
Metal-free construction of aminated isoquinoline frameworks from 2-(2-oxo-2-arylethyl) benzonitrile in an aqueous medium†
Himanshi
Sharma‡
a,
Manoj
Kumar‡
 a,
Aaftaab
Sethi
ab,
Poonam
c and
Brijesh
Rathi
*ab
a,
Aaftaab
Sethi
ab,
Poonam
c and
Brijesh
Rathi
*ab
aDepartment of Chemistry, Hansraj College, University of Delhi, Delhi 110007, India. E-mail: brijeshrathi@hrc.du.ac.in
bHeteroChem InnoTech, Hansraj College, University of Delhi, Delhi 110007, India
cDepartment of Chemistry, Miranda House, University of Delhi, Delhi 110007, India
First published on 6th December 2022
Abstract
Herein, we report a metal/additive-free protocol for the activation of nitrile towards nucleophilic addition and subsequent annulation under an aqueous medium for the first time. The protocol divulges an efficient and atom economical route for the construction of diverse aminated isoquinolines. Differently substituted primary as well as secondary amines underwent the reaction in a highly regioselective manner. The reaction is operationally simple, shows high functional group tolerance and easier modification of well-known drugs, and can be successfully extended to gram-scale synthesis.
Isoquinolines represent important structural motifs in medicinal chemistry. Apart from forming the foundation of various medicines, isoquinolines and their derivatives play an important role in materials science, functional materials, chiral ligands, natural products, dyes, paints, etc.1 Among the many known isoquinoline derivatives, aminated isoquinolines are of utmost importance owing to their ubiquity in pharmaceuticals; therefore, considerable attention has long been paid to their development (Fig. 1).2
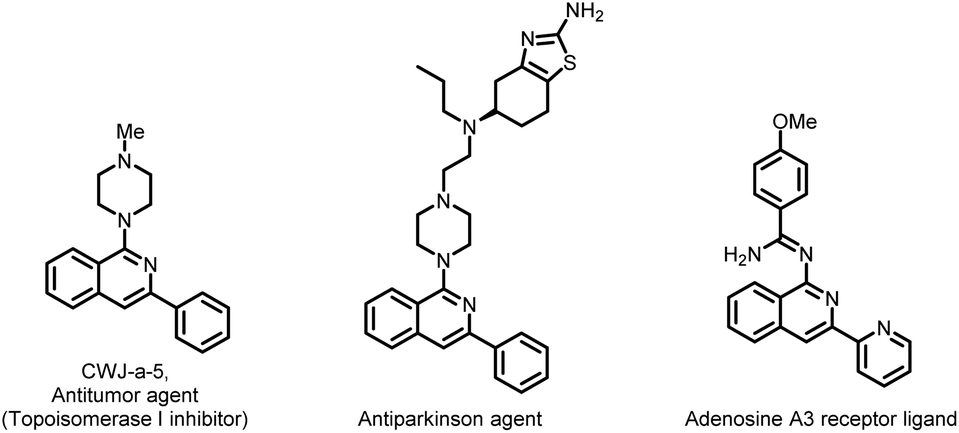 | ||
| Fig. 1 1-Aminoisoquinoline core containing bioactive molecules. | ||
The reported procedures largely rely on the transition-metal-catalyzed C–N cross-coupling reactions3 and SNAr reactions of 1-haloisoquinolines with amines.4 In the recent past, several groups have accomplished major advances in this field using C–H functionalization reactions.5 Despite showing great potential, these procedures often require multiple steps for the pre-functionalization of substrates, the use of toxic reagents and precious metal catalysts, and show region-selectivity issues. Therefore, the development of a cost-economical approach addressing the drawbacks of previous protocols is highly desirable. In the last few years, many reports have focused on the reactivity of carbonitriles. Carbonitriles lack electrophilic properties and are generally activated by transition-metal/acid/base catalysts towards nucleophilic additions and the ensuing annulation.6 Many groups have utilized differently substituted nitrile-containing functionalities for the synthesis of aryl/alkyl ketones, diketones, benzofurans, etc.7 In all such reactions the nitrogen atom was not found to be incorporated into the product due to the tendency of ketimine intermediates to undergo hydrolysis. It is imperative to develop protocols that can also make use of nitrogen of nitriles to produce N-heterocycles. Recently, a palladium-catalyzed nucleophilic addition of organoboron reagents to 2-(2-oxo-2-aryl/alkylethyl)benzonitriles was reported, which incorporates the nitrogen atom and results in the formation of isoquinolines (Scheme 1b).8 In addition, based on domino electrophilic cyclization,9 a Cu-catalysed domino reaction of 2-alkynylbenzonitriles (Scheme 1a) with secondary amines was also developed.9a More recently, Namballa and co-workers reported a Me3Al promoted synthesis of aminoisoquinolines by the domino nucleophilic addition of anilines with subsequent intramolecular cyclization on 2-(2-oxo-2-phenylethyl)benzonitriles (Scheme 1c).6g To the best of our knowledge, no metal/additive-free reaction of carbonitriles in an aqueous medium has yet been delineated. Herein, we report an efficacious and direct protocol for the synthesis of aminated isoquinoline. The method utilizes readily accessible amines and 2-(2-oxo-2-aryl/alkylethyl)benzonitrile under acid/base/metal-free conditions in an aqueous medium (Scheme 1d).
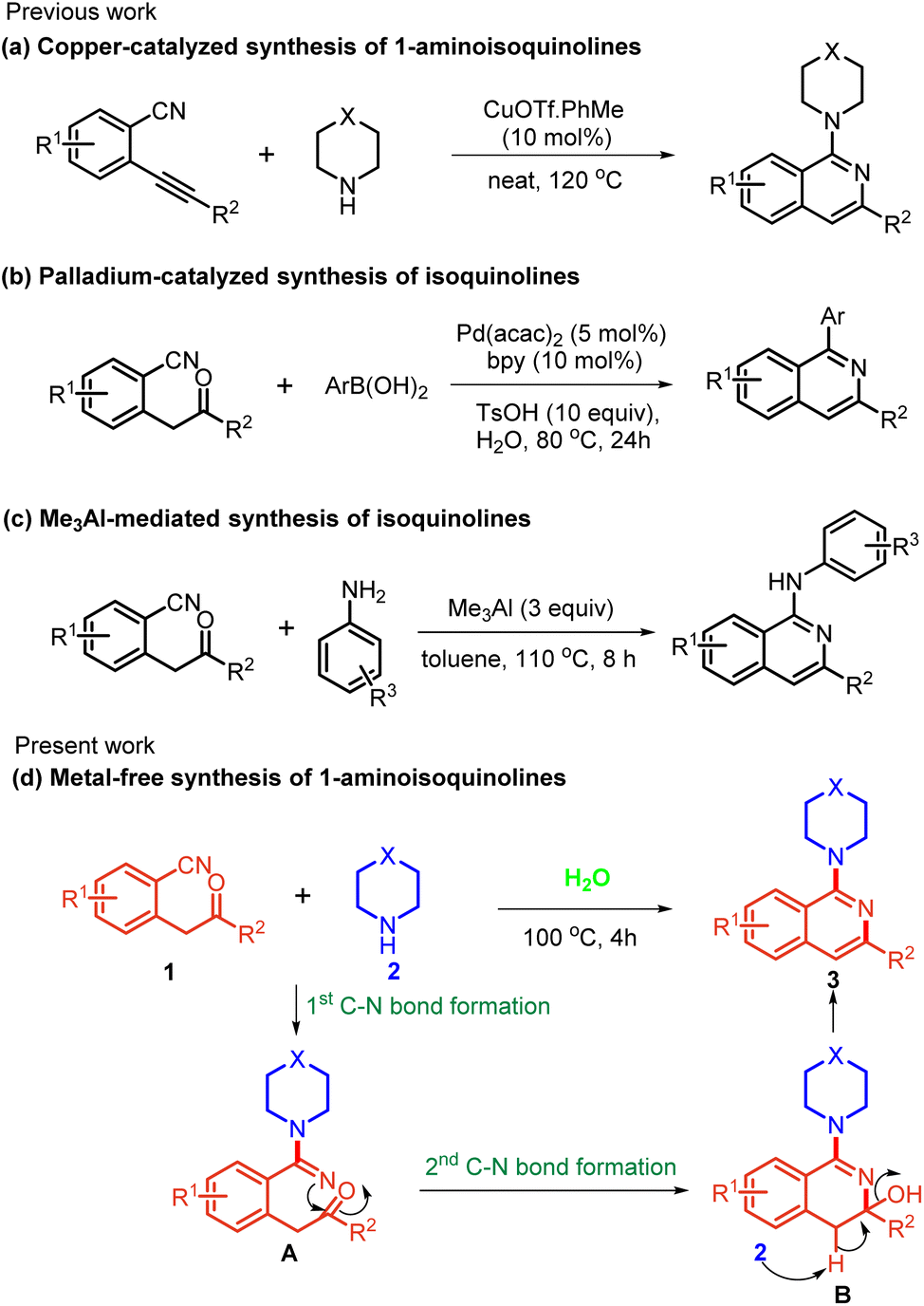 | ||
| Scheme 1 Different approaches for the activation of nitrile towards the synthesis of isoquinoline. | ||
At the outset, 2-(2-oxo-2-phenylethyl)benzonitrile 1a and 1-benzylpiperazine 2f were chosen as model substrates and allowed to react in toluene with 10 mol% of PTSA at 110 °C. To our delight, the reaction was completed in 4 h to yield 78% of the desired product 3f (Table 1, entry 1). An increase in the loading of PTSA from 10 to 20 mol% enhanced the yield of product 3f to 86% (entry 2). There was no significant improvement with increased loading of PTSA (entry 3). We next screened different solvents, i.e., THF, DCE, and MeOH, and found them to be unfavorable (entries 4–6). With DMF, the starting material was not completely consumed, giving only 56% yield of 3f (entry 7). However, we obtained a comparable yield in DMSO and water (entries 8 and 9). Interestingly, when we performed this reaction in the absence of PTSA, 3f was obtained in 86% yield (entry 10). To top that, the use of water as a solvent furnished product 3f in 92% yield (entry 11). Decreasing the temperature slightly showed a marked impact on the yield, giving 99% yield of 3f (entry 12). Further, decreasing the temperature led to a decrease in the yield (entry 13).
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Acid (mol%) | Temp. (T °C) | Yieldb (%) 3f |
| a Reactions were performed in a sealed tube using 0.5 mmol of 1a, and 0.6 mmol of 2f in 2.0 mL of the solvent. b Isolated yield. c Incomplete consumption of the starting material. d 0.5 mmol of 2f. | ||||
| 1 | Toluene | PTSA (10) | 110 | 78 |
| 2 | Toluene | PTSA (20) | 110 | 86 |
| 3 | Toluene | PTSA (30) | 110 | 85 |
| 4 | THF | PTSA (20) | 110 | n.r. |
| 5 | DCE | PTSA (20) | 110 | n.r. |
| 6 | MeOH | PTSA (20) | 110 | n.r. |
| 7c | DMF | PTSA (20) | 110 | 56 |
| 8 | DMSO | PTSA (20) | 110 | 84 |
| 9 | H2O | PTSA (20) | 100 | 90 |
| 10 | DMSO | — | 110 | 86 |
| 11 | H2O | — | 110 | 92 |
| 12 | H2O | — | 100 | 99 |
| 13c | H2O | — | 90 | 62 |
| 14d | H2O | — | 100 | 82 |
| 15c | Toluene | — | 110 | 44 |
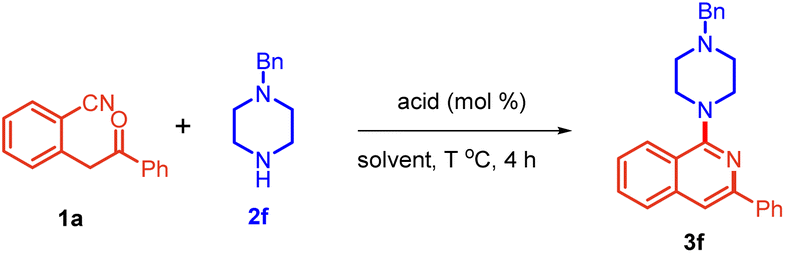
We also evaluated the effect of the loading of amine on the reaction. Decreasing its loading from 0.6 mmol to 0.5 mmol provided a comparatively lower yield of the product (entry 14). This indicated that the amine might be self-catalyzing the reaction. While conducting the reaction in toluene, the absence of PTSA gave inferior results (entry 15).
With the established reaction conditions (Table 1, entry 12), we first explored the substrate scope of various cyclic secondary amines 2a–q employing 2-(2-oxo-2-phenylethyl)benzonitrile 1a as a reacting partner (Scheme 2a). Initially, the reaction of 1a with 6-membered amines 2a–d afforded the corresponding amine-substituted isoquinolines 3a–d in excellent yields, i.e., 89–97%. 7-Membered amine (Azepane) 2e also reacted well to provide product 3e with 96% yield. Electron-rich benzyl piperazine 2f–i afforded the desired isoquinolines 3f–i in high yields (94–99%). In addition, 1-(bis(4-fluorophenyl)methyl)piperazine 2j was efficiently converted to the desired product 3j in 86% yield.
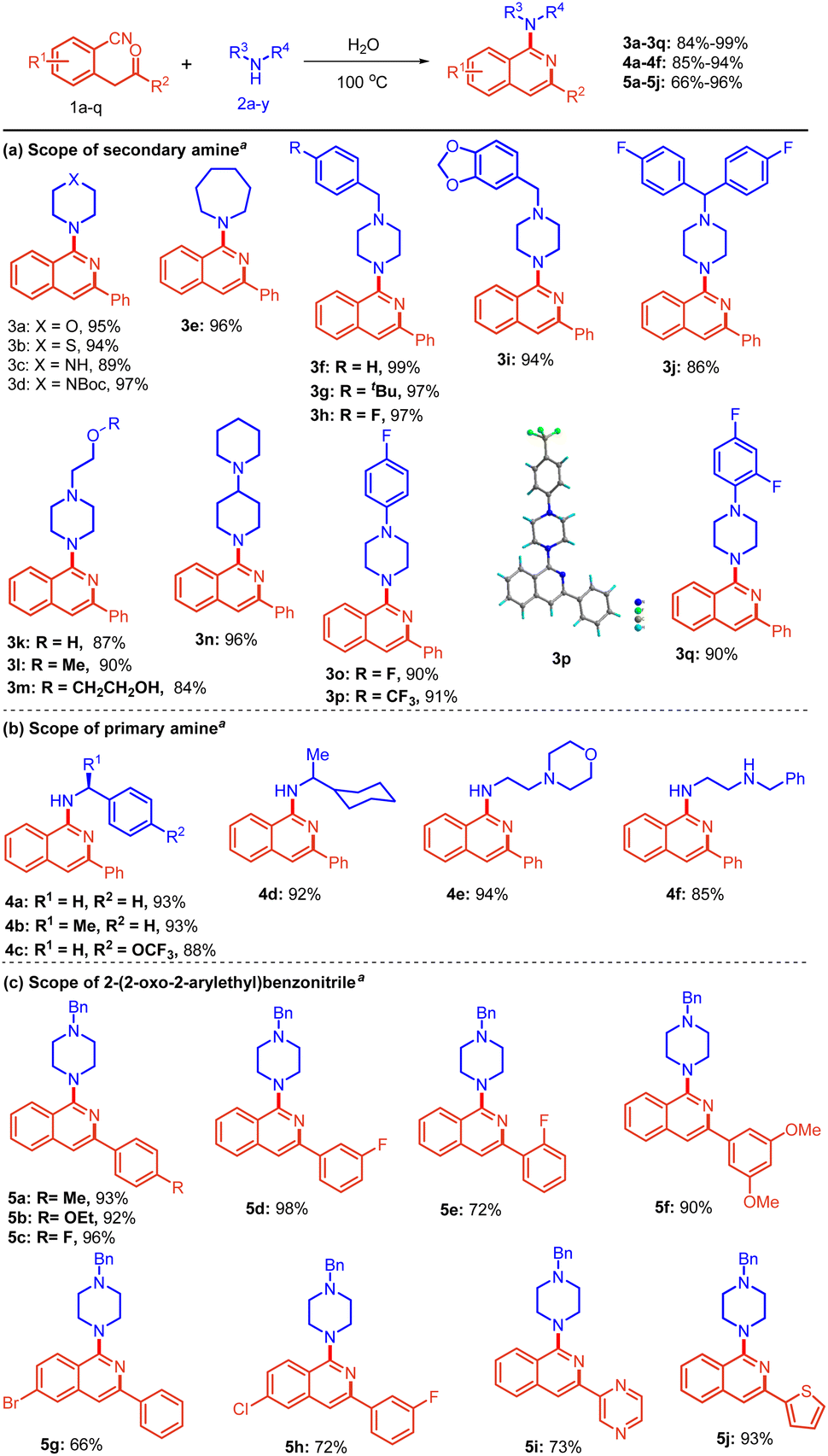 | ||
Scheme 2 Scope for the synthesis of amino isoquinolines. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reactions were performed in a sealed tube using 0.5 mmol of 1, and 0.6 mmol of 2 in 2.0 mL of distilled H2O at 100 °C for 4 h. CCDC no. 2204378†. Reactions were performed in a sealed tube using 0.5 mmol of 1, and 0.6 mmol of 2 in 2.0 mL of distilled H2O at 100 °C for 4 h. CCDC no. 2204378†. | ||
It is noteworthy that various substitutions of the N-atom of piperazine 2k–m were also well tolerated and gave the expected products 3k–m in 84–90% yields. Such scaffolds containing a free –OH group are otherwise difficult to access with other methods.
Additionally, the reaction of 1,4′-bipiperidine 2n and various phenyl-substituted piperazines 2o–q containing fluoro- and trifluoromethyl groups proceeded smoothly, affording the corresponding isoquinolines 3n–q in 90–96% yields. Compound 3p was further characterized by single crystal X-ray crystallography. Next, we focused on extending the versatility of the protocol by using primary amines 2t–y with 1a. We were pleased to obtain free NH-containing isoquinolines as the desired product (Scheme 2b).
The reaction of benzylamine 2t, (S)-1-phenylethan-1-amine 2u, and 1-(4-(trifluoromethoxy)phenyl)ethan-1-amine 2v furnished the desired product 4a–c in 88–93% yields. Likewise, 1-cyclohexylethan-1-amine 2w and 2-morpholinoethan-1-amine 2x also worked well to give 4d and 4e in 92% and 94% yields, respectively. Notably, N1-benzylethane-1,2-diamine 2y possessing secondary as well as primary amine was also amenable to the reaction conditions, giving the product 4f in 85% yield.
Thereafter, a broad range of 2-(2-oxo-2-arylethyl)benzonitriles 1b–l was investigated (Scheme 2c) with 1-benzylpiperazine 2f under the standard reaction conditions (Table 1, entry 12). It is evident from Scheme 2c that there was no profound electronic effect of electron-releasing and withdrawing substituents. Methyl, ethoxy, and fluoro 1b–d at the para position of phenyl rings were tolerated well to provide excellent yield. meta fluoro-substituted substrate 1e also reacted to give the product 5d in 98% yield. Furthermore, various substitutions (fluoro, chloro, bromo and methoxy) at the ortho, meta and para positions of 2-(2-oxo-2-arylethyl)benzonitriles 1e and 1k were also found to be compatible, providing the desired products 5e and 5h in 66% and 90% yields, respectively. Heteroaromatic substituents i.e., pyrazine and thiophene 1i–j, also underwent the reaction smoothly to provide the corresponding products 5i and 5j in 73% and 93% yields, respectively. To further extend the scope of the protocol, 2-(piperazin-1-yl)ethan-1-amine 2r, bearing a primary as well as a secondary amine, was considered. Interestingly, the reaction of 2r with substituted 2-(2-oxo-2-arylethyl)benzonitriles afforded bis-isoquinoline-containing products 6a–d in 80–94% yields (Scheme 3). In this case, both the amines acted as nucleophiles.
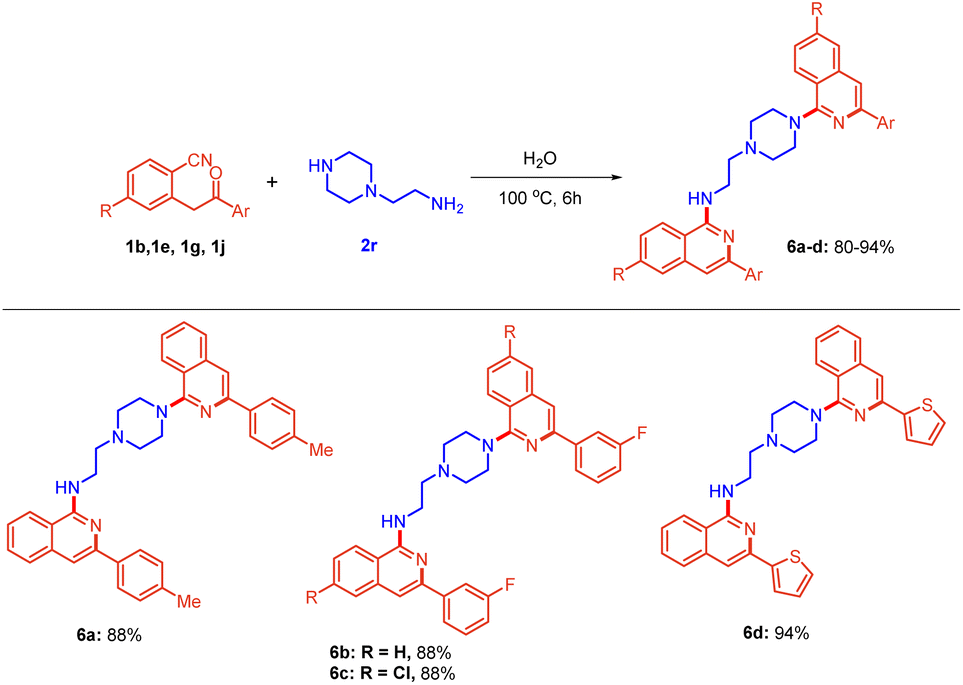 | ||
| Scheme 3 Scope for the synthesis of bis-amino isoquinolines. Reactions were performed in a sealed tube using 0.5 mmol of 1, and 0.3 mmol of 2r in 2.0 mL of distilled H2O at 100 °C for 4 h. | ||
A large number of pharmaceuticals are derivatives of isoquinolines; therefore, we attempted to derivatize some amine-based drugs utilizing the developed protocol (Scheme 4). Gratifyingly, the reaction of substrate 1a with amantadine 2z, rimantadine 2aa, and desloratadine 2ab delivered the corresponding products 7a–c in very good yields. Furthermore, nicotinic acid-derived substrates 1h with 2f furnished the target products 7d in a good yield. Similarly, substrate 1a reacted with N-methyl piperazine 2s to form compound 7e in excellent yield. It is noteworthy that compound 7e is a well-known antitumor agent (CWJ-a-5) acting as a topoisomerase I inhibitor.
 | ||
| Scheme 4 Scope of drugs containing isoquinolines. Reactions were performed in a sealed tube using 0.5 mmol of 1, and 0.6 mmol of 2 in 2.0 mL of distilled H2O at 100 °C for 4 h. | ||
Additionally, to demonstrate the synthetic potential of the strategy, substrate 1a was reacted with tert-butyl piperazine-1-carboxylate 2d at the gram scale, which yielded the target product 3d, demonstrating its applicability for large-scale synthesis (Scheme 5).
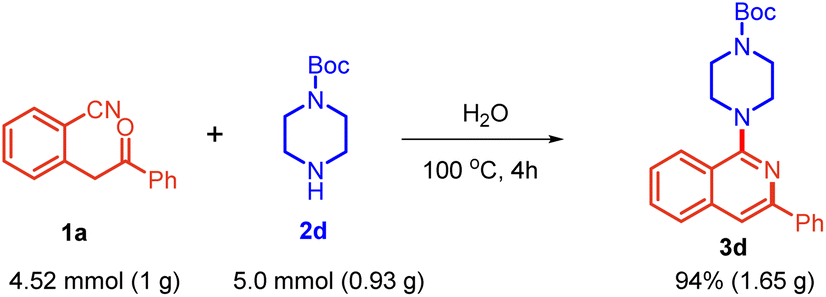 | ||
| Scheme 5 Gram-scale synthesis. | ||
Next, a series of experiments was performed to gain some mechanistic insights. Initially, the reaction of substrate 1a was conducted with 2f in the presence of TEMPO (4.0 equiv.) under identical reaction conditions (Table 1, entry 12), which did not show any profound impact on the yield of product 3f (Scheme 6a).
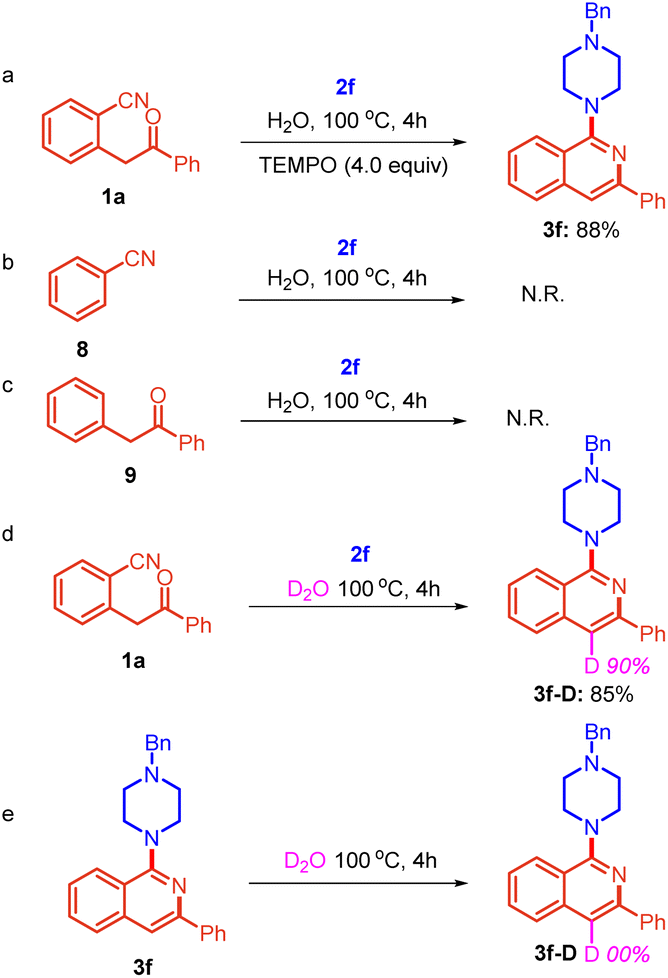 | ||
| Scheme 6 Control experiments. | ||
The result indicated that the reaction does not proceed through a radical mechanism. Furthermore, to ascertain the electrophilic center for the amine, the reaction of benzonitrile 8 and 1,2-diphenylethan-1-one 9 was also performed with 2f separately. However, no reaction was observed in both cases. This suggests that the reaction is sensitive to the relative arrangement of nitrile and 1,2-diphenylethan-1-one (Scheme 6b and c). It shows that the presence of both adjacent groups is necessary for this reaction. Further, the reaction was performed in D2O as a solvent under standard reaction conditions (Table 1, entry 12) and product 3f-D was obtained in 85% yield with 90% deuterium incorporation at the cyclised position (Scheme 6d).
Further, to confirm whether the incorporation of deuterium in product 3f-D is before the formation of 3f-D or after the formation of 3f-D, we performed a reaction using 3f as the starting substrate in D2O and found that there was no deuterium incorporation at the cyclized position (Scheme 6e). This shows that active CH2 protons present in substrate 1a were exchangeable in D2O due to keto–enol tautomerization and deuterium was incorporated before the formation of 3f-D.
Based on the results of control experiments (Scheme 6) and precedents,9,10 we have proposed a reasonable mechanistic pathway for the reaction (Scheme 7). The reaction proceeds through a nucleophilic attack of amine on the nitrile of 2-(2-oxo-2-phenylethyl)benzonitrile 1 to form intermediate A in which iminium nitrogen simultaneously attacks the carbonyl carbon, leading to the formation of intermediate B. Excess amine present in the reaction abstracts the benzylic proton from intermediate B followed by aromatization with the removal of water molecules, leading to the formation of compound 3.
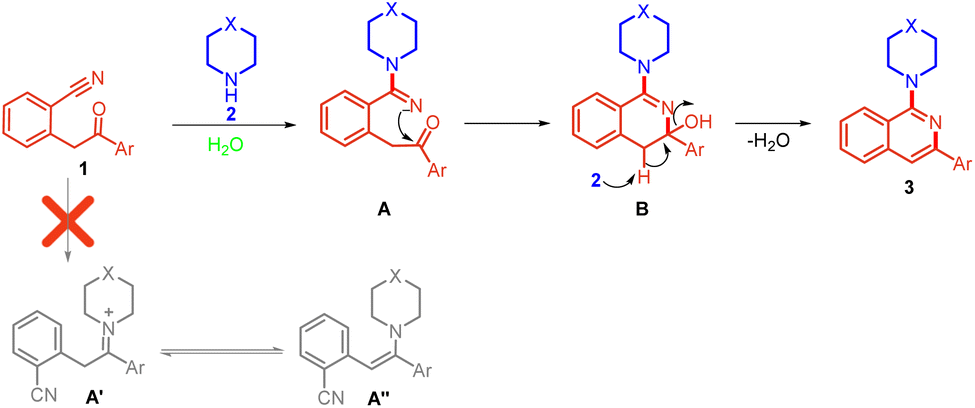 | ||
| Scheme 7 Plausible reaction mechanism. | ||
Conclusions
In summary, the utilization of metal-free activation of nitrile in an aqueous medium towards sequential nucleophilic addition and annulation provides an economical methodology for the generation of an array of diversified aminated isoquinolines in good to excellent yields. This modular synthesis shows wide functional group tolerance and is compatible with electron-rich, electron-deficient as well as hetero-aromatic moieties. The approach presented herein allows the simple synthesis of aminated isoquinolines and thus could be of high pharmaceutical interest. The proposed reaction pathway is well supported by a series of control experiments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Science & Engineering Research Board (SERB) under the core research grant scheme (CRG/2020/005800). We thank USIC, University of Delhi for providing financial support and instrumentation facilities.References
- (a) B.-A. Sweetman, B.-H. Müller and P.-J. Guiry, Tetrahedron Lett., 2005, 46, 4643–4646 CrossRef CAS; (b) F. Durola, J.-P. Sauvage and O.-S. Wenger, Chem. Commun., 2006, 171–173 RSC; (c) K.-H. Fang, L.-L. Wu, Y.-T. Huang, C.-H. Yang and I.-W. Sun, Inorg. Chim. Acta, 2006, 359, 441–450 CrossRef CAS; (d) A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S.-T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971–12979 CrossRef CAS PubMed; (e) P. Giri and G.-S. Kumar, Mini-Rev. Med. Chem., 2010, 10, 568–577 CrossRef CAS PubMed; (f) A.-Y. Khan and G.-S. Kumar, Biophys. Rev., 2015, 7, 407–420 CrossRef CAS PubMed.
- (a) A.-L. Smith, F. DeMorin, N. Paras, Q. Huang, K. Petkus, E. Doherty, T. Nixey, J. Kim, D. Whittington, L. Epstein, M. Lee, M. Rose, C. Babij, M. Fernando, K. Hess, Q. Le, P. Beltran and J. Carnahan, J. Med. Chem., 2009, 52, 6189 CrossRef CAS PubMed; (b) S. Yang, H. Van, T. Le, D. Khadka, S. Cho, K. Lee, H. Chung, S. Lee, C. Ahn, Y. Lee and W. Cho, Bioorg. Med. Chem. Lett., 2010, 20, 5277 CrossRef CAS PubMed.
- C. M. Zhu, M. Yi, D. Wei, X. Chen, Y. Wu and X. Cui, Org. Lett., 2014, 16(7), 1840–1843 CrossRef CAS PubMed.
- (a) Q. Shen, T. Ogata and J.-F. Hartwig, J. Am. Chem. Soc., 2008, 130, 6586–6596 CrossRef CAS PubMed; (b) Q. Shen, S. Shekhar, J.-P. Stambuli and J.-F. Hartwig, Angew. Chem., Int. Ed., 2005, 44, 1371–1375 CrossRef CAS PubMed.
- (a) X. Wei, M. Zhao, Z. Du and X. Li, Org. Lett., 2011, 13, 4636 CrossRef CAS PubMed; (b) J. Li, M. John and L. Ackermann, Chem. – Eur. J., 2014, 20, 5403 CrossRef CAS PubMed; (c) J. Li, M. Tang, L. Zang, X. Zhang, Z. Zhang and L. Ackermann, Org. Lett., 2016, 18(11), 2742–2745 CrossRef CAS PubMed; (d) F. Yang, J.-J. Yu, Y. Liu and J. Zhu, Org. Lett., 2017, 19, 2885–2888 CrossRef CAS PubMed; (e) Y. Zhou, Y. Wang, Y. Lou and Q. Song, Org. Lett., 2019, 21, 8869–8873 CrossRef CAS PubMed; (f) Y. Zuo, X. He, Y. Ning, Y. Wu and Y. Shang, J. Org. Chem., 2018, 83(21), 13463–13472 CrossRef CAS PubMed; (g) Y. Li, L. Gao, H. Zhu, G. Li and Z. Chen, Org. Biomol. Chem., 2014, 12, 6982–6985 RSC; (h) X. Zhang, Y. Zhou, M. Wang, Y. Chen, Y. Zhou, W. Gao, M. Liu, X. Huang and H. Wu, Chem. – Asian J., 2020, 15, 1692 CrossRef CAS PubMed; (i) J. Ren, C. Pi, X. Cui and Y. Wu, Org. Lett., 2021, 23(17), 6628–6632 CrossRef CAS PubMed.
- (a) V.-Y. Kukushkin and A.-J. Pombeiro, Chem. Rev., 2002, 102, 1771–1802 CrossRef CAS PubMed; (b) F.-F. Fleming and Q. Wang, Chem. Rev., 2003, 103, 2035–2077 CrossRef CAS PubMed; (c) X. Yang, H. Yu, Y. Xu and L. Shao, J. Org. Chem., 2018, 83, 9682–9695 CrossRef CAS PubMed; (d) V.-G. Chandrashekhar, W. Baumann, M. Beller and R.-V. Jagadeesh, Science, 2022, 376, 1433–1441 CrossRef CAS PubMed; (e) C. Zhou, T. Lei, X.-Z. Wei, C. Ye, Z. Liu, B. Chen, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2020, 142(39), 16805–16813 CrossRef CAS PubMed; (f) R. Hosseinijei, Z. H. Tejeneki, A. Nikbakht, F. Rominger and S. Balalaie, Org. Biomol. Chem., 2022, 20, 3076–3080 RSC; (g) K. M.-S. Adusumalli, L. N.-S. Konidena, H.-B. Gandham, K. Kumari, K.-R. Valluru, S. K.-R. Nidasanametla, V. R. Battula and H.-K. Namballa, Beilstein J. Org. Chem., 2021, 17, 2765–2772 CrossRef CAS PubMed.
- (a) M. Yousuf, T. Das and S. Adhikari, New J. Chem., 2015, 39, 8763 RSC; (b) Y.-C. Wong, K. Parthasarathy and C.-H. Cheng, Org. Lett., 2010, 12, 1736 CrossRef CAS PubMed; (c) G.-C. Tsui, Q. Glenadel, C. Lau and M. Lautens, Org. Lett., 2011, 13, 208 CrossRef CAS PubMed; (d) X. Wang, M. Liu, L. Xu, Q. Wang, J. Chen, J. Ding and H. Wu, J. Org. Chem., 2013, 78, 5273 CrossRef CAS PubMed; (e) J. Chen, L. Ye and W. Su, Org. Biomol. Chem., 2014, 12, 8204 RSC; (f) B. Zhao and X. Lu, Org. Lett., 2006, 8, 5987 CrossRef CAS PubMed.
- L. Qi, K. Hu, S. Yu, J. Zhu, T. Cheng, X. Wang, J. Chen and H. Wu, Org. Lett., 2017, 19(1), 218–221 CrossRef CAS PubMed.
- (a) V. Reddy, A.-S. Jadhav and R.-V. Anand, Eur. J. Org. Chem., 2016, 453–458 CrossRef CAS; (b) Y. Long, Z. She, X. Liu and Y. Chen, J. Org. Chem., 2013, 78, 2579–2588 CrossRef CAS PubMed.
- M.-L. Kuznetsov, N.-A. Bokach, D.-D. Kharlampidi, Y.-N. Medvedev, V.-Y. Kukushkin and A.-I. Dementiev, Russ. J. Gen. Chem., 2010, 80, 458–467 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2204378. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2gc04044a |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |