
Integrative analysis of the metabolome and transcriptome reveals the influence of Lactobacillus plantarum CCFM8610 on germ-free mice
Pan
Huang
ab,
Shanrong
Yi
ab,
Leilei
Yu
ab,
Fengwei
Tian
 ab,
Jianxin
Zhao
ab,
Hao
Zhang
abcd,
Wei
Chen
abc and
Qixiao
Zhai
*ab
ab,
Jianxin
Zhao
ab,
Hao
Zhang
abcd,
Wei
Chen
abc and
Qixiao
Zhai
*ab
aState Key Laboratory of Food Science and Technology, Jiangnan University, Wuxi, Jiangsu 214122, P. R China. E-mail: zhaiqixiao@sina.com; Fax: +0510-85912155; Tel: +0510-85912155
bSchool of Food Science and Technology, Jiangnan University, Wuxi, Jiangsu 214122, China
cNational Engineering Research Center for Functional Food, Jiangnan University, Wuxi, Jiangsu 214122, China
dWuxi Translational Medicine Research Center and Jiangsu Translational Medicine Research Institute Wuxi Branch, China
First published on 28th November 2022
Abstract
This study describes the influence of Lactobacillus plantarum CCFM8610 on the host by employing transcriptome and untargeted metabolomics. According to the enrichment analysis, three pathways, including the complement and coagulation cascade pathway, antigen processing and presentation pathways, and protein processing in the endoplasmic reticulum pathway, were affected by L. plantarum CCFM8610 colonization. According to partial least squares-discriminant analysis, five metabolites, L-methionine, D-tryptophan, indoleacrylic acid, DL-acetylcarnitine, and L-norleucine, were identified as key metabolites in the serum. Furthermore, integrative analysis of the metabolome and transcriptome revealed connections between enriched pathways and differential metabolites, and the regulation strategy of choline by affecting gene expression was proposed. Overall, the effects of L. plantarum CCFM8610 on host health were investigated after excluding the influence of gut microbes, which provides a valuable reference for studying the potential mechanisms of the effect of probiotics on host health.
1. Introduction
In the gastrointestinal tract, trillions of bacteria continuously interact with the host.1 These bacteria are called the gut microbiome, which is critical for host health and diseases.2 For health reasons, live microorganisms beneficial for host health when present in adequate amounts have been widely used as dietary supplements; these live microorganisms are defined as probiotics.3 Probiotic bacteria have been widely reported to alleviate the exacerbation of intestinal diseases and other diseases associated with gut microbes and drive the maturation of the immune system.4–6Among the many probiotics, Lactobacillus plantarum is a widely studied probiotic that can be easily used in probiotic products.7L. plantarum generally exists in the gastrointestinal tract transitorily.8,9 Although the persistence of L. plantarum is limited, its probiotic functions have been widely reported, including maintenance of host growth, prevention of sepsis, and resistance to pathogenic bacteria.6,10,11 In our previous studies, it has been demonstrated that L. plantarum CCFM8610 alleviates irritable bowel syndrome and resists acute cadmium toxicity.12,13 In addition, L. plantarum CCFM8610 also significantly alleviates enteritis by regulating the inflammatory response.14 However, the mechanism by which L. plantarum affects host physiology remains largely unknown.
After entering the gastrointestinal tract, probiotics must compete for ecological niches with indigenous microbiota and adapt to stressful environments to survive.9 Generally, probiotics are beneficial for host health in two ways. On the one hand, probiotics could directly contact intestinal cells and maintain homeostasis in the intestinal epithelium.15,16 On the other hand, probiotics can interact with gut microbes to indirectly alleviate disease.17,18 However, direct interactions between probiotics and the host are usually difficult to explore because of the interference of indigenous gut microbes. Therefore, the underlying mechanism of the interactions between probiotics and the host should be clarified to obviate interference from other gut microbes. For revealing the mechanisms, some omics technologies are used to illustrate the overall perspective of host changes, such as transcriptomics and metabolomics.19,20 Transcriptomics studies have been used to explore the cellular change responses to probiotics in vivo.21,22 The serum metabolome is also used to estimate the impact of probiotics on the host's physiological changes.23,24
Germ-free (GF) animals are cultivated in a sterile isolator without any bacteria, which is suitable for studying the relationship between the host's physiological and pathological development and microorganisms.25,26 GF animals are effective experimental models to evaluate the influence of probiotic strains on the host directly without the influence of the gut microbiome. Transcriptomes are typically used to detect the influence of probiotics on the host's physiological changes. After probiotics enter the gastrointestinal tract, the stressful environment can cause global transcriptional changes in probiotics.27,28 The survival of probiotics can change host physiology.29,30 These studies have demonstrated that foreign organisms can modulate host immunity and metabolism in GF animals.22,30 Hence, mono-colonization with a probiotic strain in GF animals facilitates studies illustrating the mechanisms of the interactions between the host and probiotic strains.
Metabolomics has been widely used to explore the effects of probiotics on host health. Gut microbes have been extensively studied for their ability to alter serum metabolites to improve host health.31 Probiotics have also been reported to promote host health by modulating serum metabolites.32,33 On the one hand, probiotics can produce beneficial metabolites, such as polysaccharides, fatty acids, and short-chain fatty acids, which can pass through the intestinal barrier to the serum.34,35 On the other hand, metabolites in serum could be affected by changes in the host, such as diet, lifestyle, and health conditions.36 However, whether changes in serum metabolites are caused by probiotics or gut microbes remains unclear.
In this study, transcriptomic changes in the colon and metabolomic changes in serum after mono-colonization with L. plantarum CCFM8610 in GF mice were investigated. Then, the key enriched pathways of differentially expressed genes were analyzed, and the key differential metabolites in the serum were identified. Finally, the key gene pathways and differential metabolites were identified using correlation analysis. The persistence of L. plantarum CCFM8610 in GF mice was monitored, which excluded the competition of indigenous microbiota.
2. Materials and methods
2.1. Bacteria and growth conditions
L. plantarum CCFM8610 was isolated from fermented vegetables and cultured in MRS liquid medium (Hopebio Company) at 37 °C for 24 h. L. plantarum CCFM8610 cells were harvested by centrifugation at 8000g for 10 min at 4 °C and washed twice with sterile normal saline. Subsequently, the concentration of bacterial cells was adjusted to 7 × 109 CFU mL−1 by resuspension in 0.9% sterile saline solution for intragastric administration.2.2. GF mice treatment
In this study, GF male C3H/HeJ mice (6–8 weeks old) were obtained from the Wuxi School of Medicine, Jiangnan University. All the experimental procedures were approved by the Ethics Committee of Jiangnan University, China (JN. no. 20210630c0360816[209]), followed by animal care guidelines. The GF mice were housed in separate isolators, with a 12 h light/dark cycle in a temperature-(22 °C ± 1 °C) and humidity-controlled (55% ± 10%) room. Six germ-free mice were separated into two groups. The control group was the blank germ-free mice, and the Lp group was the germ-free mice mono-colonized with L. plantarum CCFM8610. After L. plantarum gavage administration for four days, the germ-free mice were housed for thirty-one days, and the mice were sacrificed at day 35. Meanwhile, fecal samples were collected for detecting the colonization of L. plantarum during experiments at days 1–8, day 10, day 12, day 14, day 21, day 28 and day 35.2.3. Evaluation of colonization of L. plantarum CCFM8610
To verify that the mice were colonized by L. plantarum CCFM8610, a gavage was performed four times. Fresh feces were collected 15 times over the next 35 d after the first gavage. Each fecal sample (0.1 g) was suspended and homogenized in 900 μL of sterile saline, and further decimal dilutions were performed. The counts of viable L. plantarum CCFM8610 in feces were counted on MRS plates after 48 h of incubation at 37 °C, and the counts were calculated to assess colonization.2.4. Transcriptomics analysis
TRIzol reagent (Invitrogen, Carlsbad, CA, USA) was used to extract total RNA, according to the manufacturer's protocol, and RNA was subsequently converted to cDNA. RNA sequencing was performed using the Illumina HiSeq instrument by MajorBio (Shanghai, China), and bioinformatics analysis was performed. Differentially expressed genes (DEGs) were identified by a fold change ≥ 2 and a P value < 0.05, with a false discovery rate (FDR) cutoff < 0.05. For the enrichment analysis, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were analyzed.2.5. Metabolomics analysis
The samples were pretreated as previously described,37 with slight modifications. Briefly, the samples were stored at −80 °C and thawed on ice before analysis. To precipitate protein, one volume of serum was added to three volumes of methanol. Then, the mixture was homogenized for 15 s, ultrasonicated on ice for 10 min, and stored at −20 °C for 1 h. The mixture was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 15 min at 4 °C. After removing the precipitate, the supernatant (70 μl) was dried in a centrifugal vacuum evaporator, and 80% methanol was used to dissolve the samples. In addition, one sample volume was placed in a container as a quality control (QC) sample. QC samples were used to estimate the stability and reproducibility of the system.
000g for 15 min at 4 °C. After removing the precipitate, the supernatant (70 μl) was dried in a centrifugal vacuum evaporator, and 80% methanol was used to dissolve the samples. In addition, one sample volume was placed in a container as a quality control (QC) sample. QC samples were used to estimate the stability and reproducibility of the system.
A Thermo Fisher Q-Exactive Orbitrap mass spectrometer with an Acquity HSS T3 column (Waters, 1.8 μm, 2.1 × 100 mm) was used to analyze the serum metabolites. The flow rate was 0.3 mL min−1, and the injection volume was 2 μL per sample. Mobile phases A and B consisted of 0.1% aqueous formic acid and 0.5 mM ammonium acetate–acetonitrile, respectively. The gradient elution conditions were as follows: 1 min, 5% B; 10 min, 99% B; 12 min, 99% B; and 15 min, 5% B.
The MS analysis used a Thermo Fisher Q-Exactive Plus. The data range of mass (m/z) in the positive and negative ion modes was set to 151 to 2000 Da and 70 to 1050 Da, respectively. And the electrospray ionization parameters were set as described previously.37
2.6. Quantitative real-time PCR
TRIzol reagent (Invitrogen, Carlsbad, CA, USA) was used to extract total RNA, according to the manufacturer's protocol, and RNA was subsequently converted to cDNA. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed on the CFX Connect Real-Time System (Bio-Rad, USA). All primers are listed in Table 1. GAPDH was used as a housekeeping gene, and the expression of the target genes was calculated according to the formula 2−ΔΔCt.| Target gene | Primer sequences (forward and reverse, respectively) (5′ to 3′) | Amplicon size (bp) |
|---|---|---|
| Hspa1a | TGGTGCTGACGAAGATGAAG | 100 |
| CGCTGAGAGTCGTTGAAGTAG | ||
| Mup7 | TAGTGTGTGTCCATGCAGAAG | 101 |
| CTTGTCAGAGGCCAGGATAATAG | ||
| Pou1f1 | CGGATGGCTGAAGAACTGAA | 110 |
| GAGAACAGGCTCTGATTGAGAC | ||
| Clspn | GGAATTGCAGCGGAAACTAATG | 111 |
| TTCCAGCTCTCCTCGTCTTA | ||
| Dynlt2a1 | CTCAATTGCTGATGTAGGCAAAG | 99 |
| TGTGCTTGAAATTTCTTCAGTGG | ||
| Tcf19 | TGGTGACCTTCTGACCTTTG | 87 |
| TGAGGTTTAACCCGGACTTG | ||
| GAPDH | GTGGCAAAGTGGAGATTGTTG | 99 |
| CGTTGAATTTGCCGTGAGTG |
2.7. Statistical analysis
The plot and analysis of the data were performed using GraphPad Prism software (version 8.0). The comparison of bacterial counts was done using a paired t-test, and significant differences (P < 0.05) among the means were identified. Pearson's correlations between gene expression levels and differential metabolites were also evaluated. Compound Discover 3.2 software was used to process the MS data using default parameters. The supervised multivariate models were achieved by partial least squares-discriminant analysis (PLS-DA) using MetaboAnalyst 4.0 software (https://www.metaboanalyst.ca). Key metabolites were selected based on VIP values in the PLS-DA model. The metabolites were identified using the Human Metabolome Database (HMDB; https://www.hmdb.ca) based on accurate mass and mass spectrometric fragmentation patterns.3. Results
3.1. L. plantarum CCFM8610 effectively colonized the GF mice intestines
During the first four days of gavage, the counts of L. plantarum CCFM8610 were 9.4 ± 0.48, 9.6 ± 0.19, 9.4 ± 0.18, and 9.5 ± 0.08 log10 CFUs per g feces (Fig. 1a). At later time points, the bacterial numbers ranged from 9.2 ± 0.12 log10 CFUs per g feces to 9.7 ± 0.06 log10 CFUs per g feces, and Fig. 1a shows the trends of bacterial number changes. The results showed that L. plantarum could establish stable colonization in GF mice after four gavage treatments. At day 35, the mice were sacrificed to determine the bacterial distribution along the length of the gastrointestinal tract. In the stomach and duodenum, L. plantarum CCFM8610 counts were lower than in the jejunum, ileum, cecum, and colon (Fig. 1b). L. plantarum CCFM8610 had the highest density in the colon and ileum. In different parts of the gastrointestinal tract, the distribution of L. plantarum CCFM8610 showed differences.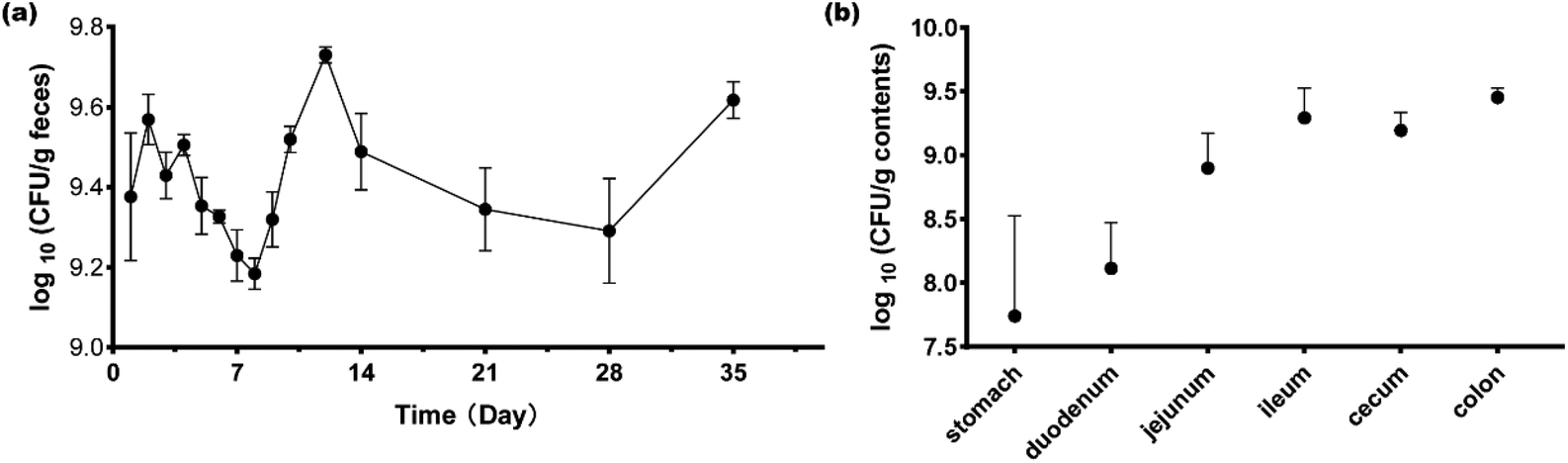 | ||
| Fig. 1 L. plantarum CCFM8610 colonized in GF mice. (a) Fecal bacterial counts of the CCFM8610 group at different time points. (b) Bacterial counts of the CCFM8610 group in the stomach, duodenum, jejunum, ileum, cecum, and colon. | ||
3.2. Colonization by L. plantarum CCFM8610 affected the gene expression in the colon
To understand the physiological changes in GF mice after L. plantarum CCFM8610 colonization in the gastrointestinal tract, the transcriptome of the colon was analyzed. The gene expression in untreated GF mice and GF mice colonized with L. plantarum CCFM8610 was compared. A total of 383 genes were identified as differentially expressed (P < 0.05; fold-changes > 2). A total of 193 and 190 genes were upregulated and downregulated, respectively (P < 0.05; fold-changes > 2) (Fig. 2a). In Fig. 2a, the DEGs are shown in the heat map, which provides a more intuitive representation of transcriptional variation. The critical differential genes are marked in Fig. 2b, and the gene expression was confirmed by qPCR data in Fig. 2c and d.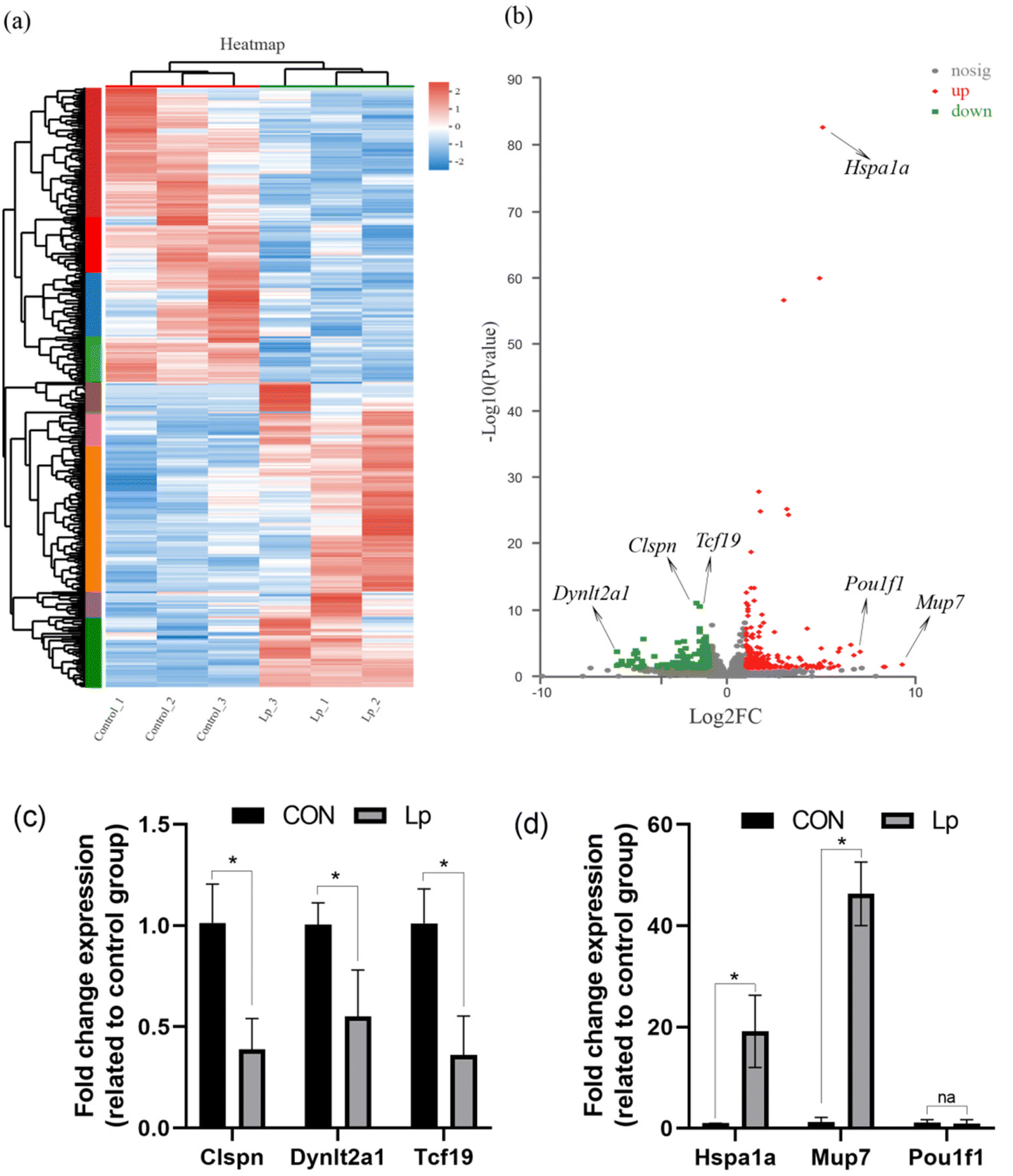 | ||
| Fig. 2 Expression profile in the colon tissue of GF mice colonized L. plantarum CCFM8610. (a) Heat map displaying hierarchical clustering of differentially expressed genes. Upregulated genes are shown in red, whereas downregulated genes are shown in green. (b) Volcano plot of microarray data. The horizontal bar represents the nominal significance level of 0.01 for Student's t-tests, and the vertical dashed bars denote ≤2-fold downregulation (left) or ≥2-fold upregulation (right). (c) and (d) The gene expression of critical differential genes. | ||
3.3. Colonization with L. plantarum CCFM8610 stimulated the immunity and protein processing in the colon
DEGs in the L. plantarum CCFM8610-colonized batch were enriched in 211 biological pathways according to the KEGG enrichment analysis. The predominant pathways were complement and coagulation cascades, protein processing in the endoplasmic reticulum, prion disease, antigen processing and presentation, enrichment pathways mainly related to the immune system, and protein modification (Fig. 3a). The related genes in the enriched pathways are shown in Fig. 3b. The KEGG enrichment analysis identified a significance threshold of P < 0.05. Besides, all genes and differential genes were annotated by GO annotation as shown in Fig. 3c and d, respectively. Generally, the number of differential genes was much smaller than the number of all genes. Cellular processes, cell sections and binding were GO terms enriched in all genes and differential genes. Besides, two GO terms, including the immune system process and the multi-organism process, were not annotated in all genes’ GO annotation, but in differential genes’ GO annotation.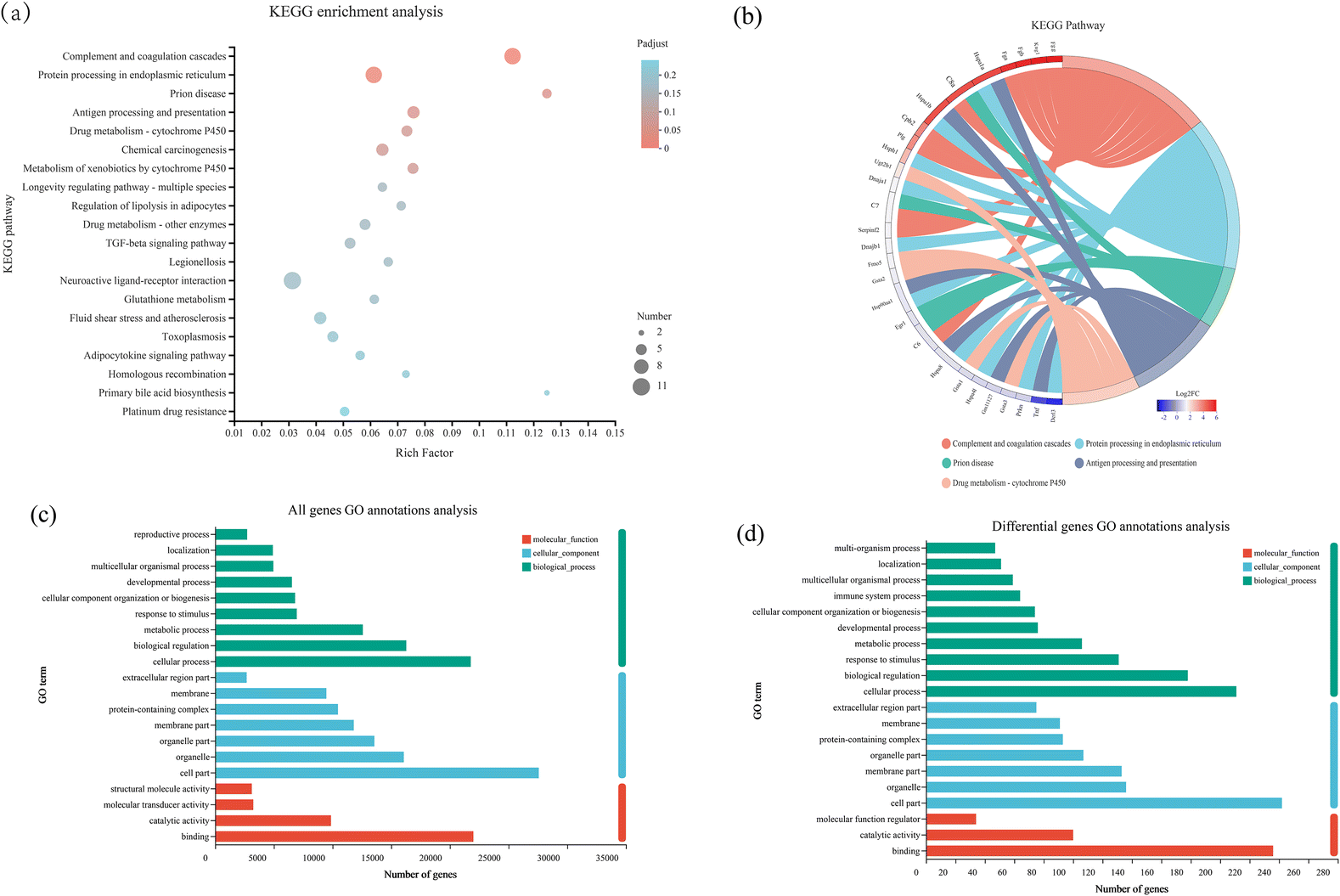 | ||
| Fig. 3 (a) Top 20 most significant pathways identified from the KEGG pathway analysis of the significantly altered differential genes between the germ-free mice with or without L. plantarum CCFM8610 colonization. (b) Circular visualization of connectivity and the expression patterns of top five pathways. (c) All gene annotations based on GO terms. (d) Differential gene annotations based on GO terms. | ||
3.4. Colonization with L. plantarum CCFM8610 affected the metabolites in the serum
The serum metabolic change response to L. plantarum CCFM8610 administration in GF mice was investigated, and the differential metabolites were identified by metabolome analysis. A total of 135 metabolites were identified. The PLS-DA score plot showed the clustering of metabolome profiles (Fig. 4a). Based on the VIP score, we identified differential metabolites between the L. plantarum CCFM8610 group and the untreated GF mice (Fig. 4b). These results showed distinct differences between the two groups. The top five differential metabolites were L-methionine, D-tryptophan, indoleacrylic acid, acetylcarnitine, and L-norleucine. Hierarchical clustering showed that 50 metabolites were most significantly differentially expressed (Fig. 4c).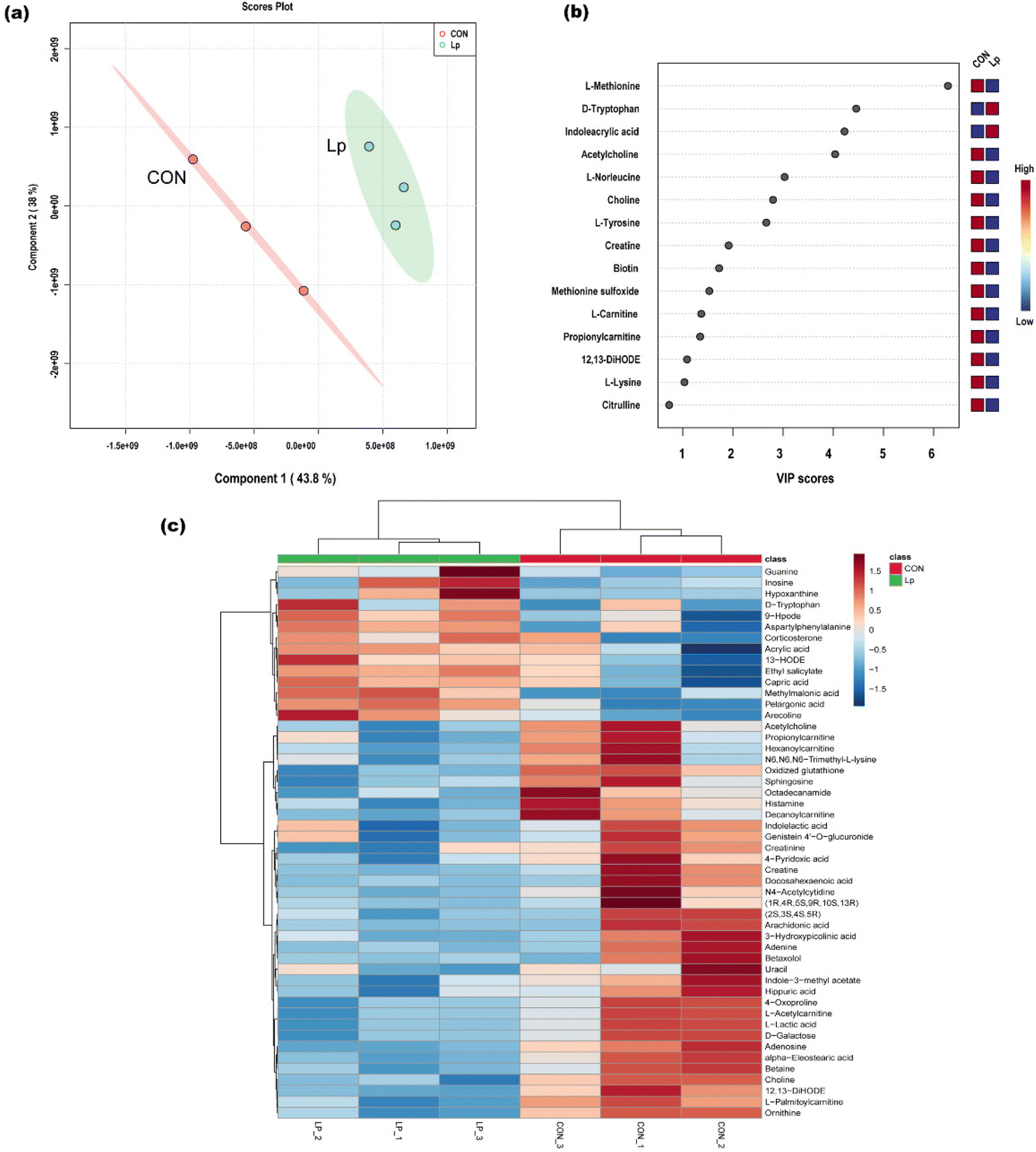 | ||
| Fig. 4 The metabolic profiles in the serum of GF mice with or without L. plantarum CCFM8610 colonization. (a) The PLS-DA plot to distinguish the differential metabolites in the serum of GF mice without L. plantarum CCFM8610 colonization (CON) and GF mice with L. plantarum CCFM8610 colonization (Lp). (b) Differential metabolites between CON and the Lp batch. (c) Heat map and hierarchical clustering of metabolic profiles in the serum of CON and the Lp batch. | ||
3.5. Gene and metabolite co-expression network construction
Integrative analysis of the metabolome and transcriptome revealed that three pathways, including protein processing in the endoplasmic reticulum pathway, the fluid shear stress and atherosclerosis pathway, and the longevity regulating pathway, were significantly correlated with metabolites. Among the metabolites, the degree of choline was highest in the network, and the concentration of choline was significantly correlated with the differential genes of the three pathways (Fig. 5).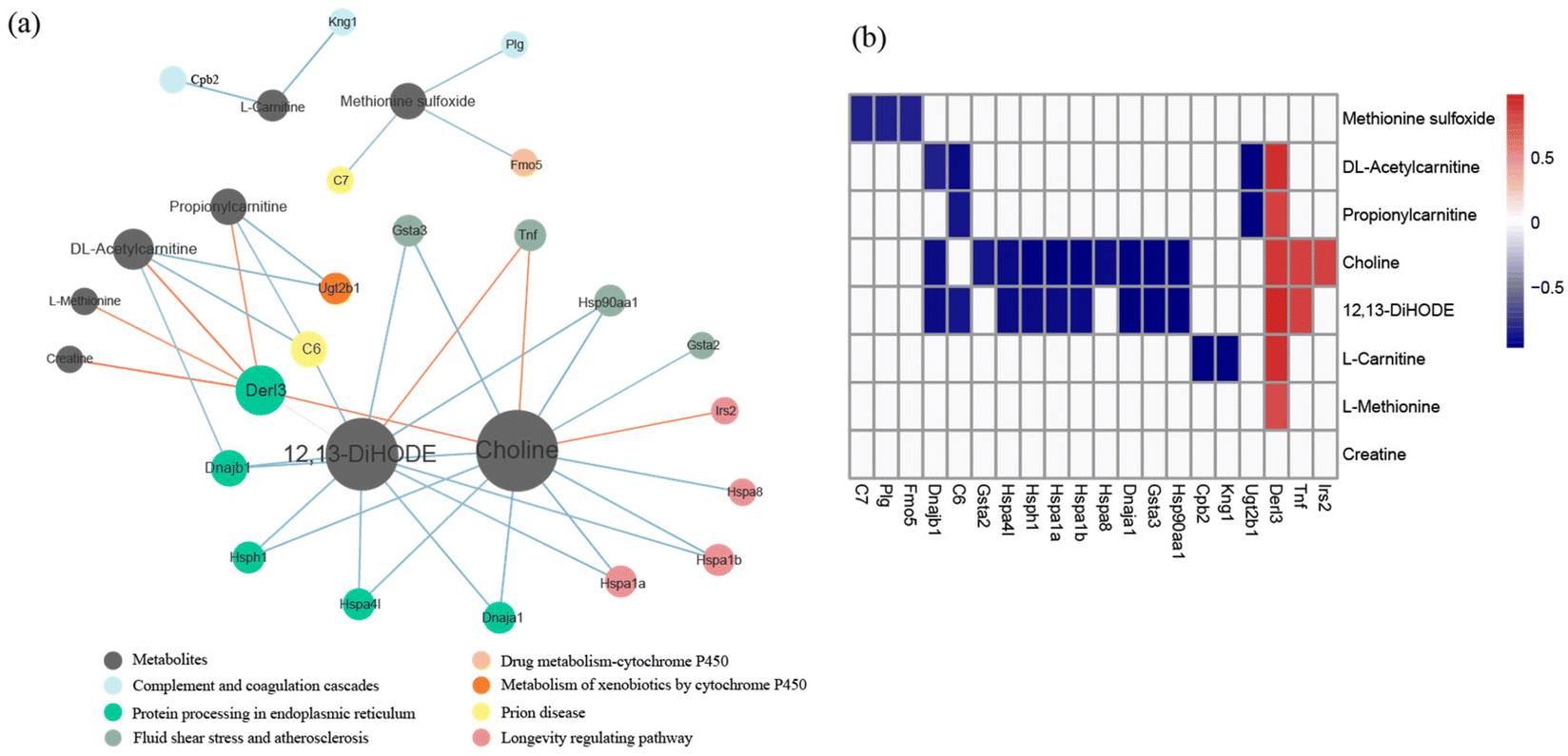 | ||
| Fig. 5 Analysis of the correlation between differential genes and metabolites. (a) Networks of interactions between differential genes and metabolites. The size of the circle represents the node connectivity, and the thickness of the lines represents the magnitude of correlation. (b) The correlation heat map of differential genes and metabolites. All correlations on display are significantly different (P < 0.05). | ||
4. Discussion
Probiotics, live microorganisms that confer health benefits to the host, have been reported to have many functions, such as ameliorating inflammatory bowel diseases and improving obesity.33,38 Our previous studies have demonstrated that L. plantarum CCFM8610 has many probiotic functions, such as alleviating enteric diseases, preventing gut microbiota dysbiosis, and protecting the gut barrier from toxins.5,12–14,39 In addition, this strain is beneficial for host health because it produces beneficial metabolites and modulates immune reactions.5,14The system consisted of numerous gut microbes that maintain homeostasis in the intestinal tract. These indigenous gut microbes are resistance to probiotic colonization.3,8 Stress in the gastrointestinal tract can cause the death of probiotic microorganisms.40–42 Hence, probiotics usually survive in the host's gut. Because of the uncontested circumstances in GF mice, the persistence of L. plantarum CCFM8610 in the intestinal tract was prolonged, consistent with previous studies.27 Usually, the colonization of healthy microbiota could decrease the pathogen colonization and the risks of diseases. Meanwhile, the homeostasis of the gut ecosystem could be enhanced because of the colonization of probiotics. Hence, illustrating the colonization of L. plantarum is helpful for clarifying the functions of probiotics. After L. plantarum stably colonizes the intestinal tract, the host generates the corresponding physiological changes. The colon contains the most abundant gut microbes, and many metabolites are produced by gut microbes.17,43 In this study, we colonized GF mice with L. plantarum CCFM8610, a functional probiotic.13,14 The strain colonized the intestinal tract within 35 days after gavage four times, and the counts were the highest in the colon. Furthermore, the transcriptional profiles of the mouse colon were determined to examine the changes in the transcriptome of a GF host following colonization by commensal intestinal bacteria.
After excluding the interference of indigenous gut microorganisms, differential gene expression may reflect the effects of probiotics on the host. According to our results, the DEGs were enriched in some specific pathways, including the complement and coagulation cascade pathway, antigen processing and presentation pathways, and protein processing in the endoplasmic reticulum pathway. Complement and coagulation cascades are believed to be associated with the overall biological activity and the disease outcome and are known to be critical for involvement in inflammatory responses.44–46 As the key sentinel of innate immunity, the complement and coagulation systems usually respond to immunostimulation by external factors.47 In addition, antigen processing and presentation is another pathway related to inflammatory responses identified from the KEGG database analysis in this study. Similarly, a GO term, the immune system process, was annotated in differential genes’ annotation. Both KEGG and GO annotation pointed to immune reactions. When L. plantarum enters the intestinal tract, the bacteria can contact some immune cells and are recognized as antigens from the environment, resulting in the corresponding inflammatory responses.48,49 In GF mice, the development of the immune system in GF mice is defective because of the absence of intestinal microbiota.50–52 In this study, the metabolic pathways related to the immune response in GF mice were upregulated, consistent with previous studies.50,52 In addition, protein processing in the endoplasmic reticulum pathway was enriched in this study. The endoplasmic reticulum (ER) is an important cytoplasmic organelle for secretory and membrane protein synthesis. Stress in the ER has been speculated to contribute to many diseases, such as Alzheimer's and Parkinson's disease.53,54 In general, the intake of exogenous bacteria, L. plantarum CCFM8610, could bring about a series of changes in GF mice at the gene transcription level, which caused some immune stimulation and cellular function changes.
The gut microbiome is connected not only to intestinal cells but also to serum metabolites.55,56 Metabolomic analysis was carried out in this study to explore the metabolic changes in serum caused by L. plantarum CCFM8610 in GF mice, and L-methionine, D-tryptophan, indoleacrylic acid, DL-acetylcarnitine, and L-norleucine were identified as the top five important differential metabolites in PLS-DA analysis. D-Tryptophan and indoleacrylic acid were recognized as important differential metabolites in serum, and their content in the L. plantarum CCFM8610 colonization group was higher than that in the untreated GF mouse group. Tryptophan catabolites of Lactobacillus in the gastrointestinal tract play a crucial role in host health, especially immune reactions.57–59 More importantly, tryptophan can pass through the gut barrier to reach the serum and even the brain, which could be beneficial for the systemic health of the host.60,61 Hence, the increasing content of D-tryptophan in the serum could bring systemic benefits to host health. In addition, tryptophan can be decomposed by L. plantarum, and the tryptophan products are mainly indole-related substances.62 Indoleacrylic acid is a product of tryptophan catabolites, which increased the serum content in this study. Both tryptophan and indoleacrylic acid have been suggested to activate the immune system by binding to the aryl hydrocarbon receptor, which may contribute to systemic homeostasis in health and disease.57,61,63 These results were consistent with the transcriptome, and the host immune reactions were activated after mono-colonization with L. plantarum CCFM8610 in GF mice. Except for tryptophan and indoleacrylic acid, other differential metabolites, L-methionine, DL-acetylcarnitine, and L-norleucine, had lower contents in the L. plantarum CCFM8610 colonization group than in the untreated GF mice group in this study. However, these metabolites are typically associated with the development of disease. The decrease in methionine and DL-acetylcarnitine content in the L. plantarum CCFM8610 colonization group was believed to be beneficial for host health. L-Methionine restriction has been suggested to be beneficial for health in some diseases.64,65 The increasing DL-acetylcarnitine content has been positively associated with some conditions.66–68L-Norleucine is an isomer of leucine that explicitly affects protein synthesis in skeletal muscle and has antiviral activity.69 The mono-colonization of GF mice by L. plantarum influenced serum metabolites. The results of the differential metabolites showed a positive effect on host health, and the influence of the primary metabolites was consistent with the transcriptome results. In addition, some differential metabolites, such as D-tryptophan and indoleacrylic acid, could be biomarkers for estimating host health after the intake of L. plantarum.
Integrative analysis of the metabolome and transcriptome provided potential relevance between gene transcription in the colon and serum metabolites, which could provide a way to regulate serum metabolites by affecting gene expression and pathway enrichment.70 The correlations between differential metabolites and genes were calculated, and three pathways, including protein processing in the ER pathway, the fluid shear stress and atherosclerosis pathway, and the longevity regulating pathway, were identified. Among the metabolites, the degree of choline was the highest in the network. Choline is critical for various metabolic processes and lipid metabolism as a precursor of acetylcholine and a methyl donor, and it is also related to immune responses and some diseases.71–73 Besides, choline is associated with shear stress and atherosclerosis.74,75 In this study, choline was associated with all three identified KEGG pathways.74–77 Based on these results, regulating the choline concentration could regulate the corresponding gene expression in the colon, which provided a new concept for indirectly improving beneficial metabolites in serum. Furthermore, the connections between serum metabolites and colon pathways were credible in GF mice mono-colonized with L. plantarum CCFM8610. These results suggest that L. plantarum CCFM8610, as a probiotic strain, could affect the physiological health of the host by directly acting on the colon and indirectly acting on the serum.
5. Conclusion
This study revealed the effects of L. plantarum CCFM8610 on host physiology using integrative analysis of the metabolome and transcriptome. Global gene expression in the colon and the profile of metabolic substances in serum were altered in GF mice with L. plantarum CCFM8610 mono-colonization. After enrichment analysis, three pathways, including the complement and coagulation cascade pathway, antigen processing and presentation pathways, and protein processing in the ER pathway, were affected by L. plantarum CCFM8610. Enrichment of these pathways is of great physiological importance to host health, particularly in immunological regulation. Using PLS-DA analysis, five metabolites, L-methionine, D-tryptophan, indoleacrylic acid, DL-acetylcarnitine, and L-norleucine, were identified as key metabolites involved in the colonization of L. plantarum CCFM8610. Specifically, the beneficial metabolites, D-tryptophan and indoleacrylic acid, were increased in GF mice with L. plantarum CCFM8610 mono-colonization, and they could have positive effects on immunological regulation on host health, which was consistent with the pathway enrichment results. Furthermore, integrative analysis of the metabolome and transcriptome revealed potential connections between enriched pathways and differential metabolites, which were difficult to find by functional analysis. The results showed that the choline concentration was significantly correlated with the expression of specific genes. The results indicated that the choline concentration could be regulated by gene expression in the colon, which provides new insight into the potential mechanisms of the effect of probiotics on host health.Author contributions
Pan Huang: conceptualization, methodology, and writing – original draft. Shanrong Yi: writing – review & editing. Leilei Yu: writing – review & editing. Fengwei Tian: visualization. Jianxin Zhao: writing – review & editing. Hao Zhang: supervision. Wei Chen: supervision and validation. Qixiao Zhai: data curation, funding acquisition, and project administration.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have influenced the work reported in this paper.Acknowledgements
This work was supported by the National Natural Science Foundation of China [No. 32122067 and 31871773], the Natural Science Foundation of Jiangsu Province [BK20200084], the Collaborative Innovation Center of Food Safety and Quality Control in Jiangsu Province and the Postgraduate Research & Practice Innovation Program of Jiangsu Province [KYCX22_2394].References
- J. M. Kinross, A. W. D. Darzi and J. K. N. Nicholson, Gut microbiome-host interactions in health and disease, Genome Med., 2011, 3(3), 1–12 CrossRef PubMed.
- E. Holmes, J. V. Li, T. Athanasiou, H. Ashrafian and J. K. Nicholson, Understanding the role of gut microbiome-host metabolic signal disruption in health and disease, Trends Microbiol., 2011, 19, 349–359 CrossRef CAS PubMed.
- C. Hill, F. Guarner, G. Reid, G. R. Gibson, D. J. Merenstein, B. Pot, L. Morelli, R. B. Canani, H. J. Flint, S. Salminen, P. C. Calder and M. E. Sanders, Expert consensus document. The international scientific association for probiotics and prebiotics consensus statement on the scope and appropriate use of the term probiotic, Nat. Rev. Gastroenterol. Hepatol., 2014, 11, 506–514 CrossRef PubMed.
- Y. Liu, M. F. Mian, K. A. McVey Neufeld and P. Forsythe, CD4(+)CD25(+) T Cells are essential for behavioral effects of lactobacillus rhamnosus jb-1 in male BALB/c mice, Brain, Behav., Immun., 2020, 88, 451–460 CrossRef CAS PubMed.
- Y. Liu, X. Yu, L. Yu, F. Tian, J. Zhao, H. Zhang, L. Qian, Q. Wang, Z. Xue, Q. Zhai and W. Chen, Lactobacillus plantarum CCFM8610 alleviates irritable bowel syndrome and prevents gut microbiota dysbiosis: a randomized, double-blind, placebo-controlled, pilot clinical trial, Engineering, 2021, 7, 376–385 CrossRef.
- P. Panigrahi, S. Parida, N. C. Nanda, R. Satpathy, L. Pradhan, D. S. Chandel, L. Baccaglini, A. Mohapatra, S. S. Mohapatra, P. R. Misra, R. Chaudhry, H. H. Chen, J. A. Johnson, J. G. Morris, N. Paneth and I. H. Gewolb, A randomized synbiotic trial to prevent sepsis among infants in rural India, Nature, 2017, 548, 407–412 CrossRef CAS PubMed.
- H. A. Seddik, F. Bendali, F. Gancel, I. Fliss, G. Spano and D. Drider, Lactobacillus plantarum and its probiotic and food potentialities, probiotics antimicrob, Proteins, 2017, 9, 111–122 Search PubMed.
- M. Derrien and J. E. van Hylckama Vlieg, Fate, activity, and impact of ingested bacteria within the human gut microbiota, Trends Microbiol., 2015, 23, 354–366 CrossRef CAS PubMed.
- N. Zmora, G. Zilberman-Schapira, J. Suez, U. Mor, M. Dori-Bachash, S. Bashiardes, E. Kotler, M. Zur, D. Regev-Lehavi, R. B. Brik, S. Federici, Y. Cohen, R. Linevsky, D. Rothschild, A. E. Moor, S. Ben-Moshe, A. Harmelin, S. Itzkovitz, N. Maharshak, O. Shibolet, H. Shapiro, M. Pevsner-Fischer, I. Sharon, Z. Halpern, E. Segal and E. Elinav, Personalized gut mucosal colonization resistance to empiric probiotics is associated with unique host and microbiome features, Cell, 2018, 174, 1388–1405 CrossRef CAS PubMed.
- M. Schwarzer, K. Makki, G. Storelli, I. S. Machuca-Gayet, D. P. Hermanova, M. E. Martino, B. Severine, T. Hudcovic, A. Heddi, J. Rieusset, H. Kozakova, H. Vidal and F. Leulier, Lactobacillus plantarum strain maintains growth of infant mice during chronic undernutrition, Science, 2016, 351, 854 CrossRef CAS PubMed.
- B. Klarin, A. Adolfsson, A. Torstensson and A. Larsson, Can probiotics be an alternative to chlorhexidine for oral care in the mechanically ventilated patient? A multicentre, prospective, randomised controlled open trial, Crit. Care, 2018, 22, 272 CrossRef PubMed.
- Z. Fang, W. Lu, J. Zhao, H. Zhang, L. Qian, Q. Wang and W. Chen, Probiotics modulate the gut microbiota composition and immune responses in patients with atopic dermatitis: a pilot study, Eur. J. Nutr., 2020, 59, 2119–2130 CrossRef CAS PubMed.
- Q. Zhai, G. Wang, J. Zhao, X. Liu, F. Tian, H. Zhang and W. Chen, Protective effects of Lactobacillus plantarum, CCFM8610 against acute cadmium toxicity in mice, Appl. Environ. Microbiol., 2013, 79, 1508–1515 CrossRef CAS PubMed.
- Y. Liu, Y. Sheng, Q. Pan, Y. Xue, L. Yu, F. Tian, J. Zhao, H. Zhang, Q. Zhai and W. Chen, Identification of the key physiological characteristics of Lactobacillus plantarum, strains for ulcerative colitis alleviation, Food Funct., 2020, 11, 1279–1291 RSC.
- Y. S. Lee, T. Y. Kim, Y. Kim, S. H. Lee, S. Kim, S. W. Kang, J. Y. Yang, I. J. Baek, Y. H. Sung, Y. Y. Park, S. W. Hwang, O. Eunju, K. S. Kim, S. Liu, N. Kamada, N. Gao and M. N. Kweon, Microbiota-derived lactate accelerates intestinal stem-cell-mediated epithelial development, Cell Host Microbe, 2018, 24, 833–846 CrossRef CAS PubMed.
- H. Kayama, R. Okumura and K. Takeda, Interaction between the microbiota, epithelia, and immune cells in the intestine, Annu. Rev. Immunol., 2020, 38, 23–48 CrossRef CAS PubMed.
- L. Zhai, J. Wu, Y. Y. Lam, H. Y. Kwan, Z. X. Bian and H. L. X. Wong, Gut-microbial metabolites, probiotics and their roles in type 2 diabetes, Int. J. Mol. Sci., 2021, 22, 12846 CrossRef CAS PubMed.
- C. Wang, Y. Xiao, L. Yu, F. Tian, J. Zhao, H. Zhang, W. Chen and Q. Zhai, Protective effects of different Bacteroides vulgatus strains against lipopolysaccharide-induced acute intestinal injury, and their underlying functional genes, J. Adv. Res., 2022, 36, 27–37 CrossRef CAS PubMed.
- R. Lowe, N. Shirley, M. Bleackley, S. Dolan and T. Shafee, Transcriptomics technologies, PLoS Comput. Biol., 2017, 13, e1005457 CrossRef PubMed.
- X. Liu and J. W. Locasale, Metabolomics: A Primer, Trends Biochem. Sci., 2017, 42, 274–284 CrossRef CAS PubMed.
- P. van Baarlen, F. Troost, C. van der Meer, G. Hooiveld, M. Boekschoten, R. J. Brummer and M. Kleerebezem, Human mucosal in vivo transcriptome responses to three lactobacilli indicate how probiotics may modulate human cellular pathways, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(Suppl 1), 4562–4569 CrossRef CAS PubMed.
- A. Kumar, A. N. Vlasova, Z. Liu, K. S. Chattha, S. Kandasamy, M. Esseili, X. Zhang, G. Rajashekara and L. J. Saif, In vivo gut transcriptome responses to Lactobacillus rhamnosus GG and Lactobacillus acidophilus, in neonatal gnotobiotic piglets, Gut Microbes, 2014, 5, 152–164 CrossRef PubMed 23.
- K. Mokkala, T. Vahlberg, N. Houttu, E. Koivuniemi, L. Lahti and K. Laitinen, Impact of combined consumption of fish oil and probiotics on the serum metabolome in pregnant women with overweight or obesity, EBioMedicine, 2021, 73, 103655 CrossRef CAS PubMed.
- A. Liu, T. Ma, N. Xu, H. Jin, F. Zhao, L. Y. Kwok, H. Zhang, S. Zhang and Z. Sun, Adjunctive Probiotics Alleviates Asthmatic Symptoms via Modulating the Gut Microbiome and Serum Metabolome, Microbiol. Spectrum, 2021, 9, e0085921 CrossRef PubMed.
- T. Uzbay, Germ-free animal experiments in the gut microbiota studies, Curr. Opin. Pharmacol., 2019, 49, 6–10 CrossRef CAS PubMed.
- M. Al-Asmakh and F. Zadjali, Use of germ-free animal models in microbiota-related research, J. Microbiol. Biotechnol., 2015, 25, 1583–1588 CrossRef PubMed.
- M. L. Marco, M. C. de Vries, M. Wels, D. Molenaar, P. Mangell, S. Ahrne, W. M. de Vos, E. E. Vaughan and M. Kleerebezem, Convergence in probiotic Lactobacillus gut-adaptive responses in humans and mice, ISME J., 2010, 4, 1481–1484 CrossRef CAS PubMed.
- M. L. Marco, T. H. Peters, R. S. Bongers, D. Molenaar, S. van Hemert, J. L. Sonnenburg, J. I. Gordon and M. Kleerebezem, Lifestyle of Lactobacillus plantarum, in the mouse caecum, Environ. Microbiol., 2009, 11, 2747–2757 CrossRef CAS PubMed.
- M. Derrien, P. Van Baarlen, G. Hooiveld, E. Norin, M. Muller and W. M. de Vos, Modulation of mucosal immune response, tolerance, and proliferation in mice colonized by the mucin-degrader Akkermansia muciniphila, Front. Microbiol., 2011, 2, 166 Search PubMed.
- J. Xia, S. Jiang, L. Lv, W. Wu, Q. Wang, Q. Xu, J. Ye, D. Fang, Y. Li, J. Wu, X. Bian, L. Yang, H. Jiang, K. Wang, R. Yan and L. Li, Modulation of the immune response and metabolism in germ-free rats colonized by the probiotic Lactobacillus salivarius, LI01, Appl. Microbiol. Biotechnol., 2021, 105, 1629–1645 CrossRef CAS PubMed.
- X. Liu, X. Tong, Y. Zou, X. Lin, H. Zhao, L. Tian, Z. Jie, Q. Wang, Z. Zhang, H. Lu, L. Xiao, X. Qiu, J. Zi, R. Wang, X. Xu, H. Yang, J. Wang, Y. Zong, W. Liu, Y. Hou, S. Zhu, H. Jia and T. Zhang, Mendelian randomization analyses support causal relationships between blood metabolites and the gut microbiome, Nat. Genet., 2022, 54, 52–61 CrossRef CAS PubMed.
- L. Lahti, A. Salonen, R. A. Kekkonen, J. Salojarvi, J. Jalanka-Tuovinen, A. Palva, M. Oresic and W. M. de Vos, Associations between the human intestinal microbiota, Lactobacillus rhamnosus, GG and serum lipids indicated by integrated analysis of high-throughput profiling data, PeerJ, 2013, 1, e32 CrossRef PubMed.
- C. Li, S. Nie, K. Zhu, T. Xiong and M. Xie, Lactobacillus plantarum, NCU116 fermented carrot juice evokes changes of metabolites in serum from type 2 diabetic rats, Food Res. Int., 2016, 80, 36–40 CrossRef CAS.
- M. Li, L. Yang, C. Mu, Y. Sun, Y. Gu, D. Chen, T. Liu and H. Cao, Gut microbial metabolome in inflammatory bowel disease: From association to therapeutic perspectives, Comput. Struct. Biotechnol. J., 2022, 20, 2402–2414 CrossRef PubMed.
- A. Trompette, J. Pernot, O. Perdijk, R. A. A. Alqahtani, J. S. Domingo, D. Camacho-Munoz, N. C. Wong, A. C. Kendall, A. Wiederkehr, L. P. Nicod, A. Nicolaou, C. von Garnier, N. D. J. Ubags and B. J. Marsland, Gut-derived short-chain fatty acids modulate skin barrier integrity by promoting keratinocyte metabolism and differentiation, Mucosal Immunol., 2022, 15, 908–926 CrossRef CAS PubMed.
- N. Bar, T. Korem, O. Weissbrod, D. Zeevi, D. Rothschild, S. Leviatan, N. Kosower, M. Lotan-Pompan, A. Weinberger, C. I. Le Roy, C. Menni, A. Visconti, M. Falchi, T. D. Spector, I. D. Consortium, J. Adamski, P. W. Franks, O. Pedersen and E. Segal, A reference map of potential determinants for the human serum metabolome, Nature, 2020, 588, 135–140 CrossRef PubMed.
- Z. Fan, R. P. Ross, C. Stanton, B. Hou, J. Zhao, H. Zhang, B. Yang and W. Chen, Lactobacillus casei CCFM1074 alleviates collagen-induced arthritis in rats via balancing treg/th17 and modulating the metabolites and gut microbiota, Front. Immunol., 2021, 12, 680073 CrossRef CAS PubMed.
- L. Xu, B. Liu, L. Huang, Z. Li, Y. Cheng, Y. Tian, G. Pan, H. Li, Y. Xu, W. Wu, Z. Cui and L. Xiea, Probiotic consortia and their metabolites ameliorate the symptoms of inflammatory bowel diseases in a colitis mouse model, Microbiol. Spectrum, 2022, 10, 4 Search PubMed.
- Q. Zhai, X. Gong, C. Wang, J. Zhao, H. Zhang, F. Tian and W. Chen, Food-borne patulin toxicity is related to gut barrier disruption and can be prevented by docosahexaenoic acid and probiotic supplementation, Food Funct., 2019, 10, 1330–1339 RSC.
- S. Drouault, G. Corthier, S. D. Ehrlich and P. Renault, Survival, physiology, and lysis of Lactococcus lactis in the digestive tract, Appl. Environ. Microbiol., 1999, 65, 4881–4886 CrossRef CAS PubMed.
- S. Mills, C. Stanton, G. F. Fitzgerald and R. P. Ross, Enhancing the stress responses of probiotics for a lifestyle from gut to product and back again, Microb. Cell Fact., 2011, 10, S19 CrossRef PubMed.
- J. Wang, J. Zhang, W. Liu, H. Zhang and Z. Sun, Metagenomic and metatranscriptomic profiling of Lactobacillus casei, Zhang in the human gut, npj Biofilms Microbiomes, 2021, 7, 55 CrossRef CAS PubMed.
- G. Wieers, L. Belkhir, R. Enaud, S. Leclercq, J. M. Philippart de Foy, I. Dequenne, P. de Timary and P. D. Cani, How probiotics affect the microbiota, Front. Cell. Infect. Microbiol., 2019, 9, 454 CrossRef PubMed.
- K. Oikonomopoulou, D. Ricklin, P. A. Ward and J. D. Lambris, Interactions between coagulation and complement–their role in inflammation, Semin. Immunopathol., 2012, 34, 151–165 CrossRef CAS PubMed.
- U. Amara, M. A. Flierl, D. Rittirsch, A. Klos, H. Chen, B. Acker, U. B. Bruckner, B. Nilsson, F. Gebhard, J. D. Lambris and M. Huber-Lang, Molecular intercommunication between the complement and coagulation systems, J. Immunol., 2010, 185, 5628–5636 CrossRef CAS PubMed.
- J. D. Lambris, Current Topics in Complement II, Springer, 2008 Search PubMed.
- M. M. Markiewski, B. Nilsson, K. N. Ekdahl, T. E. Mollnes and J. D. Lambris, Complement and coagulation: strangers or partners in crime?, Trends Immunol., 2007, 28, 184–192 CrossRef CAS PubMed.
- F. Kotsias, I. Cebrian and A. Alloatti, Antigen processing and presentation, Int. Rev. Cell Mol. Biol., 2019, 348, 69–121 CAS.
- D. Forni, R. Cagliani, C. Tresoldi, U. Pozzoli, L. De Gioia, G. Filippi, S. Riva, G. Menozzi, M. Colleoni, M. Biasin, S. Lo Caputo, F. Mazzotta, G. P. Comi, N. Bresolin, M. Clerici and M. Sironi, An evolutionary analysis of antigen processing and presentation across different timescales reveals pervasive selection, PLoS Genet., 2014, 10, e1004189 CrossRef PubMed.
- H. Tlaskalova-Hogenova, R. Stepankova, H. Kozakova, T. Hudcovic, L. Vannucci, L. Tuckova, P. Rossmann, T. Hrncir, M. Kverka, Z. Zakostelska, K. Klimesova, J. Pribylova, J. Bartova, D. Sanchez, P. Fundova, D. Borovska, D. Srutkova, Z. Zidek, M. Schwarzer, P. Drastich and D. P. Funda, The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ-free and gnotobiotic animal models of human diseases, Cell. Mol. Immunol., 2011, 8, 110–120 CrossRef CAS PubMed.
- J. J. Cebra, Influences of microbiota on intestinal immune system development, Am. J. Clin. Nutr., 1999, 69, 1046s–1051s CrossRef CAS PubMed.
- J. L. Round and S. K. Mazmanian, The gut microbiota shapes intestinal immune responses during health and disease, Nat. Rev. Immunol., 2009, 9, 313–323 CrossRef CAS PubMed.
- M. S. Forman, V. M. Y. Lee and J. Q. Trojanowski, ‘Unfolding’ pathways in neurodegenerative disease, Trends Neurosci., 2003, 26, 407–410 CrossRef CAS PubMed.
- A. Gow and R. Sharma, The unfolded protein response in protein aggregating diseases, Neuromol. Med., 2003, 4, 73–94 CrossRef CAS PubMed.
- R. Liu, J. Hong, X. Xu, Q. Feng, D. Zhang, Y. Gu, J. Shi, S. Zhao, W. Liu, X. Wang, H. Xia, Z. Liu, B. Cui, P. Liang, L. Xi, J. Jin, X. Ying, X. Wang, X. Zhao, W. Li, H. Jia, Z. Lan, F. Li, R. Wang, Y. Sun, M. Yang, Y. Shen, Z. Jie, J. Li, X. Chen, H. Zhong, H. Xie, Y. Zhang, W. Gu, X. Deng, B. Shen, X. Xu, H. Yang, G. Xu, Y. Bi, S. Lai, J. Wang, L. Qi, L. Madsen, J. Wang, G. Ning, K. Kristiansen and W. Wang, Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention, Nat. Med., 2017, 23, 859–868 CrossRef CAS PubMed.
- S. M. Lancaster, B. Lee-McMullen, C. W. Abbott, J. V. Quijada, D. Hornburg, H. Park, D. Perelman, D. J. Peterson, M. Tang, A. Robinson, S. Ahadi, K. Contrepois, C. J. Hung, M. Ashland, T. McLaughlin, A. Boonyanit, A. Horning, J. L. Sonnenburg and M. P. Snyder, Global, distinctive, and personal changes in molecular and microbial profiles by specific fibers in humans, Cell Host Microbe, 2022, 30, 848–862 CrossRef CAS PubMed.
- H. M. Roager and T. R. Licht, Microbial tryptophan catabolites in health and disease, Nat. Commun., 2018, 9, 3294 CrossRef PubMed.
- T. Zelante, R. G. Iannitti, C. Cunha, A. De Luca, G. Giovannini, G. Pieraccini, R. Zecchi, C. D'Angelo, C. Massi-Benedetti, F. Fallarino, A. Carvalho, P. Puccetti and L. Romani, Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22, Immunity, 2013, 39, 372–385 CrossRef CAS PubMed.
- J. Gao, K. Xu, H. Liu, G. Liu, M. Bai, C. Peng, T. Li and Y. Yin, Impact of the gut microbiota on intestinal immunity mediated by tryptophan metabolism, Front. Cell. Infect. Microbiol., 2018, 8, 13 CrossRef PubMed.
- A. Bosi, D. Banfi, M. Bistoletti, C. Giaroni and A. Baj, Tryptophan Metabolites Along the Microbiota-Gut-Brain Axis: An Interkingdom communication system influencing the gut in health and disease, Int. J. Tryptophan Res., 2020, 13, 1178646920928984 Search PubMed.
- S. M. O'Mahony, G. Clarke, Y. E. Borre, T. G. Dinan and J. F. Cryan, Serotonin, tryptophan metabolism and the brain-gut-microbiome axis, Behav. Brain Res., 2015, 277, 32–48 CrossRef PubMed.
- J. Shi, P. Du, Q. Xie, N. Wang, H. Li, E. E. Smith, C. Li, F. Liu, G. Huo and B. Li, Protective effects of tryptophan-catabolizing Lactobacillus plantarum KLDS 1.0386 against dextran sodium sulfate-induced colitis in mice, Food Funct., 2020, 11, 10736–10747 RSC.
- S. Heath-Pagliuso, W. J. Rogers, K. Tullis, S. N. D. Seidel, P. H. Cenijn, A. Brouwer and M. S. Denison, Activation of the ah receptor by tryptophan and tryptophan metabolites, Biochemistry, 1998, 37, 11508–11515 CrossRef CAS PubMed.
- J. Kaiser, A plateful of medicine, Science, 2021, 372, 20–23 CrossRef CAS PubMed.
- B. Ren, L. Wang, L. Shi, X. Jin, Y. Liu, R. H. Liu, F. Yin, E. Cadenas, X. Dai, Z. Liu and X. Liu, Methionine restriction alleviates age-associated cognitive decline via fibroblast growth factor 21, Redox Biol., 2021, 41, 101940 CrossRef CAS PubMed.
- Y. Ni, G. Xie and W. Jia, Metabonomics of human colorectal cancer: new approaches for early diagnosis and biomarker discovery, J. Proteome Res., 2014, 13, 3857–3870 CrossRef CAS PubMed.
- M. H. Abu Bakar and M. R. Sarmidi, Association of cultured myotubes and fasting plasma metabolite profiles with mitochondrial dysfunction in type 2 diabetes subjects, Mol. Biosyst., 2017, 13, 1838–1853 RSC.
- C. G. Costa, E. A. Struys, A. Bootsma, H. J. ten Brink, L. Dorland, I. Tavares de Almeida, M. Duran and C. Jakobs, Quantitative analysis of plasma acylcarnitines using gas chromatography chemical ionization mass fragmentography, J. Lipid Res., 1997, 38, 173–182 CrossRef CAS.
- K. J. Schott, J. Gehrmann, U. Potter and V. Neuhoff, On the role of branched-chain amino acids in protein turnover of skeletal muscle. studies in vivo with L-norleucine, Z. Naturforsch., C: J. Biosci., 1985, 40, 427–437 CrossRef CAS PubMed.
- C. A. Thaiss, M. Levy, T. Korem, L. Dohnalova, H. Shapiro, D. A. Jaitin, E. David, D. R. Winter, M. Gury-BenAri, E. Tatirovsky, T. Tuganbaev, S. Federici, N. Zmora, D. Zeevi, M. Dori-Bachash, M. Pevsner-Fischer, E. Kartvelishvily, A. Brandis, A. Harmelin, O. Shibolet, Z. Halpern, K. Honda, I. Amit, E. Segal and E. Elinav, Microbiota diurnal rhythmicity programs host transcriptome oscillations, Cell, 2016, 167, 1495–1510 CrossRef CAS PubMed.
- S. H. Zeisel and K. A. da Costa, Choline: an essential nutrient for public health, Nutr. Rev., 2009, 67, 615–623 CrossRef PubMed.
- B. K. Swain and T. S. Johri, Effect of supplemental methionine, choline and their combinations on the performance and immune response of broilers, Br. Poult. Sci., 2000, 41, 83–88 CrossRef CAS PubMed.
- A. Buchman, M. E. Ament, M. Sohel, M. Dubin, D. J. Jenden, M. Roch, H. Pownall, M. Farley, M. Awal and C. Ahn, Choline deficiency causes reversible hepatic abnormalities in patients receiving parenteral nutrition: proof of a human choline requirement: a placebo–controlled trial, JPEN, J. Parenter. Enteral Nutr., 2001, 25, 260 CrossRef CAS PubMed.
- T. Santos, R. Mesquita, J. M. E. Silva and C. Saldanha, Effects of choline on hemorheological properties and NO metabolism of human erythrocytes, Clin. Hemorheol. Microcirc., 2003, 29, 41–51 CAS.
- J. Yu, J. Fu, X. Zhang, X. Cui and M. Cheng, The integration of metabolomic and proteomic analyses revealed alterations in inflammatory-related protein metabolites in endothelial progenitor cells subjected to oscillatory shear stress, Front. Physiol., 2022, 13, 825966 CrossRef PubMed.
- E. Sanchez-Lopez, T. Zimmerman, T. Gomez del Pulgar, M. P. Moyer, J. C. Lacal Sanjuan and A. Cebrian, Choline kinase inhibition induces exacerbated endoplasmic reticulum stress and triggers apoptosis via CHOP in cancer cells, Cell Death Dis., 2013, 4, e933 CrossRef CAS PubMed.
- Z. M. Yao, H. Jamil and D. E. Vance, Choline deficiency causes translocation of CTP:phosphocholine cytidylyltransferase from cytosol to endoplasmic reticulum in rat liver, J. Biol. Chem., 1990, 265, 4326–4331 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |