | 2372.3 |
| P2d1a/1b r3 | 38.8 | 48.1 | 1629.6 | 337.3 | 354.9 | 369.9 | 35.5 | 14.8 | 1642.6 |
| P2d1a/1b r4 | 35.1 | 46.6 | 1940.3 | 311.4 | 344.7 | 369.6 | 33.6 | 6.9 | 770.0 |
| Sample | Tensile strength (MPa) | Elongation at break (%) | Young's modulus (MPa) |
|---|---|---|---|
| P1a2b | 2.6 ± 0.5 | 147.4 ± 14.9 | 2.3 ± 0.0 |
| P1a2c | 10.7 ± 2.4 | 139.0 ± 8.8 | 6.8 ± 0.1 |
| P1a2d | 13.0 ± 1.2 | 135.7 ± 5.1 | 142.8 ± 0.1 |
| P1a2a/2b r1 | 2.8 ± 0.1 | 278.2 ± 9.6 | 1.1 ± 0.1 |
| P1a2a/2c r1 | 5.3 ± 0.6 | 236.7 ± 15.3 | 1.5 ± 0.0 |
| P1a2a/2d r1 | 7.0 ± 0.4 | 167.2 ± 9.4 | 135.8 ± 8.6 |
| P1a2a/2d r2 | 4.4 ± 0.2 | 380.3 ± 6.2 | 45.1 ± 3.4 |
| P1a2a/2d r3 | 3.5 ± 0.1 | 510.2 ± 12.3 | 1.4 ± 0.1 |
| P2d1a/1b r1 | 8.3 ± 0.5 | 31.6 ± 2.9 | 62.6 ± 0.0 |
| P2d1a/1b r2 | 13.3 ± 0.3 | 59.7 ± 0.3 | 189.5 ± 9.7 |
| P2d1a/1b r3 | 24.8 ± 2.6 | 29.1 ± 3.6 | 598.7 ± 2.4 |
| P2d1a/1b r4 | 16.6 ± 1.3 | 89.5 ± 7.8 | 372.9 ± 30.0 |
The degree of curing reaction could be characterized by gel content which is a crucial factor to determine the performance of the thiol–ene networks. Thus, the gel content of those thiol–ene networks were determined after 48 h of immersion in methanol and the results are summarized in Table 1. All thiol–ene networks possessed high gel content over 95%, indicating highly cross-linked polymer networks were formed.
Dynamic mechanical properties and thermal properties
Dynamic mechanical analysis (DMA) was performed on the thermosets to investigate their viscoelastic behaviour as a function of temperature, and the curves of storage modulus (E′) and loss factor (tanδ) as a function of temperature are shown in Fig. 2. All the thiol–ene networks exhibit a similar tendency of storage modulus against temperature in the range of −50 to 100 °C. In the glassy state, thiol–ene networks cured with –4SH exhibited higher E′ than those cured with –3SH. As the temperature increased, E′ decreased rapidly and the value of E′ at 25 °C was 20.1, 609.4, and 1340.6 MPa for P1a2b, P1a2c and P1a2d, respectively. By comparison, the introducing of tetravinyl siloxane monomer 1b resulted in thiol–ene networks with much higher E′ at 25 °C than those without 1b.
 | ||
| Fig. 2 DMA curves of P1a2n (a), P1a2a/2n r1 (b), P1a2a/2d r1-3 (c) and P2d1a/1b r1-4 (d) networks. | ||
According to rubber elasticity theory, the storage modulus of the rubber platform can be used to calculate the cross-linking density (υe) as follows:
| υe = E′/3RT. | (3) |
The higher crosslinking density of P1a2c may be due to the fact that alkyl chain of monomer 2c is longer than monomer 2d, allowing it to be a more reactive cross-linking agent. When employing 2a as co-monomer, the thiol–ene network exhibited decreased υe. On the contrary, υe increased sharply when 1b was used as co-monomer. This indicates that increasing the functionality of the monomers leads to higher cross-linking density, thus resulting in higher E′.
During the DMA analysis, curves of tanδ against temperature were also obtained, where the peak value is the glass transition temperature (TgDMA) reflecting the motions of the structure. The value of TgDMA increased in the following order: P1a2b < P1a2c < P1a2d, which is related to the functionality of thiol. The TgDMA further decreased when 2a was included in the thiol–ene networks. By employing 1b, the TgDMA increased, which might be caused by the increased crosslinking density. In addition, only one tanδ peak was observed in the curves, indicating that the obtained thiol–ene networks are homogeneous without phase separation.
Fig. 3 shows the DSC heating curves of all the cross-linked products. All the thiol–ene networks exhibit only one Tg ranging from 14.5 to 43.3 °C and no melting or crystallization peak was observed, suggesting that the networks are homogeneous and amorphous. Those networks prepared from 4SH showed higher Tg than those prepared from 3SH due to the higher functionality of thiol. As for different 4SH-based networks, those containing shorter carbon chains exhibit higher Tg owning to decreased flexibility. By introducing two-functional thiols (1,2-ethanethiol) as co-monomer, the obtained thiol–ene networks showed decreased Tg. Furthermore, as the content of 1,2-ethanethiol increased the Tg decreased from 32.7 to 14.5 °C. In contrast, by introducing 1b containing four double bonds as co-monomer, the Tg of P1a2d increased from 32.7 to 43.3 °C. This might be due to the fact that 1b has more unsaturated double bonds in its structure, which leads to increased crosslink density as more 1b is introduced.
 | ||
| Fig. 3 DSC curves of P1a2n (a), P1a2a/2n r1 (b), P1a2a/2d r1-4 (c), and P2d1a/1b r1-4 (d) networks. | ||
Thermal stability
The thermal stability of vanillin-based thiol–ene networks was characterized by TGA under nitrogen. According to Fig. 4a, the thermal degradation of 1a can be divided into two stages: the first stage is in the temperature range from 200 to 350 °C. In this region, the flexible segments in 1a decomposed with associated weight loss. The second stage is in the temperature range from 350 to 500 °C with weight loss corresponding to the degradation of the hard segments.58 Due to the presence of benzene rings from vanillin, the thiol–ene networks are thermally stable without obvious weight loss (Td5%) up to 270 °C. The Td5% of P1a2d increased from 269.1 to 289.8 °C after introducing 1,2-ethanedithiol as co-monomer (Table 2). On further increasing the amount of 1,2-ethanedithiol the thermal stability increased slightly. P1a2b and P1a2c exhibited similar increased thermal stability in the presence of 1,2-ethanedithiol. Employing 1,2-ethanedithiol as –SH group donor, the proportion of multifunctional thiols containing ester groups decreased, resulting in thiol–ene networks with fewer ester linkages. Reducing the number of ester linkages, that have a tendency to be cleaved, might beneficial for the thermal stability of the crosslinked network.55 Furthermore, the Td5% of P1a2d increased sharply as 1b was included in the system. All these polymers exhibited good thermal stability with T5% over 300 °C, which is nearly 80 °C higher than P1a2d. This result may correlate with the more stable siloxane bonds and increased crosslinking density resulting from the higher functionality of 1b. Char yields of P1a2d networks at 800 °C are 21.0%. By comparison, thiol–ene networks containing silicon exhibit much higher residues ranging from 33.6% to 38.7% (Table 2), which increased as the loading of 1b increased. Char yield plays an important role in hindering the thermal degradation and combustion, and a high char yield is conducive to obtain good flame retardancy.59 | ||
| Fig. 4 TGA and DTG curves of P1a2n (a), P1a2a/2n r1 (b), P1a2a/2d r1-r-4 (c) and P2d1a/1b r1-4 (d) networks. | ||
Mechanical properties
The mechanical properties of the thiol–ene networks were evaluated on the basis of their tensile strength. Fig. 5 illustrates the tensile stress–strain curves of the thiol–ene networks, and the tensile properties are summarized in Table 3. P1a2b and P1a2c exhibit elastic deformation with elongation at break up to 147.4 and 139.0%, respectively. By comparison, P1a2d turn out to be a tough material, showing yielding behaviour in the stress-stain curve. The tensile strength and Young's modulus of the networks increased with increased crosslinking density induced by the higher functionality of thiols. In contrast, the elongation at break decreased as the functionality of the thiol increased. In thiol–ene networks, the rigid benzene ring from vanillin was used as a hard segment to provide the necessary tensile strength, while the aliphatic structure from thiol was used as a soft segment to improve the toughness and elongation at break. | ||
| Fig. 5 The strain-stress curves of P1a2n (a), P1a2a/2n r1 (b), P1a2a/2d r1-3 (c) and P2d1a/1b r1-4 (d) networks. | ||
As indicated in Table 3, the content of 2a has a substantial influence on the mechanical properties of the thiol–ene network. As the loading of 2a increased, the tensile strength of P1a2a/2d decreased from 13.0 to 3.5 MPa and the Young's modulus decreased sharply from 142.8 to 1.4 MPa. The elongation at break also increased significantly from 135.7 to 510.2%. A similar trend was observed in thiol–ene networks cured with 2b and 2c. Introducing 1b into thiol–ene network caused the tensile strength to increase while the elongation at break and Young's modulus decreased sharply (P1a2dvs.P2d1a/1b). On increasing the amount of 1b, the tensile strength and Young's modulus first increased and then decreased, reaching their highest values of 24.8 and 598.7 MPa. However, in all cases, the elongation remains lower than that of P1a2d. It was well recognized that the tensile strength and modulus were related to the crosslinking density and the stiffness of the thiol–ene networks. Compared with 1a monomer, 1b monomer possessed higher functionality of CC group. As the molar ratio of 1b monomer increased from 0 to 40 mol% of the total CC bonds, the crosslinking density of the obtained thiol–ene networks increased from 775.4 (P1a2d) to 1642.6 mol m−3 (P2d1a/1b r3), thus resulting in increased tensile strength and modulus. Further increasing the amount of 1b monomer to 80 mol% resulted in an increase in the crosslinking density to 2373.7 mol m−3. Correspondingly, the tensile strength and modulus decreased to 8.3 and 62.6 MPa, respectively, which could be caused by the decreased stiffness of the thiol–ene networks. As the molar ratio of 1b increased, the rigid benzene ring of 1a was replaced by the flexible Si–O bonds of 1b, making the thiol–ene network become more flexible. The results suggest that the impact of increasing the crosslinking density by introducing 1b was offset by the increase of overall chain flexibility. In addition, the mechanical properties of the thiol–ene networks can be regulated by selecting appropriate vinyl and thiol monomers. The wide range of tensile properties indicates that these materials have great potential in various areas. For example, materials with high elongation at break can be elastomers, which can be used for toughening polylactic acid60 and in the preparation of flexible electronic materials61etc. Polymers with high strength and high modulus, such as P2d1a/1b, with good thermal stability can be used for the preparation of flame-retardant coating.62
Optical properties
The transparency of vanillin-based thiol–ene networks was investigated by UV-vis spectroscopy. As shown in Fig. 6, all the thiol–ene networks exhibited excellent transparency with transmissivity exceeding 85% at 600 nm. In particular, the transmissivity of P2d1a/1b is higher than 90% at 600 nm. To illustrate this transparency, P1a2c film was placed in front of a mobile phone camera and the picture was almost unaffected. It is reasonable that the existence of sulfur atoms and benzene rings provides excellent transparency for the thiol–ene networks.63 The transparency of these networks is similar to that of their non-biobased counterparts.64 Below 320 nm, the transmittance of thiol–ene network is close to zero indicating good blocking properties in ultraviolet B (UVB) and ultraviolet C (UVC) regions. One reasonable explanation is that the aromatic structure of 1a has strong absorption in the UVB and UVC regions. Due to this transparency, excellent flexibility, and near body temperature Tgs, these thiol–ene have potential applications in flexible materials, sensors, and biomedical materials. | ||
| Fig. 6 (a) UV-vis spectra of P1a2n networks. (b) UV-vis spectra of P1a2a/2n networks. (c) UV-vis spectra of P2d1a/1b networks. (d) Digital photo of P1a2c network. (e) Digital photo of the same scene: without (above) and with (below) P1a2c film in front of the camera. | ||
Degradation properties
Traditionally, thiol–ene networks cannot be degraded due to their permanently cross-linked structure. However, the networks prepared in this work contain ester bonds within 1a units. Therefore, the thermosetting film is readily degraded into carboxylic acids and alcohols in water in the presence of sodium hydroxide, resulting in decomposition of the network.An initial degradation study was performed under alkaline condition with linear poly(thioether ester) prepared from 1a and 1,2-ethanethiol. 1H NMR spectra showed the structural changes before and after degradation. As indicated in Fig. S4,† the signal at 5.26 ppm is assigned to the methylene group of the benzyl benzoate units in the linear poly(thioether ester). After degradation, new resonances appear at 5.21 and 4.40 ppm corresponding to the OH and methylene attached to benzene, and the integral area of these two peaks was 1:2 demonstrating the formation of benzyl alcohol structure from the cleavage of ester bond in benzyl benzoate. This result indicate that polymers derived from 1a can be degraded under alkaline conditions owing to the inherent ester group from 1a.
The influence of different solvents on the degradation process was investigated with P1a2d. NaOH solution (1 M) was mixed with different organic solvents (organic solvent/water = 5/5, v/v), and P1a2d was immersed in those solution at room temperature until completely dissolved. As shown in Fig. 7a, the degradation time of P1a2d in pure NaOH solution was 0.9 hours. Longer exposure times of several days were required to completely degrade P1a2d in ethanol/water, DMF/water, and methanol/water solutions. Acetone-based alkaline solutions provide a rapid degradation rate for P1a2d, which can be completely degraded in 0.3 hours (Fig. S5†), but this is not conducive to environmental protection due to the use of a low boiling point solvent. The difference of degradation time in various solvents may be ascribed to different solvency of the products in solvent. As the polarity of solvent increased, the ability to dissolve the product decreased leading to prolonged degradation time. The degradation of P1a2d can also be promoted by heating even without the participation of organic solvents. It was confirmed that all the P1a2n and P1a2a/2n samples could be degraded rapidly in 1 M NaOH solution at 60 °C (Fig. 7b).
 | ||
| Fig. 7 (a) Degradation times of P1a2d in 1 M NaOH with different organic solvents (organic solvent/water = 5/5, v/v). (b) Degradation of P1a2n and P1a2a/2n in 1 M NaOH at 60 °C. | ||
After degradation, the residues were collected, partitioned between ethyl acetate and water (to give DP-EA and DP-H2O respectively) and characterized by FT-IR spectroscopy. As shown in Fig. 8a, characteristic peaks at 1703 cm−1, ascribed to carboxylic groups, and around 1500 cm−1, corresponding to benzene rings, were observed. For DP-H2O, a waxy solid was obtained after drying. As shown in Fig. 8b, the peaks at 1577 and 1420 cm−1 are assigned to the carboxylate group, due to the relatively low acidity of glacial acetic acid. The peak at 3001 cm−1 is assigned to C–H group. And the peak at 3433 cm−1 is assigned to a hydroxyl group.65 Thus, DP-H2O is a mixture of carboxylate and polyol compound.
 | ||
| Fig. 8 (a) The FT-IR spectra of DP-EA, (b) the FT-IR spectra of DP-H2O. | ||
Conclusions
In summary, a vanillin-derived α,ω-diene monomer containing an ester group was polymerized with multiple functional thiols to prepare a series of thiol–ene networks with tunable properties. UV-thermal dual curing can achieve total conversion of thiols. Thiol–ene networks derived from higher functionality thiols exhibit higher cross-link density, thus resulting in higher E′ and Tg than those derived from lower functionality thiols. Incorporation of 1,2-ethanedithiol or tetravinyl siloxane as a co-monomer was found to have a substantial influence on the properties of the obtained thiol–ene networks. Increasing the molar ratio of 2a from 0 to 80%, the Tg of thiol-Nen networks (P1a2d, P1a2a/2d r1–r4) decreased gradually from 32.7 to 14.5 °C. Increasing the molar ratio of 1b from 0 to 80%, the Tg of thiol-Nen networks (P1a2d, P2d1a/1b r4–r1) increased from 32.7 to 43.3 °C. Incorporation of 2a and 1b as co-monomer was beneficial for thermal stability, especially in the presence of 1b the thermal stability increased sharply. As the functionality of thiol increased, the tensile strength and Young's modulus increased, while the elongation at break decreased. As the loading of 2a increased, the tensile strength and Young's modulus decreased from 13.0 to 3.5 MPa and from 142.8 to 1.4 MPa, respectively. In the case of P2d1a/1b r4–r1, as the amount of 1b increased, the tensile strength and Young's modulus first increased and then decreased with the highest values of 24.8 and 598.7 MPa, respectively. Moreover, the transmissivity of the thiol–ene networks under visible light was higher than 85%. The thiol–ene networks were susceptible to degradation in aqueous alkaline condition due to their inherent ester groups. Therefore, this novel, vanillin-derived, α,ω-diene monomer possesses great potential in the preparation of degradable thermosets, particularly for biomedical materials.Author contributions
Zijun Gao: Investigation, formal analysis, writing – original draft. Yang You: Methodology, writing – review & editing. Qin Chen: Methodology, funding acquisition, writing – review & editing. Michael North: Writing – review & editing. Haibo Xie: Conceptualization, supervision, project administration, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (NSFC 22065006, 21704019); Guizhou Provincial Science and Technology Projects (Grant No. QianKeHe foundation [2020]1Z004; Platform & Talents [2016]5652, [2019]5607).References
- D. J. Fortman, J. P. Brutman, G. X. De Hoe, R. L. Snyder, W. R. Dichtel and M. A. Hillmyer, ACS Sustainable Chem. Eng., 2018, 6, 11145–11159 CrossRef CAS.
- J. S. Mahajan, R. M. O'Dea, J. B. Norris, L. T. Korley and T. H. Epps III, ACS Sustainable Chem. Eng., 2020, 8, 15072–15096 CrossRef CAS.
- F. Zareanshahraki, J. Lu, S. Yu, A. Kiamanesh, B. Shabani and V. Mannari, Prog. Org. Coat., 2020, 147, 105725 CrossRef CAS.
- Y. Guo, S. Chen, L. Sun, L. Yang, L. Zhang, J. Lou and Z. You, Adv. Funct. Mater., 2021, 31, 2009799 CrossRef CAS.
- B. G. Falzon and R. S. Pierce, in Revolutionizing Aircraft Materials and Processes, Springer, 2020, pp. 57–86 Search PubMed.
- C. Jehanno and H. Sardon, Nature, 2019, 568, 467–468 CrossRef CAS PubMed.
- C. Sánchez, Biotechnol. Adv., 2020, 40, 107501 CrossRef PubMed.
- L. Jiang, Y. Lei, Y. Xiao, X. Fu, W. Kong, Y. Wang and J. Lei, J. Mater. Chem. A, 2020, 8, 22369–22378 RSC.
- D. Lei, Y. Yang, Z. Liu, S. Chen, B. Song, A. Shen, B. Yang, S. Li, Z. Yuan and Q. Qi, Mater. Horiz., 2019, 6, 394–404 RSC.
- K. M. Zia, A. Noreen, M. Zuber, S. Tabasum and M. Mujahid, Int. J. Biol. Macromol., 2016, 82, 1028–1040 CrossRef CAS.
- Q. Cai, T. Bai, H. Zhang, X. Yao, J. Ling and W. Zhu, Mater. Today, 2021, 51, 155–164 CrossRef CAS.
- C. Zhang and F. Wang, Acc. Chem. Res., 2020, 53, 470–484 CrossRef CAS PubMed.
- M. Fache, B. Boutevin and S. Caillol, Eur. Polym. J., 2015, 68, 488–502 CrossRef CAS.
- L. Fang, Y. Tao, J. Zhou, C. Wang, M. Dai, J. Sun and Q. Fang, Polym. Chem., 2021, 12, 766–770 RSC.
- Z. Wang, P. Gnanasekar, S. Sudhakaran Nair, R. Farnood, S. Yi and N. Yan, ACS Sustainable Chem. Eng., 2020, 8, 11215–11223 CrossRef CAS.
- R. M. Cywar, N. A. Rorrer, C. B. Hoyt, G. T. Beckham and E. Y.-X. Chen, Nat. Rev. Mater., 2021, 7, 83–103 CrossRef.
- H. Niu, H. Nabipour, X. Wang, L. Song and Y. Hu, ACS Sustainable Chem. Eng., 2021, 9, 5268–5277 CrossRef CAS.
- D. Bai, Q. Chen, Y. Chai, T. Ren, C. Huang, I. D. Ingram, M. North, Q. Zheng and H. Xie, RSC Adv., 2018, 8, 34297–34303 RSC.
- C. Zhao, C. Huang, Q. Chen, I. D. Ingram, X. Zeng, T. Ren and H. Xie, Polymers, 2020, 12, 586 CrossRef CAS PubMed.
- M. Peerzada, S. Abbasi, K. T. Lau and N. Hameed, Ind. Eng. Chem. Res., 2020, 59, 6375–6390 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- L. Pezzana and M. Sangermano, Prog. Org. Coat., 2021, 157, 106295 CrossRef CAS.
- J. Dai, S. Wang, L. Zhu and S. Ma, Polymer, 2017, 108, 215–222 CrossRef CAS.
- J. Zuo, Z. Liu, C. Luo and F. Lin, Prog. Org. Coat., 2021, 151, 106048 CrossRef CAS.
- C. L. Frewin, M. Ecker, A. Joshi-Imre, J. Kamgue, J. Waddell, V. R. Danda, A. M. Stiller, W. E. Voit and J. J. Pancrazio, Polymers, 2019, 11, 902 CrossRef PubMed.
- L. Chen, Q. Wu, G. Wei, R. Liu and Z. Li, J. Mater. Chem. C, 2018, 6, 11561–11568 RSC.
- T. O. Machado, C. Sayer and P. H. Araujo, Eur. Polym. J., 2017, 86, 200–215 CrossRef CAS.
- S.-S. Kim, H. Ha and C. J. Ellison, ACS Sustainable Chem. Eng., 2018, 6, 8364–8373 CrossRef CAS.
- E. Cakmakci, F. Sen and M. V. Kahraman, ACS Sustainable Chem. Eng., 2019, 7, 10605–10615 CrossRef CAS.
- T. Liu, L. Sun, R. Ou, Q. Fan, L. Li, C. Guo, Z. Liu and Q. Wang, Chem. Eng. J., 2019, 368, 359–368 CrossRef CAS.
- J.-T. Miao, L. Yuan, Q. Guan, G. Liang and A. Gu, ACS Sustainable Chem. Eng., 2018, 6, 7902–7909 CrossRef CAS.
- K. Zhang, M. Han, L. Han and H. Ishida, Eur. Polym. J., 2019, 116, 526–533 CrossRef CAS.
- L. Pezzana, M. Mousa, E. Malmstrm, M. Johansson and M. Sangermano, Prog. Org. Coat., 2021, 150, 105986 CrossRef CAS.
- R. Ohno, K. Sugane and M. Shibata, Eur. Polym. J., 2021, 159, 110738 CrossRef CAS.
- M. Ge, J.-T. Miao, K. Zhang, Y. Wu, L. Zheng and L. Wu, Polym. Chem., 2021, 12, 564–571 RSC.
- W. An, X.-L. Wang, X. Liu, G. Wu, S. Xu and Y.-Z. Wang, Green Chem., 2022, 24, 701–712 RSC.
- A. Takahashi, T. Ohishi, R. Goseki and H. Otsuka, Polymer, 2016, 82, 319–326 CrossRef CAS.
- Q. Li, S. Ma, S. Wang, Y. Liu, M. A. Taher, B. Wang, K. Huang, X. Xu, Y. Han and J. Zhu, Macromolecules, 2020, 53, 1474–1485 CrossRef CAS.
- S. You, S. Ma, J. Dai, Z. Jia, X. Liu and J. Zhu, ACS Sustainable Chem. Eng., 2017, 5, 4683–4689 CrossRef CAS.
- S. Ma and D. C. Webster, Macromolecules, 2015, 48, 7127–7137 CrossRef CAS.
- Y. Tian, Q. Wang, J. Cheng and J. Zhang, Green Chem., 2020, 22, 921–932 RSC.
- D. D. Silva, M. Kaduri, M. Poley, O. Adir, N. Krinsky, J. Shainsky-Roitman and A. Schroeder, Chem. Eng. J., 2018, 340, 9–14 CrossRef.
- L. K. Ncube, A. U. Ude, E. N. Ogunmuyiwa, R. Zulkifli and I. N. Beas, Materials, 2020, 13, 4994 CrossRef CAS PubMed.
- M. A. Elsawy, K.-H. Kim, J.-W. Park and A. Deep, Renewable Sustainable Energy Rev., 2017, 79, 1346–1352 CrossRef CAS.
- X. Tang and E. Y.-X. Chen, Chem, 2019, 5, 284–312 CAS.
- A. M. de Ilarduya and S. M. Guerra, Polym. Chem., 2020, 11, 4850–4860 RSC.
- J. M. Sadler, F. R. Toulan, G. R. Palmese and J. J. La Scala, J. Appl. Polym. Sci., 2015, 132, 42315 CrossRef.
- A. Douka, S. Vouyiouka, L.-M. Papaspyridi and C. D. Papaspyrides, Prog. Polym. Sci., 2018, 79, 1–25 CrossRef CAS.
- K. S. Egorova and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 12150–12162 CrossRef CAS.
- T. Ren, Q. Chen, C. Zhao, Q. Zheng, H. Xie and M. North, Green Chem., 2020, 22, 1542–1547 RSC.
- Q. Li, S. Ma, N. Lu, J. Qiu, J. Ye, Y. Liu, S. Wang, Y. Han, B. Wang and X. Xu, Green Chem., 2020, 22, 7769–7777 RSC.
- T. Modjinou, D.-L. Versace, S. Abbad-Andallousi, N. Bousserrhine, J. Babinot, V. Langlois and E. Renard, ACS Sustainable Chem. Eng., 2015, 3, 1094–1100 CrossRef CAS.
- T. Modjinou, D.-L. Versace, S. Abbad-Andallousi, N. Bousserrhine, P. Dubot, V. Langlois and E. Renard, React. Funct. Polym., 2016, 101, 47–53 CrossRef CAS.
- G. Yang, S. L. Kristufek, L. A. Link, K. L. Wooley and M. L. Robertson, Macromolecules, 2015, 48, 8418–8427 CrossRef CAS.
- J. Dai, S. Ma, L. Zhu, S. Wang, L. Yang, Z. Song, X. Liu and J. Zhu, Polymer, 2017, 108, 215–222 CrossRef CAS.
- L. Jiang, Y. Tian, J. Cheng and J. Zhang, Polym. Chem., 2021, 12, 6527–6537 RSC.
- Y. Hu, Y. Tian, J. Cheng and J. Zhang, ACS Sustainable Chem. Eng., 2020, 8, 4158–4166 CrossRef CAS.
- J. Xue, X. Yang, Y. Ke, Z. Yan, X. Dong, Y. Luo and C. Zhang, Ind. Crops Prod., 2021, 171, 113956 CrossRef CAS.
- C. Liu, T. Chen, C. Yuan, C. Song, Y. Chang, G. Chen, Y. Xu and L. Dai, J. Mater. Chem. A, 2016, 4, 3462–3470 RSC.
- E. Brown, M. Abdelwahab, O. Valerio, M. Misra and A. K. Mohanty, ACS Omega, 2018, 3, 3857–3867 CrossRef CAS PubMed.
- Q. Sun, B. Qian, K. Uto, J. Chen, X. Liu and T. Minari, Biosens. Bioelectron., 2018, 119, 237–251 CrossRef CAS PubMed.
- Y. Huang, T. Ma, Q. Wang and C. Guo, ACS Sustainable Chem. Eng., 2019, 7, 14727–14738 CrossRef CAS.
- L. Fang, J. Sun, X. Chen, Y. Tao, J. Zhou, C. Wang and Q. Fang, Macromolecules, 2020, 53, 125–131 CrossRef CAS.
- H. Bao, Y. Wu, J. Liu, X. Hua, G. Lai and X. Yang, ACS Omega, 2020, 5, 29513–29519 CrossRef CAS PubMed.
- Y. Tian, Q. Wang, J. Cheng and J. Zhang, Green Chem., 2020, 22, 921–932 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra, details of the click reaction and images of the polymer degradation. See DOI: https://doi.org/10.1039/d2gc02901d |
| This journal is © The Royal Society of Chemistry 2023 |